
The role of the PI3K/AKT/mTOR pathway in brain tumor metastasis


Abstract
The PI3K/AKT/mTOR (PAM) pathway is involved in a variety of cellular functions and often contributes to oncogenesis and cancer progression. It has been recognized that this pathway is frequently activated in the most common central nervous system cancers of adults and children, malignant gliomas and medulloblastomas (MB). In these tumors, the PAM network controls key functions necessary for cell invasion and metastasis, such as cell motility. This review summarizes the current knowledge about the role of PAM signaling in cell invasion and metastasis in gliomas and MB. Current approaches to inhibit cell invasion and metastasis by targeting the PAM pathway will also be discussed.
Keywords
Introduction
Tumors of the central nervous system include a broad range of neoplasms that arise from different cell lineages. The most common variants in adult and pediatric populations are malignant gliomas and MB, respectively.
Glioblastoma (GBM) is a highly aggressive tumor that arises from different glial cell types. Based on WHO classification, GBM is a grade IV astrocytoma that either develops de novo (primary GBM) or gradually from lower grade astrocytomas (secondary GBM).[1] Due to limited therapy options, the median survival is a dismal 15 months with standard of care, which includes surgical resection, temozolomide chemotherapy and radiation.[2]
Medulloblastomas are embryonal tumors that originate from fetal tissue due to aberrant developmental signaling.[3] By using treatment protocols that combine chemotherapy, surgery and cranio-spinal radiotherapy, 70-80% of patients can be cured, albeit with debilitating long term side effects.[4]
Advances in molecular biology have led to remarkable insights into the understanding of the underlying molecular pathogenesis of malignant gliomas and MB and have revealed specific pathways and signaling networks that promote tumorigenesis in these malignancies.[5,6] These frequently feature aberrant receptor tyrosine kinase (RTK) signaling via the PI3K/AKT/mTOR (PAM) pathway.
The PAM signaling axis integrates extracellular signals via RTK and G protein-coupled receptors and regulates a host of intracellular functions, such as cell cycle, metabolism, migration and apoptosis.[7-9] Phosphatidylinositol 3-kinase (PI3K) phosphorylates the 3’-hydroxyl group of phosphatidylinositol, producing second messengers that recruit cytoplasmic proteins to the membrane. These include various modulators of small GTPase activity, TEC family tyrosine kinases and members of the AGC protein kinase family like AKT (also known as Protein Kinase B, PKB).[10] The serine-threonine kinase mTOR, a regulator of translation and protein synthesis, is activated by AKT signaling.
Since many hallmarks of malignancy are controlled by PAM signaling, genetic and epigenetic alterations in various components of this pathway are frequent events in central nervous system (CNS) cancers. These include gain-of-function mutations and amplifications in genes encoding RTKs such as epidermal growth factor receptor (EGFR), loss-of-function mutations of the phosphatase and tensin homolog deleted on the chromosome 10 (PTEN) tumor suppressor gene, and oncogenic mutations in various PI3K isoforms that lead to a constitutively activated pathway.[11,12] Aberrant PAM signaling also favors essential steps for cell invasion and metastasis in CNS malignancies [Figure 1]. The implications of aberrant PAM signaling in angiogenesis, epithelial to mesenchymal transition (EMT) and immune response modulation is currently under intense investigation.[13-15] Components of the PAM pathway are therefore being considered as potential drug targets [Table 1] to inhibit the often fatal events of metastasis and cell invasion.[16-18]
Stage of clinical development of PAM pathway inhibitors for brain tumors[138]
| Inhibitor | Target | Stage of clinical development for brain tumors |
|---|---|---|
| SF-1126 (RGDS-conjugated LY294002 prodrug) | Pan-PI3K | Phase I |
| PX-866 | Pan-PI3K | Phase II |
| Pictilisib (GDC-0941) | Pan-PI3K | Phase II |
| LY294002 | Dual PI3K/mTOR | Preclinical |
| Wortmannin | Dual PI3K/mTOR | Preclinical |
| Dactolisib (NVP-BEZ235) | Dual PI3K/mTOR | Phase II |
| Perifosine (KRX-0401) | Akt | Phase II |
| KP-372-1 | Akt | Preclinical |
| KP-372-2 | Akt | Preclinical |
| A-443654 | Akt | Preclinical |
| Bevacizumab (Avastin) | VEGF-A | Phase III |
| Aflibercept | VEGF and placental growth factor | Phase I |
| Cediranib (AZD2171) | VEGFR, Flt1/4, PDGFR, FGFR1, c-KIT | Phase I |
| Cabozantinib (XL-184) | c-MET and VEGFR2 | Phase I |
| SGX-523 | c-MET | Phase I |
| Osthole | IGF-1/IGF-1R and calcium channel blocker | Preclinical |
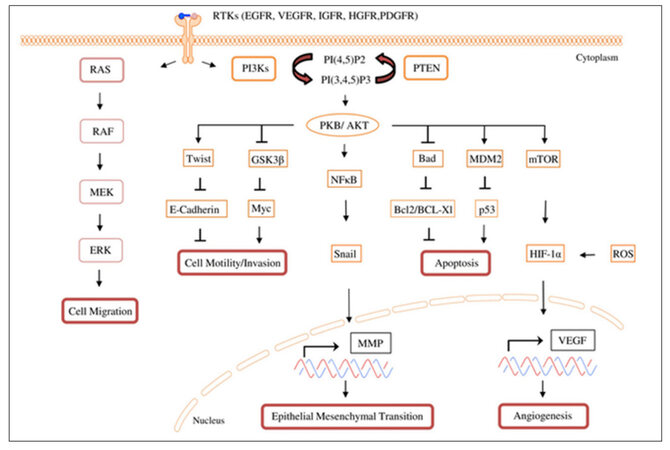
Figure 1. PAM-signaling network and effector functions associated with metastasis: In GB and MB, aberrant PAM signaling can promote tumor progression by over- inducing angiogenesis, EMT, cell migration and invasion, and also by inhibiting loss of adhesion associated apoptosis. PAM: PI3K/AKT/mTOR; VEGF: vascular endothelial growth factor; PDGFR: platelet derived growth factor receptor; IGF-1: insulin-like growth factor-1; IGFR: insulin-like growth factor receptor; NFκB = nuclear factor kappa-light-chain-enhancer of activated B cells; HIF-1α: hypoxia inducible factor 1α; PTEN: phosphatase and tensin homolog deleted on chromosome 10; PI3Ks: Phosphatidylinositol-3-kinases; MEK: mitogen-activated ERK kinase; EGFR: epidermal growth factor receptor; ERK: extracellular-signal regulated kinase
Angiogenesis
Angiogenesis is a process consisting of the generation of blood vessels and is essential for the growth of tumor mass beyond 1mm in diameter.[19] This process allows tumors to become invasive by supporting them with nutrients and oxygen. Tumor and host cells synthesize and secrete pro-angiogenic factors, such as vascular endothelial growth factor (VEGF), that activate quiescent endothelial cells and induce the formation of new blood vessels from pre-existing vascular structures.[20]
The PAM pathway plays a critical role in this neovascularization process by controlling the hypoxia-inducible factor 1-alpha (HIF-1α) mediated expression and secretion of VEGF.[21,22] In cancer cells, VEGF stimulation can be mediated by chronic stimulation by growth factors, such as insulin-like growth factor-1 (IGF-1); constitutive activation of PI3K; or constitutive activation of AKT due to inactivation of PTEN.[23,24] The important role of the PAM pathway in angiogenesis has been confirmed in various malignancies where inhibition of pan-PI3K by LY294002 and downregulation of p110α (or recently, PI3KC2α) were shown to block tumor vascularization.[15,22,25] In myeloid cells, PI3Kϒ was reported to be involved in the activation of integrin α4β1, leading to myeloid cell invasion into tumors and, in turn, to tumor angiogenesis.[26]
In GBM, the most aggressive glioma subtype, the PAM pathway also plays a crucial role in the induction of invasion, angiogenesis and the expression of VEGF in cells.[24,27] Therefore, new small molecule inhibitors targeting PI3K enzymes are being tested in this CNS malignancy. These include the PI3K inhibitors SF1126 (a RGDS-conjugated LY294002 prodrug) and PX-866, and the dual PI3K/mTOR inhibitor NVP-BEZ235.[28-30] These compounds were shown to induce a substantial inhibition of the expression of VEGF, thus reducing the invasive and angiogenic capabilities of GBM cells. In fact, PX-866 has recently entered phase II studies in patients with recurrent GBM. Unfortunately, preliminary results of this trial have shown a low overall response rate.[31]
The combined inhibition of VEGF and vascular endothelial growth factor receptor (VEGF/VEGFR) is currently thought to be an effective way to control GBM growth.[32-34] Examples of VEGF/VEGFR inhibitors are bevacizumab, already in phase III trial,[35] and aflibercept, a VEGF/VEGFR inhibitor that also targets placental growth factor.[36] Unfortunately, long-term treatment with aflibercept was reported to induce an invasive phenotype of GBM.[37,38]
