
A primer on recent developments in cancer immunotherapy, with a focus on neoantigen vaccines

Abstract
Cancer immunotherapy has now been conclusively shown to be capable of producing durable responses for a substantial number of patients. Adoptive cell transfer and checkpoint blockade therapies in particular both demonstrate that antigen-specific immune responses can be dramatically effective, even in previously refractory late stage disease. Such developments, together with advances in technology, have strongly encouraged revisiting the concept of neoantigen vaccines. Here we introduce basic ideas in the field to allow investigators from diverse backgrounds to understand these developments, grasp current issues, and contribute to further progress.
Keywords
Introduction
In the late 1800s, Coley[1] pursued investigations of cancer regression in the context of bacterial disease. It has been clear since then that the immune system plays an important role in cancer. Over the ensuing century, strong arguments were put forward for both why cancer immunotherapy should work and why it should not, occasionally by the same investigator[2]. The past decade has seen dramatic progress in cancer immunotherapies, such as checkpoint blockade, adoptive cell transfer, and vaccines. The success came on two fronts: complete durable patient response was achieved in a substantial fraction of patients in the clinic, and the mechanism of action was T-cell antigen-specific. This spurred confidence that therapy approaching a “cure” was at hand, based on a rational extrapolation of current knowledge. The immune system is inextricably linked to both the phenomenon of cancer and its treatment. This represents a paradigm shift, where cancer is no longer seen as just a collection of aberrant cells, but rather a systemic disease.
While this new vista continues to capture the public imagination worldwide, we have learned enough over the years to understand that cancer immunotherapy, in its current form, is not a panacea. The central challenge facing cancer immunotherapies and neoantigen vaccines in particular is understanding resistance.
Integrating immunology and cancer research, already two of the most complex topics in biomedicine, is an interdisciplinary effort, drawing from fields such as biology, pharmacology, chemistry, physics, engineering, statistics, and mathematics. Our main aim in this primer is to lower the barrier to entry for readers who are not specialists in immunotherapy. We focus on neoantigen vaccines, which in some ways represent T-cell based cancer immunotherapy in its most elementary form. We also address general issues, enabling readers to quickly grasp other immunotherapies and future developments.
Background on the immune system and cancer
We embark first on a brief tour of immunology, with the caveat that the specifics and even the broad outlines may shift as the field advances. Many of the features described below have bearing on possible cancer resistance mechanisms.
In brief, the requirements for an effective immune system include mechanisms to recognize foreign invaders, the means to trigger and coordinate a potentially complex attack (“expansion”), then return to equilibrium (“contraction”), while not attacking normal tissue. This rests critically on the ability to distinguish self from non-self. In vertebrates, robust response also leads to the development of immune memory. Immunotherapy can be viewed as an attempt to shift the equilibrium point in a complex system that can actively amplify or suppress its effects.
Cancer cells can evade the immune system through a variety of routes, such as being viewed as self, hijacking suppressive mechanisms that prevent damage, attacking or subverting immune system agents, or simply growing at a rate beyond the capacity of an often aged and weakened immune system.
The vertebrate immune system is broadly divided into two arms. Innate immunity[3] is encoded in the germline, while adaptive (“acquired”) immunity[4] is mediated by B and T lymphocytes that undergo processes of diversification and selection. T cell selection relies on processes of central tolerance (at the thymus) and peripheral tolerance (on mature circulating T cells)[5]. The two arms interact, with some cell types having a role in both arms.
In the adaptive system, T cells play the key role in recognizing pathology via antigens. The core of this task involves three parts: a presenter (major histocompatibility complex molecule, MHC), an antigen fragment (peptide), and a recognizer (T cell receptor, TCR). Elaborate processes of MHC expression and maturation, antigen processing, peptide MHC loading, and generation of mature naive T cells through the thymus underlie their formation and interaction[6-9].
Antigen recognition takes place when a receptor on a T cell encounters a cell presenting a cognate peptide-MHC (pMHC) complex on its surface. If a CD28 co-stimulatory receptor on the T cell simultaneously binds with CD80 or CD86 expressed on the presenting cell, an activation signal is propagated on the cytosolic side of the TCR, leading to cell proliferation, differentiation, and secretion of cytokines. A lack of a co-stimulatory signal leads to a hypo-responsive state known as T cell anergy[10]. Inhibitory checkpoint molecules “put the brakes” on adaptive immunity, where for example cytotoxic T-lymphocyte associated protein 4 (CTLA-4) competes with CD80/86 in binding CD28, thus suppressing activation[11].
The detection limit for such T cell triggering is impressively low (four pMHC per TCR cluster)[12]. Note that the vast majority of the 104 presented peptides in vivo are in fact normal “self” peptides, with only a few from foreign antigens, if any[13].
MHC molecules and T cells come in two subtype pairs [Figure 1]. MHC class I (MHC-I) is normally expressed in all nucleated cells and presents intracellular (endogenous) antigen fragments. The pMHC-I complexes are recognized by CD8+ T cells, which are then activated and differentiate into cytotoxic T cells (CTLs) with direct cell killing capability. MHC class II (MHC-II) is expressed in “professional” antigen presenting cells (APCs), including dendritic cells, and presents exogenous antigens that have been engulfed by the APC. The resulting pMHC-II complexes are recognized by CD4+ T cells, which can differentiate e.g. into T helper cells whose primary role is to activate other immune system components.
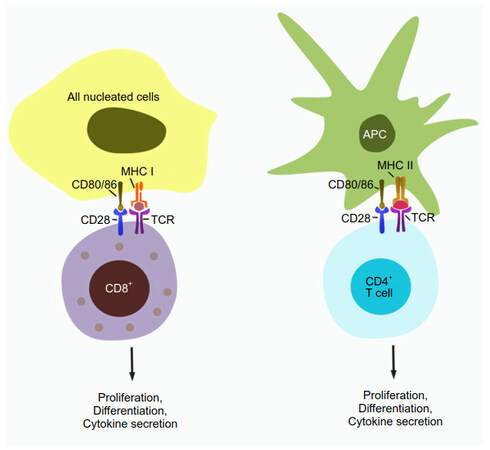
Figure 1. T cell activation. CD8+ T cells inspect the surface of cells they encounter and are activated if a T cell receptor binds to a presented pMHC-I complex, leading to downstream processes including proliferation, differentiation, and cytokine secretion. CD4+ T cells are similarly activated when binding pMHC-II complexes presented by professional APCs such as dendritic cells. A co-stimulating signal from CD28/CD80 (86) binding is required for full activation; its absence leads to T cell anergy. APC: antigen-presenting cell; MHC: major histocompatibility complex; TCR: T cell receptor
The loaded peptides in the case of MHC-I are typically 8 to 12 residues in length and are loaded into a groove that is closed on both ends. The MHC-II-loaded peptides range in length from 12 to 25 residues and are loaded into a groove that is open on both ends [Figure 2][6,7,14].
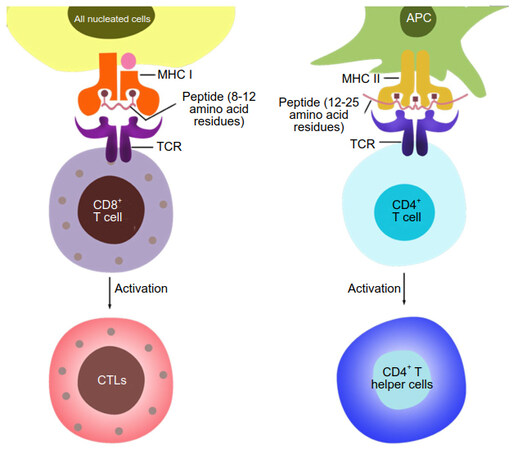
Figure 2. Differences in recognition and downstream processes between CD8+ and CD4+ T cells. CD8+ T cells recognize pMHC-I complexes, where the peptide is a fragment from an endogenous protein typically 8 to 11 AAs in length, which occupies a groove that is closed on both ends. CD4+ T cells recognize pMHC-II complexes, where the peptide is derived from cells or antigens engulfed by the APC and is typically longer, 12 to 25 AAs in length. The MHC-II groove is open on both ends. After activation, CD8+ T cells differentiate into cytotoxic T lymphocytes (CTLs), whereas CD4+ T cells differentiate e.g. into T helper cells, depending on receipt of further cytokine signals. APC: antigen-presenting cell; MHC: major histocompatibility complex; TCR: T cell receptor
Cells that do not express MHC-I on their surface are considered aberrant by the immune system. In normal environments, these are eliminated by natural killer cells, which are innate lymphoid cells with recognition receptors encoded in the germline[15].
Both the presentation (MHC) and recognition (TCR) components are highly diverse, although MHC diversity only appears at the population level. In humans, the MHC is known as the human leukocyte antigen (HLA) complex. Each individual inherits six MHC-I alleles from three loci, HLA-A, -B and -C (i.e. two parental alleles from each locus), and similarly, six MHC-II alleles from HLA-DP, -DQ, and -DR loci. Note that the HLA nomenclature was revised in 2010[16]. As of Oct. 2015, there were 10,297 class I and 3543 class II known alleles[17,18]. Hence, the pMHC binding profile (“HLA peptidome”) varies broadly between individuals.
On the recognition side, there are in principle at least 1015 possible TCR variants. The number of T-cells in any individual is on the order of 1012, and the number of clonotypes possibly around 107[19,20]. The processes of receptor diversification and negative selection for immune self-tolerance is largely completed during youth. The TCR repertoire shows a linear loss of naive T cell diversity with age[21], although more subtle characterizations can be made[22]. Such age-related changes have been hypothesized to contribute to cancer susceptibility, although their impact is not yet clear[23]. The TCR repertoire continues to be a subject of intense research, which we touch on further below. The impact of age-related changes more generally is discussed in the context of checkpoint blockade therapy by Elias et al.[24].
The idea that one clonal TCR recognizes one specific antigen has been supplanted by the notion that TCRs are cross-reactive. A discussion of how TCRs must be cross-reactive in principle is given by Sewell[25]. Indeed, TCR recognition that straddles the self/non-self boundary (e.g. between self and microbial peptides) underlies the theory of molecular mimicry, whereby bacterial antigens do not provoke attack or conversely may lead to autoimmune disease[26]. The specific mechanisms are now being worked out[27,28]. Similarly, mutant tumor proteins may avoid immunogenicity by being cross-reactive with self-proteins.
Each individual’s immune system will also have peptides that it cannot recognize, which can be characterized as “holes” or “blind spots”. These can arise both from gaps in presentation (lack of peptide-MHC binding) or recognition (absence from the TCR repertoire)[29]. A vaccine based solely on an antigen in such a hole will not work for that individual. Such phenomena are seen in the context of microbial immunity[30,31]. The concept of original antigenic sin[32] states that such a hole can paradoxically be created by initial exposure to an antigen, as the immune system does not mount a novel response when it encounters a slight variant.
Immunological research continues to reveal new features. Activated CD8+ T cells were found to require cross presentation, i.e. co-stimulation by dendritic cells that can present exogenous antigens on MHC-I, for full induction of cytotoxic response[33,34]. The CD4 lineage was resolved into four lines[35] and then a plastic set of more[36]. Some CD4+ T cells can acquire cytotoxic activity[37,38] (i.e. not only CD8+ T cells can be cytotoxic). More recently, a “second touch hypothesis”[39] suggests that the high-level picture for polarization of T cells may not yet be complete. New immune cell subtypes continue to be discovered[40]. As one consequence, mathematical modeling of the immune system is likely to remain a difficult endeavor for some time.
We mention here briefly the once-dominant view of cancer as an autonomous genetic disease, as captured by the original “hallmarks of cancer”[41]. The cancer phenotype arises as a result of selection pressure on genome mutations, leading to acquisition of limitless growth and survival potential, with genome instability as an “enabling characteristic”. These mechanisms also underlie cancer’s uncanny ability to acquire additional phenotypes such as eliciting immune tolerance and angiogenesis. A recent proposal that epigenetics alone may be sufficient to generate the hallmarks of cancer[42] may, amongst other things, alter our understanding of the time scales involved in tumor response[43].
The careful examination of tumor cell evolution and its therapeutic implications are in its beginning stages[44-47]. Principles such as antagonistic pleiotropy[48], where reproductive fitness in youth is played off against fitness in old age, are also sometimes raised as setting fundamental biological limits.
For further background on cancer and immunology, the reader can consult reference books[49,50], a three-volume series[51], and a broad history from a contrarian perspective[52].
Recent developments in cancer immunotherapy
Modalities of T-cell based immunotherapy
The design of currently popular T-cell based immunotherapies can be described as follows:
• Release the brakes: checkpoint blockade[53];
• Boost instruction, via antigens: cancer vaccines;
• Boost instruction, via cell transfer, bypassing presentation: adoptive dendritic cell therapy[54,55];
• Boost recognition, via cell transfer, bypassing instruction: adoptive T cell therapy[56];
• Boost recognition, via cell transfer, bypassing instruction and MHC restriction: adoptive chimeric antigen receptor T-cell (CAR-T) therapy[57,58].
