
Pancreatic neuroendocrine tumor with hypoglycemia and elevated insulin-like growth factor II: a case report
 0
0
Abstract
Pancreatic neuroendocrine tumors (pNETs) can be associated with different clinical syndromes. Insulinoma is the most common functioning pNET characterized by hypoglycemia and hyperinsulinemia. The authors report a case of a man presenting with hypoglycemia and biochemical features of insulinoma. A pancreatic lesion was found and growth hormone (GH) deficiency was also diagnosed associated with an empty sella present on the pituitary magnetic resonance imaging. The disappearance of hypoglycemia and normalization of GH secretion after surgical resection of the pancreatic lesion, revealed a rare pNET secreting insulin-like growth factor II.
Keywords
Introduction
Pancreatic neuroendocrine tumors (pNETs) represent 1-2% of all pancreatic tumors and 7% of NETs in general, with an incidence of 0.43 per 100,000. Epidemiological data show that pNET incidence is increasing, perhaps due to more widespread use of diagnostic imaging techniques, especially computed tomography (CT) scans, and increased physician awareness of this tumor type. Moreover, a high prevalence of pNETs is reported in autopsy studies (from 0.8% to 10%), thus suggesting that they are frequently clinically silent. A slight male predominance (55% male vs. 45% female) is reported and the median age at presentation is around 50 years.[1]
pNETs may be sporadic or part of a genetic syndrome, most commonly multiple endocrine neoplasia type 1 (MEN1), von Hippel-Lindau disease (VHL), neurofibromatosis type I (NF), or tuberous sclerosis complex (TSC). Clinically pNETs can be distinguished into two groups: functional (F-pNET) and nonfunctional (NF-pNET). The majority of pNETs are non-functional (90%) and present with symptoms due to mass effect or as incidental findings, whereas F-pNETs (10%) are characterized by hormone hypersecretion with different clinical signs and symptoms. F-pNET are distinguished according to the clinical hormonal syndrome and the hormone hypersecreted: insulinoma, gastrinoma (Zollinger-Ellison syndrome), glucagonoma, VIPoma, GHRFoma (growth hormone releasing factor secreting), ACTHoma, and somatostatinoma. Among F-pNETs, insulinoma is the most common with an estimated annual incidence of 1-4 per million patients, representing 35-40% of all F-pNETs.[1-3] Although rare, insulinoma represents the most common cause of hypoglycemia related to endogenous hyperinsulinemia, characterized by inappropriately high insulin and/or proinsulin and high C-peptide concentrations. The presence of hypoglycemia together with a pancreatic lesion is usually the clinical picture of insulinoma. Nevertheless hypoglycemia represents a relatively common biochemical finding, which may be due to many causes, thus a careful clinical history, together with biochemical and radiological tests, is essential to identify the underlying cause. Other subtypes of F-pNETs have been reported, although rarely. Diagnosis of F-pNET can be challenging, as clinical presentation may simulate other more common diseases, thus causing delay in diagnosis. Of note, the hormone-excess state in F-pNET requires both acute and long term control, since it represents a potential life threatening condition along with the treatment of the pNET itself.[4,5]
Case report
A 64-year-old man presented at our institution because of clinical findings suggestive of hypoglycemia. Past medical and family histories were unremarkable, except for arterial hypertension controlled with angiotensin-converting enzyme inhibitors; body mass index was 30 kg/m2. Hypoglycemic episodes had begun 2 years before hospitalization and were initially characterized by anxiety, irritability, sweating, palpitations and hunger. Since then, the patient had experienced a progressive worsening of symptoms, complaining of blurred vision, nausea, temporary amnesia, and episodic disorientation that took place mainly in the morning and disappeared after eating. Soon after hospitalization, hypoglycemic episodes occurred and hypoglycemia was biochemically confirmed with an average 7 h serum glucose concentration of 45 mg/dL (normal range 80-120 mg/dL). Hypoglycemic episodes fulfilled the Whipple’s triad, characterized by signs and symptoms of hypoglycemia, evidence of low plasma glucose (< 55 mg/dL) concentration and resolution of signs and symptoms after glucose administration. Liver, renal