
Growth factor receptors: promising drug targets in cancer


Abstract
Genetic, epigenetic and somatic changes deregulate the expression of growth factor receptors (GFRs), leading to cancer initiation and progression. Tumor cell growth and survival are orchestrated by clonal expansion and evasion of apoptotic signals in cancer cells. The growth of cells is further supported by angiogenesis and metastasis to distant organs. High expression of GFRs also contributes to the development of resistance. Therefore, therapeutics to target GFRs is a potentially attractive molecular approach to treat cancer more effectively. In this review, we have discussed the contribution of GFRs to cancer development and addressed molecular approaches undertaken to inhibit GFR-mediated pathways. A wide number of monoclonal antibodies (mAbs) and protein kinase inhibitors targeting these GFR-mediated functions are in clinical trials to treat human malignancies. However, most drugs that target GFRs lead to the development of drug resistance and generate adverse effects. Nucleic acid-based therapeutics, e.g. short interfering RNA (siRNA) could be harnessed to selectively silence GFR genes in cancer cells. Different polymer, liposome-based nanocarriers, and the most recently developed pH-sensitive inorganic carbonate apatite nanoparticles have been used in cell culture and preclinical trials for cytoplasmic delivery of the siRNAs targeting different GFR genes. siRNA-based therapeutics have been shown to have significant potential to suppress GFR expression and functions and thus could be developed as molecular therapeutics. Multi-targeting of tumors at different levels by combining various approaches along with chemotherapy would be a promising therapeutic approach to fight the disease. Suitable nanocarriers capable of entrapping siRNA, mAb, GFR inhibitors and classical drugs targeting GFR have potential therapeutic applications.
Keywords
Introduction
The heterogeneous nature of cancer is characterized by continuous clonal expansion and uncontrolled growth of mutated cells, intravasation and extravasation of blood and lymphatic vessels, dissemination, and finally metastasis into distant organs. In the tumor microenvironment, cells are supplied with nutrients by the formation of disorganized blood vessels with leaky vasculature by the process of angiogenesis. Growth factor receptors (GFRs), expressed on cell membranes or in the cytoplasm, have profound roles in cell growth, survival, angiogenesis and metastasis. Amplification of GFRs generates inherent and acquired resistance to classical chemotherapies and targeted molecules. Escalated growth signals cross-talk differently with death signals to inhibit apoptosis that is programmed cell death. Accordingly, signals mediated by GFRs function in collaboration to enhance the complexity of the tumor microenvironment. Here, we have discussed the involvement of GFRs at different stages of cancer progression and the molecular therapeutic approaches to target GFRs.
GFR involvement in cancer progression
Epidermal growth factor receptor
The epidermal growth factor receptor (EGFR) family encompasses four receptor proteins, namely ErbB-1/EGFR-1 to -4 (also called HER 1-4) that are expressed on cell surface and exhibit tyrosine kinase activities. These proteins have similar structures and are comprised of three domains: an extracellular domain with ligand binding site, a transmembrane domain, and an intracellular domain with kinase activity [Figure 1a]. There are 11 different growth factors, each possessing a conserved EGF domain that can bind with those four receptors. Upon ligand binding, the receptors form homo-or hetero-dimers, promoting activation, relaying signals for proliferation, survival, migration and differentiation and thus playing major roles in cancer progression [Table 1]. Overexpression and/or gene amplification of EGFR confer malignancy to diverse tissues. Moreover, constitutively active mutants of EGFR are found in different cancers, where they are often associated with poor prognosis [Table 2].
Involvement of GFRs in cancer progression
| Family | Receptors | Tumor growth | Metastasis | Angiogenesis induction | Cell survival/death | Chemoresistance |
|---|---|---|---|---|---|---|
| EGFR/ErbB/HER | ErbB1 | + | + | Pro-angiogenic | Pro-survival signals | + |
| ErbB2 | + | + | + | |||
| ErbB3 | + | + | ? | |||
| ErbB4 | + | + | − | |||
| IGFR | IGF1R | + | + | Pro-angiogenic | + | + |
| IGF2R | Suppress growth | − | − | − | − | |
| TGF-βR (TβR) | TβR I-II | Dual role (contextual) | + | + | Dual role | + |
| VEGFR | VEGFR1 | − | ? | + | + | − |
| VEGFR2 | + | + | + | + | + | |
| VEGFR3 | + | + | + | + | + | |
| PDGFR | PDGFR (α/β) | + | + | + | + | + |
| FGFR | FGFR1-4 | + | + | + | + | + |
GFR expression in cancer
| Family | Cancer | References |
|---|---|---|
| EGFR/ErbB/HER | Amplification and overexpression of Her1 gene were found in breast (14-91%), bladder, lung, glial (50%) cancer patients; HER2 gene related to poor prognosis was observed in different malignancies; aggressive metastatic breast (15-30%), gastric (10-30%), ovarian (20-30%), endometrial (1-47%), esophageal (0-83%), lung (20%), and invasive urothelial bladder (0-80%) carcinomas | [1] |
| Constitutive expression of active truncated EGFR vIII that lacks extracellular domain was found in breast cancer (20-78%) associated with aggressiveness of tumor | ||
| Mutations in HER2 gene in lung, Her3 gene (somatic) in breast, colon, gastric,Her4gene in melanoma, colorectal, gastric, lung, and breast cancer were observed in patients | ||
| IGFR | Amplification of IGF1R gene was reported in small number of breast and melanoma cases | [1] |
| Mutations in IGF2R gene was found in squamous cell carcinomas of the lung | ||
| TGF-βR | TGF-βRII gene is mutated in colon (58-82%) and pancreatic (4%) cancer, absent in prostate (24%), and down-regulated in breast and lung cancer | [2] |
| VEGFR | High expression of VEGFR1-3 genes was reported in a wide number of malignancies like bladder, brain, breast, colon, gastric, lung, ovarian, prostate, and head and neck carcinomas | [3] |
