
Muscarinic receptor signaling and colon cancer progression

Abstract
Due to the lack of effective treatments, advanced colorectal cancer (CRC) remains a leading cause of cancer death in the United States. Emerging evidence supports the observation that muscarinic receptor (MR) signaling plays a critical role in growth and progression of CRC. MR activation by acetylcholine and bile acids results in transactivation of epidermal growth factor receptors (EGFR) and post-EGFR signal transduction that enhances cell proliferation, migration, and invasion. Here, the authors review recent progress in understanding the molecular mechanisms underlying MR-mediated CRC progression and its therapeutic implications.
Keywords
Introduction
Colorectal cancer (CRC) is the second leading cause of cancer death worldwide with 1.4 million new cases and 693,900 deaths each year.[1] In the United States, CRC is currently the third leading cause of cancer death for both men and women.[2,3] Annually, approximately 140,000 people are diagnosed with CRC of which 50,000 will die, primarily from advanced disease.[2,3] Although surgical and endoscopic treatment is very effective for patients with early-stage CRC, late-stage CRC is generally resistant to chemo- and radiation- therapy.
Uncontrolled cell proliferation is an integral part of CRC progression. Cancer cell proliferation is regulated by a variety of growth factors and receptors. Epidermal growth factor (EGF) and other EGF receptor (EGFR) agonists play key roles in promoting growth of many human cancers, including CRC. As a result, biologicals that target EGFR have been approved by the Food and Drug Administration for the treatment of EGFR-positive advanced CRC; this approach enhances survival by several months.[4,5] However, the 5-year survival rate for advanced CRC remains only 10-15%.[4,5] New therapeutic approaches are urgently needed.
Muscarinic receptors in normal colon tissue and CRC
The muscarinic cholinergic family of G-protein-coupled receptors (GPCRs) consists of five muscarinic receptor (MR) subtypes designated muscarinic acetylcholine receptor subtype M1 (M1R)-M5R (for review see[6-9]). MR is expressed in many tissue types and play important roles in progression of many cancers including breast, prostate, lung and CRC.[10-12] M1R and M3R, expressed widely in the gastrointestinal (GI) tract, are coupled to Gq11, activate phospholipase C and increase cell calcium. Using reverse transcription polymerase chain reaction with primers specific to MR subtypes, radiolabeled ligand binding assays, and calcium mobilization studies, Frucht et al.[13,14] reported that 60% of colon cancer cell lines they tested expressed M3R. Subsequently, Yang and Frucht[15] reported up to 8-fold increased M3R expression in 62% of colon cancers compared to normal adjacent normal colon epithelium.
Muscarinic receptor ligands in CRC (acetylcholine and bile acids)
Role of acetylcholine
Acetylcholine (ACh) was traditionally regarded solely as a neurotransmitter that functioned exclusively in the central and peripheral nervous systems. However, over the past decade emerging evidence indicates that ACh is also produced and released by normal and neoplastic non-neuronal cells including human keratinocytes, small cell lung cancer cells, immune cells, and intestinal epithelial cells.[16-21] Choline acetyltransferase (ChAT) plays a critical catalytic role in the biosynthesis of both neuronal and non-neuronal ACh. Homozygous ChAT mutant embryos lack detectable ACh.[22] Also, ACh released from nerve endings is rapidly hydrolyzed by choline esterases thereby limiting its actions to immediately-neighboring cells. As a consequence, ACh production by non-neuronal cells can play a key role in regulating the actions of cells or tissues that are not directly innervated by cholinergic neurons.