In addition, RTK inhibitors such as cediranib (an inhibitor of VEGFR, platelet-derived growth factor receptor, fibroblast growth factor receptor 1, and v-kit Hardy-Zuckerman 4 feline sarcoma viral oncogene homolog), have also been used with promising results.[39,40] Inhibitors of c-MET such as cabozantinib are also being considered, and have been reported to induce a significant increase in overall survival of mice bearing GBM xenografts.[41]
However, anti-angiogenic therapies targeting VEGF/VEGFR have had less of an effect than expected.[42] This could be because, in highly vascularized tissues like the lung and brain, tumors can often proliferate around existing vessels and hijack them, a process called vessel co-option.[43,44] These pre-existing blood vessels circumvent the need to generate new tumor vasculature, and may explain the inefficacy of anti-proliferative therapies in GBM, the most vascularized tumor in humans.[38]
Autophagy is an evolutionarily conserved, catabolic process that maintains cellular biosynthesis through the degradation and recycling of proteins and organelles to support metabolism and survival during starvation. This process has been shown to have a complex relationship with angiogenesis induction in various malignancies. While some studies have reported that autophagy inhibits angiogenesis,[45,46] other studies have found that induction of autophagy promoted cancer and its inhibition prevented angiogenesis.[47,48] This illustrates the dual role that autophagy plays in cancer, acting as a pro-survival or pro-death mechanism depending on the tumor type and stage.[49]
Autophagy is induced by different cellular stress-mediated signaling pathways, the inputs of which are integrated by the protein kinase mammalian target of rapamycin (mTOR). The mTOR complex 1 (mTORC1) is a negative regulator of autophagy and a downstream target of the PI3K/AKT pathway.[50] Anti-cancer agents that target this pathway are able to induce autophagy, which has a cytoprotective role as well as an anti-angiogenic potential similar to the action of the dual PI3K-mTOR inhibitor NVP-BEZ235.[51-53]
High-grade gliomas have been reported to have lower expression of autophagy-related proteins than low-grade gliomas.[54] The amplification of EGFR, which is often found in these tumors, is known to suppress autophagy.[55] The progression of astrocytic tumors is associated with a decrease in autophagic capacity.[56] In most of these CNS malignancies, the modulation of autophagy sensitizes tumor cells to standard chemotherapy and radiotherapy induced cell death.
EMT, cell invasion and motility
EMT is a biological process that allows immobile epithelial cells to acquire a mobile mesenchymal phenotype, becoming detached and invasive. It was initially described in the context of embryonic differentiation.[57] In tumor cells, this process, together with the induction of neo-angiogenesis, initiates cancer metastasis, inducing enhanced migratory properties, invasiveness and resistance to apoptosis.[58,59]
During EMT, a variety of transcription factors are upregulated in metastatic cells, such as Snail, Slug, Twist and Zeb ½.[60] Snail can be activated by a number of pathways, including hypoxia, HIF-1, HIF-2, Notch, nuclear factor kappa-light-chain-enhancer of activated B cells (NF-kB), and transforming growth factor beta (TGF-β), a pro-apoptotic factor. Snail up-regulates AKT phosphorylation and Bcl-Xl, countering the induction of apoptosis,[61] and down-regulates cyclin D2, inhibiting cell cycle progression.[62]
Twist, which promotes loss of E-cadherin mediated cell-cell adhesion and cell motility,[63] has been linked to the PI3K/AKT pathway in various malignancies. This link is established by the AKT2 isoform, a Twist-mediated transcriptional regulator that activates Twist, constituting a positive feedback loop that promotes EMT.[64,65] Twist also maintains hyper-activation of the PI3K/AKT pathway in breast cancer cells, through its transcriptional target TGF-β2.[65]
AKT hyper-activation and PIK3CA knock-in can promote EMT in various human cancers.[61-66] The association between EMT and PI3K activation has also been reported in ERα-negative endometrial carcinomas.[67]
Twist overexpression has also been correlated with the induction of tumor cell invasion in GBM.[68] However, these malignancies usually do not metastasize out of the CNS, mainly due to their rapid relapse rate and poor prognosis.[69] Even so, there are reports describing GBM metastasis[70] involving the spread of GBM cells out the CNS through cerebrospinal fluid, blood or lymphatic vessels.[71,72]
Medulloblastoma, on the other hand, has a high tendency to disseminate to the spinal cord and leptomeninges of the cerebellum and forebrain. These tumors are classified into 4 molecular subgroups: wingless (WNT), sonic hedgehog (SHH), group 3 and group 4.[73] Group 3, characterized by cMYC amplification, is associated with metastatic disease.[74]
The PI3K/AKT pathway is activated in 50% of GBMs. In the case of MB, there are a number of studies concerning alterations in this pathway.[6,75,76] This pathway appears to facilitate an invasive phenotype of GBM and MB, especially in terms of motility and resistance to stress.[77]
The class IA PI3K isoform p110α is the most relevant PI3K isoform affecting cell growth and survival. The gene encoding this isoform, PIK3CA, is usually mutated in GBM (27%).[78] In this malignancy, PIK3CA mutated form plays a main role in cell growth under anchorage-independent conditions. In MB, however, this PI3K isoform is typically overexpressed,[79] promoting cell proliferation, for example, through the regulation of the leukemia inhibitory factor receptor α (LIFR α).[80] The inhibition of p110α impairs cancer cell growth, migration, and survival in these CNS malignancies.[16,79]
Other class IA PI3K isoforms are also overexpressed in brain tumors, such as p110δ, which has been reported to be overexpressed at the mRNA level in primary GBM, controlling migration in these cells.[81,82] The isoform p110ϒ, which is overexpressed in primary MB, contributes to cisplatin resistance and has emerged as a novel target for combinatorial treatments.[83] The class II PI3K isoform PI3KC2β, which is overexpressed in a variety of cancers, acts as a modulator of cell migration, survival and proliferation in leukemia and brain tumors.[84] The highly specific pan-PI3K inhibitor GDC-0941 has recently been shown to have anti-migratory, anti-proliferative and pro-apoptotic effects in MB cell lines, showing synergy with the standard chemotherapeutic drug etoposide and good clinical tolerability.[85]
Other elements of the PI3K/AKT pathway are also being considered as potential targets to inhibit cell proliferation and migration in GBM and MB. One example is AKT, which usually shows high levels of phosphorylation in these brain tumors.[86] Its inhibition by KP-372-1, KP-372-2, A-443654, or perifosine, was reported to inhibit cell growth and induce radio-sensitizing effects in GBM and MB.[87-89] Clinical trials of perifosine in GBM patients are ongoing.[90]
PTEN is a tumor suppressor usually mutated and inactivated in GBM, with an inverse correlation between its expression and glioma grade.[91] In MB, PTEN is rarely mutated but frequently downregulated, by promoter hypermethylation and/or allelic losses, inducing AKT activation.[86]
PTEN, together with the MAPK signaling pathway, has a primary role in the regulation of G1/S cell cycle checkpoint-defective astrocytoma invasion, and its deletion increases migration, invasion and resistance to apoptosis in GBM cell lines.[92] PTEN controls integrin-dependent migration through the regulation of Src family kinase activation, in a PI3K/AKT-independent manner.[93] The re-expression of PTEN in GBM cell lines increases the cellular content and activity of the p53 tumor suppressor protein inducing cell cycle arrest and increasing the sensitivity of the tumor cells to various chemotherapeutic agents such as etoposide.[94]
Upstream regulators of EMT induction, such as insulin-like growth Factor-1 receptor (IGF-1R), c-MET and the CXCR4 receptor, have been proposed as potential targets to inhibit GBM or MB invasion.