| PDGFR | Overexpression of PDGFRα gene was found in 20% of glioblastoma | [4] |
| Germline point mutation (gain of function) in PDGFRβ gene was observed in 8 families with infantile myofibromatosis | ||
| FGFR | Amplification of FGFR1-3 genes was observed in different cancer. For example, FGFR1 gene in lung (20%), breast (10%), ovarian (~5%), bladder (3%); FGFR2 gene in gastric (10%), breast (4% in triple negative); FGFR3 genein bladder and salivary adenoid cystic cancer | [5] |
| Mutations in FGFR1-4 genes were observed. For example, FGFR1 gene in melanoma (rare), gliblastoma; FGFR2 gene in endometrial (12%), lung (5%), gastric (rare); FGFR3 gene in bladder (50-60% in nonmuscle invasive, 10-15% muscle invasive), cervical (5%), prostate (3%), colorectal; FGFR4 gene in rhabdomyosarcoma (7-8%) cancer |
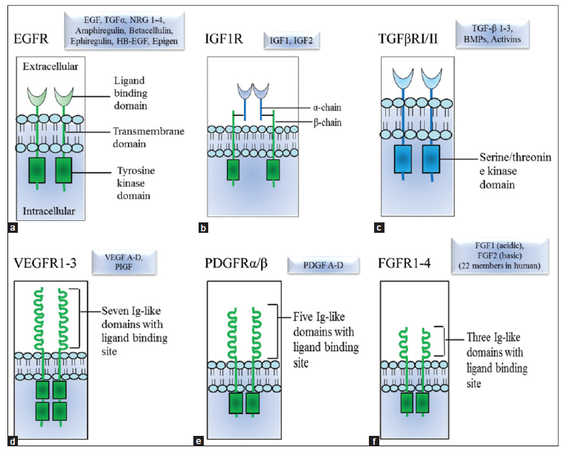
Figure 1. Schematic diagrams of membrane bound growth factor receptors and their ligands involve in cancer progression. Ligands are shown in boxes. (a) Epidermal growth factor receptor (ErbB/Her); (b) insulin-like growth factor receptor; (c) transforming growth factor-beta receptor; (d) vascular endothelial growth factor receptor; (e) platelet derived growth factor receptor, and (f) fibroblast growth factor receptor. ErbB2 (HER2) binds no known epidermal growth factor-like ligands, and ErbB3 shows no tyrosine kinase activity. They relay signals by forming heterodimer with other ErbB proteins from EGFR family. TGFβRIII does not pose any intracellular tyrosine kinase domain
Insulin-like growth factor receptor
The insulin-like growth factor receptor (IGFR) family consists of two cell membrane receptors, IGF1R and IGF2R. IGF1R (that also forms a heterodimer with the insulin receptor [IR]) binds to insulin-like growth factor 1 (IGF1) with higher affinity and IGF2 with comparatively lower affinity to elicit the growth signals required for foetal and postnatal development. The post-translationally modified IGF1R is a polypeptide containing one α- and one β-chain that are connected by a disulfide bond and expressed on the cell surface [Figure 1b]. The α-chain and portion of the β-chain comprise the extracellular domain followed by transmembrane and cytoplasmic domain in β-chain.[6-8] The mature IGF1R is a homodimer comprising the α2 and β2 chains linked by disulfide bonds. The intracellular domain has tyrosine kinase activity that auto-phosphorylates the receptor and a number of downstream proteins upon binding to the ligands. The notion of involvement of this receptor in tumorigenesis came from the studies of IGF1R-transfected cells and the effects of IGF1R gene mutation.[9-11] Overexpression of IGF1R gene is implicated in cellular proliferation, transformation, and metastasis in several carcinomas [Table 1]. Amplification of IGF1R gene in breast cancer and melanoma and overexpression of IGF1R gene in pediatric cancer has been reported [Table 2].
The relatively simpler IGF2R (also called mannose-6 phosphate receptor, M6P) comprises a single polypeptide chain, and functions as a “scavenger receptor” for IGF2. It suppresses tumor growth, modulates invasiveness, and blocks angiogenesis [Table 1].[12] Mutations in IGF2R locus have been observed in lung cells[12] and identified as an early event in hepatocellular carcinoma in different populations.[13]
Transforming growth factor-beta receptor
The transforming growth factor-beta receptor (TGF-βR) family comprises three membrane receptors (TβRI, TβRII and TβRIII) which are expressed in diverse types of cells and regulate distinct cellular functions by the signals transduced upon TGF-β ligand binding. TβR and TβRII are single pass serine/threonine kinases with N-terminal ectodomains and C-terminal kinase domains. TβRIII (also known as betaglycan) is a cell surface proteoglycan > 300 kDa in molecular mass and does not possess an intracellular kinase domain [Figure 1c]. TβIII binds with TGF-β ligands and presents them to TβRII or the ligands bind directly with TβRII depending on cell types. After binding, TβRII recruits and trans-phosphorylates TβRI, which in turn activates SMAD proteins. SMAD complexes translocate into the nucleus and function as transcription factors for TGF-β responsive genes and thus regulate cell proliferation, survival, migration and differentiation [Table 1]. TGF-βR-mediated signals play context-dependent dual roles in cell growth.[1] Under physiological conditions, TGF-β prevents cell growth, stimulates apoptosis or differentiation. During tumorigenesis, TGF-βR-mediated signals promote cell growth due to genetic and epigenetic changes. Mutations and dis-regulation of TGF-βR genes were observed in different cancers [Table 2], for example, down-regulation of TGF-βRII gene in breast and lung cancer[14,15] and different mutations in colon and pancreatic cancer.[16-18]
Vascular endothelial growth factor receptor
This family consists of three membrane receptors (VEGFR1-3), predominantly expressed on endothelial cells and few additional cell types. VEGFRs are single pass protein with seven immunoglobulin (Ig)-like domains on the extracellular site and two split tyrosine kinase domains in the intracellular site [Figure 1d]. They bind with the disulfide-linked homodimer of VEGF isoform (VEGFA-D) ligands and placenta growth factors (PIGF1 and 2) to form homodimers or heterodimers of VEGFR-1 and-2 and relay the signal inside cells. The signals transduced by VEGFR are different between these receptors. For example, VEGFR2 (also known as KDR/flk-1) induces mitogen-activated protein kinases (MAPK)-dependent cell proliferation whereas VEGFR1 (flt-1) does not induce cell growth. However, activation of VEGFR1 by VEGF stimulates cell migration, a response that is also triggered by VEGFR2 activation. These VEGF-VEGFR interactions are well-known for their key roles in vasculogenesis and angiogenesis. VEGFR3 (flt-4) that is expressed on lymphatic vessels interacts with VEGF-C and VEGF-D and is thought to promote lymphangiogensis. VEGFRs are thought to be responsible for blood and lymph vessel formation in tumor microenvironment and thus promote tumor growth and progression [Table 1]. High expression of VEGFR gene is observed in many different types of malignancies [Table 2]. Moreover, somatic mutations in VEGFR2 and VEGFR3 genes were identified in the most common infants’ malignancy, juvenile hemangioma.[19]
