
Withaferin A targeting both cancer stem cells and metastatic cancer stem cells in the UP-LN1 carcinoma cell model


Abstract
Aim: As our understanding of cancer stem cell (CSC) biology improves, search for inhibitory agents of CSCs and metastatic CSCs (mCSCs) positive for CXCR4 is warranted. Withaferin A (WA), a withanolide extracted from the medicinal plant Withania somnifera, has been shown to exhibit anti-cancer effects through multiple mechanisms. Whether WA could selectively target CSCs, mCSCs, or non-CSCs of a gastrointestinal (GI) carcinoma tumor remains unclear.
Methods: Side-population (SP) analysis, flow cytometric phenotyping and sorting, non-invasive imaging in conjunction with xenotransplantation, and immunohistology were used in this investigation.
Results: Using the lymph node metastatic GI cancer cell line UP-LN1, consisting of CD44high/CD24low floating (F) and CD44low/CD24high adherent (A) cell subsets, this study demonstrated that as compared with parental UP-LN1 cells or A cells, WA preferentially reduced F-cell proliferation, tumor sphere formation, and SP cells in vitro in greater effi ciencies by apoptosis. This action was mechanistically mediated via the down-regulation of CXCR4/CXCL12 and STAT3/interleukin-6 axes, both of which are instrumental in the acquisition of metastatic ability. Attenuation of interferon-γ-induced CXCR4 expression in F cells by knockdown with siRNA or blocking with an anti-CXCR4 antibody, followed by Western blot analysis, showed signifi cantly reduced metastatic potential in vitro. The extent of in vitro anti-invasive effect of WA on the IFN-γ-treated F cells was signifi cantly greater than on the F cells without WA treatment, or F cells treated with control siRNA or with control IgG antibody. The observed in vitro effects of WA on the CSC and mCSC targeting were validated by data obtained with non-invasive imaging in NOD/SCID mouse xenotransplantation.
Conclusion: WA could effi ciently block the formation of both CSCs and mCSCs in the UP-LN1 cell line, suggesting that WA may be considered an effective therapeutic agent for this type of GI malignancies.
Keywords
Introduction
Distant metastasis represents one of the few most challenging aspects in cancer management. Cancer cells progress from the primary lesion site and gain the ability to spread to distant organs. It has been demonstrated both experimentally and clinically that the tumor microenvironment plays a pivotal role in tumor progression, particularly in the acquisition of metastatic potential.[1] During the process of tumor metastasis, a cellular event termed epithelial-to-mesenchymal transition (EMT), is initiated and is believed to be a prerequisite for tumor dissemination.[2] Accumulating evidence from neoplastic tissues indicates the presence of self-renewing, stem-like cells within tumors called cancer stem cells (CSCs) or tumor-initiating cells. CSCs, which constitute a small subpopulation of neoplastic cells within a tumor, are defined operationally by their ability to seed new tumors.[3] Recently, a seminal study has demonstrated that disseminating cancer cells require self-renewal capability, similar to that exhibited by stem cells and has indicated that the EMT process enriched the CSC population.[4] Collectively, these studies provided important evidence and insights into CSC biology. The existence of CSCs has been shown to contribute to many aspects of tumorigenesis, especially therapy resistance[5,6] and metastasis.[7] However, studying CSCs has been a great challenge due to their rarity and the accuracy of identification methods. Therefore, a reliable tumor cell model which replicates the physiological properties of CSCs and metastatic CSCs (mCSCs) becomes a valuable tool for the understanding of CSC biology.
The UP-LN1 lymph node metastatic cell line we have established previously exhibits CSC characteristics.[8,9] UP-LN1 is a CEA-producing gastrointestinal (GI) carcinoma cell line which harbors a unique co-existence of 2 major naturally occurring cell populations, adherent (A) and floating (F) cells. Between the 2 subpopulations, the F cells were characterized to possess several CSC-like properties, including CD44high/CD24low phenotype, high expression of multiple drug resistance genes, and tumor-initiating ability in NOD/SCID mice with low cell numbers, depressed HLA class I expression,[9,10] and resistance to natural killer (NK)/lymphokine activated killer (LAK)-mediated cytolysis, relative to CD44low/CD24high A cells.[9] In addition, F and A cells were found mutually convertible with F to A cells at a faster rate. It is also conceivable that F cells may be more easily separated from the primary lesion than A cells to enter the bloodstream as circulating tumor cells and then deposit and proliferate at the new site through extravasation and intravasation, as the initial step toward metastasis. This phenomenon appears not to be restricted to GI malignancies since a similar result was recently reported with other cancer types such as breast cancer recently.[11] Within the CSC cell population, there is an even smaller subset which could be become induced to CXCR4-positive mCSCs responsible for initiating metastatic activity in or migrate toward/invade a new microenvironment where a greater CXCL12 gradient is present.[9] Moreover, in response to interferon-γ (IFN-γ) or activated NK or LAK cells, the CXCR4-positive mCSCs could only be induced from CSCs, which were harbored in the highly tumorigenic CD44high/CD24low F subset. Thus, the UP-LN1 cell line represents an ideal in vitro model for studying CSCs and screening for effective anti-CSC and anti-mCSCs agents.