All of these therapies are based on T cells. Checkpoint blockade therapy is distinguished by not targeting cancer, relying instead on the host immune system training (or having already trained) itself to target tumors. At the other end of the spectrum, CAR-T therapy does not rely on the host immune system for tumor killing. These span so-called active to passive therapies. Passive therapies do not necessarily induce immune memory, although T cell proliferation may allow extended response. The various immunotherapies can be visualized in an informative hierarchy[59]. The 2014 Society for Immunotherapy of Cancer (SITC) primer provides an unhurried perspective on many of these developments[60].
While the current wave of immunotherapies was heralded by dendritic cell therapy (sipuleucel-T)[55], the most notable breakthrough was probably the development of anti-CTLA4 checkpoint blockade, which utilizes antibodies to block receptors that inhibit T cell activation. This treatment allowed some of the first demonstrations in humans of the therapeutic efficacy of neoantigen-specific T cells[61]. Checkpoint therapies based on programmed cell death protein 1 (PD-1) and programmed death-ligand 1 (PD-L1) blockade have further demonstrated improved efficacy with reduced toxicity.
Impacts of immunotherapy on standard practice
The mainstream acceptance of cancer immunotherapy has stimulated efforts to modify clinical trial reporting[62], with the introduction of “immune-related” adverse events (irAE) and response criteria (irRC). Progression criteria must now allow for pseudo-progression, i.e. the appearance of growing or new lesions that indicate T cell infiltration. A call for “assay harmonization” seeks to reduce variability in cellular immune response reporting. Survival criteria must account for time-dependent hazard ratios, with agent-specific delays in Kaplan-Meier survival curve separation ranging from four to eight months.
Clinical trial design itself is evolving, a process that began in response to targeted therapies (precision oncology) and is now accelerating[63]. This has seen the advent of expansion cohorts, and platform, bucket, adaptive[64], and seamless trials. It will be increasingly important to understand the cohort and trial design to interpret results.
We note in passing the recent reports of hyperprogression[65]. Tumor size has been observed to dramatically increase with anti-PD-1/PD-L1 treatment, although whether this is more than a statistical fluctuation has been questioned[66]. It is nevertheless safe to say immunotherapies behave differently than previous standard therapies.
The effort to go beyond tumor cell-based staging has begun with the proposal of an Immunoscore[67], which quantifies the density of CD3+ and CD8+ T cells in solid tumors. Due to its prognostic value, it has been proposed to augment traditional tumor size/nodal status/distant metastasis (TNM) staging[68].
Recent advances have triggered a reconsideration of the effect of conventional therapies (surgery, chemotherapy, radiation) and of molecularly targeted therapies[69,70]. Oncogenes such as Myc have been found to also regulate immune response. When such oncogenes are inactivated, immune response is restored and plays a role in the subsequent “oncogene withdrawal”[71]. Chemotherapy perhaps surprisingly also appears to rely in part on the immune system for cytotoxic effect[72].
Cancer immunotherapies can in principle have much milder side effects compared to radiotherapy and chemotherapy. In practice, they are associated with their own spectrum of adverse events[73,74]. In particular, cytokine release syndrome (“cytokine storm”) can lead to organ failure and death. Both treatment efficacy and adverse events are associated with proliferative and persistent cellular responses, which can vary significantly between individuals, thus requiring careful monitoring[75]. Adverse events associated with neoantigen vaccines appear to be relatively mild, compared to adoptive cell transfer, checkpoint blockade, and tumor-associated antigen (TAA) vaccine therapies[76].
Neoantigen vaccines
Introduction
We now turn to neoantigen-based cancer vaccines. The objective of a vaccine is to introduce a small amount of material to instruct T and B cells to eliminate invaders that present the cognate antigen[77]. Vaccines in general can be prophylactic (preventative) or therapeutic (cure or control of observable disease). Current neoantigen vaccines are therapeutic, with the goal of restoring immune surveillance of a tumor that has likely already evolved to evade the immune system (e.g. through immunoediting; see below).
Cancer cells are genomically unstable[41,78], which leads to the expression of novel proteins due to non-synonymous mutations. Many of these are likely to be immunogenic and are termed neoantigens. Vaccines that precisely target such neoantigens (also known as tumor-specific antigens, or TSAs) would prime an immune response that rejects tumors while sparing normal tissues, leading to optimal therapies with mild if any toxicity. A timeline that traces the foundations of this idea back to 1943 is provided by Coulie et al.[79].
Types of antigen-based cancer vaccines
Prior to the advent of next-generation sequencing, cancer vaccines were developed based on TAAs or cancer germline antigens. These self-antigens are overexpressed in tumors, or normally expressed only during development but re-expressed in tumors. Vaccines targeting these can be produced in advance at lower cost and applied across a range of tumors that share expression of the target. As the targets are self-antigens, such vaccines are possibly limited by self-tolerance and adverse events. Tumor resistance mechanisms, many of which are shared with neoantigen vaccines, are also a prominent concern[80,81]. Another class of targets are shared tumor neoantigens, which are commonly found across a subtype of tumor. As in TAAs, the vaccine can be produced beforehand, and treatment progress can be easily followed, as the neoantigen epitopes (i.e. recognized peptide fragments) are typically well known. Such epitopes however may not be the most effective for any given tumor.
With massively parallel sequencing and MHC binding and functional prediction software tools, the key hurdle to developing personalized neoantigen vaccines can now be overcome. Vaccines custom designed for each patient represents a paradigm shift in cancer treatment[82].
Some of the strengths and weaknesses of the neoantigen vaccine approach are summarized in [Table 1] and are discussed further below.
Strengths and weakness of the neoantigen vaccine approach
| Strengths | Weaknesses |
|---|---|
| Precise targeting | Need for tumor biopsy (in general) |
| Mild adverse events | Need to overcome tumor defenses |
| Few constraints on dosage | Slow induction of immune response |
| Better profile than TAA vaccines | May not be applicable to tumors with few mutations |
| No need for T cell extraction and ex vivo growth | Unreliable epitope binding prediction algorithms |
| Many opportunities to optimize/combine formulations | Time lag from biopsy to vaccine |
| Multi-epitope designs can compensate for inaccurate binding predictions, tumor heterogeneity and evolution | Cost |
| Induction of antigen spreading and immune memory can cope with occult disease |
Vaccine formulation and administration
In addition to the selection of epitopes, a number of other considerations can strongly influence the success or failure of neoantigen vaccines. Cancer vaccines can be formulated as whole cells, peptides/proteins, RNA, DNA[80], and glycolipids[83]. Vaccines are typically formulated as peptides, due to ease of construction and low cost, although these are often observed to be weakly immunogenic. They can be modified to enhance delivery to immune cells and improve pMHC binding stability[84]. Synthetic long peptides require dendritic cell processing, argued as essential for durable response[85]. Protein vaccines are more immunogenic but have a higher risk of anaphylaxis. The robust discussion about designing and assessing peptide vaccines has been reviewed by Kumai et al.[86]. DNA vaccines introduce DNA coding for antigenic fragments into host cells, where they are expressed and lead to the presentation of epitopes via the MHC-I pathway. They are generally safe, stable, and easy to produce at low cost, although currently weakly immunogenic. The vaccine or delivery vehicle itself can be attacked by the host immune system[87]. RNA vaccines can encode several epitopes on a single molecule, can trigger the innate immune system, and are not at risk of integrating into the genome[88-90]. Whole cell vaccines that employ weakened or killed tumor cells can trigger immune response with the entire complement of tumor antigens, without specific instruction of the immune system, reducing time and expense. They may however induce immune response to self-proteins.
Other considerations are choice of carrier, delivery vehicle[91] (including bacteria[92] or viral vectors[93]), and administration route (intravenous, intratumoral, subcutaneous, intra-lymph node, nasal, ingested). Further afield, cancer vaccine engineering has emerged to offer benefits such as lymph node targeting, reduced systemic toxicity, elimination of ex vivo expansion requirements, and controlled release of immunomodulators while ignoring suppressive signals[94-96].
Neo-epitope binding prediction
Neoantigen vaccines are produced by first inspecting the patient’s tumor for immunogenic peptides, specifically epitopes[97]. TCRs recognize linear epitopes, i.e. a continuous fragment of an antigen. Note that pMHC binding is a necessary but not sufficient condition for immunogenicity.
The neo-epitope selection problem can thus be reduced to finding mutant peptides that bind well to the patient’s MHC alleles. This is amenable to computational treatment and is one of the most prominent applications of machine learning to immunology[98]. The realization that such bioinformatic approaches can reveal a “gold mine” of targets and that neoantigen vaccines were feasible can be traced back to a 2008 paper[99].
A simplified neo-epitope selection pipeline can be described as follows:
• Perform exome sequencing of tumor and normal tissue to identify non-synonymous single nucleotide variants and generate an initial list of candidate genes;
• Perform RNA-Seq to confirm expression;
• Use informatics tools to predict neoantigen-derived peptides that bind to the patient’s set of HLA alleles;
• Filter candidates based on survival or growth function (“driver genes”);
• Choose the top 10 or 20 epitopes.
Proteasomal cleavage predictions[100] can also be incorporated into the workflow, although the predictive value is rather low, due to the lack of sufficient training data[98].
Numerous excellent reviews of the available tools are available[82,98,101]. The Immune Epitope Database[102] is probably the most prominent epitope database and analysis resource, freely available on the Web. TANTIGEN[103] is a database of tumor-tissue derived antigens with experimentally validated HLA binding. Step-by-step instructions on the use of a prominent suite of tools is available[104]. Mutant Peptide Extractor and Informer[105] is a web-based tool that attempts to integrate best practices and simplify neo-epitope analysis and selection for non-bioinformaticians (limited to MHC-I epitopes). ImmunoNodes[106] is a software framework for building complex immunoinformatics workflows, such as those for neo-epitope selection.
Amongst other challenges, prediction of MHC-II peptide binding lags behind MHC-I prediction, partly due to the greater length of loaded peptides that interact in flanking regions with highly polymorphic alleles. Also, binding data does not exist for many less common MHC alleles, which has given rise to “pan MHC” algorithms with somewhat reduced performance. Therapeutic strategies based on so-called promiscuous epitopes that bind to several MHC alleles may place less stringent requirements on the accuracy of binding predictions[107].
The extent to which epitope binding scores are a good surrogate for immunogenicity remains unclear. Peptide binding stability rather than affinity has been proposed as a better predictor of immunogenicity[29]. Numerous factors can affect antigen presentation and recognition processes, such as pH, inflammation, and peptide post-translational modifications[108].
Many of the structural aspects of peptide-MHC binding and TCR recognition are reviewed by Hudrisier and Gairin[109]. Important aspects of the problem formulation can be found e.g. in the references cited by Meydan et al.[14]. A recent examination of empirical TCR-pMHC kinetic constants measured in three-dimensional assays suggests these may not accurately reflect dynamics in a two-dimensional context, such as T cell scanning of the APC surface[110]. This could suggest that some of the data underlying current epitope binding prediction algorithms needs to be re-measured.
In general, while immunogenic antigens tend to have high binding scores, the converse does not hold[111]. In addition, indels and gene fusions are typically not chosen, due to the difficulty of predicting binding. Snyder and Chan[101] caution that current prediction tools on their own are not ready for routine clinical use.
Choice of epitope candidates
In tumors with a large number of mutations, the candidate filtering step is essential to avoid being overwhelmed by false positives[112]. Mass spectrometry has been effectively used for this task by identifying MHC-bound peptides[113]. Indeed, it can be used to generate candidates on its own[114,115]. There remain possible issues with sensitivity and translation into a clinical setting[112]. Combining functional analysis and T cell detection via multimers can help in the search for tumor rejection epitopes[116]. Proximity ligation assays can assess whether an antigen is presented in situ, although this requires a mutant-specific antibody[117]. Another approach tests epitopes experimentally in MHC-transgenic mice[118,119]. Further work is necessary to validate the efficacy of such workflows[120]. An interesting suggestion is that PD-1+ peripheral blood cells are enriched in tumor neoantigens, from which candidate epitopes can be derived[121].
There is a general exhortation to prioritize genes that target essential tumor “driver” functions such as growth and survival. This however may not be too helpful, as only a small percentage of neoantigens are of this type in e.g. melanoma[112], the vast majority being “passenger” mutants not associated with cell transformation. Efforts to expand and/or refine the list of functional cancer genes may help in this regard[122,123].
Current vaccine strategy employs several epitopes to address tumor heterogeneity and reduce acquisition of resistance, while also compensating for the imperfect predictive value of pMHC binding tools. The phenomena of immunodominance[124-127] and T cell cross-reactivity[128] suggests that simply increasing the number of epitopes in a vaccine may not be advisable, as a suboptimal epitope may interfere with the others in a dominance hierarchy, and auto-immunity remains an issue. Indeed, pioneering efforts in cancer epitope selection[113,129] found possible instances of immunodominance. Initial experience with long peptides on the other hand suggested this may not be an issue[130]. Further work is required to understand how to choose the number of epitopes to include in a vaccine, which could be e.g. cancer type-specific. The thinking behind many current vaccine approaches is examined by Kumai et al.[131] who also describe four steps to developing cancer vaccines and five ways of monitoring the response.