IGF-1R is typically overexpressed in malignant GBM,[95] and its activation by IGF-1 contributes to Snail and Twist expression though PI3K/AKT signaling pathway activation.[96,97] Therefore, IGF-1R tyrosine kinase inhibitors or IGF-1 inhibitors, such as osthole, have been used to inhibit GBM proliferation, migration and EMT.[97,98] In a recent study of 218 cases of human GBM, IGF-1R overexpression was reported as an independent prognostic factor associated with shorter survival time and a less favorable response to temozolomide.[99]
C-MET expression levels correlate with tumor grade in CNS malignancies,[100] and its activation also mediates EMT-promoting signals in cancer cells via class IA PI3K.[101,102] In MB, c-MET signaling is deregulated, thus inducing tumor growth and an anaplastic histology.[103] The use of c-MET kinase inhibitors, such as SGX523, suppressed tumor growth in GBM cell lines.[104] This inhibition blocked the EMT induced by VEGF ablation in a GBM mouse model[105] and induced an effective decrease in MB cell migration and invasion.[106,107]
Stromal cell derived factor (SDF-1) or CXCL2 and its chemokine receptor CXCR4 can induce EMT in GBM via activation of PI3K/AKT and extracellular-signal-regulated kinases (ERK) pathways, and its inhibition suppressed EMT in glioma cell lines by upregulating E-cadherin.[108]
However, single agents targeting the PAM pathway have been reported to be an inefficient approach in MB and to increase invasion in the surviving fraction of GBM.[109] Therefore, new therapeutic approaches should be based on increasing the therapeutic window by targeting two different routes, namely the PAM and ERK pathways, or on combining PAM inhibitors with chemotherapeutic agents.[110]
MicroRNAs have also been shown to play an important role in various CNS malignancies, and miR-142-5p and miR-25 are upregulated in all of them.[111] In MB, miR-21 suppression inhibited tumor migration.[112] MiR-183 has a pro-tumorigenic effect in the MYC-driven MB subgroup through the inhibition of apoptosis, deregulation of the mTOR pathway and modulation of cell motility and migration.[113]
During the EMT process, malignant cells start to intravasate into the surrounding blood vessels in order to migrate to other parts of the body. To accomplish this, the extracellular matrix and basement membrane of blood vessels have to be degraded by matrix metalloproteases (MMP).[114] The most relevant metalloproteases in this invasive process are MMP-2 and MMP-9.[115]
One of the upstream pathways controlling MMP production is the PI3K/AKT pathway.[116] As a consequence, drugs like wortmannin, a drug that inhibits the secretion of MMP-2, blocks GBM invasion through the down-regulation of the PI3K/AKT/NF-kB signaling pathway.[117] Since Snail induces MMP-9 expression, EMT seems to be necessary for intravasation of lymph vessels in GBM and other cancers.[119]
PI3Ks in inflammation/microenvironment
The process of inflammation has been extensively linked to tumor progression, as it can stimulate immune suppression, angiogenesis and tumor metastasis.[119,120] In response to tumor-derived growth factors and chemokines, inflammatory cells of the immune system are recruited to the tumor microenvironment. There, cells normally involved in chronic inflammation, such as mast cells, granulocytes and monocytes, provide the tumor with angiogenic factors, enzymes for extracellular matrix (EM) remodeling and growth factors to create a favorable milieu for expansion and dissemination.[121,122]
Members of the class I PI3K family have also been implicated in tumor-associated inflammatory responses. In myeloid cells, p110γ can be activated via tumor-derived chemoattractants, such as IL-6, Il-8, TNF-α and CSF-1. Upon activation, p110γ promotes extravasation into the tumor microenvironment (TME) via integrin α4β1 and promotes inflammation-associated tumor progression.[26,123] This is in line with other reports indicating a crucial role of p110y for immune cell chemotaxis, as well as for chronic inflammation.[124]
Microglial cells are resident macrophages of the CNS. Depending on the signaling context, these cells possess a dual role in tumor biology. By secreting cytokines like IL-6, IL-10 and immune suppressive molecules, gliomas can polarize microglia into tumor supporting M2 phenotypes that participate in matrix remodeling and cell invasion.[125-127] In a recent study, PAM signaling was upregulated in microglial cells that were exposed to glioma derived factors, indicating that PAM signaling is needed to force microglial cells into a tumor supportive M2 state.[128] This result was supported by a report showing that mTOR inhibition with rapamycin polarizes microglia cells to express a tumor suppressive M1 phenotype.[129] To date, the exact molecular mechanism by which PI3K signaling contributes to M2-polarization of microglia is still unknown and should be the subject of further investigation.
The tumor microenvironment of MB is also being investigated. A recent study associated the SHH-MB subtype with high infiltration of tumor associated macrophages (TAM) and strong expression of the inflammatory genes CSF1R and CD163.[130] It has been shown that PI3K binding to CSF1R stimulates spreading and motility in macrophages and their enhancement of tumor cell invasion.[131] Inhibition of p110δ impairs CSF-1 induced macrophage spreading and their invasive capacity.[132] Hence, it may be worth investigating whether selective inhibition of PI3Ks in the SHH-MB subtype impairs TAM-driven tumor invasiveness. The CD163 gene is a surface marker that is strongly expressed by tumor promoting M2 macrophages, but it is not clear whether or not MB cells polarize surrounding TAM via PI3K to enhance tumor invasion.
Clinical trials of kinase inhibitors in glioblastoma
Oncogenic kinase signaling (e.g. via the PAM pathway) is crucial in GBM and hence attractive for targeted therapy.[133,134] Unfortunately, the overall response rate of GBMs to kinase inhibitors in clinical trials has been poor so far.[135] One reason for these disappointing results may be inadequate trial design. Systematic flaws such as small sample sizes, absent control groups and unverified drug activity have been reported in the past.[135] Therefore, various changes in study design have been proposed to improve the reliability of the results. Clinical trials enriched for patients with an aberrant kinase target are likely to give a better picture of the overall performance of a particular inhibitor.[136] In addition, the importance of monitoring target inhibition and negative feedback has been shown in a phase I trial in PTEN-deficient glioblastomas.[137] To improve the results of clinical trials using kinase inhibitors, it appears necessary to set higher requirements for preclinical models and to verify efficacy in a broader spectrum of GBM models in order to address each model’s shortcomings. Given the fact that kinase signaling pathways are often dysregulated in parallel, it may also prove worthwhile to evaluate combinations of different kinase inhibitors.
Conclusion
Aberrant PAM signaling can promote crucial metastatic events such as angiogenesis, EMT, and modulation of immune cells in both MB and GBM. Targeting the PAM network may be a useful way to inhibit these often fatal events. Understanding the molecular mechanisms and the context by which different components of the PAM pathway contribute to tumor progression is a prerequisite for the design of novel treatment strategies. Some of these mechanisms, such as the interaction between malignant CNS cells and TME, have only recently become the focus of investigation and are still incompletely understood. Further studies are necessary to elucidate these mechanisms and to determine which components of the PAM pathway should be targeted to inhibit the metastasis of CNS malignancies.
Financial support and sponsorship
Work in the author’s laboratory is supported by grants from the European Union FP7 (ASSET, project number: 259348 and LUNGTARGET, project number: 259770), the Swiss National Science Foundation (Grant 31003A-146464), the Fondation FORCE, the Novartis Stiftung für Medizinisch-Biologische Forschung, the Jubiläumsstiftung der Schweizerischen Mobiliar Genossenschaft, the Stiftung zur Krebsbekämpfung, the Huggenberger-Bischoff Stiftung zur Krebsforschung, the UniBern Forschungsstiftung, the Stiftung für klinisch-experimentelle Tumorforschung, Bern and the Berner Stiftung für krebskranke Kinder und Jugendliche.
