
A new twist to neurotransmitter receptors and cancer


Background
Nicotinic acetylcholine receptors (nAChRs) and beta-adrenergic receptors (β-ARs) are cell membrane receptors expressed in most mammalian cells where they function as the recipients of signals from the autonomic nervous system that maintains physiological homeostasis in the mammalian organism and regulates cell and organ responses to endogenous and exogenous signals. The neurotransmitter of the parasympathetic branch of the autonomic nervous system, acetylcholine, binds as an agonist to all members of the nAChR family, thus opening the ligand-gated ion channel of the receptors. The resulting depolarization of the cell membrane opens voltage-gated Ca2+-channels (VOCs), allowing influx of additional Ca2+ that triggers the release of cell type-specific intracellular products via exocytosis.[1] Influx of Ca2+ is particularly high in response to agonist binding to the homomeric (comprised of alpha subunits only) α7nAChR due to the selectivity of its ion channel for Ca2+ whereas heteromeric (comprised of alpha and non-alpha subunits) nAChRs have non-selective ion channels. The mechanisms of nAChR-mediated neurotransmitter release by the central and peripheral nervous system, their role in memory, cognition and stress responses and the nAChR-mediated mechanisms of nicotine addiction have been extensively studied.[1,2]
Beta-adrenergic receptors are coupled to the stimulatory G-protein Gs that activates the enzyme adenylyl cyclase (AC) upon binding of an agonist to the receptor, leading to the formation of intracellular cyclic adenosine monophosphate (cAMP) that activates protein kinase A (PKA) and numerous PKA-dependent and independent intracellular signaling cascades in a cell type-specific manner.[3] In addition, β1 and β2-ARs can increase intracellular Ca2+ levels by a variety of mechanisms [Figure 1], including the PKA-induced upregulation of L-type Ca2+-channels[4] and release of Ca2+ from intracellular stores that can also be induced by the cAMP binding protein exchange factor directly activated by cAMP (Epac).[5] Of particular importance for the regulation of cancer cells is the fact that activated PKA and/or cAMP stimulate the release of epidermal growth factor (EGF),[6] arachidonic acid (AA),[7,8] interleukins and vascular endothelial growth factor (VEGF),[9] which jointly stimulate the development, progression and metastasis of numerous cancers. The neurotransmitters of the sympathetic branch of the autonomic nervous system, epinephrine (Epi) and norepinephrine (Nor) are the physiological agonists for β-ARs. Epi and Nor are additionally synthesized and released by the adrenal medulla and are often referred to as “stress neurotransmitters” because psychological stress triggers their simultaneous release from the sympathetic nervous system and adrenal gland.[10,11] The release of stress neurotransmitters from the sympathetic nervous system and adrenal gland is regulated by nAChRs via Ca2+ influx that triggers their exocytosis.[12,13] The biology of β-ARs as it relates to cardio-vascular disease has been extensively studied and beta-adrenergic receptor antagonists (beta-blockers) and VOC blockers are widely used as therapeutics for this disease complex.[14-17]
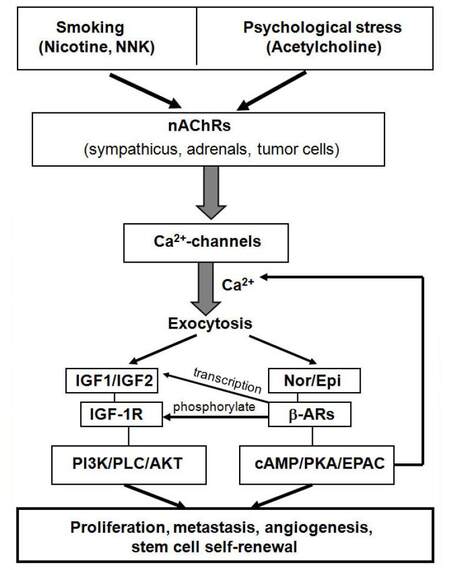
Figure 1. Working model illustrating the mechanistic interactions of nicotinic acetylcholine receptors, Ca2+-channels, beta-adrenergic receptors and the IGF pathway in cancers associated with smoking and psychological stress. NNK: nicotine-derived nitrosamine ketone; nAChRs: nicotinic acetylcholine receptors; IGF-1R: insulin-like growth factor-1 receptor; PLC: phospholipase C; AKT: protein kinase B; cAMP: cyclic adenosine monophosphate; PKA: protein kinase A