Platelet derived growth factor receptor
The platelet-derived growth factor receptor (PDGFR) family contains two receptors (PDGFR-α and-β) that are encoded by two different genes and are expressed on the membrane of different cell types. These single chain receptor proteins have five Ig-like extracellular domains and a tyrosine kinase domain [Figure 1e]. Dimerization of receptors occurs upon binding to homo/heterodimers of PDGF (A-D) ligands, leading to conformational changes in receptors, activating them to trans-phosphorylate and stimulate downstream proteins. This relays the signals into receiving cells via mainly MAPK and PI3K pathways and thus regulates cell proliferation, differentiation, growth, migration, and survival. They have roles in angiogenesis and thus support tumor growth [Table 1]. Overexpression and mutations in the PDGFR genes are associated with diverse cancers [Table 2]. Aberrant expression of PDGFR due to amplification and/or overexpression of PDGFRα and PDGFRβ genes were reported in human glioblastoma multiforme.[4,20] Moreover, mutations and genetic translocation in PDGFRα gene were observed in gastrointestinal stromal tumors and chronic leukemia respectively.[21,22] A germline point mutation (gain of function) in PDGFRβ gene was found in the most common fibrous tumor of infancy, myofibromatosis.[23]
Fibroblast growth factor receptor
The fibroblast growth factor receptor (FGFR) family consists of four closely related transmembrane proteins (FGFR1-4) and their different isoforms with altered ligand specificity due to differential splicing of FGFR mRNA. These single chain receptors contain one extracellular domain with three immunoglobulin repeats (Ig I-III) with ligand binding capacity, one transmembrane domain and one intracellular domain with kinase activity at the carboxy-terminus [Figure 1f]. There are 18 different FGF ligands that can bind to different FGF receptors. Upon binding, dimerization of FGFR leads to auto-phosphorylation and kinase activation. Phosphorylated FGFRs in turn phosphorylate a number of proteins and/or serve as molecular docking sites for many effectors, thus orchestrating context-dependent cellular functions including cell proliferation, growth, differentiation, migration, vascular repair, wound healing, and cell survival. FGF-FGFR interactions have pivotal roles in tumorigenesis [Table 1] as the downstream mitogenic growth signals (MAPK) and anti-apoptotic PI3K/AKT signals lead to uncontrolled growth and inhibition of cell death, respectively. The PLC/PKC pathway downstream of FGFRs also converges to the MAPK pathway to support cell growth.[24-26] These receptors have been shown to exert profound roles in angiogenesis both in paracrine and autocrine fashions. FGFR expression causes tumor cells to acquire resistance to several drugs, especially inhibitors targeting other growth factor receptors (EGFR, PDGFR and VEGFR) because of their extensive cross-talks. Amplification and mutations in FGFR genes that lead to constitutive activation/up-regulation of receptors are found in different types of malignancies, including breast, ovarian, gastric and lung cancers [Table 2].
Targeting GFR-mediated signals with cancer therapeutics
Accumulated understanding over the last 30 years of signaling pathways mediated by different GFRs and their relationship with cancer progression has led to the development of targeted agents for cancer treatment. There are at least 6 approaches to target these pathways: (1) monoclonal antibodies (mAbs) against GFRs; (2) protein kinase inhibitors; (3) nucleic acid-based therapeutics for gene silencing (use of antisense RNA or short interfering RNA [siRNA] to block receptor expression); (4) soluble receptors for growth factor ligands (“Traps”); (5) inhibitors of heat shock proteins and (6) antagonists of signaling pathway proteins.
Among these, the first three strategies including mAbs, protein kinase inhibitors and nucleic acid-based therapeutics are designed to target the GFRs, and thus are highlighted in this review. Radiological responses to targeted molecular agents used as monotherapy are typically more limited compared to the conventional chemotherapy and radiotherapy. However, effects on progression-free survival have been observed. Nowadays, targeted molecular agents are often combined with chemo- or radiotherapy, mainly for two reasons: combination with chemo- or radio-therapy improves efficacy and the molecular specificity of mAbs aids to target tumor selectively.
Monoclonal antibodies against GFRs
The development of hybridoma technology to produce mAbs is the first step in the process of turning the dream of “magic bullets” for targeted treatment of cancer a reality. A wide number of mAbs-based therapies have been approved by the Food and Drug Administration (FDA) for the treatment of different malignancies and many more are in clinical trials.[27] These therapeutics act by directly blocking the function of GFRs and/or by antibody-dependent cytotoxicity, mediated by Fc fragment recognizing immune cells. Antibody-drug conjugates are designed for targeted drug delivery to cells expressing their cognate GFR. A variety of mAbs are used to modulate GFR functions in different indications [Table 3]. For example, the humanized mAb, trastuzumab designed to target ErbB2 and approved by FDA in 1998, is successfully used to treat HER2+ metastatic breast cancer patients.[28] The approval of the drug was further expanded in 2006 for women with cancer in breast and lymph node region as early stage therapy after primary therapy (lumpectomy, mastectomy). In October 2010, it was approved to use for HER2-overexpressing, gastroesophageal junction adenocarcinoma in combination with either cisplatin or fluoropyrimidines in patients who have not received any prior treatment.