Conflicts of interest
There are no conflicts of interest.
REFERENCES
1. Louis DN, Ohgaki H, Wiestler OD, Cavenee WK, Burger PC, Jouvet A, Scheithauer BW, Kleihues P. The 2007 WHO classification of tumours of the central nervous system. Acta Neuropathol 2007;114:97-109.
2. Johnson DR, O'Neill BP. Glioblastoma survival in the United States before and during the temozolomide era. J Neurooncol 2012;107:359-64.
3. Grimmer MR, Weiss WA. Childhood tumors of the nervous system as disorders of normal development. Curr Opin Pediatr 2006;18:634-8.
4. Gajjar AJ, Robinson GW. Medulloblastoma-translating discoveries from the bench to the bedside. Nat Rev Clin Oncol 2014;11:714-22.
5. Cancer Genome Atlas Research Network. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature 2008;455:1061-8.
6. Northcott PA, Shih DJ, Peacock J, Garzia L, Morrissy AS, Zichner T, Stütz AM, Korshunov A, Reimand J, Schumacher SE, Beroukhim R, Ellison DW, Marshall CR, Lionel AC, Mack S, Dubuc A, Yao Y, Ramaswamy V, Luu B, Rolider A, Cavalli FM, Wang X, Remke M, Wu X, Chiu RY, Chu A, Chuah E, Corbett RD, Hoad GR, Jackman SD, Li Y, Lo A, Mungall KL, Nip KM, Qian JQ, Raymond AG, Thiessen NT, Varhol RJ, Birol I, Moore RA, Mungall AJ, Holt R, Kawauchi D, Roussel MF, Kool M, Jones DT, Witt H, Fernandez-L A, Kenney AM, Wechsler-Reya RJ, Dirks P, Aviv T, Grajkowska WA, Perek-Polnik M, Haberler CC, Delattre O, Reynaud SS, Doz FF, Pernet-Fattet SS, Cho BK, Kim SK, Wang KC, Scheurlen W, Eberhart CG, Fèvre-Montange M, Jouvet A, Pollack IF, Fan X, Muraszko KM, Gillespie GY, Di Rocco C, Massimi L, Michiels EM, Kloosterhof NK, French PJ, Kros JM, Olson JM, Ellenbogen RG, Zitterbart K, Kren L, Thompson RC, Cooper MK, Lach B, McLendon RE, Bigner DD, Fontebasso A, Albrecht S, Jabado N, Lindsey JC, Bailey S, Gupta N, Weiss WA, Bognár L, Klekner A, Van Meter TE, Kumabe T, Tominaga T, Elbabaa SK, Leonard JR, Rubin JB, Liau LM, Van Meir EG, Fouladi M, Nakamura H, Cinalli G, Garami M, Hauser P, Saad AG, Iolascon A, Jung S, Carlotti CG, Vibhakar R, Ra YS, Robinson S, Zollo M, Faria CC, Chan JA, Levy ML, Sorensen PH, Meyerson M, Pomeroy SL, Cho YJ, Bader GD, Tabori U, Hawkins CE, Bouffet E, Scherer SW, Rutka JT, Malkin D, Clifford SC, Jones SJ, Korbel JO, Pfister SM, Marra MA, Taylor MD. Subgroup-specific structural variation across 1,000 medulloblastoma genomes. Nature 2012;488:49-56.
7. Engelman JA, Luo J, Cantley LC. The evolution of phosphatidylinositol 3-kinases as regulators of growth and metabolism. Nat Rev Genet 2006;7:606-19.
9. Vanhaesebroeck B, Leevers SJ, Panayotou G, Waterfield MD. Phosphoinositide 3-kinases: a conserved family of signal transducers. Trends Biochem Sci 1997;22:267-72.
10. Lemmon MA. Membrane recognition by phospholipid-binding domains. Nat Rev Mol Cell Biol 2008;9:99-111.
11. Hatanpaa KJ, Burma S, Zhao D, Habib AA. Epidermal growth factor receptor in glioma: signal transduction, neuropathology, imaging, and radioresistance. Neoplasia 2010;12:675-84.
12. Quayle SN, Lee JY, Cheung LW, Ding L, Wiedemeyer R, Dewan RW, Huang-Hobbs E, Zhuang L, Wilson RK, Ligon KL, Mills GB, Cantley LC, Chin L. Somatic mutations of PIK3R1 promote gliomagenesis. PloS one 2012;7:e49466.
13. Castellino RC, Barwick BG, Schniederjan M, Buss MC, Becher O, Hambardzumyan D, Macdonald TJ, Brat DJ, Durden DL. Heterozygosity for Pten promotes tumorigenesis in a mouse model of medulloblastoma. PloS one 2010;5:e10849.
14. Zhao D, Besser AH, Wander SA, Sun J, Zhou W, Wang B, Ince T, Durante MA, Guo W, Mills G, Theodorescu D, Slingerland J. Cytoplasmic p27 promotes epithelial-mesenchymal transition and tumor metastasis via STAT3-mediated Twist1 upregulation. Oncogene 2015;34:5447-59.
15. Yoshioka K, Yoshida K, Cui H, Wakayama T, Takuwa N, Okamoto Y, Du W, Qi X, Asanuma K, Sugihara K, Aki S, Miyazawa H, Biswas K, Nagakura C, Ueno M, Iseki S, Schwartz RJ, Okamoto H, Sasaki T, Matsui O, Asano M, Adams RH, Takakura N, Takuwa Y. Endothelial PI3K-C2alpha, a class II PI3K, has an essential role in angiogenesis and vascular barrier function. Nat Med 2012;18:1560-9.
16. Holand K, Boller D, Hagel C, Dolski S, Treszl A, Pardo OE, Cwiek P, Salm F, Leni Z, Shepherd PR, Styp-Rekowska B, Djonov V, von Bueren AO, Frei K, Arcaro A. Targeting class IA PI3K isoforms selectively impairs cell growth, survival, and migration in glioblastoma. PloS one 2014;9:e94132.
17. Huse JT, Holland EC. Targeting brain cancer: advances in the molecular pathology of malignant glioma and medulloblastoma. Nat Rev Cancer 2010;10:319-31.
18. Dimitrova V, Arcaro A. Targeting the PI3K/AKT/mTOR signaling pathway in medulloblastoma. Curr Mol Med 2015;15:82-93.
19. Nagy JA, Dvorak HF. Heterogeneity of the tumor vasculature: the need for new tumor blood vessel type-specific targets. Clin Exp Metastasis 2012;29:657-62.
20. Schmid MC, Varner JA. Myeloid cell trafficking and tumor angiogenesis. Cancer Lett 2007;250:1-8.
21. Zhong H, Chiles K, Feldser D, Laughner E, Hanrahan C, Georgescu MM, Simons JW, Semenza GL. Modulation of hypoxia-inducible factor 1alpha expression by the epidermal growth factor/phosphatidylinositol 3-kinase/PTEN/AKT/FRAP pathway in human prostate cancer cells: implications for tumor angiogenesis and therapeutics. Cancer Res 2000;60:1541-5.
22. Xia C, Meng Q, Cao Z, Shi X, Jiang BH. Regulation of angiogenesis and tumor growth by p110 alpha and AKT1 via VEGF expression. J Cell Physiol 2006;209:56-66.
23. Brazil DP, Park J, Hemmings BA. PKB binding proteins. Getting in on the Akt. Cell 2002;111:293-303.
24. Galetic I, Andjelkovic M, Meier R, Brodbeck D, Park J, Hemmings BA. Mechanism of protein kinase B activation by insulin/insulin-like growth factor-1 revealed by specific inhibitors of phosphoinositide 3-kinase-significance for diabetes and cancer. Pharmacol Ther 1999;82:409-25.