Discoveries that nicotine induced the proliferation of human small cell lung cancer cells in vitro[18] while inhibiting apoptosis,[19] effects triggered by the nAChR-mediated release and re-uptake of the neurotransmitter 5-hydroxytryptamine (5-HT, serotonin),[20] first pointed to nAChRs as important regulators of a subset of cancers. Reports that β-AR agonists stimulated the proliferation of lung adenocarcinoma cells in vitro and that this response was inhibited by β-blockers first implicated β-ARs in the regulation of another subset of cancers.[21,22] The identification of the tobacco carcinogen nicotine-derived nitrosamine ketone (NNK) as a high affinity agonist for nAChRs[23,24] as well as β-ARs[7] subsequently provided a direct mechanistic link between the high carcinogenic potential of this agent and its interaction with neurotransmitter receptors. These studies also showed that NNK-induced β-AR signaling in lung adenocarcinoma cells and pancreatic ductal adenocarcinoma cells triggered the release of AA, resulting in the formation of cancer-stimulating AA metabolites while additionally trans-activating the epidermal growth factor receptor pathway.[7,8,25] Collectively, these early findings represented the starting point for a new domain in cancer research: the role of neurotransmitters and their receptors in the initiation, progression and drug resistance of cancer and the development of novel therapeutic and preventive strategies that target this regulatory network.[26-30]
It was initially thought that nAChRs and β-ARs expressed in non-neuronal cells and cancers derived from them were exclusively stimulated by the autonomic nervous system or by exposure to tobacco products. However, more recent studies have shown that numerous non-neuronal cells and the cancers derived from them synthesize and release their own acetylcholine[31] and are also able to synthesize and release Nor and Epi in response to acetylcholine self-stimulation or exposure to exogenous nAChR agonists.[32-36] In addition, it has been shown that polymorphisms in genes CHRNA3 (encodes the α3 nAChR subunit) and CHRNA5 (encodes the α5 nAChR subunit) as well as a copy number variation that duplicates the α7nAChR gene CHRNA7 are associated with an increased risk for lung cancer[37-39] and that single nucleotide polymorphisms in the β2-AR gene are associated with adverse clinical outcomes of pancreatic cancer.[40]
An important aspect of cancer regulation by neurotransmitters and their receptors is the significant influence of the mood on this regulatory network [Figure 1]. Preclinical investigations have thus shown that experimentally induced psychological stress or treatment with stress neurotransmitters have strong promoting effects on the majority of the most common human cancers via direct activation of cAMP-dependent intracellular signaling pathways by stress neurotransmitters downstream of β1 and β2-ARs[29,32,41-45] and the simultaneous suppression of the tumor suppressor gene p53 by beta-arrestin-1 signaling downstream of β2-ARs.[46] Moreover, chronic experimental stress suppressed the synthesis and release of the inhibitory neurotransmitter γ-aminobutyric acid (GABA).[41,42] These findings are in accord with the reported suppression of the GABA system by chronic psychological stress[47] and in anxiety disorders such as posttraumatic stress syndrome.[48,49] GABA is the main inhibitory neurotransmitter in the mammalian body and inhibits the AC-dependent formation of cAMP as well as the activation of voltage-gated Ca2+-channels[50] under physiological conditions by activating inhibitory G-protein (Gi) signaling downstream of Gi-coupled GABA-B-receptors. In light of findings that the GABA-B receptor has tumor suppressor function in pancreatic[42,51-53] and non small-cell lung cancer (NSCLC)[35,41,54,55] while GABA also inhibits the in vitro growth of breast cancer and colon cancer,[47,56] suppression of GABA by psychological stress has significant tumor promoting effects on these cancers.
Similar to chronic stress, smoking also increases the levels of cancer stimulating stress neurotransmitters[57] while suppressing cancer inhibiting GABA,[2] effects caused by the neuroadaptation of nAChRs to chronic nicotine, NNK and N’-nitrosonornicotine (NNN). In conjunction with the mutational activities of NNK and NNN at the K-ras and p53 genes,[58] the resulting prevalence of cancer stimulating beta-adrenergic receptor signaling contributes significantly to the increased cancer risk of smokers.