and thyroid profiles were within the normal limits. An insulinoma was suspected and a 72 h fasting test was performed with assessment of glycemia at the beginning and every 4 h. Serum insulin and C-Peptide concentrations were also assessed at the beginning and in case of biochemical and/or clinical hypoglycemia. Serum concentrations of glucose, insulin and C-peptide were measured by standard methods by using commercially available kits. During the test hypoglycemia occurred after 9 h (glucose 40 mg/dL). However, the insulin/glucose ratio was 0.1, revealing an appropriate insulin secretion. Moreover, a focal lesion within the pancreas was detected by endoscopic ultrasound (EUS), therefore an insulinoma was suspected. However, the evaluation of pituitary function with growth hormone-releasing hormone (GHRH) plus arginine test pointed out a growth hormone (GH) deficiency and magnetic resonance imaging (MRI) of the pituitary region revealed a partial empty sella. No other pituitary abnormalities were observed. An abdominal contrast-enhanced CT confirmed a nodular area of 18 mm × 12 mm in the body of pancreas, with altered contrast enhancement. An 111In-DTPA-D-Phe1 octreotide scintigraphy (Octreoscan) highlighted a focal epigastric uptake, corresponding to the pancreatic nodule. Surprisingly a EUS-guided fine-needle biopsy of the pancreatic lesion resulted in a cytological diagnosis of moderately differentiated adenocarcinoma. Therefore, the patient underwent surgery. Histology and immunohistochemistry of the specimen revealed a well-differentiated pNET, with Ki67 index of 1%. Immunostaining for chromogranin-A and synaptophysin was positive, while insulin immunostaining was negative. Postoperative course was uneventful and a progressive disappearance of the hypoglycemic syndrome occurred. Six months after surgery pituitary function was evaluated and only partial GH deficiency was evident. The GHRH plus arginine test was performed using GHRH (Ferring, Malmo, Sweden; 1 μg/kg, iv, at 0 min) and arginine-hydrochloride (0.5 g/kg, iv, during the first 30 min) with assessment of serum GH concentrations at times 0, 30, 45, 60, 90, 120 min. The GH peak during test was 13.6 ng/mL. Twelve months after surgery, GH response to stimulation was normal [GH peak 30.8 ng/mL; Figure 1], although the empty sella on MRI was unchanged. This led to the hypothesis that the pancreatic tumor may have been secreting insulin-like growth factor II (IGF-II), since IGF-II may suppress GH secretion with a negative feedback. To test this hypothesis IGF-II concentrations were measured on plasma collected before and after pancreatic surgery. IGF-II was assessed by using an ELISA, “two-step” sandwich type immunoassay. Before surgery, plasma IGF-II was 920 ng/mL and one month after surgery, it had decreased to 320 ng/mL (normal range 108-881 ng/mL). To further confirm these findings, immunohistochemical staining was performed for IGF-II and the pancreatic tumor specimen was positive for IGF-II. These findings were consistent with the diagnosis of a pancreatic IGF-II-secreting tumor. The patient did not experience any other hypoglycemic symptoms during follow up and completely recovered after surgery.
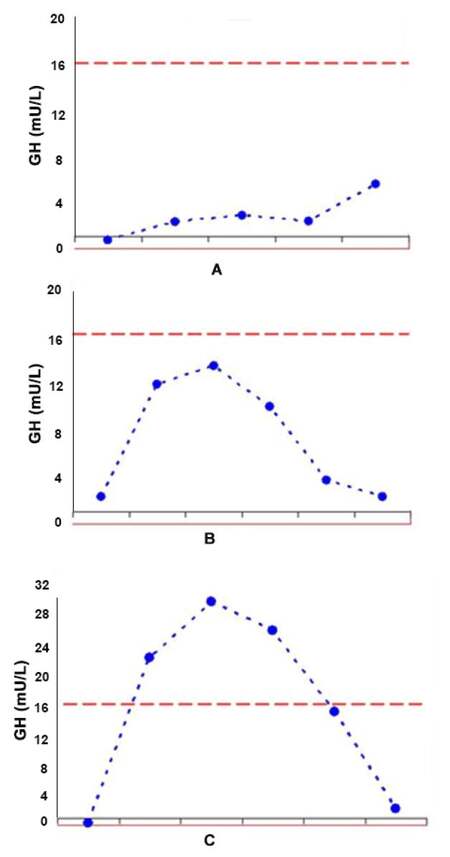
Figure 1. GHRH + Arginine test for GH: (A) basal test revealing total GH deficiency; (B) 6-month postoperative test revealing partial deficiency; (C) 12-month postoperative test revealing normal GH response. GHRH: growth hormone-releasing hormone; GH: growth hormone
Discussion
Hypoglycemia represents a relatively common biochemical finding, which may be due to many causes, such as non-islet cell tumor, drugs, organ failure, endocrine diseases, hypopituitarism, or inborn errors of metabolism. A careful clinical history, together with biochemical and radiological assessments is essential to identify the underlying cause. Although rare, insulinoma is the most frequent F-pNET. Biochemical criteria for insulinoma comprise documented hypoglycemia (plasma glucose ≤ 55 mg/dL), concomitant inappropriately high plasma insulin ≥ 3 mU/mL, C-peptide ≥ 0.6 ng/mL (≥ 0.2 nmol/L), proinsulin levels (≥ 5 pmol/L), and no detectable hypoglycemic agent levels or circulating antibodies to insulin.[6,7] The 72 h fasting test is considered the gold standard for diagnosis of insulinoma. In the present case the occurrence of hypoglycemia together with a pancreatic lesion lead to suspect an insulinoma.
