
Increased ARF6 activation correlates with HGF stimulation in non-invasive prostate cancer cells
 0
0
Abstract
Aim: The effects of hepatocyte growth factor (HGF) on a non-invasive prostate cancer cell line (CAHPV-10), expressing cMET were studied, to mimic the possible effects neo-adjuvant androgen deprivation therapy may have in promoting tumour progression.
Methods: Prostate epithelial cells and prostate cancer cells derived from cancer metastatic sites were analysed using cell culture assays, immunofluorescence, quantitative real-time polymerase chain reaction and western blotting, with or without HGF stimulation.
Results: HGF significantly enhanced cell proliferation and induced cell scattering and invasion in CAHPV-10 cells compared to untreated controls. Active adenosine diphosphate-ribosylation factor 6 (ARF6) was found to be present in all metastatic prostate cancer cells, with levels highest in the most aggressive cell line, PC-3. Following stimulation with HGF, active ARF6 expression was substantially elevated in CAHPV-10 cells.
Conclusion: These findings provide further molecular insight into the progression of prostate cancer and highlights potential issues for early prostate cancer therapeutic strategies.
Keywords
Introduction
Prostate cancer (PCa) is the most common cancer in men in the UK. Every year more than 35,000 cases of PCa are diagnosed, which equates to one man every 15 min and accounts for approximately 12% of all male deaths from cancer in the UK[1]. Androgens play a significant role in the growth and progression of PCa. The current therapy for locally advanced and metastatic disease is androgen deprivation therapy (ADT) either through orchidectomy, luteinising hormone releasing hormone or blockade through the androgen receptor (AR). For men with localised PCa, the two major treatment options are surgery by radical prostatectomy or radiotherapy. However, ADT is becoming increasingly important in the early stages[2]. ADT can be given in the neoadjuvant setting to reduce tumour size for improved excision during surgery or increase the effectiveness of radiotherapy[3]. ADT can also be given for men with localised PCa who are unfit for curative treatment (surgery or radiotherapy) or whose cancer has begun to progress and become symptomatic[4].
However, several studies have shown that following androgen suppression, expression of hepatocyte growth factor (HGF) or its receptor c-Met are elevated in prostate tissues[5,6]. Additional studies have also shown that suppression of the AR increases cMet expression in PCa cell lines[7,8] while increased c-Met expression is induced by removal of androgens in PCa cells[8]. HGF is a multifunctional cytokine, which in the prostate is secreted by prostate stromal cells and activates c-Met in a paracrine manner[9]. It is widely accepted that HGF dependent c-Met activation is involved in tumour development, including PCa, by inducing cell proliferation[10] and activating stages of the metastatic cascade by stimulating migration[10,11], cell scattering[12], invasion[13] and angiogenesis[14].
It has been reported that serum levels of HGF correlate with stage of prostate malignancy. Thus, HGF serum levels are higher in men with localised PCa compared to healthy controls and further elevated in men with metastatic PCa compared to localised disease[15-17]. Plasma levels of HGF have also been documented as predicting PCa metastasis to the lymph nodes as well as recurrence following surgery[15]. Additionally, while c-Met expression is linked to disease progression[5,18,19] elevated c-Met has been documented in localised PCa tissue when compared to healthy controls[20]. With the importance of HGF and c-Met in PCa well documented, it has been hypothesised that while current ADT potentially inhibits AR mediated cell proliferation and survival, it could also abolish its suppressive role on the HGF/c-Met pathway[7] and therefore may unintentionally drive tumour progression. In addition, while there are a plethora of studies evaluating the effects of HGF in PCa[21-24], the majority focus on metastatic cell lines and hence advanced cancer. We, however, have sought to investigate the effect of HGF on a non-invasive cell line called CAHPV-10, known to express cMET[25] with the aim of ascertaining the molecular effects that may result due to ADT therapy on early state PCa and its role in promoting tumour progression and metastasis.
Furthermore, HGF has been shown to activate adenosine diphosphate-ribosylation factor 6 (ARF6), which is a member of the Ras superfamily of GTPases[26,27]. Increased levels of activated ARF6 [ARF6-guanosine triphosphate (ARF6-GTP)] have been found to increase the invasive capacity of melanoma cells both in vitro[28] and in vivo[29], while silencing ARF6, by small-interfering RNA, has been shown to inhibit the ability of breast cancer cells to invade through an artificial basement membrane[30]. While we have recently shown that ARF proteins are over-expressed in PCa tissue compared to normal control tissue[31], published studies on the presence of ARF6 in PCa are scant. The aims of the present investigation were to determine 1) if ARF6 is up-regulated in invasive PCa cells; 2) to investigate whether HGF stimulation correlates with active ARF6 expression in cells derived from non-invasive PCa.
Methods
Cell culture
Prostate epithelial cells (PNT2) and PCa cells derived from cancer metastasis to the lymph nodes (LNCaP) and bone (PC-3) were obtained from the European Collection of Cell Cultures. PCa cells derived from non-invasive prostate cancer (CAHPV-10) and metastatic cancer to the brain (DU145) were purchased from the American Type Culture Collection. The cell lines were routinely cultured as described in Morgan et al.[32].
Gene expression analysis for ARF6
Gene expression analysis was carried out as described in Morgan et al.[32] and the following ARF6 primers were used: 5’-TGTGGGTTTCAACGTGGAGAC-3’ and 5’-CAGTGTAGTAATGCCGCCAGAG-3’. β-actin and HPRT were used as reference genes, therefore the β-actin primers used were 5’-GATGGCCACGGCTGCTTC-3’ and 5’-TGCCTCAGGGCAGCGGAA-3’. HRPT primers used were 5’-GACTGTAGATTTTATCAGACTGA-3’ and 5’-TGGATTATACTGCCTGACCAA-3’.
Western blot analysis
RIPA buffer (Sigma Aldrich, Dorset, UK) was used to extract the total protein, of which 30 μg was run a 12% tris-glycine PAGE gels (Bio-Rad Laboratories, Hemel Hempstead, UK). The western blot was carried out as described in Morgan et al.[32].