25. Hu L, Hofmann J, Jaffe RB. Phosphatidylinositol 3-kinase mediates angiogenesis and vascular permeability associated with ovarian carcinoma. Clin Cancer Res 2005;11:8208-12.
26. Schmid MC, Avraamides CJ, Dippold HC, Franco I, Foubert P, Ellies LG, Acevedo LM, Manglicmot JR, Song X, Wrasidlo W, Blair SL, Ginsberg MH, Cheresh DA, Hirsch E, Field SJ, Varner JA. Receptor tyrosine kinases and TLR/IL1Rs unexpectedly activate myeloid cell PI3kgamma, a single convergent point promoting tumor inflammation and progression. Cancer cell 2011;19:715-27.
27. Semenza GL. HIF-1 and tumor progression: pathophysiology and therapeutics. Trends Mol Med 2002;8:S62-7.
28. Su JD, Mayo LD, Donner DB, Durden DL. PTEN and phosphatidylinositol 3'-kinase inhibitors up-regulate p53 and block tumor-induced angiogenesis: evidence for an effect on the tumor and endothelial compartment. Cancer Res 2003;63:3585-92.
29. Garlich JR, De P, Dey N, Su JD, Peng X, Miller A, Murali R, Lu Y, Mills GB, Kundra V, Shu HK, Peng Q, Durden DL. A vascular targeted pan phosphoinositide 3-kinase inhibitor prodrug, SF1126, with antitumor and antiangiogenic activity. Cancer Res 2008;68:206-15.
30. Koul D, Shen R, Kim YW, Kondo Y, Lu Y, Bankson J, Ronen SM, Kirkpatrick DL, Powis G, Yung WK. Cellular and in vivo activity of a novel PI3K inhibitor, PX-866, against human glioblastoma. Neuro Oncol 2010;12:559-69.
31. Pitz MW, Eisenhauer EA, MacNeil MV, Thiessen B, Easaw JC, Macdonald DR, Eisenstat DD, Kakumanu AS, Salim M, Chalchal H, Squire J, Tsao MS, Kamel-Reid S, Banerji S, Tu D, Powers J, Hausman DF, Mason WP. Phase II study of PX-866 in recurrent glioblastoma. Neuro Oncol 2015;17:1270-4.
32. Sharma PS, Sharma R, Tyagi T. VEGF/VEGFR pathway inhibitors as anti-angiogenic agents: present and future. Curr Cancer Drug Targets 2011;11:624-53.
33. Reardon DA, Turner S, Peters KB, Desjardins A, Gururangan S, Sampson JH, McLendon RE, Herndon JE 2nd, Jones LW, Kirkpatrick JP, Friedman AH, Vredenburgh JJ, Bigner DD, Friedman HS. A review of VEGF/VEGFR-targeted therapeutics for recurrent glioblastoma. J Natl Compr Canc Netw 2011;9:414-27.
34. Kuczynski EA, Patten SG, Coomber BL. VEGFR2 expression and TGF-beta signaling in initial and recurrent high-grade human glioma. Oncology 2011;81:126-34.
35. Chinot OL, de La Motte Rouge T, Moore N, Zeaiter A, Das A, Phillips H, Modrusan Z, Cloughesy T. AVAglio: Phase 3 trial of bevacizumab plus temozolomide and radiotherapy in newly diagnosed glioblastoma multiforme. Adv Ther 2011;28:334-40.
36. Van Meir EG, Hadjipanayis CG, Norden AD, Shu HK, Wen PY, Olson JJ. Exciting new advances in neuro-oncology: the avenue to a cure for malignant glioma. CA Cancer J Clin 2010;60:166-93.
37. Hardee ME, Zagzag D. Mechanisms of glioma-associated neovascularization. Am J Pathol 2012;181:1126-41.
38. Takano S, Yamashita T, Ohneda O. Molecular therapeutic targets for glioma angiogenesis. J Oncol 2010;2010:351908.
39. di Tomaso E, Snuderl M, Kamoun WS, Duda DG, Auluck PK, Fazlollahi L, Andronesi OC, Frosch MP, Wen PY, Plotkin SR, Hedley-Whyte ET, Sorensen AG, Batchelor TT, Jain RK. Glioblastoma recurrence after cediranib therapy in patients: lack of "rebound" revascularization as mode of escape. Cancer Res 2011;71:19-28.
40. Gerstner ER, Chen PJ, Wen PY, Jain RK, Batchelor TT, Sorensen G. Infiltrative patterns of glioblastoma spread detected via diffusion MRI after treatment with cediranib. Neuro Oncol 2010;12:466-72.
41. Navis AC, Bourgonje A, Wesseling P, Wright A, Hendriks W, Verrijp K, van der Laak JA, Heerschap A, Leenders WP. Effects of dual targeting of tumor cells and stroma in human glioblastoma xenografts with a tyrosine kinase inhibitor against c-MET and VEGFR2. PloS one 2013;8:e58262.
42. Bridges E, Harris AL. Vascular-promoting therapy reduced tumor growth and progression by improving chemotherapy efficacy. Cancer cell 2015;27:7-9.
43. Donnem T, Hu J, Ferguson M, Adighibe O, Snell C, Harris AL, Gatter KC, Pezzella F. Vessel co-option in primary human tumors and metastases: an obstacle to effective anti-angiogenic treatment? Cancer Med 2013;2:427-36.
44. Qian CN. Hijacking the vasculature in ccRCC-co-option, remodelling and angiogenesis. Nat Rev Urol 2013;10:300-4.
45. Nguyen TM, Subramanian IV, Kelekar A, Ramakrishnan S. Kringle 5 of human plasminogen, an angiogenesis inhibitor, induces both autophagy and apoptotic death in endothelial cells. Blood 2007;109:4793-802.
46. Ramakrishnan S, Nguyen TM, Subramanian IV, Kelekar A. Autophagy and angiogenesis inhibition. Autophagy 2007;3:512-5.
47. Du J, Teng RJ, Guan T, Eis A, Kaul S, Konduri GG, Shi Y. Role of autophagy in angiogenesis in aortic endothelial cells. Am J Physiol Cell Physiol 2012;302:C383-91.
48. Shen W, Tian C, Chen H, Yang Y, Zhu D, Gao P, Liu J. Oxidative stress mediates chemerin-induced autophagy in endothelial cells. Free Radic Biol Med 2013;55:73-82.
49. Brech A, Ahlquist T, Lothe RA, Stenmark H. Autophagy in tumour suppression and promotion. Mol Oncol 2009;3:366-75.
51. Bhandarkar SS, Arbiser JL. Curcumin as an inhibitor of angiogenesis. Adv Exp Med Biol 2007;595:185-95.
52. Igura K, Ohta T, Kuroda Y, Kaji K. Resveratrol and quercetin inhibit angiogenesis in vitro. Cancer Lett 2001;171:11-6.
53. Lau DH, Xue L, Young LJ, Burke PA, Cheung AT. Paclitaxel (Taxol): an inhibitor of angiogenesis in a highly vascularized transgenic breast cancer. Cancer Biother Radiopharm 1999;14:31-6.
54. Pirtoli L, Cevenini G, Tini P, Vannini M, Oliveri G, Marsili S, Mourmouras V, Rubino G, Miracco C. The prognostic role of Beclin 1 protein expression in high-grade gliomas. Autophagy 2009;5:930-6.
55. Weihua Z, Tsan R, Huang WC, Wu Q, Chiu CH, Fidler IJ, Hung MC. Survival of cancer cells is maintained by EGFR independent of its kinase activity. Cancer cell 2008;13:385-93.
56. Huang X, Bai HM, Chen L, Li B, Lu YC. Reduced expression of LC3B-II and Beclin 1 in glioblastoma multiforme indicates a down-regulated autophagic capacity that relates to the progression of astrocytic tumors. J Clin Neurosci 2010;17:1515-9.
57. Micalizzi DS, Farabaugh SM, Ford HL. Epithelial-mesenchymal transition in cancer: parallels between normal development and tumor progression. J Mammary Gland Biol Neoplasia 2010;15:117-34.
58. Kalluri R, Neilson EG. Epithelial-mesenchymal transition and its implications for fibrosis. J Clin Invest 2003;112:1776-84.