Novel findings
Three publications[59-61] from the Research Institute of Pharmacological Sciences, College of Pharmacy, Seoul National University (Seoul, Republic of Korea) have recently revealed additional mechanisms of nAChR and β-AR-mediated lung cancer promotion that can potentially be exploited for the targeted prevention and therapy of lung cancer and numerous other cancers. These studies showed that NSCLC tissues from smokers expressed significantly higher levels of the phosphorylated insulin-like growth factor-1 receptor (IGF-1R) than NSCLCs from nonsmokers and that the nicotine-derived carcinogen NNK promoted NSCLC tumorigenesis in vitro and in a mouse model by inducing exocytosis of insulin-like growth factor 2 (IGF-2) that phosphorylated the IGF-1 receptor, effects inhibited by the neuronal nAChR antagonist mecamylamine, dyhydropyridine blockers of L-type VOCs as well as by antagonists for β1-and β2-ARs.[59] The investigators reported that the observed IGF-1R phosphorylation was caused by β-AR-mediated stimulation of IGF2 transcription.[61] However, the molecular mechanisms of this effect have yet to be defined. Based on the inhibitory effects of mecamylamine and VOC blockers on EGF-1R phosphorylation, nAChRs were the upstream regulators of this β-adrenergic cascade by stimulating the release of Nor and Epi. In accord with established mechanisms of stress responses (nAChR-mediated opening of VOCs causing release of stress neurotransmitters by exocytosis from the sympathetic nervous system and adrenal glands), experimental chronic stress had significant tumor promoting effects on urethane-induced mouse NSCLC and on the development of this cancer type in transgenic KrasG12D/+ mice via IGF-2-mediated activation of the IGF-1R signaling cascade.[60,61] In both animal models these effects were inhibited by the general beta-blocker propranolol or the dihydropyridine VOC blockers amlodipine or nifedipine. Propranolol also significantly prevented the development of NNK-induced lung tumors in A/J mice, an effect accompanied by suppression of phosphorylated IGF-1R.[61] The authors conclude that beta-blockers and VOC blockers should be further explored for the prevention of lung cancer, a concept that could rapidly move into clinical trials because these drugs are already widely used for the long-term management of cardiovascular disease.
Conclusions and future directions
The reported activation of the IGF-1R signaling cascade in NSCLC and their normal epithelial precursor cells by the joint actions of nAChRs, VOCs and β-ARs adds a novel aspect to the mechanisms of cancer regulation by neurotransmitter receptors. While cancer research on the regulatory function of these receptors has mostly interpreted their modulation of intracellular signaling pathways as direct events downstream of the receptors,[20,24,30,45,62,63] the cited three publications[59-61] instead take into consideration the physiological role of nAChRs and β-ARs in the release of cell type-specific products by exocytosis [Figure 1] in response to increased intracellular Ca2+. In addition to IGF-2, β-AR-I agonists also induced the release of AA, EGF, VEGF, interleukin-6 as well as several cancer stem cell markers.[36,64-66] In turn, these effects can be caused by elevated systemic levels of stress neurotransmitters in response to stress or tobacco exposure, by direct binding of NNK in tobacco products to β-ARs, or by medications that are beta-adrenergic agonists. In addition, epithelial cancer cells and their respective cancer stem cells synthesize and release their own Epi and Nor upon activation of nAChRs by nicotine or nicotine-derived nitrosamines.[33,36] The proposed re-purposing of beta-blockers and Ca2+-channel blockers for lung cancer prevention would therefore inhibit differentiated cancer cells as well as cancer stem cells.
There is an ongoing international discussion on the potential usefulness of beta-blockers for cancer intervention, with numerous preclinical studies reporting significant cancer inhibition whereas clinical investigations have generated controversial data with some even reporting cancer promoting effects.[28,29,64,67-71] The potential usefulness of beta-blockers for adjuvant cancer treatment has additionally been discussed in depth based by comprehensive reviews of published preclinical and clinical literature.[67,72,73] By contrast, the current review analyzes mechanistic aspects of Gs-coupled receptors and their physiological inhibitors and their modulating effects on cancer. The discrepancies between preclinical and clinical findings are thus not only triggered by the potential sensitization of β-ARs in response to long-term beta-blocker therapy (decades of treatment in people as opposed to a few weeks in experimental animals), but also by the potential impact of factors unrelated to β-ARs. Preclinical studies that have employed agonists of receptors coupled to the inhibitory G-protein Gi (GABA-B receptors, opioid peptide receptors) for the inhibition of β-AR-mediated progression of adenocarcinoma of the lungs and pancreas in vitro and in vivo have repeatedly shown that increases in intracellular cAMP and the associated activation of its downstream effectors are key molecular events that activate β-AR-driven development and progression of both cancers and can be successfully inhibited by agonists of Gi-coupled receptors that inhibit the formation of cAMP by blocking the activation of adenylyl cyclase.[41,42,51,54,55,66,74-76] A host of non-β-AR receptors coupled to the stimulatory G-protein Gs increase intracellular cAMP,[3,77] a reaction not inhibited by beta-blockers but effectively counteracted by agonist-induced signaling of Gi-coupled receptors. There is also a host of non-beta-adrenergic agents that increase intracellular cAMP directly. Among such agents are caffeine, theophylline and theobromine contained in numerous beverages, weight loss medications, sweets and candies. These naturally occurring phosphodiesterase inhibitors block the enzymatic breakdown of cAMP which then accumulates inside the cells. In addition, pharmacological phosphodiesterase inhibitors are widely used for the therapy of chronic obstructive pulmonary disease because of their anti-inflammatory and broncho-dilating properties. None of the clinical investigations on beta-blockers and cancer conducted to date have adjusted their data to exclude the cancer promoting effects of such non-beta-adrenergic agents.