ACh can be released by enteric neurons or produced by colon cancer cells. Although ACh release from intestinal non-neuronal cells was identified more than a decade ago,[23] its physiological relevance remains poorly understood. We showed that human colon cancer cells produce and release ACh that acts as an autocrine growth factor to stimulate cell proliferation.[24] We demonstrated that: (1) basal colon cancer cell proliferation is inhibited by the cholinergic antagonist atropine, cholinesterases, and inhibitors of choline transport; (2) ChAT is expressed at low levels by mouse and human intestinal mucosa, and at high levels by mouse colon tumors, a majority of human colon cancer cell lines, and CRC surgical specimens; and (3) human colon cancer cell lines produce and release ACh as demonstrated by high-performance liquid chromatography with electrochemical detection. These findings strongly support a role for ACh in CRC progression.
We proposed an important role for MR activation in ultrarapid growth of CRC in a patient with pheochromocytoma.[25] This elderly man with long-standing, unresectable pheochromocytoma experienced rapid development of rectal adenocarcinoma despite close endoscopic surveillance. We determined that the patient’s CRC overexpressed M3R, whereas his pheochromocytoma expressed ChAT. These findings suggested that ACh release from the pheochromocytoma stimulated rapid growth of the rectal neoplasm. As proof-of-principle we found that culture media conditioned by pheochromocytoma cells stimulates proliferation of a human colon cancer cell line, an effect attenuated by adding the MR antagonist atropine.
Role of bile acids
Besides ACh, bile acids and its derivatives are also important MR ligands. In 1998, we made the serendipitous observation that bile acids interact functionally with muscarinic receptors on gastric epithelial cells.[26] Subsequently, we made several novel observations: (1) conjugated secondary bile acids interact selectively and functionally with choline containing compounds (Cho) cells expressing muscarinic receptors;[27] (2) molecular modeling revealed a strikingly similar structural alignment of the geometry and surface electrostatic charges of bile acids and ACh;[27] (3) bile acid binding triggers appropriate post-M3R signaling;[27] (4) hybrid molecules created from bile acids and ACh are MR ligands;[28] and (5) lithocholic and deoxycholic acid conjugates interact with M3R on human colon cancer cells, thereby stimulating post-receptor signaling and cell proliferation.[29] Because muscarinic effects on colon cancer cell proliferation are mediated by transactivation of EGFR,[30,31] results of bile acid interaction with M3R depend on the cell type examined. In Cho cells that express M3R but not EGFR, deoxycholic acid conjugates are MR antagonists.[32] In H508 and HT-29 colon cancer cells that express both M3R and EGFR deoxycholyltaurine (DCT) is a MR agonist whose effects are mediated by transactivation of EGFR.[30,33] To our knowledge, no endogenous mammalian cholinergic agonists other than ACh and bile acids have been identified.
The observation that bile acids interact selectively and functionally with plasma membrane muscarinic receptors prompted us to examine their actions on intestinal epithelial cells. In particular, we studied H508 human colon cancer cells that co-express M3R and EGFR, and SNU-C4 cells that express EGFR but not muscarinic receptors.[14] DCT caused dose-dependent increases in M3R signaling and H508 cell proliferation that were not observed in SNU-C4 cells.[30] These proliferative effects of bile acids are mediated by interaction with plasma membrane M3R, not by interaction with bile acid nuclear receptors (i.e. the farnesoid X receptor) that regulate bile acid metabolism.