Initial effort e.g. in adoptive cell transfer was focused on MHC-I restricted epitopes to elicit direct tumor cell killing. Attention has now shifted to MHC-II restricted epitopes, in part due to the fuller realization that CTLs require CD4+ help[132-135]. Indeed, adoptive cell transfer of CD4+ T cells was enough to induce tumor regression in a mouse model of melanoma[136] and in a human patient[137].
Initial human trials
The personalized neoantigen vaccine strategy has begun to reach the clinic with the recent reports from two Phase I trials[76,138]. These trials, on patients with advanced melanoma, demonstrate that such therapies are safe and induce a targeted neoantigen-specific response as designed. Ott et al.[76] enrolled 6 patients who had no evidence of disease after surgery, with 4 remaining tumor-free after 25 months. Sahin et al.[138] enrolled 13 patients, and 8 patients remained tumor free after 23 months. The time to develop personalized vaccines (weeks to months) remains a key obstacle, especially for patients with advanced disease.
In an apparent pattern, both trials utilized MHC-I binding scores to select neo-epitopes (Sahin et al.[138] combined these predictions with MHC-II binding scores). The vaccines were then seen to activate CD4+ T cells, possibly because MHC-II binding is less restrictive[139]. In more detail, Ott et al.[76] selected neo-epitopes using predicted binding to HLA-A and HLA-B, and employed long peptides (15-30 amino acids) in several pools targeting up to 20 neoantigens for five priming and two boosting vaccinations injected subcutaneously. They observed CD4+ response almost immediately and a peak response generally at 16 weeks, and found two to four immunogenic peptides per patient. Sahin et al.[138] ranked mutant epitopes on a combination of predicted MHC-I and MHC-II binding, plus high expression and variant allele frequency, and chose 10 epitopes per patient. Two synthetic RNAs were used to encode five 27mer peptides with the single nucleotide variant (SNV) at position 14. Patients were treated with at least eight and up to twenty neo-epitope vaccine doses injected into inguinal lymph nodes, and T cells were developed against at least three mutations per patient, with the majority being exclusively CD4+ responses. Neo-epitope specific CD8+ T cells expanded within two to four weeks.
Who benefits?
We now return to a more general discussion of immunotherapies. The seminal studies in checkpoint blockade therapy have primarily been done in melanoma and lung cancer. A survey of solid tumor types that have been studied with immunotherapy, with an emphasis on understudied cancers, has been made by Young[140]. In terms of number of clinical trials, breast cancer tops the list.
In an emerging consensus, the cancers that are best indicated for immunotherapy are slow growing, exhibit high mutational load and low burden at the start of therapy, and are inflamed[141]. This suggests immunotherapies may be more effective in the early stage disease setting. A correlation has been observed in checkpoint blockade between somatic mutations per megabase and objective response rates[111]. Mutagen induced cancers such as melanoma and lung cancer subtypes with high mutational loads were some of the first to show durable complete response. Estimating mutational load using custom reduced gene panels[142] or pre-existing ones[143] may aid quick assessments within the clinic. Cancer types are characterized by a wide range of mutational loads[144].
Recent work has sharpened focus on mismatch-repair deficiency as a biomarker to identify patients who can benefit from PD-1 blockade, independent of tissue type[145]. The immunological phenotype of microsatellite instability-high (MSI-H) colorectal tumors in particular may be unique[146]. In a literature review of anti-PD-1 clinical trials, atypical responses appeared in all cancer types except tumors with mismatch-repair deficiency and head and neck squamous cell carcinoma[147].
While checkpoint inhibitors are often presumed to exacerbate the symptoms of patients with inflammatory/autoimmune diseases, anecdotal reports suggest this may not be the case[74].
Resistance and escape
The fact that immunotherapies fail to produce durable responses in a majority of patients has spurred intensive investigations of resistance. Both primary resistance, where no beneficial response is observed, and secondary resistance, where initial benefit is followed by relapse, are observed.
Before proceeding, we first ask why neoantigen vaccines work at all, as they would appear catastrophically prone to failure due to antigen loss. Such loss has been seen in checkpoint blockade therapy, where both chromosomal deletion of clonal neoantigens and negative selection of tumor subclones were observed[148].
Accumulating evidence suggests part of the answer lies in the phenomenon of antigen spreading, aka cascade[149]. T cells are initially “instructed” by the vaccine epitopes, but effector activity need not be limited to these. This hypothesis suggests that the role of the vaccine is to nucleate immunity to an iteratively growing cascade of antigens, the epitopes of which are then committed to T cell memory. This could be key to a durable response that can also target new tumor mutations. The time required to generate such a cascade could also explain the lag often seen between vaccine administration and objective response. A related idea suggests that the initial vaccine-induced attack reverses immuno-suppressive mechanisms, allowing preexisting CTLs that already recognize other tumor neoantigens to be unleashed in a cascade[79].
Cancers can resist therapy by circumvention of each of the immune system roles mentioned previously:
• Disrupt presentation:
• Inhibit MHC-I expression[150-152];
• Disrupt dendritic cell trafficking to lymph nodes (i.e. T cell priming);
• Disrupt peptide processing;
• Disrupt recognition (prevent T-cell trafficking from lymph nodes back to tumor[153], exploit holes in TCR repertoire);
• Exploit immune suppressive mechanisms.
In addition, tumor cells employ explicit defense mechanisms, e.g. downregulation of pro-apoptotic pathways, and counterattack by secreting FasL death ligands, resulting in CTL death[153].
The tumor microenvironment (TME), i.e. the nearby cellular, vascular, and extracellular matrix environment remodeled by the tumor, is the focus of much research into resistance[154]. Here tumors are seen to induce host self-protective mechanisms, through recruitment of suppressive cells such as MDSCs[155], regulatory T cells[156], and tumor associated macrophages[157]. The tumor creates an immune privileged site, akin to the eye and brain, that excludes T cells[158]. Recent work provides a detailed picture of effector T cell exclusion based on a β-catenin signaling mechanism[159].
The TME is a metabolically demanding place, with competition for oxygen and nutrients[160-162]. Tumor cells can outlast T cells through the induction of T cell anergy or exhaustion, part of a class of phenomena termed T cell dysfunction[10,162,163]. There also remains the possibility that the tumor simply grows faster than an often aged and weakened immune system can eliminate it. A careful 2011 discussion of the TME in which CTLA4 operates is given by Quezada et al.[164].
As antigen-specific vaccines seek to activate the adaptive system, harnessing the innate immune system, and in particular natural killer (NK) cells[165-167], would appear to be an attractive complementary approach. Unlike naive CD8+ T cells that require time to acquire cytotoxic activity, NK cells are “ready to kill”[168]. NK and dendritic cells engage in mutual activation, and the former can “edit” the latter population[169]. Inhibitory receptors that bind MHC-I allow NK cells to recognize and eliminate cells that do not present MHC-I, thus closing one avenue of tumor cell escape. The activating receptor NKG2D has attracted particular interest, as its ligands (NKG2DL) are commonly expressed by tumors. Tumor cells however can also express NKG2D and hijack NKG2DL signaling to drive stem-cell like behavior[170]. NK cells participate in tumor-induced polarization, acquiring a pro-tumorigenic and pro-angiogenic phenotype[171].
Dammeijer et al.[172] provide a thorough review of primary and secondary resistance mechanisms and treatment options for re-sensitizing tumors. Guo et al.[80] provide a compact review of the wide variety of immunosuppressive mechanisms employed by tumors. Chen and Mellman[173] describe these mechanisms in the context of the cancer immunity cycle. A concise table of many elements that underlie tumor escape is given by Accolla and Tosi[174]. Seliger[150] and Seliger et al.[175] review MHC-I and MHC-II-based evasion mechanisms, respectively. A report of HLA allele-specific risk of metastasis in papillary thyroid cancer[176] provides evidence that MHC allele status impacts cancer progression.
Frameworks for understanding tumor-immune system interactions
Reducing therapeutic resistance is closely tied to our understanding of how cancer arises in the context of the immune system, which we briefly discuss here. The primary framework for understanding the interplay between cancer and the immune system is known as immunoediting[177]. This posits that selection pressure from the immune system “edits” the tumor, forcing it to find a custom response to the local and systemic state of the immune system in order to escape immune pressure after many years of genetic changes.
Therapeutic success is often defined by reduction in the incidence and impact of metastatic cancer. The origin and nature of metastases is a dynamic research area, with much remaining to be discovered. Do metastases represent dissemination of cancer cells from a primary tumor in late stage disease (Halsted-Meyer theory)[178], or do they reflect the outgrowth of pre-existing cells that disseminated early on [Figure 3][179]? TNM staging[180] encourages the former perspective. Weichselbaum and Hellman[181] posit the existence of cancers with intermediate metastatic potential. The hypothesis of cancer dormancy posits that tumor cells may disseminate early and are forced into dormancy in order to survive immune surveillance[182,183]. From this perspective, one goal of immunotherapy is to keep such cells dormant, as opposed to attempting to eliminate them all[184,185]. Such topics have been covered in a chapter-length review, including the different kinds of dormancy, the role of circulating tumor cells and of innate and adaptive immune cells, and ideas for keeping dormant tumor cell indolent[186]. It is evident that this research area is both difficult and in its early stages.
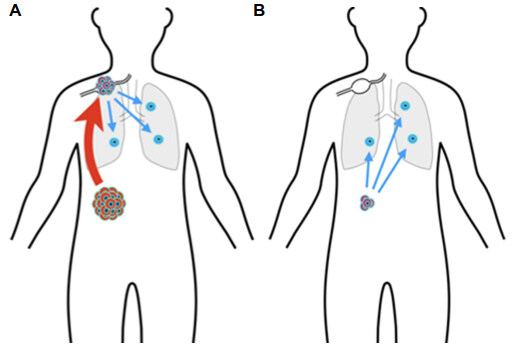
Figure 3. Origin of metastases. (A) TNM staging, closedly related to Halsted-Meyer theory of breast cancer progression, suggests that remote metastases arise late in the progression of the primary tumor, with disseminating cells first traveling to the lymph nodes; (B) the cancer dormancy hypothesis suggests that tumor cells disseminate early to remote sites and are then forced into a dormant state by immune surveillance. These two alternatives can be distinguished in part by examining cell genomes to trace cell lineages
Next steps
A number of authors have sought to identify the most urgent and interesting near-term trends. Whiteside et al.[187] and Hoos[188] foresee a focus on, amongst other topics, understanding PD-1 nonresponders, targeting the tumor microenvironment, improving therapy of tumors with few mutations or low tumor infiltrating lymphocyte count, better tumor and patient assessments, combination therapies, and biomarkers of response. This includes the proposed acknowledgment that stable chronic disease (“functional cure”) is a worthwhile endpoint.
Combination therapy is hailed as possibly the best way to increase the percentage of responders. Current examples of combination therapy tend to have a reactive character, applying an additional therapy in response to failure of an initial one. A strong call was issued in 2015 for increased funding of trials to study how to combine molecularly targeted and immuno-therapies[189].
In general, it is hoped that progress can be made through examination of rational combinations[172]. The meaning of “rational synergy” has been dissected in the context of cytotoxic drug combinations[190]. The diversity of currently available modalities may allow combinations where treatments are carefully scheduled to act as e.g. “mutual adjuvants”. Adjuvants[191-193] continue to be topics of active research, with the line between adjuvant and therapeutic agent blurring. New adjuvants and combinations thereof show promise and are proposed as the focus of clinical trials[194].
Improved outcomes are being reported for combined checkpoint therapies, though at the cost of more adverse events[195,196]. Even those withdrawing from combination treatment due to severe adverse events may still be receiving benefit[197]. Emens et al.[198] present a list of clinical development priorities to push forward the state of the cancer immunotherapy art.
The literature on combination therapies is expanding rapidly. Dunn and Rao[199] have reviewed the combination of epigenetics and immunotherapies. Increasing attention is being paid to traditional targets of the innate immune system. Expression of endogenous viral peptides[200] and dsRNA have been shown to lead to innate and adaptive responses. The role of microbes, both as commensal microbiota that are modulatory targets[201] and as therapeutic agents[202], are subjects of active research. The universe of immunomodulator targets is rapidly growing[203], expanding the scope of possible combinations.
We note that therapy modalities in combination are not necessarily additive, e.g. the combination of chemotherapy (tyrosine kinase inhibitor) and a TAA vaccine required careful scheduling to avoid failure in a mouse model[204]. A literature review of combination (mostly targeted) therapies in metastatic renal cancer expresses both the promise and challenges of extracting benefit from such studies[205]. Careful reporting as captured e.g. in the Consolidated Standards of Reporting Trials (CONSORT) guidelines[206] will be essential for trial data to have maximum value.
Monitoring and modeling
Immunological research is increasingly driven by the ability to gather data, often in a high throughput manner. This opens new vistas that will allow therapy to be properly targeted and monitored, and may eventually alter the character of therapy itself.