Beta-blockers should not be used for the general prevention/therapy of cancer because they are selectively effective only in cancers that are stimulated by beta-adrenergic agonists. In fact, without prior testing of patients for increased stress neurotransmitter and cAMP levels, beta-blocker treatment is contra-indicated because it can promote certain cancers due to the fact that cAMP functions as a tumor promoter in some cancers while acting as a tumor suppressor in others. It has thus been shown that cAMP inhibits the growth/progression of squamous cell carcinoma,[78] small cell lung carcinoma,[79,80] medulloblastoma and basal cell carcinoma.[81] The arbitrary use of Ca2+-channel blockers for cancer prevention and therapy is equally ill advised. While preclinical investigations have identified cancer preventive effects of Ca2+-channel blockers in a large spectrum of cancers,[82-84] these agents not only suppress molecular targets studied in these cancers but additionally inhibit the release of Nor and Epi from sympathetic nerves,[85] thereby suppressing the beta-adrenergic receptor-mediated formation of cAMP. In turn, this effect can selectively promote the development and progression of cancers in which cAMP has tumor suppressor function.
In summary, successful cancer prevention and improved therapeutic outcomes can be achieved by strategies that aim to maintain/restore cAMP homeostasis. Too much cAMP will promote the development and progression of cAMP-driven cancers (e.g. adenocarcinoma of the lungs, pancreas, colon, stomach and prostate) while too low cAMP levels will increase the risk for development and progression of cancers in which cAMP has tumor suppressor function (e.g. small cell lung cancer, squamous cell carcinoma, medulloblastoma, basal cell carcinoma). In analogy to the long-term management of diabetes by insulin injections that are based on blood glucose testing, this approach requires routine testing of cAMP levels. Beta-blockers will only be beneficial if elevated levels of Nor/Epi indicate hyperactive β-AR signaling.
Authors’ contributions
H.M. Schuller contributed solely to the paper.
Financial support and sponsorship
None.
Conflicts of interest
There are no conflicts of interest.
Patient consent
Not applicable.
Ethics approval
Not applicable.
REFERENCES
1. Sudhof TC. Calcium control of neurotransmitter release. Cold Spring Harb Perspect Biol 2012;4:a011353.
2. D'Souza MS. Neuroscience of nicotine for addiction medicine: novel targets for smoking cessation medications. Prog Brain Res 2016;223:191-214.
3. Lefkowitz RJ. Seven transmembrane receptors: something old, something new. Acta Physiol (Oxf) 2007;190:9-19.
4. Reuter H. Calcium channel modulation by beta-adrenergic neurotransmitters in the heart. Experientia 1987;43:1173-5.
5. Yang Z, Kirton HM, MacDougall DA, Boyle JP, Deuchars J, Frater B, Ponnambalam S, Hardy ME, White E, Calaghan SC, Peers C, Steele DS. The Golgi apparatus is a functionally distinct Ca2+ store regulated by the PKA and Epac branches of the beta1-adrenergic signaling pathway. Sci Signal 2015;8:ra101.
6. Grau M, Soley M, Ramirez I. Interaction between adrenaline and epidermal growth factor in the control of liver glycogenolysis in mouse. Endocrinology 1997;138:2601-9.
7. Schuller HM, Tithof PK, Williams M, Plummer H 3rd. The tobacco-specific carcinogen 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone is a beta-adrenergic agonist and stimulates DNA synthesis in lung adenocarcinoma via beta-adrenergic receptor-mediated release of arachidonic acid. Cancer Res 1999;59:4510-5.
8. Weddle DL, Tithoff P, Williams M, Schuller HM. Beta-adrenergic growth regulation of human cancer cell lines derived from pancreatic ductal carcinomas. Carcinogenesis 2001;22:473-9.
9. Madden KS, Szpunar MJ, Brown EB. beta-Adrenergic receptors (beta-AR) regulate VEGF and IL-6 production by divergent pathways in high beta-AR-expressing breast cancer cell lines. Breast Cancer Res Treat 2011;130:747-58.
10. Ziegler MG, Milic M. Sympathetic nerves and hypertension in stress, sleep apnea, and caregiving. Curr Opin Nephrol Hypertens 2017;26:26-30.
11. McEwen BS. The neurobiology of stress: from serendipity to clinical relevance. Brain Res 2000;886:172-89.
12. Sala F, Nistri A, Criado M. Nicotinic acetylcholine receptors of adrenal chromaffin cells. Acta Physiol (Oxf) 2008;192:203-12.