We demonstrated that efficacious concentrations of pro-proliferative bile acids are achieved in the intestine.[34] Because H508 cells derive from a moderately well-differentiated cecal adenocarcinoma, cecal contents were obtained immediately post-mortem from 19 persons. Using internal controls, bile acid spectrum and concentration were determined by an enzymatic assay and gas-chromatography/mass spectrometry. Total 3α-hydroxy bile acids were 400 ± 200 μmol/L (mean ± SD) and deoxycholic acid conjugates were 12 ± 28 μmol/L (maximum, 104 μmol/L).[33] Overall, in one-third of subjects, cecal conjugated deoxycholic acid achieved levels (10-100 μmol/L) that stimulate colon cancer cell proliferation in vitro.[30,33,34] Cecal bile acid concentrations in persons with ileal disease, ileal resection, and colon cancer are not known. Additional factors suggest that bile acid interaction with GI epithelial cell muscarinic receptors can regulate cell proliferation: (1) fecal bile acids are in contact with intestinal epithelial cells for many years (average age for developing colon cancer is > 50 years);[35] (2) bile acids lack an ester linkage and are not hydrolyzed by tissue cholinesterases that rapidly inactivate ACh;[28] (3) lipophilic lithocholic acid derivatives have access to muscarinic receptors in the lipid bilayer of cell membranes (i.e. the novel bile acid: ACh hybrid molecule, lithocholylcholine, interacts with muscarinic receptors on rat aortic strips);[28] (4) neoplastic cells commonly lose polarity, thereby expanding expression of muscarinic receptors, usually restricted to the basolateral membrane, to the entire plasma membrane; and (5) increased tight junction permeability between neoplastic cells allows access of luminal bile acids to basolateral membrane receptors.[36] We believe that this collective evidence makes a compelling argument that muscarinic receptors and MR ligands play an important role in intestinal epithelial cell proliferation and CRC progression.
Although experimental studies in rodents suggest that bile acids are intestinal tumor promoters,[37] the role of endogenous bile acids in colon carcinogenesis remains poorly understood. Dawson et al.[38] showed that in mice deficient in the ileal apical sodium-dependent bile acid transporter (ASBT, encoded by SLC10A2), fecal bile acid excretion increased more than 10-fold. To examine the development of aberrant crypt foci (ACF), the earliest histological marker of colon neoplasia, we treated WT and Asbt-deficient [Slc10a2 (-/-)] male mice with azoxymethane (AOM), an intestine-selective carcinogen.[39] We also used a combination of AOM and dextran sodium sulfate to induce colon tumorigenesis. Compared to littermate controls, we found that Asbt-deficient mice demonstrated significant increases in ACF, as well as colon tumor number and size. Also, Asbt-deficient mice had a two-fold increase in the number of colon adenocarcinomas. Finally, in murine colon neoplasia, increased fecal bile acids were associated with increased expression of M3R and EGFR, and activation of post-EGFR signaling. These observations indicate that endogenous bile acids also promote intestinal tumorigenesis.
MR signaling in CRC and disease progression
Transactivation of EGFR
EGFR is commonly over-expressed in many epithelial malignancies and this feature often indicates a more aggressive phenotype.[40] Likewise, as observed with M3R, EGFR is frequently over-expressed in colon cancer (in 25-77% of tumors compared to adjacent normal mucosa).[40,41] Co-expression of M3R and EGFR in many colon cancer cell lines, and over-expression of these receptors in the majority of colon cancers suggests that the functional interaction observed between M3R and EGFR is important for regulating colon cancer cell proliferation.[31]
Previously, we found that in H508 cells which over-express both M3R and EGFR, but not in SNU-C4 cells that express EGFR but not M3R, ACh stimulated cell proliferation by approximately 200% compared to control.[31] In H508 cells, both ACh and EGF stimulated calcium-dependent EGFR activation (tyrosine phosphorylation) and activation of extracellular signal-regulated kinase 1 and 2 (ERK1/2); MR antagonists and inhibitors of the mitogen-activated protein kinase (MAPK) phosphorylation blocked these effects. In addition, ACh- and EGF-induced phosphorylation of ERK1/2 MAPK and cell proliferation were abolished by EGFR inhibitors. However, in Cho cells transfected with rat M3R, which lack EGFR, ACh-induced ERK1/2 MAPK phosphorylation was not altered by EGFR inhibitors. It was concluded that, in H508 cells, cholinergic ligand interaction with M3R results in transactivation of EGFR, thereby stimulating cell proliferation.[31] These results indicate that EGFR transactivation is a key mechanism underlying MR-mediated intestinal tumorigenesis and cancer progression.