Primary mouse antibody to ARF6 (1:1000), as well as horseradish peroxidase conjugated secondary anti-mouse were used (1:1000) (supplied in the Active ARF6 pull down and detection. β-actin (1:1000) (New England Biolabs, Hertfordshire, UK) was a control for protein loading.
Immunofluorescence for localisation of ARF6
All prostate cells were seeded at 1 × 105 cells/mL onto sterile microscope slides. Slide preparation was carried out as previously described by Morgan et al.[32]. Immunofluorescence of ARF6 expression was performed by first incubating the slides with rabbit anti-ARF6 (1:1000 in 6% BSA/PBS) (abcam, Cambridge Science Park, Cambridge, UK) for 2 h at 37 °C. Following the washing steps to remove the primary antibody, secondary antibody anti-rabbit (Qdot525 Invitrogen, Paisley UK) (1:100 in 6% BSA/PBS) was added and the sample incubated for 1 h at room temperature. The slides were again washed in water and fixed in ethanol (70%, 85% and 95%) for 2 min each. The AxioCam fluorescent microscope (Carl Zeiss Ltd, Hertfordshire, UK) was then used to analyse the slides. Negative controls (to rule out autofluorescence) were carried out by substituting the primary antibody for 1% BSA/PBS.
GST pull down assay for activated ARF6
All prostate cells were seeded in a T-175 cm3 flask and grown to 80% confluence. Total protein was extracted and ARF6-GTP isolated using the Active ARF6 pull down and detection kit (Thermo Scientific Langenselbold, Germany) according to manufacturer’s instructions. Briefly, cells were lysed using 1 mL lysis buffer and agitated on ice for 5 min before being scraped into a 2 mL tube and sheared through a 20 gauge needle 5 times and centrifuged at 16,000 g at 4 °C for 15 min.
The supernatant containing the total lysate was transferred to a new tube. An aliquot of the cell lysate was reserved for protein quantification using the Pierce BCA (Thermo Scientific, Langenselbold, Germany) and for detection of total ARF6 by western blot analysis.
For detection of activated ARF6, the remaining lysate was added to a spin column containing GST-GGA3-beads and incubated for 1 h at 4 °C with gentle rocking. The column was then centrifuged at 6000 g for 10-30 s and washed with 400 µL of Lysis/Binding/Wash Buffer before centrifugation for a second time at 6000 g for 10-30 s. Twenty five microlitres of the elute (ARF6-GTP) and 25 µL of total protein lystate (total ARF6) were used for western blot analysis, as described above and experiments were performed in triplicate.
HGF induced cell proliferation
CAHPV-10 cells were seeded at 2000 cells/100 µL in 96 well plates and incubated at 37 °C for 24 h to allow for cell adherence and growth. Cells were then washed twice in phosphate buffered saline (PBS) (Invitrogen, Paisley UK) and the cells grown for a further 24 h in serum starved medium (0.5% FCS). Hepatocyte growth factor (R&D systems, Minneapolis, USA) was then added to the cells at 10, 25, 50 and 100 ng/mL for 24 and 48 h. PBS/0.1 BSA (Bovine Serum Albumen, Sigma Aldrich, Dorset, UK) was used as a negative control. Cell proliferation was assessed using the Cell 96® AQueous One Solution Cell Proliferation Assay (Promega, Southampton, UK) according to the manufacturers’ instructions. Four replicates for each HGF concentration were performed and the experiment repeated in triplicate.
HGF induced cell invasion
The invasive potential of CAHPV-10 cells following HGF treatment were assessed using a commercial cell invasion assay kit (Innocyte Cell Invasion Assay kit, Calbiochem, Merk, Middlesex, UK) utilising 8 µmol/L pore transwell inserts pre-coated with basement membrane extract. Briefly, the basement membrane extract was rehydrated by adding 300-400 µL of warm, serum-free medium for 30-60 min at room temperature. Following incubation the serum free medium was removed and 350 µL of a 1 × 106 cell suspension was added to each insert. Five hundred microlitres of medium containing either 10-100 ng/mL HGF or 0.1% BSA/PBS vehicle control were added to both the upper and lower and the chambers of the transwell plate and incubated for 48 h. Following incubation, the inserts were removed and placed into unused 24-well plates containing 500 µL of cell staining solution. Cells that had migrated through the basement membrane to the underside of the insert were then dislodged by tapping the insert against the bottom of the well before incubating for a further 30 min. Following incubation the inserts were removed and the wells containing the dislodged cells was incubated for an additional 30 min. Finally, 200 µL of the dislodged cell suspension were added to triplicate wells of a black 96-well plate (Thermo Scientific Langenselbold, Germany) and the fluorescence measured using an excitation wavelength of 485 nm and an emission wavelength of 520 nm. This experiment was performed in triplicate.
HGF induced cell scattering
CAHPV-10 and DU145 (positive control) cells were seeded at a density of 1 × 104 cells/mL of culture medium in 6-well plates. Cells were incubated until small colonies formed. Cells were then serum starved (0.5% FCS) for 24 h prior to stimulation with 10-100 ng/mL HGF for 24 and 48 h. Changes in cell morphology were observed using a light microscope (Carl Zeiss Ltd, Hertfordshire, UK) and representative colonies were photographed using a Canon Powershot A640. Negative controls using 0.1% BSA/PBS were also performed. The experiment was performed in triplicate.
HGF stimulation of CAHPV-10 cells for activation of ARF6
It was observed that CAHPV-10 cells, having very low levels of activated ARF6, were used to ascertain whether HGF stimulation could activate ARF6 in a non-invasive cell line. CAHPV10 cells were seeded as above and grown to 80% confluence. Cells were then treated with either 50 ng/mL HGF or 0.1% BSA/PBS vehicle control for 48 h. GST pull down assay was performed as described above.