Hypoglycemia may also occur in large tumors of mesenchymal, epithelial, or hematopoietic origin.[8] These tumors often secrete incompletely processed IGF-II, a hormone with higher molecular weight, capable of activating the insulin receptor, thus causing hypoglycemia with consequent suppression of β cell secretion, lipolysis and ketogenesis. The IGF-II in serum is usually synthesized in the liver and then it is processed into a mature form that is secreted. The incompletely processed IGF-II is a smaller complex that can interact with insulin receptors in the liver, muscle, and adipocytes, leading to suppression of GH and insulin secretion.
Incompletely processed IGF-II affects the hypothalamic-pituitary axis suppressing GH secretion with a negative feedback, with subsequent lowering of GH-dependent IGF-I and IGF binding proteins secreted by the liver. Therefore, tumors secreting incompletely processed IGF-II are characterized by an increased total IGF-II to IGF-I ratio, suppressed insulin and C peptide, and inappropriately low GH.[9] The production of IGF-II represents a very rare cause of hypoglycemia. To date this is the second case reported of hypoglycemia due to production of IGF-II by a pNET.[10] This unusual case highlights the importance of taking into account the production of IGF-II in case of hypoglycemia and pancreatic lesion when clinical, biochemical, and immunohistochemical data are not consistent with insulinoma. In our patient, the finding of empty sella could have justified GH deficiency, so IGF-II was not immediately evaluated. Although insulin was not suppressed in our case, the finding of negative insulin and positive IGF-II at immunostaining support the hypothesis of an IGF-II secreting tumor. Furthermore the prompt resolution of signs and symptoms of hypoglycemia soon after the resection of the pNET may be attributed to the normalization of serum IGF-II levels.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
Patient consent
Obtained.
Ethics approval
The patient was treated within the standards of our institute and the report was approved.
REFERENCES
1. McKenna LR, Edil BH. Update on pancreatic neuroendocrine tumors. Gland Surg 2014;3:258-75.
2. Fraenkel M, Kim M, Faggiano A, de Herder WW, Valk GD, Knowledge NETwork. Incidence of gastroenteropancreatic neuroendocrine tumours: a systematic review of the literature. Endocr Relat Cancer 2014;21:153-63.
3. Okabayashi T, Shima Y, Sumiyoshi T, Kozuki A, Ito S, Ogawa Y, Kobayashi M, Hanazaki K. Diagnosis and management of insulinoma. World J Gastroenterol 2013;19:829-37.
4. Service FJ, McMahon MM, O'Brien PC, Ballard DJ. Functioning insulinoma – incidence, recurrence, and long-term survival of patients: a 60-year study. Mayo Clin Proc 1991;66:711-9.
5. De Herder WW, Niederle B, Scoazec JY, Pauwels S, Klöppel G, Falconi M, Kwekkeboom DJ, Öberg K, Eriksson B, Wiedenmann B, Rindi G, O'Toole D, Ferone D and all other Frascati Consensus Conference participants. Well-Differentiated Pancreatic Tumor/Carcinoma: Insulinoma. Neuroendocrinology 2006;84:183-8.
6. Cryer PE, Axelrod L, Grossman AB, Heller SR, Montori VM, Seaquist ER, Service FJ. Evaluation and management of adult hypoglycemic disorders: an endocrine society clinical practice guideline. J Clin Endocrinol Metab 2009;94:709-28.
9. Marks V, Teale JD. Glucocorticoid therapy suppresses abnormal secretion of big IGF-II by non-islet cell tumours inducing hypoglycaemia (NICTH). Clin Endocrinol (Oxf) 1998;49:491-8.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Modica R, Sarno AD, Colao A, Faggiano A. Pancreatic neuroendocrine tumor with hypoglycemia and elevated insulin-like growth factor II: a case report. J Cancer Metastasis Treat 2016;2:345-7. http://dx.doi.org/10.20517/2394-4722.2016.44
AMA Style
Modica R, Sarno AD, Colao A, Faggiano A. Pancreatic neuroendocrine tumor with hypoglycemia and elevated insulin-like growth factor II: a case report. Journal of Cancer Metastasis and Treatment. 2016; 2: 345-7. http://dx.doi.org/10.20517/2394-4722.2016.44
Chicago/Turabian Style
Modica, Roberta, Antonella Di Sarno, Annamaria Colao, Antongiulio Faggiano. 2016. "Pancreatic neuroendocrine tumor with hypoglycemia and elevated insulin-like growth factor II: a case report" Journal of Cancer Metastasis and Treatment. 2: 345-7. http://dx.doi.org/10.20517/2394-4722.2016.44
ACS Style
Modica, R.; Sarno AD.; Colao A.; Faggiano A. Pancreatic neuroendocrine tumor with hypoglycemia and elevated insulin-like growth factor II: a case report. J. Cancer. Metastasis. Treat. 2016, 2, 345-7. http://dx.doi.org/10.20517/2394-4722.2016.44
About This Article
Special Issue
Copyright

Data & Comments
Data
0

 Cite This Article 9 clicks
Cite This Article 9 clicks

 Like This Article 0
likes
Like This Article 0
likes






Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.