Cell proliferation and tumorigenesis
Uncontrolled cell proliferation is a hallmark of malignancies. We showed that in human colon cancer cells ACh-induced activation of M3R stimulates robust but selective matrix metalloproteinase (MMP) gene expression.[42] In H508 human colon cancer cells, ACh caused a striking dose- and time- dependent increase in mRNA and protein levels of MMP1, 7, and 10 by upregulating gene transcription. As a consequence, ACh stimulated MMP7-dependent cell proliferation by transactivating EGFR.
Using in vivo models, we showed that genetic ablation of M3R in AOM-treated mice attenuates epithelial cell proliferation, and the number of adenomas and adenocarcinomas per mouse colon (65% reduction in the number of adenocarcinomas/colon).[43] Whereas 50% of AOM-treated wild-type (WT) animals had multiple adenocarcinomas/colon, this was not the case with any M3R-deficient animal. Moreover, in M3R-deficient mice the overall colon tumor volume was reduced by 60% compared to that in WT animals. Collectively, these observations suggest that M3R may play a role in both tumor initiation and promotion; that is, both the number and size of tumors was reduced in M3R-deficient animals.
MR antagonists are potential therapeutics in CRC. We showed that M3R gene ablation and treatment with scopolamine butylbromide, a non-subtype-selective MR inhibitor, attenuated small intestinal neoplasia in ApcMin/+ mice with aberrant beta-catenin signaling.[44] Compared with ApcMin/+ mice, ApcMin/+M3R-/- mice showed 70% and 81% reductions in tumor number and volume, respectively. After 8 weeks of continuous treatment, scopolamine butylbromide-treated mice had a significant reduction in both tumor number and volume as compared with control mice. Overall, these findings indicate that the interplay of M3R and beta-catenin signaling is important for intestinal mucosal differentiation and neoplasia.
Because intestines express both M1R and M3R receptors, it is important to determine whether these two receptors have distinct functions. We showed divergent effects of MR subtype gene ablation on murine colon tumorigenesis.[45] Although AOM-treated M3R-deficient mice had fewer and smaller colon tumors than control WT mice, reductions in colon tumor number and size were not observed in M1R-deficient and dual M1R/M3R-deficient mice. Microarray and real-time PCR analyses revealed a possible role for zinc finger protein (Zfp) 277 expression in mediating these different phenotypes. However, the molecular mechanism underlying the MR-dependent regulation of Zfp 277 requires further investigation.
We demonstrated cholinergic MR activation augments murine intestinal epithelial cell proliferation and tumorigenesis in vivo.[46] Mice treated with the MR agonist bethanechol, provided in drinking water, had increased AOM-induced colon tumor numbers and size compared to AOM-treated mice drinking untreated water. Cell proliferation in both normal mucosa and adenocarcinomas was increased in bethanechol-treated compared to control mice. Also, in tumors, bethanechol treatment was associated with increased expression of M3R, EGFR and post-EGFR signaling molecules Myc and cyclin D1. Bethanechol treatment also increased normal colon mucosal thickness and stimulated expression of selected MMP genes, including MMP7, MMP10, and MMP13. These findings confirm that MR agonists are intestinal tumor promoters.
Cell migration and invasion
Cell migration is a key mechanism of cancer invasion. Using three distinct in vitro models, we showed that MR activation enhances cell migration and invasion. Using a soft agar colony formation assay, we showed that ACh enhanced anchorage- and MMP-dependent growth of H508 human CRC cells. In addition, in H508 and HT29 human CRC cells, using in vitro wound closure and Matrigel invasion models,[47] we showed that ACh treatment increased cell migration that was was blocked by inhibiting RhoA and Rho kinase, key proteins that interact with the actin cytoskeleton. Lastly, using an electrical cell impedance sensing invasion assay, we showed that ACh stimulated MMP1-dependent invasion of H508 cells.[48]
Conclusion
Muscarinic receptors are expressed in normal colon epithelial cells and overexpressed in colon tumors and colon cancer cell lines. Primary MR Ligands include ACh, deriving from both neuronal and non-neuronal tissues, and secondary bile acids. MR activation enhances colon cancer cell proliferation, cell migration, and invasion by transactivating EGFR, thereby initiating post-EGFR signaling [Figure 1]. MR antagonists and other agents that block MR activation or subsequent post-MR signal transduction have promise for CRC therapeutics.