The ease with which data can now be generated highlights the responsibility to employ best practices in experimental and trial design, data acquisition (including patient metadata), and downstream analysis. The accumulation of high quality datasets should ideally go hand in hand with the ability to model the data, with the ultimate goal of defining optimal interventions to reach a desired outcome (e.g. disease stabilization or cure)[77].
An initial goal is the discovery of prognostic and predictive biomarkers. These can be used for treatment selection[207], e.g. high PD-L1 expression level for pembrolizumab treatment[208]. At a more rigorous level, biomarkers indicate system state of the immune system, cancer, or both. The importance of prospective studies for data collection and analysis has long been emphasized. REMARK guidelines (REporting recommendations for tumour MARKer prognostic studies) attempt to capture the minimal information needed to objectively assess the import of a given biomarker study[209]. This baseline however is still commonly not met[210].
The field of immune system-related prognostic and predictive biomarkers is complex and rapidly advancing. Urgent efforts are now being made to translate current knowledge and capabilities to the understanding of baseline immunity and response monitoring, and thence the choice of predictive biomarkers[68,211]. Magnetic resonance imaging (MRI) based biomarkers of response to immunotherapy have been recently proposed[212]. Systemic immune response coordinated across tissues has been observed to be essential to tumor rejection[213]. The fraction of tumor-infiltrating partially exhausted cytotoxic T lymphocytes (peCTLs) correlates with response to anti-PD-1 monotherapy, with a low fraction indicating the use of combination checkpoint blockade therapy[214]. A possible implication is that checkpoint blockade therapy is most effective when the immune system has already mounted a tumor-specific if suppressed response.
TCR repertoire profiling shows promise in immune monitoring and perhaps response prediction[215,216]. Checkpoint blockade is seen to induce diversification of T cell receptor repertoire[217], which has been suggested as a biomarker for PD-1 inhibitor disease stabilization[218]. The assessment of TCR repertoire diversity is becoming increasingly accessible[219]. Important choices such as library preparation method, in-house versus service provider, output data type (raw and/or analyzed), and the use or not of unique molecular identifiers must first be matched to project goals[220]. Basic features of the T cell receptor repertoire are still being revealed, e.g. unexpectedly biased distributions of TCR receptors (CDR3 sequence similarity networks) that change in stereotyped ways with aging, immunization, and antigen selection[221]. Progress has been reported in developing statistical means of “reading” T cell memory, as relates e.g. to cytomegalovirus status and HLA typing[222].
As we dissect components and interactions in more detail, the research enterprise can begin to embrace variation to learn better from animal models[223,224] and humans[225,226], including with respect to age[227]. Data sharing can help ensure technical advances are employed towards broad evidence-based progress[228]. In this regard, standards for reporting neo-antigens, HLA alleles, and TCR repertoires may need to be developed.
Adoption of high throughput technologies such as massively parallel sequencing, immunosequencing, microarrays, mass cytometry, and DNA-barcoded pMHC multimers has led to the advent of systems immunology[20]. Rather than dissect mechanistic relationships between a few actors, systems methods attempt to capture the behavior of the immune system as a whole. The resulting descriptions tend to have a multi-scale (hierarchical) character in both space and time[229,230]. The wide variety of available modeling formalisms and applications has been surveyed by Narang et al.[231].
Mathematical modeling has begun to impact the clinic through efforts to optimize dosage and timing (“schedule optimization”), which have gained a foothold in chemotherapy[232,233] and radiotherapy[234,235]. There is now a rich literature on the modeling of immunotherapy[236,237]. As an example, modeling the kinetics of the immune response[238] reveals the possibility that a proper choice of schedule can summon a robust T cell response, overcoming what appears to be tumor resistance. To increase their impact, such models may need to integrate into Bayesian adaptive trials[63].
One theme borrowed from the physics community is to develop simplified abstract models. Such models can generate powerful predictions, derived from the concept of universality[239]. The observation of unexpected dynamical patterns in the immune system such as oscillations over several days[240] suggests that phenomenological models have an important role to play.
Detailed mechanistic models employ knowledge at the molecular or cell level to explain and predict phenomena[241]. A recent attempt to model the cancer-immune system interaction using 12 immune cell types and 13 cytokines plus cancer cells finds steady state “basins” corresponding to escape, elimination, and equilibrium phases in immunoediting, while also finding oscillatory states[242]. The interested reader is referred to volumes focused on cancer[243] and the immune system[244]. A textbook on computational immunology has recently been released[245].
From an artificial intelligence perspective, therapy can be viewed as planning in the presence of uncertainty. The idea that the immune system can be “steered” has been demonstrated by proof-of-concept in silico work[246]. Cancer cells can be treated as adversaries in a game theory context[247]. In the clinical trials arena, reinforcement learning approaches promise a model-free approach to sequential treatment selection[248,249].
Conclusion
After a long history of doubt and failure, checkpoint blockade therapy has opened the door for cancer immunotherapy[250]. With this key modality now accepted, the full weight of technological progress can be brought to bear. New tools provide windows through which the process of disease and treatment can be viewed. Their integration will allow increasingly sophisticated descriptions of immune system and tumor state. Neoantigen cancer vaccines in particular are beneficiaries of this new environment and are poised to lead the way to more precise and effective therapies.
While neoantigen vaccines can now be created with workflows that are increasingly standardized and almost routine, many challenges lie ahead to elicit their true potential. Foremost is gaining a better understanding of primary and secondary resistance. This can be viewed in the light of theories in which cancer cells and the immune system train each other, for better or worse, over decades.
Combination therapies are now pursued as the next step forwards. The examination of all possible protocols may however become infeasible or at least inefficient. Principled methods will need to be developed to systematically identify promising approaches and learn from both successes and failures. This complexity is also an opportunity to formalize therapy as a strategy and not simply an application of magic bullets. Over the longer term, this promises growth in novel interventions that integrate technology, data, models, and algorithms as part of an interdisciplinary biomedical science.
Declarations
AcknowledgmentsThe authors wish to express their gratitude to Rong (Shirly) Li, who created Figures 1 and 2. They also wish to thank Qiubin Lin, Bin Yu, Gang Fang, Antoine Danchin, Chenli Liu, and Chung-I Wu for enlightening conversations.
Authors’ contributionsConception: Huang JD
Design: Tokuyasu TA, Huang JD
Manuscript writing: Tokuyasu TA
Manuscript editing: Huang JD
Read and approved the final manuscript: Tokuyasu TA, Huang JD
Financial support and sponsorshipThis work was supported by a National Basic Research Program of China (973 Program, No. 2014CB745202); the Shenzhen Peacock Team Project (No. KQTD2015033117210153); the Shenzhen Science and Technology Innovation Committee Basic Science Research Grants (No. JCYJ20170413154523577; No. JCYJ20150629151046896); the Guangdong Natural Science Funds for Distinguished Young Scholar Grant (No. S2013050016987).
Conflicts of interestThere are no conflicts of interest.
Patient consentNot applicable.
Ethics approvalNot applicable.
Copyright© The Author(s) 2018.
REFERENCES
1. Coley WB. The treatment of malignant tumors by repeated inoculations of erysipelas. With a report of ten original cases. 1893. Clin Orthop Relat Res 1991;(262):3-11.
2. Parish CR. Cancer immunotherapy: the past, the present and the future. Immunol Cell Biol 2003;81:106-13.
3. Riera Romo M, Pérez-Martínez D, Castillo Ferrer C. Innate immunity in vertebrates: an overview. Immunology 2016;148:125-39.
4. Litman GW, Rast JP, Fugmann SD. The origins of vertebrate adaptive immunity. Nat Rev Immunol 2010;10:543-53.
5. Xing Y, Hogquist KA. T-Cell tolerance: central and peripheral. Cold Spring Harb Perspect Biol 2012;4:a006957.
6. Whitton JL, editor. Antigen Presentation. Berlin, Heidelberg: Springer; 1998; doi: 10.1007/978-3-642-72045-1.
7. van Endert P, editor. Antigen Processing: Methods and Protocols. Totowa, NJ: Humana Press; 2013; doi: 10.1007/978-1-62703-218-6.
8. Roche PA, Furuta K. The ins and outs of MHC class II-mediated antigen processing and presentation. Nat Rev Immunol 2015;15:203-16.
9. Shah DK. T-cell development in thymus. British Society for Immunology. Available from:https://www.immunology.org/public-information/bitesized-immunology/immune-development/t-cell-development-in-thymus. [Last accessed on 29 Jul 2017].
10. Crespo J, Sun H, Welling TH, Tian Z, Zou W. T cell anergy, exhaustion, senescence, and stemness in the tumor microenvironment. Curr Opin Immunol 2013;25:214-21.
11. Walker LSK, Sansom DM. Confusing signals: recent progress in CTLA-4 biology. Trends Immunol 2015;36:63-70.
12. Manz BN, Jackson BL, Petit RS, Dustin ML, Groves J. T-cell triggering thresholds are modulated by the number of antigen within individual T-cell receptor clusters. Proc Natl Acad Sci U S A 2011;108:9089-94.
13. Reinherz EL. αβ TCR-mediated recognition: relevance to tumor-antigen discovery and cancer immunotherapy. Cancer Immunol Res 2015;3:305-12.
14. Meydan C, Otu HH, Sezerman OU. Prediction of peptides binding to MHC class I and II alleles by temporal motif mining. BMC Bioinformatics 2013;14 Suppl 2:S13.
16. Marsh SGE, Albert ED, Bodmer WF, Bontrop RE, Dupont B, Erlich HA, Fernández-Viña M, Geraghty DE, Holdsworth R, Hurley CK, Lau M, Lee KW, Mach B, Maiers M, Mayr WR, Müller CR, Parham P, Petersdorf EW, Sasazuki T, Strominger JL, Svejgaard A, Terasaki PI, Tiercy JM, Trowsdale J. Nomenclature for factors of the HLA system, 2010. Tissue Antigens 2010;75:291-455.
17. Ghattaoraya GS, Dundar Y, González-Galarza FF, Maia MHT, Santos EJM, da Silva ALS, McCabe A, Middleton D, Alfirevic A, Dickson R, Jones AR. A web resource for mining HLA associations with adverse drug reactions: HLA-ADR. Database (Oxford) 2016;2016:baw069.
18. Robinson J, Soormally AR, Hayhurst JD, Marsh SGE. The IPD-IMGT/HLA database -new developments in reporting HLA variation. Hum Immunol 2016;77:233-7.
19. Arstila TP, Casrouge A, Baron V, Even J, Kanellopoulos J, Kourilsky P. A direct estimate of the human αβ T cell receptor diversity. Science 1999;286:958-61.
20. Davis MM, Tato CM, Furman D. Systems immunology: just getting started. Nat Immunol 2017;18:725-32.
21. Britanova OV, Putintseva EV, Shugay M, Merzlyak EM, Turchaninova MA, Staroverov DB, Bolotin DA, Lukyanov S, Bogdanova EA, Mamedov IZ, Lebedev YB, Chudakov DM. Age-related decrease in TCR repertoire diversity measured with deep and normalized sequence profiling. J Immunol 2014;192:2689-98.
22. Qi Q, Liu Y, Cheng Y, Glanville J, Zhang D, Lee JY, Olshen RA, Weyand CM, Boyd SD, Goronzy JJ. Diversity and clonal selection in the human T-cell repertoire. Proc Natl Acad Sci U S A 2014;111:13139-44.
23. Derhovanessian E, Solana R, Larbi A, Pawelec G. Immunity, ageing and cancer. Immun Ageing 2008;5:11.
24. Elias R, Karantanos T, Sira E, Hartshorn KL. Immunotherapy comes of age: immune aging & checkpoint inhibitors. J Geriatr Oncol 2017;8:229-35.
26. Wucherpfenning KW, Strominger JL. Molecular mimicry in T cell-mediated autoimmunity: viral peptides activate human T cell clones specific for myelin basic protein. Cell 1995;80:695-705.
27. Cole DK, Bulek AM, Dolton G, Schauenberg AJ, Szomolay B, Rittase W, Trimby A, Jothikumar P, Fuller A, Skowera A, Rossjohn J, Zhu C, Miles JJ, Peakman M, Wooldridge L, Rizkallah PJ, Sewell AK. Hotspot autoimmune T cell receptor binding underlies pathogen and insulin peptide cross-reactivity. J Clin Invest 2016;126:2191-204.
28. Kumar A, Delogu F. Dynamical footprint of cross-reactivity in a human autoimmune T-cell receptor. Sci Rep 2017;7:42496.
29. Harndahl M, Rasmussen M, Roder G, Dalgaard Pedersen I, Sørensen M, Nielsen M, Buus S. Peptide-MHC class I stability is a better predictor than peptide affinity of CTL immunogenicity. Eur J Immunol 2012;42:1405-16.
30. Yager EJ, Ahmed M, Lanzer K, Randall TD, Woodland DL, Blackman MA. Age-associated decline in T cell repertoire diversity leads to holes in the repertoire and impaired immunity to influenza virus. J Exp Med 2008;205:711-23.