13. Li YF, LaCroix C, Freeling J. Specific subtypes of nicotinic cholinergic receptors involved in sympathetic and parasympathetic cardiovascular responses. Neurosci Lett 2009;462:20-3.
14. Szentmiklosi AJ, Szentandrassy N, Hegyi B, Horvath B, Magyar J, Banyasz T, Nanasi PP. Chemistry, physiology, and pharmacology of beta-adrenergic mechanisms in the heart. Why are beta-blocker antiarrhythmics superior? Curr Pharm Des 2015;21:1030-41.
15. Ferguson SS, Feldman RD. Beta-adrenoceptors as molecular targets in the treatment of hypertension. Can J Cardiol 2014;30:S3-8.
16. Cameron AC, Lang NN, Touyz RM. Drug treatment of hypertension: focus on vascular health. Drugs 2016;76:1529-50.
17. Fares H, DiNicolantonio JJ, O'Keefe JH, Lavie CJ. Amlodipine in hypertension: a first-line agent with efficacy for improving blood pressure and patient outcomes. Open Heart 2016;3:e000473.
18. Schuller HM. Cell type specific, receptor-mediated modulation of growth kinetics in human lung cancer cell lines by nicotine and tobacco-related nitrosamines. Biochem Pharmacol 1989;38:3439-42.
19. Maneckjee R, Minna JD. Opioid and nicotine receptors affect growth regulation of human lung cancer cell lines. Proc Natl Acad Sci U S A 1990;87:3294-8.
20. Cattaneo MG, Codignola A, Vicentini LM, Clementi F, Sher E. Nicotine stimulates a serotonergic autocrine loop in human small-cell lung carcinoma. Cancer Res 1993;53:5566-8.
21. Schuller HM, Cole B. Regulation of cell proliferation by beta-adrenergic receptors in a human lung adenocarcinoma cell line. Carcinogenesis 1989;10:1753-5.
22. Park PG, Merryman J, Orloff M, Schuller HM. Beta-adrenergic mitogenic signal transduction in peripheral lung adenocarcinoma: implications for individuals with preexisting chronic lung disease. Cancer Res 1995;55:3504-8.
23. Schuller HM, Orloff M. Tobacco-specific carcinogenic nitrosamines. Ligands for nicotinic acetylcholine receptors in human lung cancer cells. Biochem Pharmacol 1998;55:1377-84.
24. Arredondo J, Chernyavsky AI, Grando SA. Nicotinic receptors mediate tumorigenic action of tobacco-derived nitrosamines on immortalized oral epithelial cells. Cancer Biol Ther 2006;5:511-7.
25. Askari MD, Tsao MS, Schuller HM. The tobacco-specific carcinogen, 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone stimulates proliferation of immortalized human pancreatic duct epithelia through beta-adrenergic transactivation of EGF receptors. J Cancer Res Clin Oncol 2005;131:639-48.
26. Schuller HM. Neurotransmission and cancer: implications for prevention and therapy. Anticancer Drugs 2008;19:655-71.
27. Schuller HM. Is cancer triggered by altered signalling of nicotinic acetylcholine receptors? Nat Rev Cancer 2009;9:195-205.
28. Tang J, Li Z, Lu L, Cho CH. β-Adrenergic system, a backstage manipulator regulating tumour progression and drug target in cancer therapy. Semin Cancer Biol 2013;23:533-42.
29. Chang A, Kim-Fuchs C, Le CP, Hollande F, Sloan EK. Neural regulation of pancreatic cancer: a novel target for intervention. Cancers (Basel) 2015;7:1292-312.
30. Cesario A, Russo P, Nastrucci C, Granone P. Is alpha7-nAChR a possible target for lung cancer and malignant pleural mesothelioma treatment? Curr Drug Targets 2012;13:688-94.
31. Wessler I, Kirkpatrick CJ. Acetylcholine beyond neurons: the non-neuronal cholinergic system in humans. Br J Pharmacol 2008;154:1558-71.
32. Shin VY, Wu WK, Chu KM, Koo MW, Wong HP, Lam EK, Tai EK, Cho CH. Functional role of beta-adrenergic receptors in the mitogenic action of nicotine on gastric cancer cells. Toxicol Sci 2007;96:21-9.
33. Wong HP, Yu L, Lam EK, Tai EK, Wu WK, Cho CH. Nicotine promotes cell proliferation via alpha7-nicotinic acetylcholine receptor and catecholamine-synthesizing enzymes-mediated pathway in human colon adenocarcinoma HT-29 cells. Toxicol Appl Pharmacol 2007;221:261-7.
34. Al-Wadei HA, Al-Wadei MH, Masi T, Schuller HM. Chronic exposure to estrogen and the tobacco carcinogen NNK cooperatively modulates nicotinic receptors in small airway epithelial cells. Lung Cancer 2010;69:33-9.