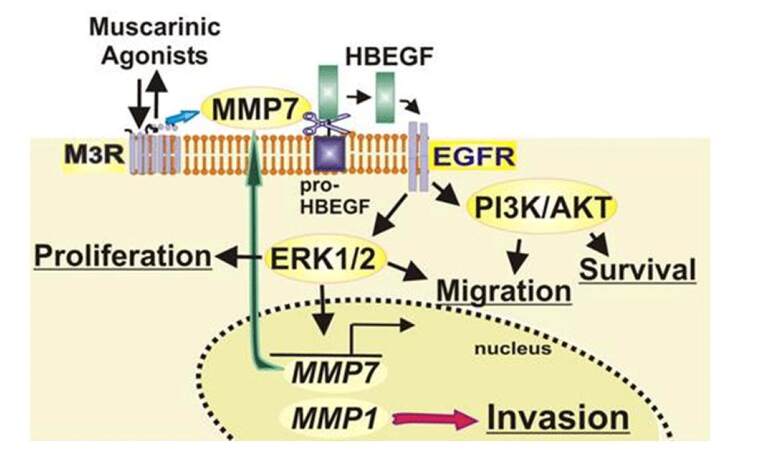
Figure 1. Muscarinic acetylcholine receptor subtype M3 (M3R) agonist-induced signaling in human colon cancer cells. Post-M3R matrix metalloproteinase 7 (MMP7) activation releases heparin binding epidermal growth factor (EGF) like growth factor (HBEGF), thereby activating EGF receptors (EGFR) signaling which promotes cell proliferation, survival and migration downstream extracellular signal-regulated kinase (ERK) activation induces MMP7, which replenishes MMP7 (green arrow), and MMP1, which promotes colon cancer cell invasion (red arrow)
Future directions
It is clear that muscarinic receptors and ligands play key roles in CRC progression. Unfortunately, there are no published translational and clinical studies at this time. Future studies focused on determining overexpression of muscarinic receptors in different patient cohorts, relation to CRC staging, tumor differentiation and prognosis will shed more lights in the importance of muscarinic receptor signaling in CRC.
Another important aspect of MR signaling in CRC is therapeutic potential of MR-based therapy. Because M1R and M3R are both expressed abundantly in the intestinal epithelium, buy have distinct functions,[45] it is critical to develop M1R- and M3R-specifc ligands, and test their pharmacological and therapeutic characteristics both in vitro and in vivo. These studies will lay important foundations for future human clinical trials that are focused on MR-based therapy. In addition, colon-specific drug delivery systems[49] should be utilized to reduce unwanted systemic side-effects.
Financial support and sponsorship
This work was supported by Merit Review Awards from the United States Department of Veterans Affairs Program (G. Xie and J. P. Raufman) and NIH R21-DK093406 and T32 DK067872 (J. P. Raufman).
Conflicts of interest
There are no conflicts of interest.
REFERENCES
1. Torre LA, Bray F, Siegel RL, Ferlay J, Lortet-Tieulent J, Jemal A. Global cancer statistics, 2012. CA Cancer J Clin 2015;65:87-108.
2. Siegel R, Desantis C, Jemal A. Colorectal cancer statistics, 2014. CA Cancer J Clin 2014;64:104-17.
4. Modjtahedi H, Essapen S. Epidermal growth factor receptor inhibitors in cancer treatment: advances, challenges and opportunities. Anticancer Drugs 2009;20:851-5.