31. Wölfl M, Rutebemberwa A, Mosbruger T, Mao Q, Li H, Netski D, Ray SC, Pardoll D, Sidney J, Sette A, Allen T, Kuntzen T, Kavanagh DG, Kuball J, Greenberg PD, Cox AL. Hepatitis C virus immune escape via exploitation of a hole in the T cell repertoire. J Immunol 2008;181:6435-46.
32. Vatti A, Monsalve DM, Pacheco Y, Chang C, Anaya JM, Gershwin ME. Original antigenic sin: a comprehensive review. J Autoimmun 2017;83:12-21.
33. Joffre OP, Segura E, Savina A, Amigorena S. Cross-presentation by dendritic cells. Nat Rev Immunol 2012;12:557-69.
34. Fehres CM, Unger WWJ, Garcia-Vallejo JJ, van Kooyk Y. Understanding the biology of antigen cross-presentation for the design of vaccines against cancer. Front Immunol 2014;5:149.
36. Caza T, Landas S. Functional and phenotypic plasticity of CD4+ T cell subsets. Biomed Res Int 2015;2015:521957.
37. van de Berg PJ, van Leeuwen EM, ten Berge IJ, van Lier R. Cytotoxic human CD4+ T cells. Curr Opin Immunol 2008;20:339-43.
38. Takeuchi A, Saito T. CD4 CTL, a cytotoxic subset of CD4+ T cells, their differentiation and function. Front Immunol 2017;8:194.
39. Ley K. The second touch hypothesis: T cell activation, homing and polarization. version 2. F1000 Research 2014;3:37.
40. Crome SQ, Nguyen LT, Lopez-Verges S, Yang SYC, Martin B, Yam JY, Johnson DJ, Nie J, Pniak M, Yen PH, Milea A, Sowamber R, Katz SR, Bernardini MQ, Clarke BA, Shaw PA, Lang PA, Berman HK, Pugh TJ, Lanier LL, Ohashi PS. A distinct innate lymphoid cell population regulates tumor-associated T cells. Nat Med 2017;23:368-75.
42. Flavahan WA, Gaskell E, Bernstein BE. Epigenetic plasticity and the hallmarks of cancer. Science 2017;357:eaal2380.
43. Thomas F, Fisher D, Fort P, Marie J-P, Daoust S, Roche B, Grunau C, Cosseau C, Mitta G, Baghdiguian S, Rousset F, Lassus P, Assenat E, Grégoire D, Missé D, Lorz A, Billy F, Vainchenker W, Delhommeau F, Koscielny S, Itzykson R, Tang R, Fava F, Ballesta A, Lepoutre T, Krasinska L, Dulic V, Raynaud P, Blache P, Quittau-Prevostel C, Vignal E, Trauchessec H, Perthame B, Clairambault J, Volpert V, Solary E, Hibner U, Hochberg ME. Applying ecological and evolutionary theory to cancer: a long and winding road. Evol Appl 2013;6:1-10.
44. Davis A, Gao R, Navin N. Tumor evolution: linear, branching, neutral or punctuated? Biochim Biophys Acta 2017;1867:151-61.
45. Paguirigan AL, Smith J, Meshinchi S, Carroll M, Maley C, Radich JP. Single-cell genotyping demonstrates complex clonal diversity in acute myeloid leukemia. Sci Transl Med 2015;7:281re2.
46. Sun R, Hu Z, Sottoriva A, Graham TA, Harpak A, Ma Z, Fischer JM, Shibata D, Curtis C. Between-region genetic divergence reflects the mode and tempo of tumor evolution. Nat Genet 2017;49:1015-24.
47. Aktipis CA, Kwan VSY, Johnson KA, Neuberg SL, Maley CC. Overlooking evolution: a systematic analysis of cancer relapse and therapeutic resistance research. PLoS One 2011;6:e26100.
49. Weinberg RA. The biology of cancer. Second edition. New York: Garland Science, Taylor & Francis Group; 2014.
50. Abbas AK, Lichtman AH, Pillai S. Cellular and molecular immunology. Philadelphia, PA: Elsevier Saunders; 2015.
51. Rezaei N, editor. Cancer Immunology. Berlin, Heidelberg: Springer Berlin Heidelberg; 2015.
52. Sell S. Cancer immunotherapy: breakthrough or "deja vu, all over again"? Tumor Biol 2017;39:1010428317707764.
53. Korman AJ, Peggs KS, Allison JP. Checkpoint blockade in cancer immunotherapy. Adv Immunol 2006;90:297-339.
54. Palucka K, Banchereau J. Cancer immunotherapy via dendritic cells. Nat Rev Cancer 2012;12:265-77.
55. Gardner T, Elzey B, Hahn NM. Sipuleucel-T (Provenge) autologous vaccine approved for treatment of men with asymptomatic or minimally symptomatic castrate-resistant metastatic prostate cancer. Hum Vaccines Immunother 2012;8:534-9.
56. Rosenberg SA, Restifo NP. Adoptive cell transfer as personalized immunotherapy for human cancer. Science 2015;348:62-8.
58. Gross G, Waks T, Eshhar Z. Expression of immunoglobulin-T-cell receptor chimeric molecules as functional receptors with antibody-type specificity. Proc Natl Acad Sci U S A 1989;86:10024-8.
59. Longoria TC, Eskander RN. Immunotherapy in endometrial cancer - an evolving therapeutic paradigm. Gynecol Oncol Res Pract 2015;2:11.
60. Page DB, Bourla AB, Daniyan A, Naidoo J, Smith E, Smith M, Friedman C, Khalil DN, Funt S, Shoushtari AN, Overwijk WW, Sharma P, Callahan MK. Tumor immunology and cancer immunotherapy: summary of the 2014 SITC primer. J Immunother Cancer 2015;3:25.
61. van Rooij N, van Buuren MM, Philips D, Velds A, Toebes M, Heemskerk B, van Dijk LJ, Behjati S, Hilkmann H, El Atmioui D, Nieuwland M, Stratton MR, Kerkhoven RM, Kesmir C, Haanen JB, Kvistborg P, Schumacher TN. Tumor exome analysis reveals neoantigen-specific t-cell reactivity in an ipilimumab-responsive melanoma. J Clin Oncol 2013;31:e439-42.
62. Hoos A, Eggermont AMM, Janetzki S, Hodi FS, Ibrahim R, Anderson A, Humphrey R, Blumenstein B, Old L, Wolchok J. Improved endpoints for cancer immunotherapy trials. J Natl Cancer Inst 2010;102:1388-97.
63. Bates SE, Berry DA, Balasubramaniam S, Bailey S, LoRusso PM, Rubin EH. Advancing clinical trials to streamline drug development. Clin Cancer Res 2015;21:4527-35.
65. Kato S, Goodman A, Walavalkar V, Barkauskas DA, Sharabi A, Kurzrock R. Hyperprogressors after immunotherapy: analysis of genomic alterations associated with accelerated growth rate. Clin Cancer Res 2017;23:4242-50.
66. Tedeschi B. Cancer researchers worry immunotherapy may hasten growth of tumors in some patients. Available from: https://www.statnews.com/2017/04/03/immunotherapy-cancer-tumor-growth/. [Last accessed on 15 Dec 2017].
67. Galon J, Pagès F, Marincola FM, Angell HK, Thurin M, Lugli A, Zlobec I, Berger A, Bifulco C, Botti G, Tatangelo F, Britten CM, Kreiter S, Chouchane L, Delrio P, Arndt H, Asslaber M, Maio M, Masucci GV, Mihm M, Vidal-Vanaclocha F, Allison JP, Gnjatic S, Hakansson L, Huber C, Singh-Jasuja H, Ottensmeier C, Zwierzina H, Laghi L, Grizzi F, Ohashi PS, Shaw PA, Clarke BA, Wouters BG, Kawakami Y, Hazama S, Okuno K, Wang E, O'Donnell-Tormey J, Lagorce C, Pawelec G, Nishimura MI, Hawkins R, Lapointe R, Lundqvist A, Khleif SN, Ogino S, Gibbs P, Waring P, Sato N, Torigoe T, Itoh K, Patel PS, Shukla SN, Palmqvist R, Nagtegaal ID, Wang Y, D'Arrigo C, Kopetz S, Sinicrope FA, Trinchieri G, Gajewski TF, Ascierto PA, Fox BA. Cancer classification using the Immunoscore: a worldwide task force. J Transl Med 2012;10:205.
68. Gnjatic S, Bronte V, Brunet LR, Butler MO, Disis ML, Galon J, Hakansson LG, Hanks BA, Karanikas V, Khleif SN, Kirkwood JM, Miller LD, Schendel DJ, Tanneau I, Wigginton JM, Butterfield LH. Identifying baseline immune-related biomarkers to predict clinical outcome of immunotherapy. J Immunother Cancer 2017;5:44.
69. Zitvogel L, Galluzzi L, Smyth MJ, Kroemer G. Mechanism of action of conventional and targeted anticancer therapies: reinstating immunosurveillance. Immunity 2013;39:74-88.
70. O'Donnell JS, Massi D, Teng MWL, Mandala M. PI3K-AKT-mTOR inhibition in cancer immunotherapy, redux. Semin Cancer Biol 2017; doi: 10.1016/j.semcancer.2017.04.015.
71. Casey SC, Baylot V, Felsher DW. MYC: master regulator of immune privilege. Trends Immunol 2017;38:298-305.
72. Bracci L, Schiavoni G, Sistigu A, Belardelli F. Immune-based mechanisms of cytotoxic chemotherapy: implications for the design of novel and rationale-based combined treatments against cancer. Cell Death Differ 2014;21:15-25.
73. Tey SK. Adoptive T-cell therapy: adverse events and safety switches. Clin Transl Immunol 2014;3:e17.
74. Bertrand A, Kostine M, Barnetche T, Truchetet ME, Schaeverbeke T. Immune related adverse events associated with anti-CTLA-4 antibodies: systematic review and meta-analysis. BMC Med 2015;13:211.
75. Villadolid J, Amin A. Immune checkpoint inhibitors in clinical practice: update on management of immune-related toxicities. Transl Lung Cancer Res 2015;4:560-75.
76. Ott PA, Hu Z, Keskin DB, Shukla SA, Sun J, Bozym DJ, Zhang W, Luoma A, Giobbie-Hurder A, Peter L, Chen C, Olive O, Carter TA, Li S, Lieb DJ, Eisenhaure T, Gjini E, Stevens J, Lane WJ, Javeri I, Nellaiappan K, Salazar AM, Daley H, Seaman M, Buchbinder EI, Yoon CH, Harden M, Lennon N, Gabriel S, Rodig SJ, Barouch DH, Aster JC, Getz G, Wucherpfennig K, Neuberg D, Ritz J, Lander ES, Fritsch EF, Hacohen N, Wu CJ. An immunogenic personal neoantigen vaccine for patients with melanoma. Nature 2017;547:217-21.
77. Pappalardo F, Chiacchio F, Motta S. Cancer vaccines: state of the art of the computational modeling approaches. Biomed Res Int 2013;2013:106407.
78. Andor N, Maley CC, Ji HP. Genomic instability in cancer: teetering on the limit of tolerance. Cancer Res 2017;77:2179-85.
79. Coulie PG, Van den Eynde BJ, van der Bruggen P, Boon T. Tumour antigens recognized by T lymphocytes: at the core of cancer immunotherapy. Nat Rev Cancer 2014;14:135-46.
80. Guo C, Manjili MH, Subjeck JR, Sarkar D, Fisher PB, Wang XY. Therapeutic cancer vaccines: past, present, and future. Adv Cancer Res 2013;119:421-75.
81. Melief CJM, Hall T van, Arens R, Ossendorp F, van der Burg SH. Therapeutic cancer vaccines. J Clin Invest 2015;125:3401-12.
82. Gubin MM, Artyomov MN, Mardis ER, Schreiber RD. Tumor neoantigens: building a framework for personalized cancer immunotherapy. J Clin Invest 2015;125:3413-21.
83. Slovin SF, Keding SJ, Ragupathi G. Carbohydrate vaccines as immunotherapy for cancer. Immunol Cell Biol 2005;83:418-28.
84. Neefjes J, Ovaa H. A peptide's perspective on antigen presentation to the immune system. Nat Chem Biol 2013;9:769-75.
85. Melief CJM, van der Burg SH. Immunotherapy of established (pre)malignant disease by synthetic long peptide vaccines. Nat Rev Cancer 2008;8:351-60.
86. Kumai T, Kobayashi H, Harabuchi Y, Celis E. Peptide vaccines in cancer—old concept revisited. Curr Opin Immunol 2017;45:1-7.
88. Sayour EJ, Mitchell DA. Manipulation of innate and adaptive immunity through cancer vaccines. J Immunol Res 2017;2017:3145742.
89. Kramps T, Elbers K, editors. RNA Vaccines. New York, NY: Springer New York; 2017; doi: 10.1007/978-1-4939-6481-9.
91. Beitelshees M, Li Y, Pfeifer BA. Enhancing vaccine effectiveness with delivery technology. Curr Opin Biotechnol 2016;42:24-9.