35. Al-Wadei HA, Al-Wadei MH, Schuller HM. Cooperative regulation of non-small cell lung carcinoma by nicotinic and beta-adrenergic receptors: a novel target for intervention. PLoS One 2012;7:e29915.
36. Al-Wadei MH, Al-Wadei HA, Schuller HM. Pancreatic cancer cells and normal pancreatic duct epithelial cells express an autocrine catecholamine loop that is activated by nicotinic acetylcholine receptors alpha3, alpha5, and alpha7. Mol Cancer Res 2012;10:239-49.
37. Sun H, Pan Y, He B, Deng Q, Ying H, Chen J, Liu X, Wang S. Different effects of the three polymorphisms on 15q25.1 onlung cancer risk: evidence from published literatures. J Cancer Res Ther 2016;12:12-9.
38. Xu ZW, Wang GN, Dong ZZ, Li TH, Cao C, Jin YH. CHRNA5 rs16969968 polymorphism association with risk of lung cancer -- evidence from 17,962 lung cancer cases and 77,216 control subjects. Asian Pac J Cancer Prev 2015;16:6685-90.
39. Zhou W, Geng T, Wang H, Xun X, Feng T, Zou H, Kang L, Jin T, Chen C. CHRNA3 genetic polymorphism and the risk of lung cancer in the Chinese Han smoking population. Tumour Biol 2015;36:4987-92.
40. Wenjuan Y, Yujun L, Ceng Y. Association of single nucleotide polymorphisms of beta2-adrenergic receptor gene with clinicopathological features of pancreatic carcinoma. Acta Histochem 2013;115:198-203.
41. Al-Wadei HA, Plummer HK 3rd, Ullah MF, Unger B, Brody JR, Schuller HM. Social stress promotes and gamma-aminobutyric acid inhibits tumor growth in mouse models of non-small cell lung cancer. Cancer Prev Res (Phila) 2012;5:189-96.
42. Schuller HM, Al-Wadei HA, Ullah MF, Plummer HK 3rd. Regulation of pancreatic cancer by neuropsychological stress responses: a novel target for intervention. Carcinogenesis 2012;33:191-6.
43. Sood AK, Bhatty R, Kamat AA, Landen CN, Han L, Thaker PH, Li Y, Gershenson DM, Lutgendorf S, Cole SW. Stress hormone-mediated invasion of ovarian cancer cells. Clin Cancer Res 2006;12:369-75.
44. Palm D, Lang K, Niggemann B, Drell TLt, Masur K, Zaenker KS, Entschladen F. The norepinephrine-driven metastasis development of PC-3 human prostate cancer cells in BALB/c nude mice is inhibited by beta-blockers. Int J Cancer 2006;118:2744-9.
45. Huang XY, Wang HC, Yuan Z, Huang J, Zheng Q. Norepinephrine stimulates pancreatic cancer cell proliferation, migration and invasion via beta-adrenergic receptor-dependent activation of P38/MAPK pathway. Hepatogastroenterology 2012;59:889-93.
46. Hara MR, Sachs BD, Caron MG, Lefkowitz RJ. Pharmacological blockade of a beta(2)AR-beta-arrestin-1 signaling cascade prevents the accumulation of DNA damage in a behavioral stress model. Cell Cycle 2013;12:219-24.
47. Hu W, Zhang M, Czeh B, Flugge G, Zhang W. Stress impairs GABAergic network function in the hippocampus by activating nongenomic glucocorticoid receptors and affecting the integrity of the parvalbumin-expressing neuronal network. Neuropsychopharmacology 2010;35:1693-707.
48. Bandelow B, Baldwin D, Abelli M, Bolea-Alamanac B, Bourin M, Chamberlain SR, Cinosi E, Davies S, Domschke K, Fineberg N, Grunblatt E, Jarema M, Kim YK, Maron E, Masdrakis V, Mikova O, Nutt D, Pallanti S, Pini S, Strohle A, Thibaut F, Vaghi MM, Won E, Wedekind D, Wichniak A, Woolley J, Zwanzger P, Riederer P. Biological markers for anxiety disorders, OCD and PTSD: a consensus statement. Part II: neurochemistry, neurophysiology and neurocognition. World J Biol Psychiatry 2017;18:162-214.
49. Kelmendi B, Adams TG, Yarnell S, Southwick S, Abdallah CG, Krystal JH. PTSD: from neurobiology to pharmacological treatments. Eur J Psychotraumatol 2016;7:31858.
50. Padgett CL, Slesinger PA. GABAB receptor coupling to G-proteins and ion channels. Adv Pharmacol 2010;58:123-47.
51. Schuller HM, Al-Wadei HA, Majidi M. GABA B receptor is a novel drug target for pancreatic cancer. Cancer 2008;112:767-78.