59. Kalluri R, Weinberg RA. The basics of epithelial-mesenchymal transition. J Clin Invest 2009;119:1420-8.
60. Boutet A, Esteban MA, Maxwell PH, Nieto MA. Reactivation of Snail genes in renal fibrosis and carcinomas: a process of reversed embryogenesis? Cell cycle 2007;6:638-42.
61. Grille SJ, Bellacosa A, Upson J, Klein-Szanto AJ, van Roy F, Lee-Kwon W, Donowitz M, Tsichlis PN, Larue L. The protein kinase Akt induces epithelial mesenchymal transition and promotes enhanced motility and invasiveness of squamous cell carcinoma lines. Cancer Res 2003;63:2172-8.
62. Vega S, Morales AV, Ocana OH, Valdes F, Fabregat I, Nieto MA. Snail blocks the cell cycle and confers resistance to cell death. Genes Dev 2004;18:1131-43.
63. Yang J, Mani SA, Donaher JL, Ramaswamy S, Itzykson RA, Come C, Savagner P, Gitelman I, Richardson A, Weinberg RA. Twist, a master regulator of morphogenesis, plays an essential role in tumor metastasis. Cell 2004;117:927-39.
64. Cheng GZ, Zhang W, Wang LH. Regulation of cancer cell survival, migration, and invasion by Twist: AKT2 comes to interplay. Cancer Res 2008;68:957-60.
65. Xue G, Restuccia DF, Lan Q, Hynx D, Dirnhofer S, Hess D, Rüegg C, Hemmings BA. Akt/PKB-mediated phosphorylation of Twist1 promotes tumor metastasis via mediating cross-talk between PI3K/Akt and TGF-beta signaling axes. Cancer Discov 2012;2:248-59.
66. Wallin JJ, Guan J, Edgar KA, Zhou W, Francis R, Torres AC, Haverty PM, Eastham-Anderson J, Arena S, Bardelli A, Griffin S, Goodall JE, Grimshaw KM, Hoeflich KP, Torrance C, Belvin M, Friedman LS. Active PI3K pathway causes an invasive phenotype which can be reversed or promoted by blocking the pathway at divergent nodes. PloS one 2012;7:e36402.
67. Wik E, Raeder MB, Krakstad C, Trovik J, Birkeland E, Hoivik EA, Mjos S, Werner HM, Mannelqvist M, Stefansson IM, Oyan AM, Kalland KH, Akslen LA, Salvesen HB. Lack of estrogen receptor-alpha is associated with epithelial-mesenchymal transition and PI3K alterations in endometrial carcinoma. Clin Cancer Res 2013;19:1094-105.
68. Elias MC, Tozer KR, Silber JR, Mikheeva S, Deng M, Morrison RS, Manning TC, Silbergeld DL, Glackin CA, Reh TA, Rostomily RC. TWIST is expressed in human gliomas and promotes invasion. Neoplasia 2005;7:824-37.
69. Dunn GP, Bruce AT, Ikeda H, Old LJ, Schreiber RD. Cancer immunoediting: from immunosurveillance to tumor escape. Nat Immunol 2002;3:991-8.
70. Shahideh M, Fallah A, Munoz DG, Loch Macdonald R. Systematic review of primary intracranial glioblastoma multiforme with symptomatic spinal metastases, with two illustrative patients. J Clin Neurosci 2012;19:1080-6.
71. Lawton CD, Nagasawa DT, Yang I, Fessler RG, Smith ZA. Leptomeningeal spinal metastases from glioblastoma multiforme: treatment and management of an uncommon manifestation of disease. J Neurosurg Spine 2012;17:438-48.
72. Kalokhe G, Grimm SA, Chandler JP, Helenowski I, Rademaker A, Raizer JJ. Metastatic glioblastoma: case presentations and a review of the literature. J Neurooncol 2012;107:21-7.
73. Taylor MD, Northcott PA, Korshunov A, Remke M, Cho YJ, Clifford SC, Eberhart CG, Parsons DW, Rutkowski S, Gajjar A, Ellison DW, Lichter P, Gilbertson RJ, Pomeroy SL, Kool M, Pfister SM. Molecular subgroups of medulloblastoma: the current consensus. Acta Neuropathol 2012;123:465-72.
74. Gajjar A, Bowers DC, Karajannis MA, Leary S, Witt H, Gottardo NG. Pediatric Brain Tumors: Innovative Genomic Information Is Transforming the Diagnostic and Clinical Landscape. J Clin Oncol 2015;33:2986-98.
75. Robinson G, Parker M, Kranenburg TA, Lu C, Chen X, Ding L, Phoenix TN, Hedlund E, Wei L, Zhu X, Chalhoub N, Baker SJ, Huether R, Kriwacki R, Curley N, Thiruvenkatam R, Wang J, Wu G, Rusch M, Hong X, Becksfort J, Gupta P, Ma J, Easton J, Vadodaria B, Onar-Thomas A, Lin T, Li S, Pounds S, Paugh S, Zhao D, Kawauchi D, Roussel MF, Finkelstein D, Ellison DW, Lau CC, Bouffet E, Hassall T, Gururangan S, Cohn R, Fulton RS, Fulton LL, Dooling DJ, Ochoa K, Gajjar A, Mardis ER, Wilson RK, Downing JR, Zhang J, Gilbertson RJ. Novel mutations target distinct subgroups of medulloblastoma. Nature 2012;488:43-8.
76. Blom T, Roselli A, Hayry V, Tynninen O, Wartiovaara K, Korja M, Nordfors K, Haapasalo H, Nupponen NN. Amplification and overexpression of KIT, PDGFRA, and VEGFR2 in medulloblastomas and primitive neuroectodermal tumors. J Neurooncol 2010;97:217-24.
77. Holand K, Salm F, Arcaro A. The phosphoinositide 3-kinase signaling pathway as a therapeutic target in grade IV brain tumors. Curr Cancer Drug Targets 2011;11:894-918.
78. Hartmann C, Bartels G, Gehlhaar C, Holtkamp N, von Deimling A. PIK3CA mutations in glioblastoma multiforme. Acta Neuropathol 2005;109:639-42.
79. Guerreiro AS, Fattet S, Fischer B, Shalaby T, Jackson SP, Schoenwaelder SM, Grotzer MA, Delattre O, Arcaro A. Targeting the PI3K p110alpha isoform inhibits medulloblastoma proliferation, chemoresistance, and migration. Clin Cancer Res 2008;14:6761-9.
80. Salm F, Dimitrova V, von Bueren AO, Cwiek P, Rehrauer H, Djonov V, Anderle P, Arcaro A. The Phosphoinositide 3-Kinase p110alpha Isoform Regulates Leukemia Inhibitory Factor Receptor Expression via c-Myc and miR-125b to Promote Cell Proliferation in Medulloblastoma. PloS one 2015;10:e0123958.
81. Mueller W, Mizoguchi M, Silen E, D'Amore K, Nutt CL, Louis DN. Mutations of the PIK3CA gene are rare in human glioblastoma. Acta Neuropathol 2005;109:654-5.
82. Luk SK, Piekorz RP, Nurnberg B, Tony To SS. The catalytic phosphoinositol 3-kinase isoform p110delta is required for glioma cell migration and invasion. Eur J Cancer 2012;48:149-57.
83. Guerreiro AS, Fattet S, Kulesza DW, Atamer A, Elsing AN, Shalaby T, Jackson SP, Schoenwaelder SM, Grotzer MA, Delattre O, Arcaro A. A sensitized RNA interference screen identifies a novel role for the PI3K p110gamma isoform in medulloblastoma cell proliferation and chemoresistance. Mol Cancer Res 2011;9:925-35.
84. Boller D, Doepfner KT, De Laurentiis A, Guerreiro AS, Marinov M, Shalaby T, Depledge P, Robson A, Saghir N, Hayakawa M, Kaizawa H, Koizumi T, Ohishi T, Fattet S, Delattre O, Schweri-Olac A, Höland K, Grotzer MA, Frei K, Spertini O, Waterfield MD, Arcaro A. Targeting PI3KC2beta impairs proliferation and survival in acute leukemia, brain tumours and neuroendocrine tumours. Anticancer Res 2012;32:3015-27.