5. Overman MJ, Hoff PM. EGFR-targeted therapies in colorectal cancer. Dis Colon Rectum 2007;50:1259-70.
6. Ilyas M. Wnt signalling and the mechanistic basis of tumour development. J Pathol 2005;205:130-44.
7. Wess J. Molecular biology of muscarinic acetylcholine receptors. Crit Rev Neurobiol 1996;10:69-99.
8. Wess J. Muscarinic acetylcholine receptor knockout mice: novel phenotypes and clinical implications. Annu Rev Pharmacol Toxicol 2004;44:423-50.
9. Shah N, Khurana S, Cheng K, Raufman JP. Muscarinic receptors and ligands in cancer. Am J Physiol Cell Physiol 2009;296:C221-32.
10. Spindel ER. Muscarinic receptor agonists and antagonists: effects on cancer. Handb Exp Pharmacol 2012;208:451-68.
12. Kruse AC, Kobilka BK, Gautam D, Sexton PM, Christopoulos A, Wess J. Muscarinic acetylcholine receptors: novel opportunities for drug development. Nat Rev Drug Discov 2014;13:549-60.
13. Frucht H, Gazdar AF, Park JA, Oie H, Jensen RT. Characterization of functional receptors for gastrointestinal hormones on human colon cancer cells. Cancer Res 1992;52:1114-22.
14. Frucht H, Jensen RT, Dexter D, Yang WL, Xiao Y. Human colon cancer cell proliferation mediated by the M3 muscarinic cholinergic receptor. Clin Cancer Res 1999;5:2532-9.
15. Yang WL, Frucht H. Cholinergic receptor up-regulates COX-2 expression and prostaglandin E(2) production in colon cancer cells. Carcinogenesis 2000;21:1789-93.
16. Grando SA. Biological functions of keratinocyte cholinergic receptors. J Investig Dermatol Symp Proc 1997;2:41-8.
17. Schlereth T, Birklein F, an Haack K, Schiffmann S, Kilbinger H, Kirkpatrick CJ, Wessler I. in vivo release of non-neuronal acetylcholine from the human skin as measured by dermal microdialysis: effect of botulinum toxin. Br J Pharmacol 2006;147:183-7.
18. Song P, Sekhon HS, Jia Y, Keller JA, Blusztajn JK, Mark GP, Spindel ER. Acetylcholine is synthesized by and acts as an autocrine growth factor for small cell lung carcinoma. Cancer Res 2003;63:214-21.
19. Tsuji S, Anglade P. Acetylcholine in neurons and paraneurons: a histochemical study. Arch Histol Cytol 1989;52:75-83.
20. Keely SJ. Epithelial acetylcholine -- a new paradigm for cholinergic regulation of intestinal fluid and electrolyte transport. J Physiol 2011;589:771-2.
21. Rosas-Ballina M, Olofsson PS, Ochani M, Valdés-Ferrer SI, Levine YA, Reardon C, Tusche MW, Pavlov VA, Andersson U, Chavan S, Mak TW, Tracey KJ. Acetylcholine-synthesizing T cells relay neural signals in a vagus nerve circuit. Science 2011;334:98-101.
22. Brandon EP, Lin W, D'Amour KA, Pizzo DP, Dominguez B, Sugiura Y, Thode S, Ko CP, Thal LJ, Gage FH, Lee KF. Aberrant patterning of neuromuscular synapses in choline acetyltransferase-deficient mice. J Neurosci 2003;23:539-49.
23. Klapproth H, Reinheimer T, Metzen J, Münch M, Bittinger F, Kirkpatrick CJ, Höhle KD, Schemann M, Racké K, Wessler I. Non-neuronal acetylcholine, a signalling molecule synthezised by surface cells of rat and man. Naunyn Schmiedebergs Arch Pharmacol 1997;355:515-23.
24. Cheng K, Samimi R, Xie G, Shant J, Drachenberg C, Wade M, Davis RJ, Nomikos G, Raufman JP. Acetylcholine release by human colon cancer cells mediates autocrine stimulation of cell proliferation. Am J Physiol Gastrointest Liver Physiol 2008;295:G591-7.