92. Yang Y, Hou J, Lin Z, Zhuo H, Chen D, Zhang X, Chen Y, Sun B. Attenuated listeria monocytogenes as a cancer vaccine vector for the delivery of CD24, a biomarker for hepatic cancer stem cells. Cell Mol Immunol 2014;11:184-96.
93. Cawood R, Hills T, Wong SL, Alamoudi AA, Beadle S, Fisher KD, Fisher KD, Seymour LW. Recombinant viral vaccines for cancer. Trends Mol Med 2012;18:564-74.
94. Swartz MA, Hirosue S, Hubbell JA. Engineering approaches to immunotherapy. Sci Transl Med 2012;4:148rv9.
95. Mehta NK, Moynihan KD, Irvine DJ. Engineering new approaches to cancer vaccines. Cancer Immunol Res 2015;3:836-43.
96. Francis DM, Thomas SN. Progress and opportunities for enhancing the delivery and efficacy of checkpoint inhibitors for cancer immunotherapy. Adv Drug Deliv Rev 2017;114:33-42.
98. Backert L, Kohlbacher O. Immunoinformatics and epitope prediction in the age of genomic medicine. Genome Med 2015;7:119.
99. Segal NH, Parsons DW, Peggs KS, Velculescu V, Kinzler KW, Vogelstein B, Allison JP. Epitope landscape in breast and colorectal cancer. Cancer Res 2008;68:889-92.
100. Nielsen M, Lundegaard C, Lund O, Keşmir C. The role of the proteasome in generating cytotoxic T-cell epitopes: insights obtained from improved predictions of proteasomal cleavage. Immunogenetics 2005;57:33-41.
101. Snyder A, Chan TA. Immunogenic peptide discovery in cancer genomes. Curr Opin Genet Dev 2015;30:7-16.
102. Vita R, Overton JA, Greenbaum JA, Ponomarenko J, Clark JD, Cantrell JR, Wheeler DK, Gabbard JL, Hix D, Sette A, Peters B. The immune epitope database (IEDB) 3.0. Nucleic Acids Res 2015;43:D405-12.
103. Olsen LR, Tongchusak S, Lin H, Reinherz EL, Brusic V, Zhang GL. TANTIGEN: a comprehensive database of tumor T cell antigens. Cancer Immunol Immunother 2017;66:731-5.
104. Lund O, Karosiene E, Lundegaard C, Larsen MV, Nielsen M. Bioinformatics identification of antigenic peptide: predicting the specificity of major MHC class I and II pathway players. In: van Endert P, editor. Antigen processing: methods and protocols. Humana Press, Totowa, NJ; 2013. pp. 247-60.
105. Bjerregaard AM, Nielsen M, Hadrup SR, Szallasi Z, Eklund AC. MuPeXI: prediction of neo-epitopes from tumor sequencing data. Cancer Immunol Immunother 2017;66:1123-30.
106. Schubert B, de la Garza L, Mohr C, Walzer M, Kohlbacher O. ImmunoNodes - graphical development of complex immunoinformatics workflows. BMC Bioinformatics 2017;18:242.
107. Kobayashi H, Wood M, Song Y, Appella E, Celis E. Defining promiscuous MHC class II helper T-cell epitopes for the HER2/neu tumor antigen. Cancer Res 2000;60:5228-36.
108. Stern LJ, Santambrogio L. The melting pot of the MHC II peptidome. Curr Opin Immunol 2016;40:70-7.
109. Hudrisier D, Gairin JE. Peptide-major histocompatibility complex class I complex: from the structural and molecular basis to pharmacological principles and therapeutic applications. In: Whitton JL, editor. Antigen presentation. Berlin, Heidelberg: Springer; 1998. pp. 75-97.
110. Faro J, Castro M, Molina-París C. A unifying mathematical framework for experimental TCR-pMHC kinetic constants. Sci Rep 2017;7:46741.
111. Yarchoan M, Johnson Iii BA, Lutz ER, Laheru DA, Jaffee EM. Targeting neoantigens to augment antitumour immunity. Nat Rev Cancer 2017;17:209-22.
113. Yadav M, Jhunjhunwala S, Phung QT, Lupardus P, Tanguay J, Bumbaca S, Franci C, Cheung TK, Fritsche J, Weinschenk T, Modrusan Z, Mellman I, Lill JR, Delamarre L. Predicting immunogenic tumour mutations by combining mass spectrometry and exome sequencing. Nature 2014;515:572-6.
114. Kowalewski DJ, Schuster H, Backert L, Berlin C, Kahn S, Kanz L, Salih HR, Rammensee HG, Stevanovic S, Stickel JS. HLA ligandome analysis identifies the underlying specificities of spontaneous antileukemia immune responses in chronic lymphocytic leukemia (CLL). Proc Natl Acad Sci U S A 2015;112:E166-75.
115. Bassani-Sternberg M, Bräunlein E, Klar R, Engleitner T, Sinitcyn P, Audehm S, Straub M, Weber J, Slotta-Huspenina J, Specht K, Martignoni ME, Werner A, Hein R, H Busch D, Peschel C, Rad R, Cox J, Mann M, Krackhardt AM. Direct identification of clinically relevant neoepitopes presented on native human melanoma tissue by mass spectrometry. Nat Commun 2016;7:13404.
116. Bentzen AK, Marquard AM, Lyngaa R, Saini SK, Ramskov S, Donia M, Such L, Furness AJ, McGranahan N, Rosenthal R, Straten PT, Szallasi Z, Svane IM, Swanton C, Quezada SA, Jakobsen SN, Eklund AC, Hadrup SR. Large-scale detection of antigen-specific T cells using peptide-MHC-I multimers labeled with DNA barcodes. Nat Biotechnol 2016;34:1037-45.
117. Bunse L, Schumacher T, Sahm F, Pusch S, Oezen I, Rauschenbach K, Gonzalez M, Solecki G, Osswald M, Capper D, Wiestler B, Winkler F, Herold-Mende C, von Deimling A, Wick W, Platten M. Proximity ligation assay evaluates IDH1R132H presentation in gliomas. J Clin Invest 2015;125:593-606.
118. Schumacher T, Bunse L, Pusch S, Sahm F, Wiestler B, Quandt J, Menn O, Osswald M, Oezen I, Ott M, Keil M, Balß J, Rauschenbach K, Grabowska AK, Vogler I, Diekmann J, Trautwein N, Eichmüller SB, Okun J, Stevanović S, Riemer AB, Sahin U, Friese MA, Beckhove P, von Deimling A, Wick W, Platten M. A vaccine targeting mutant IDH1 induces antitumour immunity. Nature 2014;512:324-7.
119. Scheikl-Gatard T, Tosch C, Lemonnier F, Rooke R. Identification of new MUC1 epitopes using HLA-transgenic animals: implication for immunomonitoring. J Transl Med 2017;15:154.
120. Schmidt J, Guillaume P, Dojcinovic D, Karbach J, Coukos G, Luescher I. In silico and cell-based analyses reveal strong divergence between prediction and observation of T-cell-recognized tumor antigen T-cell epitopes. J Biol Chem 2017;292:11840-9.
121. Gros A, Parkhurst MR, Tran E, Pasetto A, Robbins PF, Ilyas S, Prickett TD, Gartner JJ, Crystal JS, Roberts IM, Trebska-McGowan K, Wunderlich JR, Yang JC, Rosenberg SA. Prospective identification of neoantigen-specific lymphocytes in the peripheral blood of melanoma patients. Nat Med 2016;22:433-8.
122. Tsherniak A, Vazquez F, Montgomery PG, Weir BA, Kryukov G, Cowley GS, Gill S, Harrington WF, Pantel S, Krill-Burger JM, Meyers RM, Ali L, Goodale A, Lee Y, Jiang G, Hsiao J, Gerath WFJ, Howell S, Merkel E, Ghandi M, Garraway LA, Root DE, Golub TR, Boehm JS, Hahn WC. Defining a cancer dependency map. Cell 2017;170:564-76.e16.
123. Uhlen M, Zhang C, Lee S, Sjöstedt E, Fagerberg L, Bidkhori G, Benfeitas R, Arif M, Liu Z, Edfors F, Sanli K, von Feilitzen K, Oksvold P, Lundberg E, Hober S, Nilsson P, Mattsson J, Schwenk JM, Brunnström H, Glimelius B, Sjöblom T, Edqvist PH, Djureinovic D, Micke P, Lindskog C, Mardinoglu A, Ponten F. A pathology atlas of the human cancer transcriptome. Science 2017;357:eaan2507.
124. Van Waes C, Monach PA, Urban JL, Wortzel RD, Schreiber H. Immunodominant deters the response to other tumor antigens thereby favoring escape: prevention by vaccination with tumor variants selected with cloned cytolytic T cells in vitro. Tissue Antigens 1996;47:399-407.
125. Kim A, Sadegh-Nasseri S. Determinants of immunodominance for CD4 T cells. Curr Opin Immunol 2015;34:9-15.
126. Macdonald IK, Harkiolaki M, Hunt L, Connelley T, Carroll AV, MacHugh ND, Graham SP, Jones EY, Morrison WI, Flower DR, Ellis SA. MHC class I bound to an immunodominant theileria parva epitope demonstrates unconventional presentation to T cell receptors. PLoS Pathog 2010;6:e1001149.
127. Hunt JD, Brown LE, Jackson DC. Antigenic competition. Chichester, UK: John Wiley & Sons, Ltd; 2001; doi: 10.1038/npg.els.0000948.
129. Palmowski M, Salio M, Dunbar RP, Cerundolo V. The use of HLA class I tetramers to design a vaccination strategy for melanoma patients. Immunol Rev 2002;188:155-63.
130. Slingluff CL. The present and future of peptide vaccines for cancer: single or multiple, long or short, alone or in combination? Cancer J 2011;17:343-50.
131. Kumai T, Fan A, Harabuchi Y, Celis E. Cancer immunotherapy: moving forward with peptide T cell vaccines. Curr Opin Immunol 2017;47:57-63.
132. Baxevanis CN, Voutsas IF, Tsitsilonis OE, Gritzapis AD, Sotiriadou R, Papamichail M. Tumor-specific CD4+ T lymphocytes from cancer patients are required for optimal induction of cytotoxic T cells against the autologous tumor. J Immunol 2000;164:3902-12.
133. Nakanishi Y, Lu B, Gerard C, Iwasaki A. CD8+ T lymphocyte mobilization to virus-infected tissue requires CD4+ T-cell help. Nature 2009;462:510-3.
134. Sun Z, Chen F, Meng F, Wei J, Liu B. MHC class II restricted neoantigen: a promising target in tumor immunotherapy. Cancer Lett 2017;392:17-25.
136. Quezada SA, Simpson TR, Peggs KS, Merghoub T, Vider J, Fan X, Blasberg R, Yagita H, Muranski P, Antony PA, Restifo NP, Allison JP. Tumor-reactive CD4+ T cells develop cytotoxic activity and eradicate large established melanoma after transfer into lymphopenic hosts. J Exp Med 2010;207:637-50.
137. Tran E, Turcotte S, Gros A, Robbins PF, Lu YC, Dudley ME, Wunderlich JR, Somerville RP, Hogan K, Hinrichs CS, Parkhurst MR, Yang JC, Rosenberg SA. Cancer immunotherapy based on mutation-specific CD4+ T cells in a patient with epithelial cancer. Science 2014;344:641-5.
138. Sahin U, Derhovanessian E, Miller M, Kloke B-P, Simon P, Löwer M, Bukur V, Tadmor AD, Luxemburger U, Schrörs B, Omokoko T, Vormehr M, Albrecht C, Paruzynski A, Kuhn AN, Buck J, Heesch S, Schreeb KH, Müller F, Ortseifer I, Vogler I, Godehardt E, Attig S, Rae R, Breitkreuz A, Tolliver C, Suchan M, Martic G, Hohberger A, Sorn P, Diekmann J, Ciesla J, Waksmann O, Brück AK, Witt M, Zillgen M, Rothermel A, Kasemann B, Langer D, Bolte S, Diken M, Kreiter S, Nemecek R, Gebhardt C, Grabbe S, Höller C, Utikal J, Huber C, Loquai C, Türeci Ö. Personalized RNA mutanome vaccines mobilize poly-specific therapeutic immunity against cancer. Nature 2017;547:222-6.
139. Kreiter S, Vormehr M, van de Roemer N, Diken M, Löwer M, Diekmann J, Boegel S, Schrörs B, Vascotto F, Castle JC, Tadmor AD, Schoenberger SP, Huber C, Türeci Ö, Sahin U. Mutant MHC class II epitopes drive therapeutic immune responses to cancer. Nature 2015;520:692-6.
140. Young MRI. Cancer immunology with a focus on understudied cancers as targets for immunotherapy. Int J Mol Sci 2017;18:E127.
141. Baxevanis CN, Papamichail M, Perez SA. Prostate cancer vaccines: the long road to clinical application. Cancer Immunol Immunother 2015;64:401-8.