Some mAbs against GFRs for treating cancer
| Name of mAB | Target | Targeted stages | Indication | Mechanism of action | Resistance (known mechanism) | Status (highest level) |
|---|---|---|---|---|---|---|
| Cetuximab (human IgG1) (use as single or conjunction with radiotherapy) | ErbB1 | Suppresses cell growth and metastasis | Metastatic colorectal; head and neck carcinoma | Inhibition EGFR signaling (down regulates active EGFRVII) and ADCC | Yes (mutations in a number of diverse genes are blamable for intrinsic resistance) | Approved |
| Panitumumab (human IgG2) (use as single agent) | ErbB1 | Suppresses cell growth | Metastatic colorectal cancer | Prevents EGFR activation | Approved | |
| Trastuzumab (humanized IgG1) (use as single or as adjuvant for chemotherapy) | ErbB2 | Suppresses cell growth and angiogenesis, induces cell death | HER2+ metastatic breast cancer | Inhibition ErbB2 signaling and ADCC | Yes (overexpression of membrane associated glycoprotein MUC4; increase IGF1R signaling are some of the reasons that confer resistance) | Approved |
| Ganitumab (human IgG1) (use as single or combined with different neoplastic drugs) | IGF1R | Inhibits cell growth, delays tumor progression | Non-Hodgkin lymphoma, metastatic pancreatic cancer, metastatic Ewing family of tumors | Blocks IGF-1 and-2 binding to IGF1R without crosslinking with IR, inhibits activation of IGF1R homodimer and IGF1R/IR heterodimer | Yes (calcium dependent proliferation effects acquire resistance in prostate cancer cells) | Clinical trials (passed phase II) |
| Cixutumumab (human IgG1) (use as single or combined with different neoplastic drugs) | IGF1R | Induces cancer cell apoptosis, decreases cell proliferation | Solid tumors, Ewing sarcoma family tumors | Prevents IGF1 binding to receptor and subsequent activation of PI3K/AKT survival pathway, mediates receptor internalization and degradation | NA | Clinical trials (phase I-II) |
| PF03446962/Anti-Alk1 (human) | TGF-βR | Prevents angiogenesis (dose dependent) | Transitional cell carcinoma of bladder | Disrupts co-localization of endothelial cells with perivascular cells and reduces blood flow | NA | Clinical trials (phase II) |
| Ramucirumab (human IgG1): use as single or with neoplastic drugs | VEGFR2 | Inhibits tumor angiogenesis and growth | Hepatocellular, renal cell, and ovarian carcinomas | Blocks VEGF binding to the receptor and thus VEGF-signaling and subsequently angiogenesis | Yes (VEGF-axis dependent pathway is involved for resistance) | Clinical trials (phase II) |
| 1B3 (used in combination with mAB against antitumor/anti-angiogenic agent) | Mouse PDGFRβ | Inhibits angiogenesis | Pancreatic and a nonsmall cell lung tumor xenograft models | Blocks PDGFR binding with receptor, ligand-stimulated activation of PDGFRβ and downstream signaling molecules in tumor cells | NA | Preclinical trial |
| IMC-2C5 (human) | Both mouse and human PDGFRβ | Delays growth (cell specific), inhibits angiogenesis | NA | Preclinical trial |
As VEGFR plays a key role in new blood vessel formation, different antibodies against VEGFR are designed to prevent angiogenesis and growth in cancer.[29-32] The combination of mAbs with different antineoplastic drugs often increases effectiveness. Diverse combinations of mAbs conjugated with different types of drugs are currently in clinical trials.
Development of resistance against GFR-targeted mAbs is the foremost limitation to their clinical use. Resistance to these antibodies can be either primary or acquired which develops within few months to years of treatment.[33] Patients treated with trastuzumab monotherapy showed intrinsic (66-88%) and acquired resistance within one year (15%) which resulted in loss of effectiveness.[34] Although in most cases the underlying mechanisms of resistance remain poorly understood, in few cases resistance have been linked to the compensatory pathways mediated by other GFRs or mutations in downstream signaling pathways. For example, enhancement of IGF1R-mediated signal in anti-erbB2/HER2 (trastuzumab) antibody-treated breast cancer patients confers resistance to treatment.[35] Overexpression of membrane-associated glycoprotein MUC4, PTEN-PI3K signaling pathways, and elevated HER2 extracellular domain in serum are also involved in trastuzumab resistance.[34] Mutations in K-Ras are responsible for primary resistance against anti-EGFR antibodies in colorectal cancer.[36] Treatment regimen containing multiple mAbs against different GFRs may conceivably increase resistance but may be unacceptably toxic. For example, cross-talk between EGF- and IGF-mediated signaling pathways plays major roles in acquired resistance. Preclinical data suggest that simultaneous blockade of the two pathways could be advantageous in treatment.[37-40] However, a phase II trial in colorectal cancer patients treated with cetuximab and cixitumumab failed to show any additional antitumor activity, and so this treatment regimen was eliminated from consideration in colorectal cancer patients refractory to EGFR inhibitors.[41,42] Mechanism-based combinations of GFR mAbs will require case-by-case validation in preclinical and pilot clinical studies.
Protein kinase inhibitors
Structural and functional analyses have paved the way to discovery and development of numerous protein kinase inhibitors, especially tyrosine kinase inhibitors (TKIs) that inhibit the cytoplasmic kinase activity of growth receptors and subsequently their downstream signaling cascades into the cells. Most of these compounds are hydrophobic in nature and, therefore, are orally bioavailable. The majority of these agents rapidly cross cell plasma membranes and compete with phosphate donor adenosine tyrosine phosphate (ATP), phosphorylation substrates, or both.[43] Many such compounds have shown cytostatic effects in cancer cells and animal models.[44] Because of the “druggability” of kinases, 30% of new efforts by pharmaceutical companies are dedicated to develop new kinase inhibitors with many already approved or in clinical trials. Few examples of TKIs for GFRs are enlisted in Table 4. Chemo- or radio-therapeutic agents are often combined with TKIs to increase therapeutic efficacy. The IGF1R inhibitor, BMS-754807 showed higher efficacy when combined with cytotoxic, hormonal or targeted agents. BMS-754807 in combination with docetaxel showed more than additive efficacy in triple negative breast cancer.[45] There are now several clinical trials (phase I and II) ongoing with BMS-754807-including combinations. Co-targeting of IGF1R and IR with OSI-906 inhibitor showed superior antitumor activity compared to targeting IGF1R alone by mAbs[46] and enhanced the antitumor effect of doxorubicin in a colorectal cancer model.[47]
Some TKI for GFRs for treating cancer
| Name of TKI | Target | Targeted stages | Indication | Mechanism of action | Resistance | Status (highest level) |
|---|---|---|---|---|---|---|
| Gefitinib | ErbB-1, -2 and -3 | Inhibits anti-apoptotic signals, induces anti-angiogenic activity | Breast and lung cancers (effective in cancers with mutant and overactive EGFR) | Binds reversible to the ATP binding site of receptor and inhibits formation of phosphytyrosine residues in receptor | Yes (overexpression FGF2/FGFR1 signal was found to be accountable for resistance in NSCLC cell lines) | Approved |
| Erlotinib | ErbB1 | Induces cell cycle arrest and apoptosis, inhibits angiogenesis | NSCLC, pancreatic cancer (more effective in cancers with mutant and overactive EGFR) | Approved | ||
| BMS-754807 (single or coupled with cytotoxic/hormonal/targeted agent) | IGF1R (and insulin receptor) | Inhibits growth of tumors, induces apoptosis, plays significant role in mitogenesis, angiogenesis and tumor cell survival, enhances therapeutic efficacies of attached drugs | Neoplasms, breast cancer, advanced metastatic solid tumors | Reversibly inhibits IGF1R phosphorylation | Yes (overexpression of PDGFRα is found to be responsible for acquiring resistance) | Clinical trials (phase I-II) |