Statistical analysis
To ascertain if our data was normally distributed a one sample Kolmogorov-Smirnov statistical test was performed. The resulting P > 0.05, indicated the data had a normal distribution. Parametric analysis was carried out including ANOVA test, Dunnett’s post hoc analysis to compare HGF treated with control cells and Tukey post hoc analysis was performed for multiple group comparisons. Statistical significance was considered for P < 0.05. For the invasion assay, Student T-test was performed and a P < 0.05 was considered significant.
Results
HGF stimulation induced cell proliferation and invasion
We wished to assess whether HGF could increase cell proliferation and invasion in a cell line derived from a non-invasive tumour. The results [Figure 1A and B] show PCa cell proliferation was significantly enhanced by HGF at 24 (P < 0.001) and 48 h (P < 0.001) when compared to untreated control cells, yet interestingly there was no significant difference between concentrations of HGF or time points, suggesting that the c-Met receptor or downstream signaling pathways have reached saturation point at 10 ng/mL.
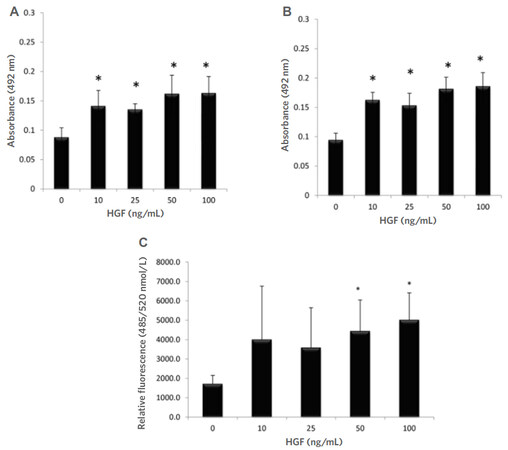
Figure 1. HGF significantly increased the proliferation of CAHPV-10 non-invasive prostate cancer cells over (A) 24 h and (B) 48 h. CAHPV-10 cells were serum starved for 24 h before exposure to HGF. The data are expressed as mean ± standard deviation (SD) (from 4 replicates repeated in triplicate). *Denotes significant difference between treated and control cells (P < 0.05); C: HGF significantly increased the invasive capacity of CAHPV-10 cells. Cells were seeded onto an artificial basement membrane extract in transwell inserts and serum starved for 24 h before treatment with HGF for 48 h. The data are expressed as mean (from triplicate experiments) ± SD. *Denotes significant difference between treated and control cells (P < 0.05). HGF: hepatocyte growth factor
It can also be seen from Figure 1C that 48 h HGF stimulation at 50 and 100 ng/mL significantly increased CAHPV-10 invasion through an artificial basement membrane (P < 0.042 and P < 0.019 respectively) with an approximate 2-fold increase in cell numbers crossing the artificial membrane compared to untreated controls. These results show that exposing PCa cells, derived from a localised tumour, to HGF increases not only their proliferation but also their invasive capacity.
HGF induced cell scattering in non-invasive cancer cells
To determine whether HGF stimulation could induce cell scattering in a non-invasive cell line, we exposed CAHPV-10 cells (grown in small colonies) to various concentrations of HGF over 24 and 48 h time period. As many studies have shown HGF induce cell scattering in DU145 PCa cells, we also used DU145 cells with and without HGF exposure as a positive and negative controls.
Concentrations of HGF ranged from 0-100 ng/mL. CAHPV-10 cells treated over a 24 h period did not show any significant signs of cell dissociation, spreading or motility. In contrast, following 48 h exposure to HGF, cell dissociation, spreading and motility became evident at 50 and 100 ng/mL HGF [Figure 2].
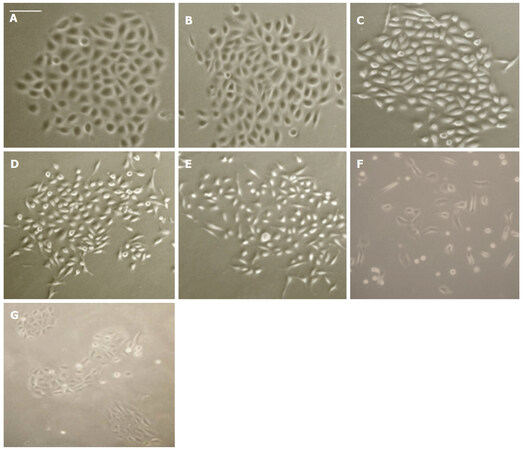
Figure 2. CAHPV-10 non-invasive prostate cancer cells treated with (A) control (PBS + medium); B: 10 ng/mL HGF; C: 25 ng/mL HGF; D: 50 ng/mL HGF; E: 100 ng/mL HGF for 48 h; F: DU145 cells treated with 10 ng/mL for 48 h as a positive control; G: DU145 cells without HGF for 48 h as a negative control. Scale bar (shown in A) represents 25 µmol/L for (A-C) and 50 µmol/L for D-G
ARF6 is expressed in PCa cells
A role for the GTPase ARF6 has been implicated in several cancers but information pertaining to its role in PCa is scant. Gene expression analysis on prostate cells derived from normal prostate epithelium through to aggressive, metastatic disease revealed ARF6 to be present in prostate cells [Figure 3A]. There was no significant difference in ARF6 levels when CAHPV-10 were compared to the normal prostate cell line PNT2 but there was a significant difference between non-invasive and invasive cancer cells. ARF6 levels were significantly higher in LNCaP (3.9-fold), DU145 (3.3-fold) and PC-3 (4.3-fold) cells compared to CAHPV-10 (P < 0.04, 0.032 and 0.009 respectively). There was no significant difference between the invasive cell lines regardless of their differing invasive capacities.