142. Roszik J, Haydu LE, Hess KR, Oba J, Joon AY, Siroy AE, Karpinets TV, Stingo FC, Baladandayuthapani V, Tetzlaff MT, Wargo JA, Chen K, Forget MA, Haymaker CL, Chen JQ, Meric-Bernstam F, Eterovic AK, Shaw KR, Mills GB, Gershenwald JE, Radvanyi LG, Hwu P, Futreal PA, Gibbons DL, Lazar AJ, Bernatchez C, Davies MA, Woodman SE. Novel algorithmic approach predicts tumor mutation load and correlates with immunotherapy clinical outcomes using a defined gene mutation set. BMC Med 2016;14:168.
143. Campesato LF, Barroso-Sousa R, Jimenez L, Correa BR, Sabbaga J, Hoff PM, Reis LF, Galante PA, Camargo AA. Comprehensive cancer-gene panels can be used to estimate mutational load and predict clinical benefit to PD-1 blockade in clinical practice. Oncotarget 2015;6:34221-7.
144. Alexandrov LB, Nik-Zainal S, Wedge DC, Aparicio SAJR, Behjati S, Biankin AV, Bignell GR, Bolli N, Borg A, Børresen-Dale AL, Boyault S, Burkhardt B, Butler AP, Caldas C, Davies HR, Desmedt C, Eils R, Eyfjörd JE, Foekens JA, Greaves M, Hosoda F, Hutter B, Ilicic T, Imbeaud S, Imielinski M, Jäger N, Jones DT, Jones D, Knappskog S, Kool M, Lakhani SR, López-Otín C, Martin S, Munshi NC, Nakamura H, Northcott PA, Pajic M, Papaemmanuil E, Paradiso A, Pearson JV, Puente XS, Raine K, Ramakrishna M, Richardson AL, Richter J, Rosenstiel P, Schlesner M, Schumacher TN, Span PN, Teague JW, Totoki Y, Tutt AN, Valdés-Mas R, van Buuren MM, van 't Veer L, Vincent-Salomon A, Waddell N, Yates LR; Australian Pancreatic Cancer Genome Initiative; ICGC Breast Cancer Consortium; ICGC MMML-Seq Consortium; ICGC PedBrain, Zucman-Rossi J, Futreal PA, McDermott U, Lichter P, Meyerson M, Grimmond SM, Siebert R, Campo E, Shibata T, Pfister SM, Campbell PJ, Stratton MR. Signatures of mutational processes in human cancer. Nature 2013;500:415-21.
145. Le DT, Durham JN, Smith KN, Wang H, Bartlett BR, Aulakh LK, Lu S, Kemberling H, Wilt C, Luber BS, Wong F, Azad NS, Rucki AA, Laheru D, Donehower R, Zaheer A, Fisher GA, Crocenzi TS, Lee JJ, Greten TF, Duffy AG, Ciombor KK, Eyring AD, Lam BH, Joe A, Kang SP, Holdhoff M, Danilova L, Cope L, Meyer C, Zhou S, Goldberg RM, Armstrong DK, Bever KM, Fader AN, Taube J, Housseau F, Spetzler D, Xiao N, Pardoll DM, Papadopoulos N, Kinzler KW, Eshleman JR, Vogelstein B, Anders RA, Diaz LA Jr. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science 2017;357:409-13.
146. Kloor M, Michel S, von Knebel Doeberitz M. Immune evasion of microsatellite unstable colorectal cancers. Int J Cancer 2010;127:1001-10.
147. Queirolo P, Spagnolo F. Atypical responses in patients with advanced melanoma, lung cancer, renal-cell carcinoma and other solid tumors treated with anti-PD-1 drugs: a systematic review. Cancer Treat Rev 2017;59:71-8.
148. Anagnostou V, Smith KN, Forde PM, Niknafs N, Bhattacharya R, White J, Zhang T, Adleff V, Phallen J, Wali N, Hruban C, Guthrie VB, Rodgers K, Naidoo J, Kang H, Sharfman W, Georgiades C, Verde F, Illei P, Li QK, Gabrielson E, Brock MV, Zahnow CA, Baylin SB, Scharpf RB, Brahmer JR, Karchin R, Pardoll DM, Velculescu VE. Evolution of neoantigen landscape during immune checkpoint blockade in non-small cell lung cancer. Cancer Discov 2017;7:264-76.
149. Gulley JL, Madan RA, Pachynski R, Mulders P, Sheikh NA, Trager J, Drake CG. Role of antigen spread and distinctive characteristics of immunotherapy in cancer treatment. J Natl Cancer Inst 2017:109.
150. Seliger B. Molecular mechanisms of HLA class I-mediated immune evasion of human tumors and their role in resistance to immunotherapies. HLA 2016;88:213-20.
151. Khanna R. Tumour surveillance: missing peptides and MHC molecules. Immunol Cell Biol 1998;76:20-6.
152. Thor Straten P, Garrido F. Targetless T cells in cancer immunotherapy. J Immunother Cancer 2016;4:23.
153. Mahmoud F, Shields B, Makhoul I, Avaritt N, Wong HK, Hutchins LF, Shalin S, Tackett AJ. Immune surveillance in melanoma: from immune attack to melanoma escape and even counterattack. Cancer Biol Ther 2017;18:451-69.
154. Gajewski TF, Woo S-R, Zha Y, Spaapen R, Zheng Y, Corrales L, Spranger S. Cancer immunotherapy strategies based on overcoming barriers within the tumor microenvironment. Curr Opin Immunol 2013;25:268-76.
155. Dilek N, Vuillefroy de Silly R, Blancho G, Vanhove B. Myeloid-derived suppressor cells: mechanisms of action and recent advances in their role in transplant tolerance. Front Immunol 2012;3:208.
157. Yang L, Zhang Y. Tumor-associated macrophages: from basic research to clinical application. J Hematol Oncol 2017;10:58.
158. Joyce JA, Fearon DT. T cell exclusion, immune privilege, and the tumor microenvironment. Science 2015;348:74-80.
159. Spranger S, Dai D, Horton B, Gajewski TF. Tumor-residing Batf3 dendritic cells are required for effector T cell trafficking and adoptive T cell therapy. Cancer Cell 2017;31:711-23.e4.
160. Buck MD, Sowell RT, Kaech SM, Pearce EL. Metabolic instruction of immunity. Cell 2017;169:570-86.
161. Chang C-H, Pearce EL. Emerging concepts of T cell metabolism as a target of immunotherapy. Nat Immunol 2016;17:364-8.
162. Schietinger A, Philip M, Krisnawan VE, Chiu EY, Delrow JJ, Basom RS, Lauer P, Brockstedt DG, Knoblaugh SE, Hämmerling GJ, Schell TD, Garbi N, Greenberg PD. Tumor-specific T cell dysfunction is a dynamic antigen-driven differentiation program initiated early during tumorigenesis. Immunity 2016;45:389-401.
163. Schietinger A, Greenberg PD. Tolerance and exhaustion: defining mechanisms of T cell dysfunction. Trends Immunol 2014;35:51-60.
164. Quezada SA, Peggs KS, Simpson TR, Allison JP. Shifting the equilibrium in cancer immunoediting: from tumor tolerance to eradication. Immunol Rev 2011;241:104-18.
166. Marcus A, Gowen BG, Thompson TW, Iannello A, Ardolino M, Deng W, Wang L, Shifrin N, Raulet DH. Recognition of tumors by the innate immune system and natural killer cells. Adv Immunol 2014;122:91-128.
167. Dahlberg CIM, Sarhan D, Chrobok M, Duru AD, Alici E. Natural killer cell-based therapies targeting cancer: possible strategies to gain and sustain anti-tumor activity. Front Immunol 2015;6:605.
168. Morvan MG, Lanier LL. NK cells and cancer: you can teach innate cells new tricks. Nat Rev Cancer 2016;16:7-19.
169. Ferlazzo G, Morandi B. Cross-talks between natural killer cells and distinct subsets of dendritic cells. Front Immunol 2014;5:159.
170. Cai X, Caballero-Benitez A, Gewe MM, Jenkins IC, Drescher CW, Strong RK, Spies T, Groh V. Control of tumor initiation by NKG2D naturally expressed on ovarian cancer cells. Neoplasia N Y N 2017;19:471-82.
171. Bruno A, Ferlazzo G, Albini A, Noonan DM. A think tank of TINK/TANKs: tumor-infiltrating/tumor-associated natural killer cells in tumor progression and angiogenesis. J Natl Cancer Inst 2014;106:dju200.
172. Dammeijer F, Lau SP, van Eijck CHJ, van der Burg SH, Aerts JGJV. Rationally combining immunotherapies to improve efficacy of immune checkpoint blockade in solid tumors. Cytokine Growth Factor Rev 2017;36:5-15.
173. Chen DS, Mellman I. Oncology meets immunology: the cancer-immunity cycle. Immunity 2013;39:1-10.
174. Accolla RS, Tosi G. Optimal MHC-II-restricted tumor antigen presentation to CD4+ T helper cells: the key issue for development of anti-tumor vaccines. J Transl Med 2012;10:154.
175. Seliger B, Kloor M, Ferrone S. HLA class II antigen-processing pathway in tumors: molecular defects and clinical relevance. Oncoimmunology 2017;6:e1171447.
176. Ehlers M, Kuebart A, Hautzel H, Enczmann J, Reis AC, Haase M, Allelein S, Dringenberg T, Schmid C, Schott M. Epitope-specific antitumor immunity suppresses tumor spread in papillary thyroid cancer. J Clin Endocrinol Metab 2017;102:2154-61.
177. Vesely MD, Schreiber RD. Cancer immunoediting: antigens, mechanisms, and implications to cancer immunotherapy. Ann N Y Acad Sci 2013;1284:1-5.
178. Zurrida S, Bassi F, Arnone P, Martella S, Del Castillo A, Ribeiro Martini R, Semenkiw ME, Caldarella P. The changing face of mastectomy (from mutilation to aid to breast reconstruction). Int J Surg Oncol 2011;2011:980158.
179. Dittmer J. Mechanisms governing metastatic dormancy in breast cancer. Semin Cancer Biol 2017;44:72-82.
182. Sosa MS, Bragado P, Aguirre-Ghiso JA. Mechanisms of disseminated cancer cell dormancy: an awakening field. Nat Rev Cancer 2014;14:611-22.
183. Eyles J, Puaux A-L, Wang X, Toh B, Prakash C, Hong M, Tan TG, Zheng L, Ong LC, Jin Y, Kato M, Prévost-Blondel A, Chow P, Yang H, Abastado JP. Tumor cells disseminate early, but immunosurveillance limits metastatic outgrowth, in a mouse model of melanoma. J Clin Invest 2010;120:2030-9.
184. Romero I, Garrido F, Garcia-Lora AM. Metastases in immune-mediated dormancy: a new opportunity for targeting cancer. Cancer Res 2014;74:6750-7.
185. Manjili MH. Tumor dormancy and relapse: from a natural byproduct of evolution to a disease state. Cancer Res 2017;77:2564-9.
186. Barkan D, Chambers AF. Prevention of conversion of tumor dormancy into proliferative metastases. In: Cote RJ, Datar RH, editors. Circulating tumor cells. Current cancer research. New York, NY: Springer New York; 2016. pp. 121-37.
187. Whiteside TL, Demaria S, Rodriguez-Ruiz ME, Zarour HM, Melero I. Emerging opportunities and challenges in cancer immunotherapy. Clin Cancer Res 2016;22:1845-55.
188. Hoos A. Development of immuno-oncology drugs—from CTLA4 to PD1 to the next generations. Nat Rev Drug Discov 2016;15:235-47.
189. Sharma P, Allison JP. Immune checkpoint targeting in cancer therapy: toward combination strategies with curative potential. Cell 2015;161:205-14.
190. Hucl T, Gallmeier E, Kern SE. Distinguishing rational from irrational applications of pharmacogenetic synergies from the bench to clinical trials. Cell Cycle 2007;6:1336-41.
191. Khong H, Overwijk WW. Adjuvants for peptide-based cancer vaccines. J Immunother Cancer 2016;4:56.
192. Saxena M, Bhardwaj N. Turbocharging vaccines: emerging adjuvants for dendritic cell based therapeutic cancer vaccines. Curr Opin Immunol 2017;47:35-43.
193. Circelli L, Tornesello M, Buonaguro FM, Buonaguro L. Use of adjuvants for immunotherapy. Hum Vaccines Immunother 2017;13:1774-7.
194. Temizoz B, Kuroda E, Ishii KJ. Vaccine adjuvants as potential cancer immunotherapeutics. Int Immunol 2016;28:329-38.
195. Hodi FS, Chesney J, Pavlick AC, Robert C, Grossmann KF, McDermott DF, Linette GP, Meyer N, Giguere JK, Agarwala SS, Shaheen M, Ernstoff MS, Minor DR, Salama AK, Taylor MH, Ott PA, Horak C, Gagnier P, Jiang J, Wolchok JD, Postow MA. Combined nivolumab and ipilimumab versus ipilimumab alone in patients with advanced melanoma: 2-year overall survival outcomes in a multicentre, randomised, controlled, phase 2 trial. Lancet Oncol 2016;17:1558-68.