| Axitinib (single or combined with drugs) | VEGFR 1-3 (thought to act on PDGFR also) | Inhibits cell growth in xenograft models, enables to inhibit angiogenesis, vascular permeability and blood flow, decreases metastasis | Breast cancer, renal cell carcinoma, hepatocellular carcinoma, advanced pancreatic cancer, glioblastomamultiforme | Binds with VEGFR and inhibits receptor activation through phosphorylation | Yes (multi-drug transporter proteins, ABCB1, and ABCG2 may play role) | Approved for metastatic renal cell carcinoma and advanced pancreatic cancer, clinical trials (phase I-II) are ongoing for different malignancies |
| Sunitinib/SU11248 (single or attached with drugs) | VEGFR; PDGFRβ (also target some other RTK) | Inhibits cell growth and angiogenesis, delays tumor progression | Renal cell carcinoma, gastrointestinal tumor, colorectal neoplasm, metastatic breast cancer | Selectively inhibits VEGFR2 and PDGFRβ phosphorylation (in a time- and dose-dependent manner) | Yes (activation of sphingosine kinase 1 is account for acquired resistance in renal cell carcinoma) | Approved for renal cell carcinoma, clinical trials (phase I-II) for other cancer |
| TKI258 | VEGFR; PDGFR; FGFR | Inhibits cell motility and growth, delays established tumor growth, inhibits metastasis, suppresses angiogenesis | Mammary tumors, multiple myeloma, colon cancer, pancreatic cancer | Competes with ATP for the binding site, inhibits GFR-mediated signals | NA | Preclinical |
However, the development of resistance to TKIs makes their therapeutic use quite challenging.[48] Overexpression of PDGFα in cells has been found to be responsible for acquiring resistance against BMS-754807. Different mechanisms have been identified which account for TKI resistance (both acquired and inherent) in cancer cells. These are: (1) somatic, genetic or epigenetic mutations within kinase domains; (2) overexpression and amplification of GFRs genes to overrule the inhibitors’ function; (3) modifications in signaling pathways to bypass the signal mediated by specific receptor; and (4) overexpression of ATP-binding cassette transporters proteins (ABC-transporters) which transport TKIs outside of cells, limiting achievable intracellular concentrations.
TKIs with broad spectrum activity that inhibit a number of GFRs are less specific but often more effective compared to highly specific inhibitors. For example, a multi-targeted TKI against VEGFR, PDGFR and FGFR (TKI258) is more potent in inhibiting angiogenesis in pancreatic cancer cells as the signals mediated by these three receptors are crucial for the blood vessels formation.[49] This inhibitor is efficacious in delaying cancer growth and inhibiting metastasis in a pancreatic cancer model[49] and clinically used for advanced renal cell carcinoma and breast cancer.[50,51] Broad spectrum TKIs would be less susceptible to acquired resistance.
Nucleic acid-based therapeutics to block GFR expression
The clinical applications of current chemotherapeutic drugs are often limited by their toxic effects on healthy dividing cells. Dose reductions due to toxicity can limit efficacy and select drug-resistant cancer cell clones. Advances in cancer molecular and cell biology have led to the identification of numerous potentially actionable genes, not all of which encode druggable targets. These genes and their transcripts are potential targets for nucleic acid therapeutics. Gene silencing both at transcriptional and translational levels is a promising tool to treat cancer more effectively.[52] Among available technologies, RNA interference (RNAi) using double stranded siRNA or short hairpin RNA (shRNA) is a promising candidate technology, provided that pharmacokinetic obstacles to quantitative delivery are overcome.
RNAi is a biological posttranscriptional regulatory process in which small endogenous RNA (microRNA) inhibit gene expression by hybridizing with mRNAs and either causing their degradation or preventing translation. Mimicking physiological RNAi, siRNAs are designed exogenously to deliver to cancer cells for selective mRNA targeting. There are two fundamental techniques of executing RNAi: nuclear delivery of gene expression constructs to express shRNA and cytoplasmic delivery of siRNA. Silencing by synthetic siRNA, RNA oligonucleotides 21-23 nucleotides long, is more expedient than shRNA due to the difficulty of constructing shRNA expression systems[53] and the requirement for nuclear delivery.[54] The potential gene silencing ability of siRNAs in animal models has made them promising investigational drug candidates and some siRNAs are in clinical trials. However, no siRNA against GFRs have been approved yet for cancer treatment. There are few siRNAs against GFRs, which have been used in cell culture and animal models [Table 5]. The primary challenge to the clinical use of RNAi is the need to deliver a relatively small molecule in sufficient quantities to tumor cells after systemic administration. Nucleic acid therapeutics delivery is an area of very active investigation.
Some siRNAs designed against GFR genes for treating cancer. The vehicles to carry these siRNAs, their functionality in tumor progression and the experimented cell lines are listed
| Targeted genes | Delivery vehicle | Functionality in tumor progression | Cell/animal models | References |
|---|---|---|---|---|
| EGFRvII (encapsulated with Erlotinib or SAHA) | Cyclodextrin-modifi ed dendritic polyamines (DexAMs) (cationic biodegradable polymer) | Decreases cell proliferation, induces apoptosis | Brain cancer cells (U78 glioblastoma) | [55] |
| ErbB2 (encapsulated with wild p53 gene) | Carbonate apatite nanoparticles (inorganic pH sensitive) | Decreases cell viability, delays tumor growth | 4T1 cells induced breast cancer mouse | [56] |
| ErbB2 | Folate linked lipid-based nanoparticles (cationic liposome) | Inhibits tumor growth | KB (cervical adenocarcinoma-overexpresses folate receptors) cells induced nasopharyngeal cancer mouse | [57] |
| IGFR1R (encapsulated with wild p53 gene and anti Bcl-2 siRNA) | Carbonate apatite nanoparticles (inorganic pH sensitive) | Inhibits cellular growth/proliferation, regresses tumor growth, enhances drugs’ sensitivity | 4T1 cells induced breast cancer mouse | [58] |
| IGF1R | Lipofectamine 2000 (cationic liposome) | Lowers cell proliferation increases sensitivity of drug (adriamycin), induces apoptosis | Liver cell lines (HepG2 and Huh7 cells) | [59] |
| PDGFRα | siLentFect (cationic liposome) | Sensitizes cells to AKI treatment | Pancreatic cancer cells | [60] |
| FGFR1 | Lipofectamine/oligofectamine (cationic liposome) | Reduces cell viability, increases sensitivity of cells to drug (4-hydroxytamoxifen) | Breast cancer cells (MDA-MB-134) | [61, 62] |
| EGFR1, IGF1R | Lipofectamine | Induces cell death, increases sensitivity of adriamycin | Liver cell lines (Huh7 cells with mutated p53 gene) | [63] |
Concerns and Future Perspectives
The anionic nature of siRNA prevents it diffusion through cellular membrane posing a difficulty in delivering siRNA into cells. Moreover, systemically administered naked siRNA is subjected to degradation by endogenous nucleases, renal clearance, and non- specific bio-distribution. Accordingly, a smart carrier is essential for functional delivery of siRNAs into the system. A wide number of genetically engineered viral vectors or synthetic polymer/liposome-based nanovectors are in use to deliver siRNAs in different cells and animal models. However, there remain concerns surrounding the safety and efficacy of these nanovectors. The successful clinical application of siRNAs will require nanosized cargos with higher binding affinity for siRNAs and possibly other drugs, fast release of bound siRNA in the cytoplasm, versatility to be engineered for targeting tumors, in vivo stability, lack of immunogenicity and minimal toxicity. A pH-sensitive inorganic carbonate apatite nanocarrier system has recently been developed that could provide an attractive solution to the challenges presented by other carriers. This carrier has been used to transport siRNAs against ErbB2, IGF1R, and Bcl-2 genes as well as wild-type p53 gene that inhibited the growth of established tumors in syngeneic mouse models.[56,58] This platform could be used to target one or more GFRs in tumors.