25. von Rosenvinge EC, Cheng K, Drachenberg CB, Fowler CB, Evers DL, Xie G, Raufman JP. Bedside to bench: role of muscarinic receptor activation in ultrarapid growth of colorectal cancer in a patient with pheochromocytoma. Mayo Clin Proc 2013;88:1340-6.
26. Raufman JP, Zimniak P, Bartoszko-Malik A. Lithocholyltaurine interacts with cholinergic receptors on dispersed chief cells from guinea pig stomach. Am J Physiol 1998;274:G997-1004.
27. Raufman JP, Chen Y, Cheng K, Compadre C, Compadre L, Zimniak P. Selective interaction of bile acids with muscarinic receptors: a case of molecular mimicry. Eur J Pharmacol 2002;457:77-84.
28. Cheng K, Khurana S, Chen Y, Kennedy RH, Zimniak P, Raufman JP. Lithocholylcholine, a bile acid/acetylcholine hybrid, is a muscarinic receptor antagonist. J Pharmacol Exp Ther 2002;303:29-35.
29. Cheng K, Chen Y, Zimniak P, Raufman JP, Xiao Y, Frucht H. Functional interaction of lithocholic acid conjugates with M3 muscarinic receptors on a human colon cancer cell line. Biochim Biophys Acta 2002;1588:48-55.
30. Cheng K, Raufman JP. Bile acid-induced proliferation of a human colon cancer cell line is mediated by transactivation of epidermal growth factor receptors. Biochem Pharmacol 2005;70:1035-47.
31. Cheng K, Zimniak P, Raufman JP. Transactivation of the epidermal growth factor receptor mediates cholinergic agonist-induced proliferation of H508 human colon cancer cells. Cancer Res 2003;63:6744-50.
32. Raufman JP, Chen Y, Zimniak P, Cheng K. Deoxycholic acid conjugates are muscarinic cholinergic receptor antagonists. Pharmacology 2002;65:215-21.
33. Cheng K, Xie G, Raufman JP. Matrix metalloproteinase-7-catalyzed release of HB-EGF mediates deoxycholyltaurine-induced proliferation of a human colon cancer cell line. Biochem Pharmacol 2007;73:1001-12.
34. Hamilton JP, Xie G, Raufman JP, Hogan S, Griffin TL, Packard CA, Chatfield DA, Hagey LR, Steinbach JH, Hofmann AF. Human cecal bile acids: concentration and spectrum. Am J Physiol Gastrointest Liver Physiol 2007;293:G256-63.
35. Winawer S, Fletcher R, Rex D, Bond J, Burt R, Ferrucci J, Ganiats T, Levin T, Woolf S, Johnson D, Kirk L, Litin S, Simmang C; Gastrointestinal Consortium Panel. Colorectal cancer screening and surveillance: clinical guidelines and rationale-update based on new evidence. Gastroenterology 2003;124:544-60.
36. Soler AP, Miller RD, Laughlin KV, Carp NZ, Klurfeld DM, Mullin JM. Increased tight junctional permeability is associated with the development of colon cancer. Carcinogenesis 1999;20:1425-31.
37. Flynn C, Montrose DC, Swank DL, Nakanishi M, Ilsley JN, Rosenberg DW. Deoxycholic acid promotes the growth of colonic aberrant crypt foci. Mol Carcinog 2007;46:60-70.
38. Dawson PA, Haywood J, Craddock AL, Wilson M, Tietjen M, Kluckman K, Maeda N, Parks JS. Targeted deletion of the ileal bile acid transporter eliminates enterohepatic cycling of bile acids in mice. J Biol Chem 2003;278:33920-7.
39. Raufman JP, Dawson PA, Rao A, Drachenberg CB, Heath J, Shang AC, Hu S, Zhan M, Polli JE, Cheng K. Slc10a2-null mice uncover colon cancer-promoting actions of endogenous fecal bile acids. Carcinogenesis 2015;36:1193-200.