85. Ehrhardt M, Craveiro RB, Holst MI, Pietsch T, Dilloo D. The PI3K inhibitor GDC-0941 displays promising in vitro and in vivo efficacy for targeted medulloblastoma therapy. Oncotarget 2015;6:802-13.
86. Hartmann W, Digon-Sontgerath B, Koch A, Waha A, Endl E, Dani I, Denkhaus D, Goodyer CG, Sörensen N, Wiestler OD, Pietsch T. Phosphatidylinositol 3'-kinase/AKT signaling is activated in medulloblastoma cell proliferation and is associated with reduced expression of PTEN. Clin Cancer Res 2006;12:3019-27.
87. Koul D, Shen R, Bergh S, Sheng X, Shishodia S, Lafortune TA, Lu Y, de Groot JF, Mills GB, Yung WK. Inhibition of Akt survival pathway by a small-molecule inhibitor in human glioblastoma. Mol Cancer Ther 2006;5:637-44.
88. Gallia GL, Tyler BM, Hann CL, Siu IM, Giranda VL, Vescovi AL, Brem H, Riggins GJ. Inhibition of Akt inhibits growth of glioblastoma and glioblastoma stem-like cells. Mol Cancer Ther 2009;8:386-93.
89. Kumar A, Fillmore HL, Kadian R, Broaddus WC, Tye GW, Van Meter TE. The alkylphospholipid perifosine induces apoptosis and p21-mediated cell cycle arrest in medulloblastoma. Mol Cancer Res 2009;7:1813-21.
90. Becher OJ, Hambardzumyan D, Walker TR, Helmy K, Nazarian J, Albrecht S, Hiner RL, Gall S, Huse JT, Jabado N, MacDonald TJ, Holland EC. Preclinical evaluation of radiation and perifosine in a genetically and histologically accurate model of brainstem glioma. Cancer Res 2010;70:2548-57.
91. Knobbe CB, Reifenberger G. Genetic alterations and aberrant expression of genes related to the phosphatidyl-inositol-3'-kinase/protein kinase B (Akt) signal transduction pathway in glioblastomas. Brain Pathol 2003;13:507-18.
92. Vitucci M, Karpinich NO, Bash RE, Werneke AM, Schmid RS, White KK, McNeill RS, Huff B, Wang S, Van Dyke T, Miller CR. Cooperativity between MAPK and PI3K signaling activation is required for glioblastoma pathogenesis. Neuro Oncol 2013;15:1317-29.
93. Dey N, Crosswell HE, De P, Parsons R, Peng Q, Su JD, Durden DL. The protein phosphatase activity of PTEN regulates SRC family kinases and controls glioma migration. Cancer Res 2008;68:1862-71.
94. Mayo LD, Dixon JE, Durden DL, Tonks NK, Donner DB. PTEN protects p53 from Mdm2 and sensitizes cancer cells to chemotherapy. J Biol Chem 2002;277:5484-9.
95. Merrill MJ, Edwards NA. Insulin-like growth factor-I receptors in human glial tumors. J Clin Endocrinol Metab 1990;71:199-209.
96. Sinha S, Koul N, Dixit D, Sharma V, Sen E. IGF-1 induced HIF-1alpha-TLR9 cross talk regulates inflammatory responses in glioma. Cell Signal 2011;23:1869-75.
97. Lin YC, Lin JC, Hung CM, Chen Y, Liu LC, Chang TC, Kao JY, Ho CT, Way TD. Osthole inhibits insulin-like growth factor-1-induced epithelial to mesenchymal transition via the inhibition of PI3K/Akt signaling pathway in human brain cancer cells. J Agric Food Chem 2014;62:5061-71.
98. Yin S, Girnita A, Stromberg T, Khan Z, Andersson S, Zheng H, Ericsson C, Axelson M, Nistér M, Larsson O, Ekström TJ, Girnita L. Targeting the insulin-like growth factor-1 receptor by picropodophyllin as a treatment option for glioblastoma. Neuro Oncol 2010;12:19-27.
99. Maris C, D'Haene N, Trepant AL, Le Mercier M, Sauvage S, Allard J, Rorive S, Demetter P, Decaestecker C, Salmon I. IGF-IR: a new prognostic biomarker for human glioblastoma. Br J Cancer 2015;113:729-37.
100. Kong DS, Song SY, Kim DH, Joo KM, Yoo JS, Dong SM, Suh YL, Lee JI, Park K, Kim JH, Nam DH. Prognostic significance of c-Met expression in glioblastomas. Cancer 2009;115:140-8.
101. Fixman ED, Fournier TM, Kamikura DM, Naujokas MA, Park M. Pathways downstream of Shc and Grb2 are required for cell transformation by the tpr-Met oncoprotein. J Biol Chem 1996;271:13116-22.
102. Graziani A, Gramaglia D, Cantley LC, Comoglio PM. 1991. The tyrosine-phosphorylated hepatocyte growth factor/scatter factor receptor associates with phosphatidylinositol 3-kinase. J Biol Chem 1991;266:22087-90.
103. Li Y, Lal B, Kwon S, Fan X, Saldanha U, Reznik TE, Kuchner EB, Eberhart C, Laterra J, Abounader R. The scatter factor/hepatocyte growth factor: c-met pathway in human embryonal central nervous system tumor malignancy. Cancer Res 2005;65:9355-62.
104. Martens T, Schmidt NO, Eckerich C, Fillbrandt R, Merchant M, Schwall R, Westphal M, Lamszus K. A novel one-armed anti-c-Met antibody inhibits glioblastoma growth in vivo. Clin Cancer Res 2006;12:6144-52.
105. Lu KV, Chang JP, Parachoniak CA, Pandika MM, Aghi MK, Meyronet D, Isachenko N, Fouse SD, Phillips JJ, Cheresh DA, Park M, Bergers G. VEGF inhibits tumor cell invasion and mesenchymal transition through a MET/VEGFR2 complex. Cancer cell 2012;22:21-35.
106. Kongkham PN, Onvani S, Smith CA, Rutka JT. Inhibition of the MET Receptor Tyrosine Kinase as a Novel Therapeutic Strategy in Medulloblastoma. Transl Oncol 2010;3:336-43.
107. Faria CC, Golbourn BJ, Dubuc AM, Remke M, Diaz RJ, Agnihotri S, Luck A, Sabha N, Olsen S, Wu X, Garzia L, Ramaswamy V, Mack SC, Wang X3 Leadley M, Reynaud D, Ermini L, Post M, Northcott PA, Pfister SM, Croul SE, Kool M, Korshunov A, Smith CA, Taylor MD, Rutka JT. Foretinib is effective therapy for metastatic sonic hedgehog medulloblastoma. Cancer Res 2015;75:134-46.
108. Lv B, Yang X, Lv S, Wang L, Fan K, Shi R, Wang F, Song H, Ma X, Tan X, Xu K, Xie J, Wang G, Feng M, Zhang L. CXCR4 Signaling Induced Epithelial-Mesenchymal Transition by PI3K/AKT and ERK Pathways in Glioblastoma. Mol Neurobiol 2015;52:1263-8.
109. Kil WJ, Tofilon PJ, Camphausen K. Post-radiation increase in VEGF enhances glioma cell motility in vitro. Radiat Oncol 2012;7:25.
110. Westhoff MA, Karpel-Massler G, Bruhl O, Enzenmuller S, La Ferla-Bruhl K, Siegelin MD, Nonnenmacher L, Debatin KM. A critical evaluation of PI3K inhibition in Glioblastoma and Neuroblastoma therapy. Mol Cell Ther 2014;2:32.
111. Birks DK, Barton VN, Donson AM, Handler MH, Vibhakar R, Foreman NK. Survey of MicroRNA expression in pediatric brain tumors. Pediatr Blood Cancer 2011;56:211-6.