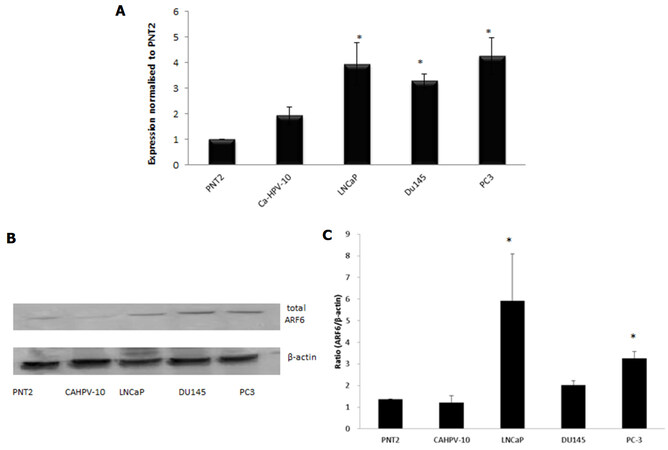
Figure 3. Gene expression analysis shows ARF6 is expressed in prostate cells derived from normal tissue (PNT2), localised prostate cancer (CAHVP-10), weakly metastatic (LNCaP), moderately (DU145) and aggressively (PC-3) metastatic prostate cancer tissue. ARF6 expression was similar between PNT2 and CAHPV-10. A significant increase in expression was observed in LNCaP, DU145 and PC-3 compared to CAHPV-10. The data are expressed as mean (from triplicate experiments), relative to PNT2, ± SD. *Denotes significant difference compared to PNT2 (P < 0.05); B: western blot analysis for total ARF6 protein; C: densitometry data is presented relative to the level of β-actin. The data are expressed as mean (from triplicate experiments) ± SD
To determine whether mRNA levels were translated through to the protein level, we then performed western blot analysis. Protein bands correlating to ARF6 were evident in all samples [Figure 3A] but densitometry [Figure 3C] revealed total levels of ARF6 were not significantly different between PNT2 (1.37) and CAHPV-10 cells (1.21) yet were significantly increased in the metastatic cell lines LNCaP, 5.92 and PC-3, 3.26 (P < 0.001 and P < 0.034 respectively).
Immunofluorescence shows ARF6 localises to the plasma membrane in aggressive cancer cells
Inactive ARF6 (ARF-GDP) localises to the cytosol and endosomes and when activated (ARF-GTP) translocates to the plasma membrane. Therefore, we carried out immunofluorescence to ascertain the localisation of ARF6 in all our PCa cells. Figure 4 shows that staining for ARF6 was predominantly localised to the cell periphery in all cell types. In PNT2 cells, ARF6 staining intensity was very weak but intensity increased with progression to an aggressive phenotype suggesting higher levels of the activated form of ARF6 is present at the cell membrane in cells with an invasive phenotype compared to cells which are derived from normal prostate epithelium or non-invasive cancer tissue.
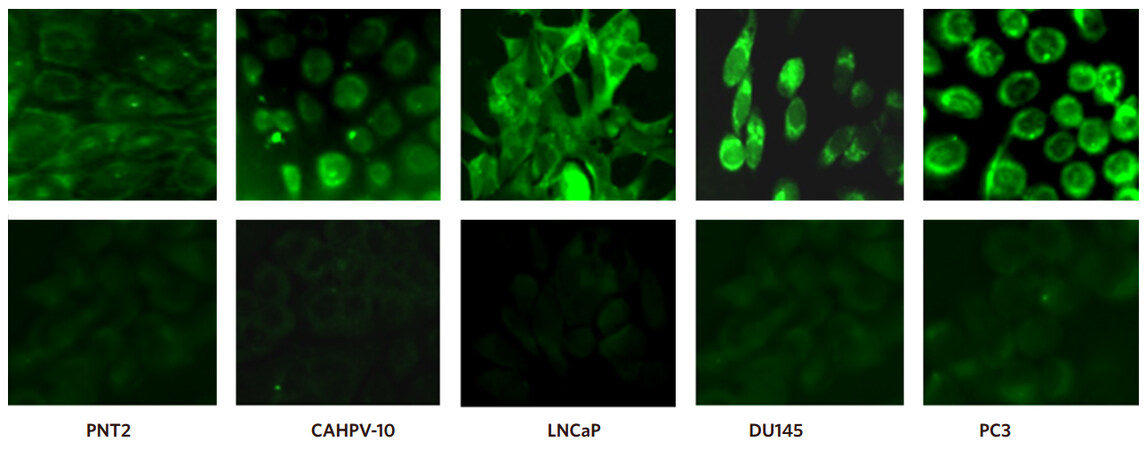
Figure 4. Inactive ARF6 localises to the cytosol and endosomes and when activated translocates to the plasma membrane. Representative immunofluorescence images show cells stained with rabbit anti-ARF6 exhibit a stronger signal intensity at the plasma membrane in the metastatic cell lines (LNCaP, DU145, PC3) than those derived from normal tissue (PNT2) and localised prostate cancer (CAHPV-10). Corresponding negative controls are shown in the bottom row
Active ARF6 can be detected in aggressive PCa cells
To quantify levels of ARF-GTP, we performed a GTP pull down assay, which utilises a fusion protein GST-GGA3 that specifically interacts with the GTP bound form of ARF6. This was performed on all prostate cells representing normal through to aggressive metastatic disease. Western blot analysis of ARF6-GTP and total ARF6 can be seen in Figure 5A, which shows ARF6-GTP to be evident in the invasive cell lines whilst very low levels were observed in the non-invasive cells. Densitometry was performed and a ratio between ARF6-GTP and total levels of ARF6 were calculated. Figure 5b shows levels of ARF6-GTP were significantly higher in LNCaP (P < 0.033) and PC3 (P < 0.034) compared to non-cancerous prostate cells (PNT2). Furthermore, ARF6-GTP levels in LNCaP and PC-3 were significantly higher (> 2.5 fold) than in the localised prostate cells CAHPV10 (P < 0.044 and 0.001 respectively).