196. Wu Y, Shi H, Jiang M, Qiu M, Jia K, Cao T, Shang Y, Shi L, Jiang K, Wu H. The clinical value of combination of immune checkpoint inhibitors in cancer patients: a meta-analysis of efficacy and safety. Int J Cancer 2017;141:2562-70.
197. Schadendorf D, Wolchok JD, Hodi FS, Chiarion-Sileni V, Gonzalez R, Rutkowski P, Grob JJ, Cowey CL, Lao CD, Chesney J, Robert C, Grossmann K, McDermott D, Walker D, Bhore R, Larkin J, Postow MA. Efficacy and safety outcomes in patients with advanced melanoma who discontinued treatment with nivolumab and ipilimumab because of adverse events: a pooled analysis of randomized phase II and III trials. J Clin Oncol 2017;35:3807-14.
198. Emens LA, Ascierto PA, Darcy PK, Demaria S, Eggermont AMM, Redmond WL, Seliger B, Marincola FM. Cancer immunotherapy: opportunities and challenges in the rapidly evolving clinical landscape. Eur J Cancer 2017;81:116-29.
199. Dunn J, Rao S. Epigenetics and immunotherapy: the current state of play. Mol Immunol 2017;87:227-39.
200. Probst P, Kopp J, Oxenius A, Colombo MP, Ritz D, Fugmann T, Neri D. Sarcoma eradication by doxorubicin and targeted TNF relies upon CD8+ T-cell recognition of a retroviral antigen. Cancer Res 2017;77:3644-54.
201. Fessler JL, Gajewski TF. The microbiota: a new variable impacting cancer treatment outcomes. Clin Cancer Res 2017;23:3229-31.
202. Li CX, Yu B, Shi L, Geng W, Lin QB, Ling CC, Yang M, Ng KT, Huang JD, Man K. "Obligate" anaerobic Salmonella strain YB1 suppresses liver tumor growth and metastasis in nude mice. Oncol Lett 2017;13:177-83.
203. Mahoney KM, Rennert PD, Freeman GJ. Combination cancer immunotherapy and new immunomodulatory targets. Nat Rev Drug Discov 2015;14:561-84.
204. Jaini R, Rayman P, Cohen PA, Finke JH, Tuohy VK. Combination of sunitinib with anti-tumor vaccination inhibits T cell priming and requires careful scheduling to achieve productive immunotherapy. Int J Cancer 2014;134:1695-705.
205. Albiges L, Choueiri T, Escudier B, Galsky M, George D, Hofmann F, Lam T, Motzer R, Mulders P, Porta C, Powles T, Sternberg C, Bex A. A systematic review of sequencing and combinations of systemic therapy in metastatic renal cancer. Eur Urol 2015;67:100-10.
206. Moher D, Hopewell S, Schulz KF, Montori V, Gøtzsche PC, Devereaux PJ, Elbourne D, Egger M, Altman DG; Consolidated Standards of Reporting Trials Group. CONSORT 2010 explanation and elaboration: updated guidelines for reporting parallel group randomised trials. J Clin Epidemiol 2010;63:e1-37.
207. Simon R. Development and validation of biomarker classifiers for treatment selection. J Stat Plan Inference 2008;138:308-20.
208. Gridelli C, Ardizzoni A, Barberis M, Cappuzzo F, Casaluce F, Danesi R, Troncone G, De Marinis F. Predictive biomarkers of immunotherapy for non-small cell lung cancer: results from an Experts Panel Meeting of the Italian Association of Thoracic Oncology. Transl Lung Cancer Res 2017;6:373-86.
209. Altman DG, McShane LM, Sauerbrei W, Taube SE. Reporting Recommendations for Tumor Marker Prognostic Studies (REMARK): explanation and elaboration. PLoS Med 2012;9:e1001216.
210. Sekula P, Mallett S, Altman DG, Sauerbrei W. Did the reporting of prognostic studies of tumour markers improve since the introduction of REMARK guideline? A comparison of reporting in published articles. PLoS One 2017;12:e0178531.
211. Stroncek DF, Butterfield LH, Cannarile MA, Dhodapkar MV, Greten TF, Grivel JC, Kaufman DR, Kong HH, Korangy F, Lee PP, Marincola F, Rutella S, Siebert JC, Trinchieri G, Seliger B. Systematic evaluation of immune regulation and modulation. J Immunother Cancer 2017;5:21.
212. Haris M, Bagga P, Hariharan H, McGettigan-Croce B, Johnson LA, Reddy R. Molecular imaging biomarkers for cell-based immunotherapies. J Transl Med 2017;15:140.
213. Spitzer MH, Carmi Y, Reticker-Flynn NE, Kwek SS, Madhireddy D, Martins MM, Gherardini PF, Prestwood TR, Chabon J, Bendall SC, Fong L, Nolan GP, Engleman EG. Systemic immunity is required for effective cancer immunotherapy. Cell 2017;168:487-502.e15.
214. Loo K, Tsai KK, Mahuron K, Liu J, Pauli ML, Sandoval PM, Nosrati A, Lee J, Chen L, Hwang J, Levine LS, Krummel MF, Algazi AP, Alvarado MD, Rosenblum MD, Daud AI. Partially exhausted tumor-infiltrating lymphocytes predict response to combination immunotherapy. JCI Insight 2017;2:93433.
216. Gros A, Robbins PF, Yao X, Li YF, Turcotte S, Tran E, Wunderlich JR, Mixon A, Farid S, Dudley ME, Hanada K, Almeida JR, Darko S, Douek DC, Yang JC, Rosenberg SA. PD-1 identifies the patient-specific CD8+ tumor-reactive repertoire infiltrating human tumors. J Clin Invest 2014;124:2246-59.
217. Robert L, Tsoi J, Wang X, Emerson R, Homet B, Chodon T, Mok S, Huang RR, Cochran AJ, Comin-Anduix B, Koya RC, Graeber TG, Robins H, Ribas A. CTLA4 blockade broadens the peripheral t-cell receptor repertoire. Clin Cancer Res 2014;20:2424-32.
218. Akyüz N, Brandt A, Stein A, Schliffke S, Mährle T, Quidde J, Goekkurt E, Loges S, Haalck T, Ford CT, Asemissen AM, Thiele B, Radloff J, Thenhausen T, Krohn-Grimberghe A, Bokemeyer C, Binder M. T-cell diversification reflects antigen selection in the blood of patients on immune checkpoint inhibition and may be exploited as liquid biopsy biomarker. Int J Cancer 2017;140:2535-44.
219. Dziubianau M, Hecht J, Kuchenbecker L, Sattler A, Stervbo U, Rödelsperger C, Nickel P, Neumann AU, Robinson PN, Mundlos S, Volk HD, Thiel A, Reinke P, Babel N. TCR repertoire analysis by next generation sequencing allows complex differential diagnosis of T cell-related pathology. Am J Transplant 2013;13:2842-54.
220. Rosati E, Dowds CM, Liaskou E, Henriksen EKK, Karlsen TH, Franke A. Overview of methodologies for T-cell receptor repertoire analysis. BMC Biotechnol 2017;17:61.
221. Madi A, Poran A, Shifrut E, Reich-Zeliger S, Greenstein E, Zaretsky I, Arnon T, Laethem FV, Singer A, Lu J, Sun PD, Cohen IR, Friedman N. T cell receptor repertoires of mice and humans are clustered in similarity networks around conserved public CDR3 sequences. Elife 2017;6:e22057.
222. Emerson RO, DeWitt WS, Vignali M, Gravley J, Hu JK, Osborne EJ, Desmarais C, Klinger M, Carlson CS, Hansen JA, Rieder M, Robins HS. Immunosequencing identifies signatures of cytomegalovirus exposure history and HLA-mediated effects on the T cell repertoire. Nat Genet 2017;49:659-65.
223. Garner JP, Gaskill BN, Weber EM, Ahloy-Dallaire J, Pritchett-Corning KR. Introducing Therioepistemology: the study of how knowledge is gained from animal research. Lab Anim (NY) 2017;46:103-13.
224. Klevorn LE, Teague RM. Adapting cancer immunotherapy models for the real world. Trends Immunol 2016;37:354-63.
227. Hurez V, Padrón áS, Svatek RS, Curiel TJ. Considerations for successful cancer immunotherapy in aged hosts. Clin Exp Immunol 2017;187:53-63.
228. Institute of Medicine (US). Sharing clinical research data: workshop summary. Washington (DC): National Academies Press (US); 2013.
229. Klinke DJ 2nd. A multiscale systems perspective on cancer, immunotherapy, and interleukin-12. Mol Cancer 2010;9:242.
230. Palsson S, Hickling TP, Bradshaw-Pierce EL, Zager M, Jooss K, O'Brien PJ, Spilker ME, Palsson BO, Vicini P. The development of a fully-integrated immune response model (FIRM) simulator of the immune response through integration of multiple subset models. BMC Syst Biol 2013;7:95.
231. Narang V, Decraene J, Wong SY, Aiswarya BS, Wasem AR, Leong SR, Gouaillard A. Systems immunology: a survey of modeling formalisms, applications and simulation tools. Immunol Res 2012;53:251-65.
232. Scharovsky OG, Mainetti LE, Rozados VR. Metronomic chemotherapy: changing the paradigm that more is better. Curr Oncol 2009;16:7-15.
233. Romiti A, Falcone R, Roberto M, Marchetti P. Current achievements and future perspectives of metronomic chemotherapy. Invest New Drugs 2017;35:359-74.
235. Michor F, Beal K. Improving cancer treatment via mathematical modeling: surmounting the challenges is worth the effort. Cell 2015;163:1059-63.
236. dePillis LG, Eladdadi A, Radunskaya AE. Modeling cancer-immune responses to therapy. J Pharmacokinet Pharmacodyn 2014;41:461-78.
237. Konstorum A, Vella AT, Adler AJ, Laubenbacher RC. Addressing current challenges in cancer immunotherapy with mathematical and computational modelling. J R Soc Interface 2017;14:20170150.
238. Radunskaya A, Hook S. Modeling the kinetics of the immune response. In: d'Onofrio A, Cerrai P, Gandolfi A, editors. New challenges for cancer systems biomedicine. Milano: Springer Milan; 2012. pp. 267-82.
241. Transtrum MK, Qiu P. Bridging mechanistic and phenomenological models of complex biological systems. PLoS Comput Biol 2016;12:e1004915.
242. Wenbo L, Wang J. Uncovering the underlying mechanism of cancer tumorigenesis and development under an immune microenvironment from global quantification of the landscape. J R Soc Interface 2017;14:20170105.
243. d'Onofrio A, Gandolfi A, editors. Mathematical Oncology 2013. New York, NY: Springer New York; 2014.
244. Molina-París C, Lythe G, editors. Mathematical models and immune cell biology. New York, NY: Springer New York; 2011.
245. Bassaganya-Riera J, editor. Computational immunology. Cambridge, MA: Academic Press; 2016.
246. Kroer C, Sandholm T. Sequential planning for steering immune system adaptation. New York, USA: AAAI Press/International Joint Conferences on Artificial Intelligence; 2016.
247. Sandholm T. Steering evolution strategically: computational game theory and opponent exploitation for treatment planning, drug design, and synthetic biology. 2015. Available from: https://www.aaai.org/ocs/index.php/AAAI/AAAI15/paper/view/10047. [Last accessed on 15 Dec 2017].
248. Zhao Y, Zeng D, Socinski MA, Kosorok MR. Reinforcement learning strategies for clinical trials in nonsmall cell lung cancer. Biometrics 2011;67:1422-33.
249. Wallace MP, Moodie EEM, Stephens DA. SMART thinking: a review of recent developments in sequential multiple assignment randomized trials. Curr Epidemiol Rep 2016;3:225-32.
250. Littman DR. releasing the brakes on cancer immunotherapy. Cell 2015;162:1186-90.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Tokuyasu TA, Huang JD. A primer on recent developments in cancer immunotherapy, with a focus on neoantigen vaccines. J Cancer Metastasis Treat 2018;4:2. http://dx.doi.org/10.20517/2394-4722.2017.52
AMA Style
Tokuyasu TA, Huang JD. A primer on recent developments in cancer immunotherapy, with a focus on neoantigen vaccines. Journal of Cancer Metastasis and Treatment. 2018; 4: 2. http://dx.doi.org/10.20517/2394-4722.2017.52
Chicago/Turabian Style
Tokuyasu, Taku A., Jian-Dong Huang. 2018. "A primer on recent developments in cancer immunotherapy, with a focus on neoantigen vaccines" Journal of Cancer Metastasis and Treatment. 4: 2. http://dx.doi.org/10.20517/2394-4722.2017.52
ACS Style
Tokuyasu, TA.; Huang J.D. A primer on recent developments in cancer immunotherapy, with a focus on neoantigen vaccines. J. Cancer. Metastasis. Treat. 2018, 4, 2. http://dx.doi.org/10.20517/2394-4722.2017.52
About This Article
Special Issue
Copyright

Data & Comments
Data


 Cite This Article 13 clicks
Cite This Article 13 clicks

 Like This Article 32
likes
Like This Article 32
likes






Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.