Conclusion
Targeting multiple GFRs offers significant therapeutic promise in cancer therapy. As overexpression of GFRs is also responsible for resistance to different drugs, combination regimens may prevent or alleviate resistance. Nanoparticles-mediated siRNA delivery may have significant clinical applications once clinically suitable delivery platforms are identified and validated.
Financial support and sponsorship
The work was supported by a research grant (FRGS/2/2013/SG05/MUSM/02/2) of the Ministry of Higher Education (MOHE), Malaysia.
Conflicts of interest
There are no conflicts of interest.
REFERENCES
1. Witsch E, Sela M, Yarden Y. Roles for growth factors in cancer progression. Physiology (Bethesda) 2010;25:85-101.
2. Elliott RL, Blobe GC. Role of transforming growth factor Beta in human cancer. J Clin Oncol 2005;23:2078-93.
4. Demoulin JB, Essaghir A. PDGF receptor signaling networks in normal and cancer cells. Cytokine Growth Factor Rev 2014;25:273-83.
5. Dieci MV, Arnedos M, Andre F, Soria JC. Fibroblast growth factor receptor inhibitors as a cancer treatment: from a biologic rationale to medical perspectives. Cancer Discov 2013;3:264-79.
6. Pilch PF, Czech MP. The subunit structure of the high affinity insulin receptor. Evidence for a disulfide-linked receptor complex in fat cell and liver plasma membranes. J Biol Chem 1980;255:1722-31.
7. Jacobs S, Kull FC Jr, Cuatrecasas P. Monensin blocks the maturation of receptors for insulin and somatomedin C: identification of receptor precursors. Proc Natl Acad Sci U S A 1983;80:1228-31.
8. Ullrich A, Gray A, Tam AW, Yang-Feng T, Tsubokawa M, Collins C, Henzel W, Le Bon T, Kathuria S, Chen E. Insulin-like growth factor I receptor primary structure: comparison with insulin receptor suggests structural determinants that define functional specificity. EMBO J 1986;5:2503-12.
9. Kaleko M, Rutter WJ, Miller AD. Overexpression of the human insulinlike growth factor I receptor promotes ligand-dependent neoplastic transformation. Mol Cell Biol 1990;10:464-73.
10. Valentinis B, Baserga R. IGF-I receptor signalling in transformation and differentiation. Mol Pathol 2001;54:133-7.
11. Prager D, Li HL, Asa S, Melmed S. Dominant negative inhibition of tumorigenesis in vivo by human insulin-like growth factor I receptor mutant. Proc Natl Acad Sci U S A 1994;91:2181-5.
12. Kong FM, Anscher MS, Washington MK, Killian JK, Jirtle RL. M6P/IGF2R is mutated in squamous cell carcinoma of the lung. Oncogene 2000;19:1572-8.
13. Oka Y, Waterland RA, Killian JK, Nolan CM, Jang HS, Tohara K, Sakaguchi S, Yao T, Iwashita A, Yata Y, Takahara T, Sato S, Suzuki K, Masuda T, Jirtle RL. M6P/IGF2R tumor suppressor gene mutated in hepatocellular carcinomas in Japan. Hepatology 2002;35:1153-63.
14. Lücke CD, Philpott A, Metcalfe JC, Thompson AM, Hughes-Davies L, Kemp PR, Hesketh R. Inhibiting mutations in the transforming growth factor beta type 2 receptor in recurrent human breast cancer. Cancer Res 2001;61:482-5.
15. Kim WS, Park C, Jung YS, Kim HS, Han J, Park CH, Kim K, Kim J, Shim YM, Park K. Reduced transforming growth factor-beta type II receptor (TGF-beta RII) expression in adenocarcinoma of the lung. Anticancer Res 1999;19:301-6.
16. Grady WM, Myeroff LL, Swinler SE, Rajput A, Thiagalingam S, Lutterbaugh JD, Neumann A, Brattain MG, Chang J, Kim SJ, Kinzler KW, Vogelstein B, Willson JK, Markowitz S. Mutational inactivation of transforming growth factor beta receptor type II in microsatellite stable colon cancers. Cancer Res 1999;59:320-4.
17. Parsons R, Myeroff LL, Liu B, Willson JK, Markowitz SD, Kinzler KW, Vogelstein B. Microsatellite instability and mutations of the transforming growth factor beta type II receptor gene in colorectal cancer. Cancer Res 1995;55:5548-50.
18. Goggins M, Shekher M, Turnacioglu K, Yeo CJ, Hruban RH, Kern SE. Genetic alterations of the transforming growth factor beta receptor genes in pancreatic and biliary adenocarcinomas. Cancer Res 1998;58:5329-32.
19. Walter JW, North PE, Waner M, Mizeracki A, Blei F, Walker JW, Reinisch JF, Marchuk DA. Somatic mutation of vascular endothelial growth factor receptors in juvenile hemangioma. Genes Chromosomes Cancer 2002;33:295-303.
20. Fleming TP, Saxena A, Clark WC, Robertson JT, Oldfield EH, Aaronson SA, Ali IU. Amplification and/or overexpression of platelet-derived growth factor receptors and epidermal growth factor receptor in human glial tumors. Cancer Res 1992;52:4550-3.