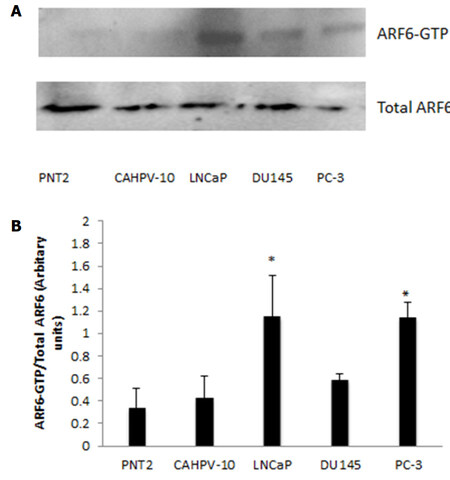
Figure 5. A: GST pull down assay for ARF6-GTP was performed on prostate cells from normal prostate (PNT2), localised prostate cancer (CAHVP-10), weakly metastatic (LNCaP), moderately (DU145) and aggressively (PC-3) metastatic prostate cancer. Twenty five microlitres of GST assay elute was used in western blot analysis for ARF6-GTP detection. A: corresponding 25 µL aliquot from total protein lysis was used for detection of total ARF6 protein; B: band density was measured by densitometry and the ratios of ARF6-GTP to total ARF6 are shown. The data are expressed as mean (from triplicate experiments) ± SD. *Denotes significant difference compared to PNT2 (P < 0.05)
HGF induces the activation of ARF6 in non-invasive cells
HGF is known to activate ARF6, therefore we wished to determine whether HGF stimulation could activate ARF6 in the non-invasive CAHPV-10 cells. Fifty nanograms per millilitre was the lowest concentration that produced a significant effect on cell invasion and a discernable effect on cell scattering, thus, we chose these parameters to stimulate the CAHPV10 cells.
Western blot analysis revealed that the active form of ARF6 was substantially elevated in HGF treated cells compared to BSA/PBS treated control cells so much so that when not stimulated with HGF no discernible band could be detected but after HGF stimulation ARF6 detection was abundantly expressed [Figure 6].
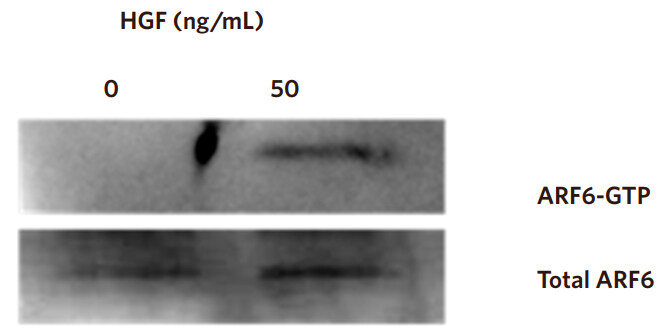
Figure 6. HGF activates ARF6. CAHPV-10 cells were serum starved for 24 h before being exposed to 50 ng/mL HGF for 48 h. Active ARF6 pull down assay and western blot analysis shows activated ARF6 to be present in HGF treated cells but not in untreated control cells
Discussion
While not standard of care, the use of ADT as an early stage treatment option is becoming increasingly important[2]. To mimic the possible effect of neo-adjuvant ADT in early PCa we stimulated a noninvasive cell line, known to express c-Met, with HGF. Our results revealed that HGF stimulation induced cell proliferation, scattering and cell invasion as well as activating ARF6. All of these processes are associated in one way or another with enhancing the aggressive nature of cancer cells and/or are essential components of the metastatic cascade. Cell proliferation was significantly increased in HGF stimulated cells when compared to untreated cells following both 24 and 48 h exposure. This finding is not surprising given that HGF plays an essential role in embryonic development and wound healing[33]. However, HGF stimulation did not produce a time or concentration dependent effect. We hypothesise that this effect is due to the c-MET receptor, or downstream pathways, reaching saturation point at 10 ng/mL and thus, any increases in concentration or length of time has no additional effect on cell proliferation. This is supported by other studies that have shown that changes in HGF and c-MET levels do not always invoke a concentration-dependent response either due to variants in HGF or c-Met or that down-stream signalling pathways become saturated and can no longer be phosphorylated[34].
Interestingly HGF also caused these non-invasive cells to scatter when compared to untreated control cells. HGF has been shown to cause the disruption of a variety of normal epithelial cells resulting in cell migration[35] necessary for wound healing but it is also an essential attribute in the metastatic phenotype. HGF has been shown to induce cell scattering by inhibiting E-cadherin function resulting in cell-cell dissociation[36], the disassembly of cell-cell adhesion complexes[37] and activation of the Ras/MAPK[38] and PI-3 kinase[39] pathways. Our results showed that HGF only induced cell dissociation, spreading and motility at the higher concentrations (50 and 100 ng/mL at 48 h). HGF is typically a paracrine factor, expressed by mesenchyme to activate c-MET in the neighbouring epithelia. Studies have shown that stromal cells secrete HGF in the range of 14-24 ng/mL[34]. Studies have also reported that serum levels increase as PCa progresses with one study showing that serum HGF levels in metastatic cancer patients were 2 times that of localised PCa patients[17]. Thus, in vitro HGF stimulation at high concentrations could possibly mimic the higher levels seen in the tumour microenvironment as a result of ADT and account for the observed phenotypic effects on cell dissociation.
It has been well documented that HGF stimulation enhances prostate tumour cell invasion in vitro[9,19,40,41] while blocking the expression of c-Met reverses the invasive properties of PCa cells[40,42]. Using 50 ng/mL HGF for 48 h we observed a significant increase in the invasive capacity of the CAHPV-10 cells compared to un-stimulated control cells. It may be suggested that an increase in cell proliferation may account for the increased number of cells invading through an artificial basement membrane matrix. However, in our cell proliferation studies, significant increases in cell proliferation were seen at 10 ng/mL of HGF while the number of cells invading through the artificial membrane only significantly increased at 50 ng/mL. Thus, we believe that if the results of the invasion assay could be influenced by an increase in cell proliferation then we would have observed a significant number of cells invading the basement membrane at 10 ng/mL Thus, unlike in previous studies that use cell lines that already have metastatic capabilities, we have shown, for the first time that treating non-invasive PCa cells with HGF can induce an invasive phenotype. Building on from this finding future work would will need to expanded the range of cell types used to include more non-invasive PCa cell lines to achieve a more rounded representation of “PCa”[43].