Withaferin A (WA), a cell-permeable steroidal lactone extracted from the Indian winter cherry, Withania somnifera, has been cited for its anti-cancer effects via multiple mechanisms.[12-16] For instance, WA has been shown to elicit oxidative stress reactive oxygen species (ROS) and mitochondrial dysfunction in leukemia cells leading to apoptosis.[14] In breast cancer, WA-induced apoptosis via the induction of Bim-s and Bim-L in estrogen-responsive MCF-7 cells and in triple-negative MDA-MB-231 cells.[17] In another study, WA has been shown to exhibit anti-tumor and anti-angiogenesis activity by binding to the intermediate filaments vimentin and F-actin.[18] More importantly, WA at low dosages appeared to eliminate cells expressing breast CSC markers including CD44, CD24, CD34, CD117 and Oct 4, and to down-regulate Notch1, Hes1 and Hey1 expression,[19] suggesting the potential of WA as a CSC-targeting compound. Together, these findings provide the rationale to further explore the anti-cancer effects of WA on the UP-LN1 cell line in terms of the mechanisms involved in blocking the formation of CSCs and/or mCSCs.
In this study, we also used side-population (SP) method[20,21] to enrich the CSC subpopulation from UP-LN1 cells, and then showed that F cells harbored the highest percentage of CSC-like cells with an elevated expression of few selected stemness-related genes. Subsequently, we demonstrated that WA treatment could inhibit the formation of tumor aggregates/spheres and induce apoptosis in F cells. More importantly, WA treatment could lead to the down-regulation of CXCR4/CXCL12 and STAT3/IL-6 axes, both being key members of a metastatic signaling pathway. Finally, using non-invasive bioluminescence imaging technique, we demonstrated that after treatment with WA, both the tumor burden and dissemination ability were significantly suppressed in NOD/SCID mice implanted with F cells.
Methods
Chemicals and reagents
WA was purchased from Sigma-Aldrich (St. Louis, MO, USA), and its purity was > 95%. Primary monoclonal antibodies (mAbs) to Oct4, Sox2, c-Myc, Nanog, vimentin, Fas receptor, caspase-3, caspase-8, caspase-9, poly ADP-ribose polymerase (PARP), Bcl-2, survivin, Akt, ERK, GRK3/2, STAT3, and β-actin were purchased from Cell Signaling Technology (Boston, MA, USA). Additional mAbs used were as follows: Mouse anti-human CXCL12 (clone 79018, R and D Systems, Minneapolis, MN, USA), and CXCR4 (clone 15G5, R and D Systems, Minneapolis, MN, USA), Fluorescein isothiocyanate (FITC) or phycoerythrin-conjugated goat-antimouse IgG (Biolegend, San Diego, USA) were used as the secondary Ab for tracing the primary mAb. For multiple color-phenotyping, 5 × 105 cells were directly incubated with FITC-conjugated mouse anti-human CD44 (clone G44-26, BD-Pharmingen, Franklin Lake, NJ, USA), and allophycocyanin-conjugated mouse anti-human CD24 (clone ML10, Biolegend, San Diego, USA) mAbs according to the manufactural instructions. Labeled cells were then washed 3 times by phosphate-buffered saline (PBS) plus 2% fetal bovine serum (FBS) followed by fixation with 1% paraformaldehyde. The fixed samples were then analyzed cytofluorometrically.
Cell line, subsets and culture conditions
The UP-LN1 cell line and its A and F subsets were used in this study. Unless specified, all the cell lines were maintained in the condition described previously.[8] For the separation of A cells, we discarded all floating cells in the culture supernatant and then harvested only the adherent cells by light trypsinization to set up new cultures for A cells. To obtain F cells, we only collected the floating cells in the culture supernatant of UP-LN1 culture for the subsequent culture passage. Each of these 2 protocols was used for the enrichment of A or F cells when subculturing for 10 consecutive rounds. To maintain the parental (P) UP-LN1 cells (termed P cells), floating cells and trypsinized adherent cells were washed and pooled, then set up in a new culture. Trypan blue dye exclusion was used to determine cell viability. A and F cells were maintained in Roswell Park Memorial Institute (RPMI)-1640 medium supplemented with 10% FBS.