52. Al-Wadei HA, Al-Wadei MH, Ullah MF, Schuller HM. Celecoxib and GABA cooperatively prevent the progression of pancreatic cancer in vitro and in xenograft models of stress-free and stress-exposed mice. PLoS One 2012;7:e43376.
53. Al-Wadei MH, Al-Wadei HA, Schuller HM. Gamma-amino butyric acid (GABA) prevents the induction of nicotinic receptor-regulated signaling by chronic ethanol in pancreatic cancer cells and normal duct epithelia. Cancer Prev Res (Phila) 2013;6:139-48.
54. Schuller HM, Al-Wadei HA, Majidi M. Gamma-aminobutyric acid, a potential tumor suppressor for small airway-derived lung adenocarcinoma. Carcinogenesis 2008;29:1979-85.
55. Banerjee J, John AM, Al-Wadei MH, Schuller HM. Prevention of pancreatic cancer in a hamster model by cAMP decrease. Oncotarget 2016;7:44430-41.
56. Joseph J, Niggemann B, Zaenker KS, Entschladen F. The neurotransmitter gamma-aminobutyric acid is an inhibitory regulator for the migration of SW 480 colon carcinoma cells. Cancer Res 2002;62:6467-9.
57. Pomerleau OF. Nicotine and the central nervous system: biobehavioral effects of cigarette smoking. Am J Med 1992;93:2-7S.
58. Hecht SS. Progress and challenges in selected areas of tobacco carcinogenesis. Chem Res Toxicol 2008;21:160-71.
59. Boo HJ, Min HY, Jang HJ, Yun HJ, Smith JK, Jin Q, Lee HJ, Liu D, Kweon HS, Behrens C, Lee JJ, Wistuba II, Lee E, Hong WK, Lee HY. The tobacco-specific carcinogen-operated calcium channel promotes lung tumorigenesis via IGF2 exocytosis in lung epithelial cells. Nat Commun 2016;7:12961.
60. Jang HJ, Boo HJ, Lee HJ, Min HY, Lee HY. Chronic stress facilitates lung tumorigenesis by promoting exocytosis of IGF2 in lung epithelial cells. Cancer Res 2016;76:6609-19.
61. Min HY, Boo HJ, Lee HJ, Jang HJ, Yun HJ, Hwang SJ, Smith JK, Lee HJ, Lee HJ. Smoking-associated lung cancer prevention by blockade of the beta-adrenergic receptor-mediated insulin-like growth factor receptor activation. Oncotarget 2016;7:70936-47.
62. Dasgupta P, Kinkade R, Joshi B, Decook C, Haura E, Chellappan S. Nicotine inhibits apoptosis induced by chemotherapeutic drugs by up-regulating XIAP and survivin. Proc Natl Acad Sci U S A 2006;103:6332-7.
63. Dasgupta P, Rizwani W, Pillai S, Kinkade R, Kovacs M, Rastogi S, Banerjee S, Carless M, Kim E, Coppola D, Haura E, Chellappan S. Nicotine induces cell proliferation, invasion and epithelial-mesenchymal transition in a variety of human cancer cell lines. Int J Cancer 2009;124:36-45.
64. Schuller HM. Effects of tobacco constituents and psychological stress on the beta-adrenergic regulation of non-small cell lung cancer and pancreatic cancer: implications for intervention. Cancer Biomark 2013;13:133-44.
65. Schuller HM, Al-Wadei HA. Beta-adrenergic signaling in the development and progression of pulmonary and pancreatic adenocarcinoma. Curr Cancer Ther Rev 2012;8:116-27.
66. Banerjee J, Papu John AM, Schuller HM. Regulation of nonsmall-cell lung cancer stem cell like cells by neurotransmitters and opioid peptides. Int J Cancer 2015;137:2815-24.
67. Coelho M, Soares-Silva C, Brandao D, Marino F, Cosentino M, Ribeiro L. β-Adrenergic modulation of cancer cell proliferation: available evidence and clinical perspectives. J Cancer Res Clin Oncol 2017;143:275-91.
68. Ishida J, Konishi M, Ebner N, Springer J. Repurposing of approved cardiovascular drugs. J Transl Med 2016;14:269.
69. Melhem-Bertrandt A, Chavez-Macgregor M, Lei X, Brown EN, Lee RT, Meric-Bernstam F, Sood AK, Conzen SD, Hortobagyi GN, Gonzalez-Angulo AM. Beta-blocker use is associated with improved relapse-free survival in patients with triple-negative breast cancer. J Clin Oncol 2011;29:2645-52.
