


Journal of Cancer Metastasis and Treatment







Xuan Zhou , ... Xiaoping Zhang
, ... Xiaoping Zhang
, ... Xiaoping Zhang
Views: Downloads:
Views: Downloads:
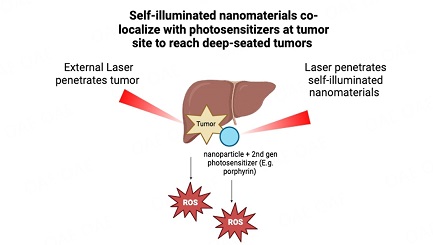
Diego Fernández-Lázaro, ... Enrique Roche
, ... Enrique Roche
Views: Downloads:
Views: Downloads:
Hongshuang Shi, ... Xiaofang Che
Views: Downloads:
Polyxeni Vafopoulou, Malamati Kourti
Views: Downloads:
Views: Downloads:
Views: Downloads:







Data
2261
Authors
1359
Reviewers
2015
Published Since
3,318,217
Article Views
563,733
Article Downloads
For Reviewers
For Readers
Add your e-mail address to receive forthcoming Issues of this journal:
Themed Collections
Cancer Treatment
Metastasis
Microenvironment
Immunotherapy
Breast Cancer Metastasis
Bone Metastasis
Metastatic Renal Cell Carcinoma
Lymph node metastasis
Mesothelioma
Hematological Malignancies
Thyroid Cancer
Liquid Biopsies
Early Diagnosis
Lung Cancer
Brain Tumors
Lymphoma
Oncolytic Virus
Cervical Cancer
Cancer Stem Cells
Related Journals

Related Journals
Data
2261
Authors
1359
Reviewers
2015
Published Since
3,318,217
Article Views
563,733
Article Downloads