ARF6 is a member of the Ras superfamily and can be activated by various growth factors, in particular, by HGF[28]. It functions in a range of biological activities and has be shown to play roles in adherens junction disassembly[26], cell migration[44] and cell proliferation[45]. While there is very little information on the presence or activation of ARF6 in PCa we have recently shown that ARF proteins are over-expressed in PCa tissue compared to normal control tissue[31]. In this study, gene expression analysis revealed that ARF6 is expressed in all the prostate cells. Analysis showed that there was no significant difference in expression between cells derived from normal prostate epithelium and localised, non-invasive PCa but a significant difference in expression between non-invasive and invasive cancer cells was evident. ARF6 levels were significantly higher in LNCaP, DU145 and PC-3 cells compared to CAHPV-10 but there was no significant difference between the invasive cell lines regardless of their differing invasive capacities. Protein analysis confirmed the gene expression data and showed that ARF6 protein was detectable in all prostate cells. Like the majority of GTPases, ARF6 cycles between an inactive GDP-bound form and an active GTP-bound form[28]. Inactive ARF6 localises to the cytosol and endosomes and when activated it translocates to the plasma membrane[46]. Immunofluorescence revealed a strong defined signal at the periphery of the cells in both DU145 and PC-3 indicating a close association of ARF6 and the cell membrane while staining intensity appeared to be markedly reduced in both CAHPV-10, and PNT2. This data would appear to suggest that activated ARF6 increases with the aggressiveness of the cancer. To validate the immunofluorescence data, Western blotting and densitometry analysis of ARF6-GTP and total ARF6 were performed. ARF6-GTP was shown to be evident in the invasive cell lines but very low levels in the non-invasive cell lines. The ratio between ARF6-GTP and total levels of ARF6 were also calculated and highlighted the fact that levels of ARF6-GTP were higher in all the cells derived from invasive tumours. In particular, in the cell line PC-3, which is a cell line derived from a highly aggressive PCa that has metastasised to the bone and is representative of the main metastatic site for PCa, ARF6-GTP levels were found to be 3.5 and 2.7 times higher than the levels found in normal and non-invasive cells respectively.
It has been documented that HGF can activate ARF6 in epithelial cells[26]. As neo-adjuvant ADT can increase the levels of HGF, we wanted to determine whether HGF stimulation could activate ARF6 in the non-invasive cell line CAHPV-10. Using 50 ng/mL for 48 h we found that activated ARF6 levels increased compared to untreated CAHPV-10 cells. ARF6 plays an essential role in epithelial and endothelial cell migration and elevated levels of activated ARF6 have been found to increase the invasive capacity of melanoma cells both in vitro[28] and in vivo[29], while silencing ARF6 has been shown to inhibit the ability of breast cancer cells to invade through an artificial basement membrane[30]. Whether HGF stimulated activation of ARF6 is responsible for the increased proliferation, motility and invasiveness observed following HGF stimulation of CAHPV-10 cells now needs to be determined.
In conclusion, ADT is increasingly being used in the neo-adjuvant setting. With the importance of HGF and cMet in PCa documented, it has been hypothesised that while current ADT may inhibit AR mediated cell proliferation and survival, it may abolish its suppressive role on the HGF/c-Met pathway, unintentionally driving tumour progression. We have shown that HGF enhanced cell proliferation, induced scattering and cell invasion in a non-invasive cell line. We have shown that ARF6 is expressed in non-invasive PCa cells and that HGF stimulation correlates with increased levels of activated ARF6. All of these processes are associated with enhancing the aggressive nature of cancer cells. While further work is now needed to determine whether HGF activation of ARF6 is responsible for driving the cellular changes observed in this study, and on additional non-invasive cell lines, these findings provide further molecular insight into the progression of PCa and would suggest that ADT is unintentionally driving PCa through increasing levels of active ARF6.
Declarations
Authors’ contributionsPerformed the experiments: Morgan C, Whiteland H
Helped plan the experiments: Doak SH
Drafted the manuscript: Morgan C, Swithenbank LJ
Reviewed the manuscript: all authors
Availability of data and materialsData can be individually requested and information on materials is provided in the methods section.
Financial support and sponsorshipThis work was funded by The St David’s Medical Foundation and Prostate Action (grant reference number G2009/27).
Conflicts of interestAll authors declare that there are no conflicts of interest.
Ethical approval and consent to participateNot applicable.
Consent for publicationNot applicable.
Copyright© The Author(s) 2018.
REFERENCES
1. Anonymous. Cancer Research UK [Internet]. Available from: http://www.cancerresearchuk.org/health-professional/cancer-statistics/statistics-by-cancer-type/prostate-cancer/incidence. [Last accessed on 14 Aug 2017].
2. Payne H, Mason M. Androgen deprivation therapy as adjuvant/neoadjuvant to radiotherapy for high-risk localised and locally advanced prostate cancer: recent developments. Br J Cancer 2011;105:1628-34.
3. Shelley MD, Kumar S, Coles B, Wilt T, Staffurth J, Mason MD. A systematic review and meta-analysis of randomised trials of neo-adjuvant hormone therapy for localised and locally advanced prostate carcinoma. Cancer Treat Rev 2009;35:9-17.
4. Heidenreich A, Aus G, Bolla M, Joniau S, Matveev VB, Schmid HP, Zattoni F; European Association of Urology. EAU guidelines on prostate cancer. Eur Urol 2008;53:68-80.
5. Humphrey PA, Zhu X, Zarnegar R, Swanson PE, Ratliff TL, Vollmer RT, Day ML. Hepatocyte growth factor and its receptor (c-MET) in prostatic carcinoma. Am J Pathol 1995;147:386-96.