70. Watkins JL, Thaker PH, Nick AM, Ramondetta LM, Kumar S, Urbauer DL, Matsuo K, Squires KC, Coleman RL, Lutgendorf SK, Ramirez PT, Sood AK. Clinical impact of selective and nonselective beta-blockers on survival in patients with ovarian cancer. Cancer 2015;121:3444-51.
71. Powe DG, Voss MJ, Zanker KS, Habashy HO, Green AR, Ellis IO, Entschladen F. Beta-blocker drug therapy reduces secondary cancer formation in breast cancer and improves cancer specific survival. Oncotarget 2010;1:628-38.
72. Wang T, Li Y, Lu HL, Meng QW, Cai L, Chen XS. Beta-adrenergic receptors: new target in breast cancer. Asian Pac J Cancer Prev 2015;16:8031-9.
73. Colucci R, Moretti S. The role of stress and beta-adrenergic system in melanoma: current knowledge and possible therapeutic options. J Cancer Res Clin Oncol 2016;142:1021-9.
74. Al-Wadei HA, Al-Wadei MH, Ullah MF, Schuller HM. Gamma-amino butyric acid inhibits the nicotine-imposed stimulatory challenge in xenograft models of non-small cell lung carcinoma. Curr Cancer Drug Targets 2012;12:97-106.
75. Al-Wadei MH, Banerjee J, Al-Wadei HA, Schuller HM. Nicotine induces self-renewal of pancreatic cancer stem cells via neurotransmitter-driven activation of sonic hedgehog signalling. Eur J Cancer 2016;52:188-96.
76. Zagon IS, Hytrek SD, Smith JP, McLaughlin PJ. Opioid growth factor (OGF) inhibits human pancreatic cancer transplanted into nude mice. Cancer Lett 1997;112:167-75.
77. Godinho RO, Duarte T, Pacini ES. New perspectives in signaling mediated by receptors coupled to stimulatory G protein: the emerging significance of cAMP efflux and extracellular cAMP-adenosine pathway. Front Pharmacol 2015;6:58.
78. Al-Wadei HA, Schuller HM. Non-genomic inhibitory signaling of beta-carotene in squamous cell carcinoma of the lungs. Int J Oncol 2009;34:1093-8.
79. Shafer SH, Phelps SH, Williams CL. Reduced DNA synthesis and cell viability in small cell lung carcinoma by treatment with cyclic AMP phosphodiesterase inhibitors. Biochem Pharmacol 1998;56:1229-36.
80. Plummer HK 3rd, Dhar MS, Cekanova M, Schuller HM. Expression of G-protein inwardly rectifying potassium channels (GIRKs) in lung cancer cell lines. BMC Cancer 2005;5:104.
81. Rao R, Salloum R, Xin M, Lu QR. The G protein Galphas acts as a tumor suppressor in sonic hedgehog signaling-driven tumorigenesis. Cell Cycle 2016;15:1325-30.
82. Schuller HM, Correa E, Orloff M, Reznik GK. Successful chemotherapy of experimental neuroendocrine lung tumors in hamsters with an antagonist of Ca2+/calmodulin. Cancer Res 1990;50:1645-9.
83. Leanza L, Manago A, Zoratti M, Gulbins E, Szabo I. Pharmacological targeting of ion channels for cancer therapy: in vivo evidences. Biochim Biophys Acta 2016;1863:1385-97.
84. Woods N, Trevino J, Coppola D, Chellappan S, Yang S, Padmanabhan J. Fendiline inhibits proliferation and invasion of pancreatic cancer cells by interfering with ADAM10 activation and beta-catenin signaling. Oncotarget 2015;6:35931-48.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Schuller HM. A new twist to neurotransmitter receptors and cancer. J Cancer Metastasis Treat 2017;3:71-7. http://dx.doi.org/10.20517/2394-4722.2017.18
AMA Style
Schuller HM. A new twist to neurotransmitter receptors and cancer. Journal of Cancer Metastasis and Treatment. 2017; 3: 71-7. http://dx.doi.org/10.20517/2394-4722.2017.18
Chicago/Turabian Style
Schuller, Hildegard M.. 2017. "A new twist to neurotransmitter receptors and cancer" Journal of Cancer Metastasis and Treatment. 3: 71-7. http://dx.doi.org/10.20517/2394-4722.2017.18
ACS Style
Schuller, HM. A new twist to neurotransmitter receptors and cancer. J. Cancer. Metastasis. Treat. 2017, 3, 71-7. http://dx.doi.org/10.20517/2394-4722.2017.18
About This Article
Copyright

Data & Comments
Data


 Cite This Article 9 clicks
Cite This Article 9 clicks

 Like This Article 28
likes
Like This Article 28
likes






Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.