40. Salomon DS, Brandt R, Ciardiello F, Normanno N. Epidermal growth factor-related peptides and their receptors in human malignancies. Crit Rev Oncol Hematol 1995;19:183-232.
41. Ozğul C, Karaöz E, Erdoğan D, Dursun A. Expression of epidermal growth factor receptor in normal colonic mucosa and in adenocarcinomas of the colon. Acta Physiol Hung 1997;85:121-8.
42. Xie G, Cheng K, Shant J, Raufman JP. Acetylcholine-induced activation of M3 muscarinic receptors stimulates robust matrix metalloproteinase gene expression in human colon cancer cells. Am J Physiol Gastrointest Liver Physiol 2009;296:G755-63.
43. Raufman JP, Samimi R, Shah N, Khurana S, Shant J, Drachenberg C, Xie G, Wess J, Cheng K. Genetic ablation of M3 muscarinic receptors attenuates murine colon epithelial cell proliferation and neoplasia. Cancer Res 2008;68:3573-8.
44. Raufman JP, Shant J, Xie G, Cheng K, Gao XM, Shiu B, Shah N, Drachenberg CB, Heath J, Wess J, Khurana S. Muscarinic receptor subtype-3 gene ablation and scopolamine butylbromide treatment attenuate small intestinal neoplasia in Apcmin/+ mice. Carcinogenesis 2011;32:1396-402.
45. Cheng K, Xie G, Khurana S, Heath J, Drachenberg CB, Timmons J, Shah N, Raufman JP. Divergent effects of muscarinic receptor subtype gene ablation on murine colon tumorigenesis reveals association of M3R and zinc finger protein 277 expression in colon neoplasia. Mol Cancer 2014;13:77.
46. Peng Z, Heath J, Drachenberg C, Raufman JP, Xie G. Cholinergic muscarinic receptor activation augments murine intestinal epithelial cell proliferation and tumorigenesis. BMC Cancer 2013;13:204.
47. Belo A, Cheng K, Chahdi A, Shant J, Xie G, Khurana S, Raufman JP. Muscarinic receptor agonists stimulate human colon cancer cell migration and invasion. Am J Physiol Gastrointest Liver Physiol 2011;300:G749-60.
48. Raufman JP, Cheng K, Saxena N, Chahdi A, Belo A, Khurana S, Xie G. Muscarinic receptor agonists stimulate matrix metalloproteinase 1-dependent invasion of human colon cancer cells. Biochem Biophys Res Commun 2011;415:319-24.
49. Philip AK, Philip B. Colon targeted drug delivery systems: a review on primary and novel approaches. Oman Med J 2010;25:79-87.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Xie G, Raufman JP. Muscarinic receptor signaling and colon cancer progression. J Cancer Metastasis Treat 2016;2:195-200. http://dx.doi.org/10.20517/2394-4722.2016.05
AMA Style
Xie G, Raufman JP. Muscarinic receptor signaling and colon cancer progression. Journal of Cancer Metastasis and Treatment. 2016; 2: 195-200. http://dx.doi.org/10.20517/2394-4722.2016.05
Chicago/Turabian Style
Xie, Guofeng, Jean-Pierre Raufman. 2016. "Muscarinic receptor signaling and colon cancer progression" Journal of Cancer Metastasis and Treatment. 2: 195-200. http://dx.doi.org/10.20517/2394-4722.2016.05
ACS Style
Xie, G.; Raufman J.P. Muscarinic receptor signaling and colon cancer progression. J. Cancer. Metastasis. Treat. 2016, 2, 195-200. http://dx.doi.org/10.20517/2394-4722.2016.05
About This Article
Copyright

Data & Comments
Data


 Cite This Article 3 clicks
Cite This Article 3 clicks

 Like This Article 0
likes
Like This Article 0
likes






Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.