6. Tsuka H, Mori H, Li B, Kanamaru H, Matsukawa S, Okada K. Enhanced hepatocyte growth factor level in human prostate cancer treated with endocrine therapy. Int J Oncol 1998;13:169-76.
7. Maeda A, Nakashiro K, Hara S, Sasaki T, Miwa Y, Tanji N, Yokoyama M, Hamakawa H, Oyasu R. Inactivation of AR activates HGF/c-Met system in human prostatic carcinoma cells. Biochem Biophys Res Commun 2006;347:1158-65.
8. Verras M, Lee J, Xue H, Li TH, Wang Y, Sun Z. The androgen receptor negatively regulates the expression of c-Met: implications for a novel mechanism of prostate cancer progression. Cancer Res 2007;67:967-75.
9. Nishimura K, Kitamura M, Takada S, Nonomura N, Tsujimura A, Matsumiya K, Miki T, Matsumoto K, Okuyama A. Regulation of invasive potential of human prostate cancer cell lines by hepatocyte growth factor. Int J Urol 1998;5:276-81.
10. Takahashi M, Ota S, Shimada T, Hamada E, Kawabe T, Okudaira T, Matsumura M, Kaneko N, Terano A, Nakamura T, et al. Hepatocyte growth factor is the most potent endogenous stimulant of rabbit gastric epithelial cell proliferation and migration in primary culture. J Clin Invest 1995;95:1994-2003.
11. Dignass AU, Lynch-Devaney K, Podolsky DK. Hepatocyte growth factor/scatter factor modulates intestinal epithelial cell proliferation and migration. Biochem Biophys Res Commun 1994;202:701-9.
12. Jeffers M, Rong S, Vande Woude GF. Enhanced tumorigenicity and invasion-metastasis by hepatocyte growth factor/scatter factor-met signalling in human cells concomitant with induction of the urokinase proteolysis network. Mol Cell Biol 1996;16:1115-25.
13. Weidner KM, Hartmann G, Naldini L, Comoglio PM, Sachs M, Fonatsch C, Rieder H, Birchmeier W. Molecular characteristics of HGF-SF and its role in cell motility and invasion. EXS 1993;65:311-28.
14. Silvagno F, Follenzi A, Arese M, Prat M, Giraudo E, Gaudino G, Camussi G, Comoglio PM, Bussolino F. In vivo activation of met tyrosine kinase by heterodimeric hepatocyte growth factor molecule promotes angiogenesis. Arterioscler Thromb Vasc Biol 1995;15:1857-65.
15. Gupta A, Karakiewicz PI, Roehrborn CG, Lotan Y, Zlotta AR, Shariat SF. Predictive value of plasma hepatocyte growth factor/scatter factor levels in patients with clinically localized prostate cancer. Clin Cancer Res 2008;14:7385-90.
16. Nagakawa O, Yamagishi T, Fujiuchi Y, Junicho A, Akashi T, Nagaike K, Fuse H. Serum hepatocyte growth factor activator (HGFA) in benign prostatic hyperplasia and prostate cancer. Eur Urol 2005;48:686-90.
17. Naughton M, Picus J, Zhu X, Catalona WJ, Vollmer RT, Humphrey PA. Scatter factor-hepatocyte growth factor elevation in the serum of patients with prostate cancer. J Urol 2001;165:1325-8.
18. Tsuka H, Mori H, Li B, Kanamaru H, Matsukawa S, Okada K. Expression of c-MET/HGF receptor mRNA and protein in human non-malignant and malignant prostate tissues. Int J Oncol 1998;13:927-34.
19. van Leenders G, van Balken B, Aalders T, Hulsbergen-van de Kaa C, Ruiter D, Schalken J. Intermediate cells in normal and malignant prostate epithelium express c-MET: implications for prostate cancer invasion. Prostate 2002;51:98-107.
20. Pisters LL, Troncoso P, Zhau HE, Li W, von Eschenbach AC, Chung LW. c-met proto-oncogene expression in benign and malignant human prostate tissues. J Urol 1995;154:293-8.
21. Miura H, Nishimura K, Tsujimura A, Matsumiya K, Matsumoto K, Nakamura T, Okuyama A. Effects of hepatocyte growth factor on E-cadherin-mediated cell-cell adhesion in DU145 prostate cancer cells. Urology 2001;58:1064-9.
22. Sanders AJ, Parr C, Mason MD, Jiang WG. Suppression of hepatocyte growth factor activator inhibitor-1 leads to a more aggressive phenotype of prostate cancer cells in vitro. Int J Mol Med 2007;20:613-9.
23. Wells CM, Ahmed T, Masters JR, Jones GE. Rho family GTPases are activated during HGF-stimulated prostate cancer-cell scattering. Cell Motil Cytoskeleton 2005;62:180-94.
24. Yue D, Wang Y, Ma P, Li YY, Chen H, Wang P, Ren CS. Effects of transferred NK4 gene on proliferation, migration, invasion and apoptosis of human prostate cancer DU145 cells. Asian J Androl 2010;12:381-9.
25. Davies G, Jiang WG, Mason MD. Cell-cell adhesion molecules and signaling intermediates and their role in the invasive potential of prostate cancer cells. J Urol 2000;163:985-92.
26. Palacios F, Price L, Schweitzer J, Collard JG, D'Souza-Schorey C. An essential role for ARF6-regulated membrane traffic in adherens junction turnover and epithelial cell migration. EMBO J 2001;20:4973-86.
27. Hongu T, Funakoshi Y, Fukuhara S, Suzuki T, Sakimoto S, Takakura N, Ema M, Takahashi S, Itoh S, Kato M, Hasegawa H, Mochizuki N, Kanaho Y. Arf6 regulates tumour angiogenesis and growth through HGF-induced endothelial β1 integrin recycling. Nat Commun 2015;6:7925.