21. Tornillo L, Terracciano LM. An update on molecular genetics of gastrointestinal stromal tumours. J Clin Pathol 2006;59:557-63.
22. Gotlib J, Cools J, Malone JM 3rd, Schrier SL, Gilliland DG, Coutre SE. The FIP1L1-PDGFRalpha fusion tyrosine kinase in hypereosinophilic syndrome and chronic eosinophilic leukemia: Implications for diagnosis, classification, and management. Blood 2004;103:2879-91.
23. Cheung YH, Gayden T, Campeau PM, LeDuc CA, Russo D, Nguyen VH, Guo J, Qi M, Guan Y, Albrecht S, Moroz B, Eldin KW, Lu JT, Schwartzentruber J, Malkin D, Berghuis AM, Emil S, Gibbs RA, Burk DL, Vanstone M, Lee BH, Orchard D, Boycott KM, Chung WK, Jabado N. A recurrent PDGFRB mutation causes familial infantile myofibromatosis. Am J Hum Genet 2013;92:996-1000.
24. Haugsten EM, Wiedlocha A, Olsnes S, Wesche J. Roles of fibroblast growth factor receptors in carcinogenesis. Mol Cancer Res 2010;8:1439-52.
25. Turner N, Grose R. Fibroblast growth factor signalling: from development to cancer. Nat Rev Cancer 2010;10:116-29.
26. Grose R, Dickson C. Fibroblast growth factor signaling in tumorigenesis. Cytokine Growth Factor Rev 2005;16:179-86.
28. Vogel CL, Cobleigh MA, Tripathy D, Gutheil JC, Harris LN, Fehrenbacher L, Slamon DJ, Murphy M, Novotny WF, Burchmore M, Shak S, Stewart SJ, Press M. Efficacy and safety of trastuzumab as a single agent in first-line treatment of HER2-overexpressing metastatic breast cancer. J Clin Oncol 2002;20:719-26.
29. Kunkel P, Ulbricht U, Bohlen P, Brockmann MA, Fillbrandt R, Stavrou D, Westphal M, Lamszus K. Inhibition of glioma angiogenesis and growth in vivo by systemic treatment with a monoclonal antibody against vascular endothelial growth factor receptor-2. Cancer Res 2001;61:6624-8.
30. Fernandez M, Vizzutti F, Garcia-Pagan JC, Rodes J, Bosch J. Anti-VEGF receptor-2 monoclonal antibody prevents portal-systemic collateral vessel formation in portal hypertensive mice. Gastroenterology 2004;126:886-94.
31. Brekken RA, Overholser JP, Stastny VA, Waltenberger J, Minna JD, Thorpe PE. Selective inhibition of vascular endothelial growth factor (VEGF) receptor 2 (KDR/Flk-1) activity by a monoclonal anti-VEGF antibody blocks tumor growth in mice. Cancer Res 2000;60:5117-24.
32. Prewett M, Huber J, Li Y, Santiago A, O'Connor W, King K, Overholser J, Hooper A, Pytowski B, Witte L, Bohlen P, Hicklin DJ. Antivascular endothelial growth factor receptor (fetal liver kinase 1) monoclonal antibody inhibits tumor angiogenesis and growth of several mouse and human tumors. Cancer Res 1999;59:5209-18.
33. Hynes NE, Lane HA. ERBB receptors and cancer: the complexity of targeted inhibitors. Nat Rev Cancer 2005;5:341-54.
34. Nahta R, Esteva FJ. HER2 therapy: molecular mechanisms of trastuzumab resistance. Breast Cancer Res 2006;8:215.
35. Nahta R, Yuan LX, Zhang B, Kobayashi R, Esteva FJ. Insulin-like growth factor-I receptor/human epidermal growth factor receptor 2 heterodimerization contributes to trastuzumab resistance of breast cancer cells. Cancer Res 2005;65:11118-28.
36. Sartore-Bianchi A, Di Nicolantonio F, Nichelatti M, Molinari F, De Dosso S, Saletti P, Martini M, Cipani T, Marrapese G, Mazzucchelli L, Lamba S, Veronese S, Frattini M, Bardelli A, Siena S. Multi-determinants analysis of molecular alterations for predicting clinical benefit to EGFR-targeted monoclonal antibodies in colorectal cancer. PLoS One 2009;4:e7287.
37. Wu JD, Odman A, Higgins LM, Haugk K, Vessella R, Ludwig DL, Plymate SR. In vivo effects of the human type I insulin-like growth factor receptor antibody A12 on androgen-dependent and androgen-independent xenograft human prostate tumors. Clin Cancer Res 2005;11:3065-74.
38. Morgillo F, Kim WY, Kim ES, Ciardiello F, Hong WK, Lee HY. Implication of the insulin-like growth factor-IR pathway in the resistance of non-small cell lung cancer cells to treatment with gefitinib. Clin Cancer Res 2007;13:2795-803.
39. Barnes CJ, Ohshiro K, Rayala SK, El-Naggar AK, Kumar R. Insulin-like growth factor receptor as a therapeutic target in head and neck cancer. Clin Cancer Res 2007;13:4291-9.
40. Slomiany MG, Black LA, Kibbey MM, Tingler MA, Day TA, Rosenzweig SA. Insulin-like growth factor-1 receptor and ligand targeting in head and neck squamous cell carcinoma. Cancer Lett 2007;248:269-79.
41. Reidy DL, Vakiani E, Fakih MG, Saif MW, Hecht JR, Goodman-Davis N, Hollywood E, Shia J, Schwartz J, Chandrawansa K, Dontabhaktuni A, Youssoufian H, Solit DB, Saltz LB. Randomized, phase II study of the insulin-like growth factor-1 receptor inhibitor IMC-A12, with or without cetuximab, in patients with cetuximab- or panitumumab-refractory metastatic colorectal cancer. J Clin Oncol 2010;28:4240-6.
42. Arcaro A. Targeting the insulin-like growth factor-1 receptor in human cancer. Front Pharmacol 2013;4:30.
43. Workman P, Brunton VG, Robins DJ. Tyrosine kinase inhibitors. Semin Cancer Biol 1992;3:369-81.
44. Arora A, Scholar EM. Role of tyrosine kinase inhibitors in cancer therapy. J Pharmacol Exp Ther 2005;315:971-9.
45. Carboni JM, Wittman M, Yang Z, Lee F, Greer A, Hurlburt W, Hillerman S, Cao C, Cantor GH, Dell-John J, Chen C, Discenza L, Menard K, Li A, Trainor G, Vyas D, Kramer R, Attar RM, Gottardis MM. BMS-754807, a small molecule inhibitor of insulin-like growth factor-1R/IR. Mol Cancer Ther 2009;8:3341-9.