112. Pang JC, Kwok WK, Chen Z, Ng HK. Oncogenic role of microRNAs in brain tumors. Acta Neuropathol 2009;117:599-611.
113. Weeraratne SD, Amani V, Teider N, Pierre-Francois J, Winter D, Kye MJ, Sengupta S, Archer T, Remke M, Bai AH, Warren P, Pfister SM, Steen JA, Pomeroy SL, Cho YJ. Pleiotropic effects of miR-183~96~182 converge to regulate cell survival, proliferation and migration in medulloblastoma. Acta Neuropathol 2012;123:539-52.
114. Stamenkovic I. Matrix metalloproteinases in tumor invasion and metastasis. Semin Cancer Biol 2000;10:415-33.
115. Vu TH, Werb Z. Matrix metalloproteinases: effectors of development and normal physiology. Genes Dev 2000;14:2123-33.
116. Chen JS, Wang Q, Fu XH, Huang XH, Chen XL, Cao LQ, Chen LZ, Tan HX, Li W, Bi J, Zhang LJ. Involvement of PI3K/PTEN/AKT/mTOR pathway in invasion and metastasis in hepatocellular carcinoma: Association with MMP-9. Hepatol Res 2009;39:177-86.
117. Jiao Y, Li H, Liu Y, Guo A, Xu X, Qu X, Wang S, Zhao J, Li Y, Cao Y. Resveratrol Inhibits the Invasion of Glioblastoma-Initiating Cells via Down-Regulation of the PI3K/Akt/NF-kappaB Signaling Pathway. Nutrients 2015;7:4383-402.
118. Greenspoon JN, Sharieff W, Hirte H, Overholt A, Devillers R, Gunnarsson T, Whitton A. Fractionated stereotactic radiosurgery with concurrent temozolomide chemotherapy for locally recurrent glioblastoma multiforme: a prospective cohort study. Onco Targets Ther 2014;7:485-90.
119. Kim S, Takahashi H, Lin WW, Descargues P, Grivennikov S, Kim Y, Luo JL, Karin M. Carcinoma-produced factors activate myeloid cells through TLR2 to stimulate metastasis. Nature 2009;457:102-6.
120. Du R, Lu KV, Petritsch C, Liu P, Ganss R, Passegué E, Song H, Vandenberg S, Johnson RS, Werb Z, Bergers G. HIF1alpha induces the recruitment of bone marrow-derived vascular modulatory cells to regulate tumor angiogenesis and invasion. Cancer cell 2008;13:206-20.
121. Mantovani A, Allavena P, Sica A, Balkwill F. Cancer-related inflammation. Nature 2008;454:436-44.
122. Coussens LM. Zitvogel L, Palucka AK. Neutralizing tumor-promoting chronic inflammation: A magic bullet? (vol 339, pg 286, 2013). Science 2013;339:286-91.
123. Schmid MC, Franco I, Kang SW, Hirsch E, Quilliam LA, Varner JA. PI3-kinase gamma promotes Rap1a-mediated activation of myeloid cell integrin alpha4beta1, leading to tumor inflammation and growth. PloS one 2013;8:e60226.
124. Rommel C, Camps M, Ji H. PI3K delta and PI3K gamma: partners in crime in inflammation in rheumatoid arthritis and beyond? Nature reviews. Immunology 2007;7:191-201.
125. Kettenmann H, Hanisch UK, Noda M, Verkhratsky A. Physiology of microglia. Physiol Rev 2011;91:461-553.
126. Wei J, Gabrusiewicz K, Heimberger A. The controversial role of microglia in malignant gliomas. Clin Dev Immunol 2013;2013:285246.
127. Markovic DS, Vinnakota K, Chirasani S, Synowitz M, Raguet H, Stock K, Sliwa M, Lehmann S, Kälin R, van Rooijen N, Holmbeck K, Heppner FL, Kiwit J, Matyash V, Lehnardt S, Kaminska B, Glass R, Kettenmann H. Gliomas induce and exploit microglial MT1-MMP expression for tumor expansion. Proc Natl Acad Sci U S A 2009;106:12530-5.
128. Ellert-Miklaszewska A, Dabrowski M, Lipko M, Sliwa M, Maleszewska M, Kaminska B. Molecular definition of the pro-tumorigenic phenotype of glioma-activated microglia. Glia 2013;61:1178-90.
129. Lisi L, Laudati E, Navarra P, Dello Russo C. The mTOR kinase inhibitors polarize glioma-activated microglia to express a M1 phenotype. J Neuroinflammation 2014;11:125.
130. Margol AS, Robison NJ, Gnanachandran J, Hung LT, Kennedy RJ, Vali M, Dhall G, Finlay JL, Erdreich-Epstein A, Krieger MD, Drissi R, Fouladi M, Gilles FH, Judkins AR, Sposto R, Asgharzadeh S. Tumor-associated macrophages in SHH subgroup of medulloblastomas. Clin Cancer Res 2015;21:1457-65.
131. Sampaio NG, Yu W, Cox D, Wyckoff J, Condeelis J, Stanley ER, Pixley FJ. Phosphorylation of CSF-1R Y721 mediates its association with PI3K to regulate macrophage motility and enhancement of tumor cell invasion. J Cell Sci 2011;124:2021-31.
132. Mouchemore KA, Sampaio NG, Murrey MW, Stanley ER, Lannutti BJ, Pixley FJ. Specific inhibition of PI3K p110delta inhibits CSF-1-induced macrophage spreading and invasive capacity. FEBS J 2013;280:5228-36.
133. Carrasco-Garcia E, Saceda M, Martinez-Lacaci I. Role of receptor tyrosine kinases and their ligands in glioblastoma. Cells 2014;3:199-235.
134. Chi AS, Wen PY. Inhibiting kinases in malignant gliomas. Expert Opin Ther Targets 2007;11:473-96.
135. De Witt Hamer PC. Small molecule kinase inhibitors in glioblastoma: a systematic review of clinical studies. Neuro Oncol 2010;12:304-16.
136. Arteaga CL, Baselga J. Tyrosine kinase inhibitors: why does the current process of clinical development not apply to them? Cancer cell 2004;5:525-31.
137. Cloughesy TF, Yoshimoto K, Nghiemphu P, Brown K, Dang J, Zhu S, Hsueh T, Chen Y, Wang W, Youngkin D, Liau L, Martin N, Becker D, Bergsneider M, Lai A, Green R, Oglesby T, Koleto M, Trent J, Horvath S, Mischel PS, Mellinghoff IK, Sawyers CL. Antitumor activity of rapamycin in a Phase I trial for patients with recurrent PTEN-deficient glioblastoma. PLoS Med 2008;5:e8.
138. Trail data from ClinicalTrails.gov [Interent]. New York: U.S. National Institutes of Health. Available from: https://clinicaltrials.gov/.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Crespo* S, Kind* M, Arcaro A. The role of the PI3K/AKT/mTOR pathway in brain tumor metastasis. J Cancer Metastasis Treat 2016;2:80-9. http://dx.doi.org/10.20517/2394-4722.2015.72
AMA Style
Crespo* S, Kind* M, Arcaro A. The role of the PI3K/AKT/mTOR pathway in brain tumor metastasis. Journal of Cancer Metastasis and Treatment. 2016; 2: 80-9. http://dx.doi.org/10.20517/2394-4722.2015.72
Chicago/Turabian Style
Crespo*, Silvia, Marcus Kind*, Alexandre Arcaro. 2016. "The role of the PI3K/AKT/mTOR pathway in brain tumor metastasis" Journal of Cancer Metastasis and Treatment. 2: 80-9. http://dx.doi.org/10.20517/2394-4722.2015.72
ACS Style
Crespo*, S.; Kind* M.; Arcaro A. The role of the PI3K/AKT/mTOR pathway in brain tumor metastasis. J. Cancer. Metastasis. Treat. 2016, 2, 80-9. http://dx.doi.org/10.20517/2394-4722.2015.72
About This Article
Copyright

Author Biographies

Data & Comments
Data


 Cite This Article 33 clicks
Cite This Article 33 clicks

 Like This Article 9
likes
Like This Article 9
likes






Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.