28. Tague SE, Muralidharan V, D'Souza-Schorey C. ADP-ribosylation factor 6 regulates tumor cell invasion through the activation of the MEK/ERK signaling pathway. Proc Natl Acad Sci U S A 2004;101:9671-6.
29. Muralidharan-Chari V, Hoover H, Clancy J, Schweitzer J, Suckow MA, Schroeder V, Castellino FJ, Schorey JS, D'Souza-Schorey C. ADP-ribosylation factor 6 regulates tumorigenic and invasive properties in vivo. Cancer Res 2009;69:2201-9.
30. Hashimoto S, Onodera Y, Hashimoto A, Tanaka M, Hamaguchi M, Yamada A, Sabe H. Requirement for Arf6 in breast cancer invasive activities. Proc Natl Acad Sci U S A 2004;101:6647-52.
31. Morgan C, Lewis PD, Hopkins L, Burnell S, Kynaston H, Doak SH. Increased expression of ARF GTPases in prostate cancer tissue. Springerplus 2015;4:342.
32. Morgan C, Jenkins SA, Kynaston HG, Doak SH. The role of adhesion molecules as biomarkers for the aggressive prostate cancer phenotype. PLoS One 2013;8:e81666.
33. Stephens P, Hiscox S, Cook H, Jiang WG, Zhiquiang W, Thomas DW. Phenotypic variation in the production of bioactive hepatocyte growth factor/scatter factor by oral mucosal and skin fibroblasts. Wound Repair Regen 2001;9:34-43.
34. Tate A, Isotani S, Bradley MJ, Sikes RA, Davis R, Chung LW, Edlund M. Met-Independent Hepatocyte Growth Factor-mediated regulation of cell adhesion in human prostate cancer cells. BMC Cancer 2006;6:197.
35. Stoker M, Gherardi E, Perryman M, Gray J. Scatter factor is a fibroblast-derived modulator of epithelial cell mobility. Nature 1987;327:239-42.
36. Pasdar M, Li Z, Marreli M, Nguyen BT, Park M, Wong K. Inhibition of junction assembly in cultured epithelial cells by hepatocyte growth factor/scatter factor is concomitant with increased stability and altered phosphorylation of the soluble junctional molecules. Cell Growth Differ 1997;8:451-62.
37. Hiscox S, Jiang WG. Association of the HGF/SF receptor, c-met, with the cell-surface adhesion molecule, E-cadherin, and catenins in human tumor cells. Biochem Biophys Res Commun 1999;261:406-11.
38. Herrera R. Modulation of hepatocyte growth factor-induced scattering of HT29 colon carcinoma cells. Involvement of the MAPK pathway. J Cell Sci 1998;111:1039-49.
39. Potempa S, Ridley AJ. Activation of both MAP kinase and phosphatidylinositide 3-kinase by ras is required for hepatocyte growth factor/scatter factor-induced adherens junction disassembly. Mol Biol Cell 1998;9:2185-200.
40. Dai Y, Siemann DW. BMS-777607, a small-molecule met kinase inhibitor, suppresses hepatocyte growth factor-stimulated prostate cancer metastatic phenotype in vitro. Mol Cancer Ther 2010;9:1554-61.
41. Fujiuchi Y, Nagakawa O, Murakami K, Fuse H, Saiki I. Effect of hepatocyte growth factor on invasion of prostate cancer cell lines. Oncol Rep 2003;10:1001-6.
42. Davies G, Watkins G, Mason MD, Jiang WG. Targeting the HGF/SF receptor c-met using a hammerhead ribozyme transgene reduces in vitro invasion and migration in prostate cancer cells. Prostate 2004;60:317-24.
43. Maitland NJ. Getting closer to prostate cancer in patients - what scientists should want from clinicians. J Cancer Metastasis Treat 2017;3:262-70.
44. Kondo A, Hashimoto S, Yano H, Nagayama K, Mazaki Y, Sabe H. A new paxillin-binding protein, PAG3/Papalpha/KIAA0400, bearing an ADP-ribosylation factor GTPase-activating protein activity, is involved in paxillin recruitment to focal adhesions and cell migration. Mol Biol Cell 2000;11:1315-27.
45. Li M, Wang J, Ng SS, Chan CY, He ML, Yu F, Lai L, Shi C, Chen Y, Yew DT, Kung HF, Lin MC. Adenosine diphosphate-ribosylation factor 6 is required for epidermal growth factor-induced glioblastoma cell proliferation. Cancer 2009;115:4959-72.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Morgan C, Swithenbank LJ, Whiteland H, Doak SH. Increased ARF6 activation correlates with HGF stimulation in non-invasive prostate cancer cells. J Cancer Metastasis Treat 2018;4:46. http://dx.doi.org/10.20517/2394-4722.2018.19
AMA Style
Morgan C, Swithenbank LJ, Whiteland H, Doak SH. Increased ARF6 activation correlates with HGF stimulation in non-invasive prostate cancer cells. Journal of Cancer Metastasis and Treatment. 2018; 4: 46. http://dx.doi.org/10.20517/2394-4722.2018.19
Chicago/Turabian Style
Morgan, Claire, Lucy J. Swithenbank, Helen Whiteland, Shareen H. Doak. 2018. "Increased ARF6 activation correlates with HGF stimulation in non-invasive prostate cancer cells" Journal of Cancer Metastasis and Treatment. 4: 46. http://dx.doi.org/10.20517/2394-4722.2018.19
ACS Style
Morgan, C.; Swithenbank LJ.; Whiteland H.; Doak SH. Increased ARF6 activation correlates with HGF stimulation in non-invasive prostate cancer cells. J. Cancer. Metastasis. Treat. 2018, 4, 46. http://dx.doi.org/10.20517/2394-4722.2018.19
About This Article
Copyright

Data & Comments
Data
0

 Cite This Article 14 clicks
Cite This Article 14 clicks

 Like This Article 30
likes
Like This Article 30
likes






Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.