46. Buck E, Gokhale PC, Koujak S, Brown E, Eyzaguirre A, Tao N, Rosenfeld-Franklin M, Lerner L, Chiu MI, Wild R, Epstein D, Pachter JA, Miglarese MR. Compensatory insulin receptor (IR) activation on inhibition of insulin-like growth factor-1 receptor (IGF-1R): rationale for cotargeting IGF-1R and IR in cancer. Mol Cancer Ther 2010;9:2652-64.
47. Flanigan SA, Pitts TM, Eckhardt SG, Tentler JJ, Tan AC, Thorburn A, Leong S. The insulin-like growth factor I receptor/insulin receptor tyrosine kinase inhibitor PQIP exhibits enhanced antitumor effects in combination with chemotherapy against colorectal cancer models. Clin Cancer Res 2010;16:5436-46.
48. Chen YF, Fu LW. Mechanisms of acquired resistance to tyrosine kinase inhibitors. Acta Pharm Sin B 2011;1:197-207.
49. Taeger J, Moser C, Hellerbrand C, Mycielska ME, Glockzin G, Schlitt HJ, Geissler EK, Stoeltzing O, Lang SA. Targeting FGFR/PDGFR/VEGFR impairs tumor growth, angiogenesis, and metastasis by effects on tumor cells, endothelial cells, and pericytes in pancreatic cancer. Mol Cancer Ther 2011;10:2157-67.
50. Escudier B, Grünwald V, Ravaud A, Ou YC, Castellano D, Lin CC, Gschwend JE, Harzstark A, Beall S, Pirotta N, Squires M, Shi M, Angevin E. Phase II results of Dovitinib (TKI258) in patients with metastatic renal cell cancer. Clin Cancer Res 2014;20:3012-22.
51. André F, Bachelot T, Campone M, Dalenc F, Perez-Garcia JM, Hurvitz SA, Turner N, Rugo H, Smith JW, Deudon S, Shi M, Zhang Y, Kay A, Porta DG, Yovine A, Baselga J. Targeting FGFR with dovitinib (TKI258): preclinical and clinical data in breast cancer. Clin Cancer Res 2013;19:3693-702.
52. Kalota A, Shetzline SE, Gewirtz AM. Progress in the development of nucleic acid therapeutics for cancer. Cancer Biol Ther 2004;3:4-12.
53. Lu PY, Xie F, Woodle MC. In vivo application of RNA interference: from functional genomics to therapeutics. Adv Genet 2005;54:117-42.
54. Chowdhury EH. Nuclear targeting of viral and non-viral DNA. Expert Opin Drug Deliv 2009;6:697-703.
55. Kim C, Shah BP, Subramaniam P, Lee KB. Synergistic induction of apoptosis in brain cancer cells by targeted codelivery of siRNA and anticancer drugs. Mol Pharm 2011;8:1955-61.
56. Kunnath AP, Tiash S, Fatemian T, Morshed M, Mohamed SM, Chowdhury EH. Intracellular delivery of ERBB2 siRNA and p53 gene synergistically inhibits the growth of established tumour in an immunocompetent mouse. J Cancer Sci Ther 2014;6:99-104.
57. Yoshizawa T, Hattori Y, Hakoshima M, Koga K, Maitani Y. Folate-linked lipid-based nanoparticles for synthetic siRNA delivery in KB tumor xenografts. Eur J Pharm Biopharm 2008;70:718-25.
58. Kunnath AP, Kamaruzman NI, Chowdhury EH. Nanoparticle-facilitated intratumoral delivery of Bcl-2/IGF-1R siRNAs and p53 Gene synergistically inhibits tumor growth in immunocompetent mice. J Nanomedicine Nanotechnol 2014;S9:001.
59. Niu J, Xu Z, Li XN, Han Z. siRNA-mediated type 1 insulin-like growth factor receptor silencing induces chemosensitization of a human liver cancer cell line with mutant P53. Cell Biol Int 2007;31:156-64.
60. Xie L, Kassner M, Munoz RM, Que QQ, Kiefer J, Zhao Y, Mousses S, Yin HH, Von Hoff DD, Han H. Kinome-wide siRNA screening identifies molecular targets mediating the sensitivity of pancreatic cancer cells to Aurora kinase inhibitors. Biochem Pharmacol 2012;83:452-61.
61. Reis-Filho JS, Simpson PT, Turner NC, Lambros MB, Jones C, Mackay A, Grigoriadis A, Sarrio D, Savage K, Dexter T, Iravani M, Fenwick K, Weber B, Hardisson D, Schmitt FC, Palacios J, Lakhani SR, Ashworth A. FGFR1 emerges as a potential therapeutic target for lobular breast carcinomas. Clin Cancer Res 2006;12:6652-62.
62. Turner N, Pearson A, Sharpe R, Lambros M, Geyer F, Lopez-Garcia MA, Natrajan R, Marchio C, Iorns E, Mackay A, Gillett C, Grigoriadis A, Tutt A, Reis-Filho JS, Ashworth A. FGFR1 amplification drives endocrine therapy resistance and is a therapeutic target in breast cancer. Cancer Res 2010;70:2085-94.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Tiash S, Chowdhury EH. Growth factor receptors: promising drug targets in cancer. J Cancer Metastasis Treat 2015;1:190-200. http://dx.doi.org/10.4103/2394-4722.163151
AMA Style
Tiash S, Chowdhury EH. Growth factor receptors: promising drug targets in cancer. Journal of Cancer Metastasis and Treatment. 2015; 1: 190-200. http://dx.doi.org/10.4103/2394-4722.163151
Chicago/Turabian Style
Tiash, Snigdha, Ezharul Hoque Chowdhury. 2015. "Growth factor receptors: promising drug targets in cancer" Journal of Cancer Metastasis and Treatment. 1: 190-200. http://dx.doi.org/10.4103/2394-4722.163151
ACS Style
Tiash, S.; Chowdhury EH. Growth factor receptors: promising drug targets in cancer. J. Cancer. Metastasis. Treat. 2015, 1, 190-200. http://dx.doi.org/10.4103/2394-4722.163151
About This Article
Copyright

Data & Comments
Data


 Cite This Article 44 clicks
Cite This Article 44 clicks

 Like This Article 4
likes
Like This Article 4
likes






Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.