Characterization of UP-LN1 cells by SP analysis
To examine the existence of CSCs in the UP-LN1 carcinoma cell line, the SP cells were isolated by flow cytometry and cell sorting techniques. SP cells have been shown to express an elevated level of ATP-binding cassette transporters (ABCG2), which enhance their ability to pump out Hoechst 33342 dye. This efflux activity of Hoechst dye is similar to many drug-resistant cancer cells and can be sorted by FACSAria flow cytometry. Therefore, we utilized SP analysis as one of the characteristics to demonstrate and analyze the population of cancer stem-like cells in our unique UP-LN1 cells. UP-LN1 cells were labeled with 2.5 µg/mL Hoechst 33342 (Sigma-Aldrich, Chemie GmbH, Munich, Germany) for 30 min at 37 °C. The control cells were incubated in the presence of 50 µmol/L verapamil (Sigma-Aldrich, Chemie GmbH, Munich, Germany). Propidium iodine (PI) 1 µg/mL was added to identify dead cells. Analysis and sorting were performed on FACSAria flow cytometry (Becton Dickinson, San Jose, CA, USA), similar to that described by Patrawala et al.[20] After sorting, SP sphere cells of UP-LN1 were placed at a density of 1,000 cells/mL under stem cell conditions by resuspension in tumor sphere medium consisting of serum-free HEScGRO medium, N2 supplement (Invitrogen, Carlsbad, CA, USA), 10 ng/mL human recombinant bFGF (Invitrogen, Carlsbad, CA, USA), and 10 ng/mL EGF (Invitrogen, Carlsbad, CA, USA), followed by culturing in ultra-low attachment plates (Corning, NY, USA) for about 1 week.
Assessment of the growth of UP-LN1 cells and subsets following WA treatment
Sulforhodamine B (SRB) dye (Sigma-Aldrich, Chemie GmbH, Munich, Germany) was used to test the effects of selective inhibitors on cell growth and viability of SP cells. The WA was dissolved in dimethyl sulfoxide (DMSO) before diluting with growth medium to a final DMSO concentration of 0.05%. The P, A, and F cells were seeded into 96 well plates in growth medium at 3,000 cells/well. After 24 h, the medium was replaced with fresh growth medium containing the WA. The cells were incubated for another 48 h. The cells were fixed with trichloroacetic acid (TCA) by gently adding 50 µL TCA (50%) to each well to a final TCA concentration of 10% with subsequent incubation for 1 h at 4 °C. The plates were then washed 5 times with tap water and air dried. The dried plates were stained with 100 µL of 0.4% (w/v) SRB prepared in 1% (v/v) acetic acid for 10 min at room temperature. The plates were rinsed quickly 4 times with 1% acetic acid to remove unbound dye and were then air dried until no moisture was visible. The bound dye was solubilized in 20 mmol/L Tris-base (100 µL/well) for 5 min on a shaker. Optical densities were read on a microplate reader (Molecular Devices, Sunnyvale, CA, USA) at 562 nm.
Apoptosis assay
Apoptosis was assessed by staining the cells with Annexin V-FITC (BD-Pharmingen, Franklin Lakes, NJ, USA), and PI and analyzing stained cells via flow cytometry. Briefly, the UP-LN1 P, A, and F cells (1 × 106 cells/mL) were grown in RPMI medium alone or in the same medium supplemented with either WA or DMSO. After 48 h, cells were washed twice with ice-cold PBS and then re-suspended in 100 µL binding buffer containing 2 µL of FITC-conjugated Annexin V and 2 µL of PI for 15 min. Following the incubation, without washing the cells with excess reagents, 400 µL binding buffer was added. Samples were then analyzed by flow cytometry. Data acquisition and analysis were done using CellQuest™ software (Becton Dickinson, San Jose, CA, USA).
Knockdown of CXCR4 by siRNA
UP-LN1 cells were transfected with Validated MISSION® siRNA (SASI_Hs01_00084886, Sigma-Aldrich Taiwan, Linkou, New Taipei City, China) according to the vendor’s instructions. Transfected cells were lyzed and subjected to both total RNA extraction and Western blot analysis 48 h post-transfection. CXCR4 expression was confirmed using Western blot and anti-CXCR4 antibody (SAB3500383, Sigma-Aldrich, China).
CDy1 immunofluorescence staining
UP-LN1 P, A, and F cells were cultured in a 60-mm culture dish for 24 h in the presence of 500 nmol/L CDy1.[22] CDy1 was a generous gift from Dr. YT Chang, Laboratory of Bioimaging Probe Development, Singapore Bioimaging Consortium Agency for Science, Technology, and Research, Singapore through Dr. Gi-Min Lai (Wan-Fan Hospital, Taipei, China). The cells were harvested by trypsin treatment, washed with PBS, and re-suspended in PBS. The cells were fixed with 4% paraformaldehyde for 10 min, permeabilized with 0.1% Triton X-100/PBS for 10 min, and blocked with 2% bovine serum albumin/PBS for 1 h. After incubation in dark at room temperature for 15 min, the cells were rinsed with PBS. The fluorescence images of the cells were acquired using fluorescence microscope (Nikon, Lewisville, TX, USA).
In vitro cell migration and invasion assays
A Boyden chamber system was used to measure the invasive ability of UP-LN1 cells. Briefly, UP-LN1 P, A, and F cells were harvested, washed with PBS, and re-suspended in a serum-free RPMI medium (5 × 104 cells/200 µL) in the presence or absence of WA. The cells were then seeded into the upper chambers of Matrigel-coated filter inserts. A serum-containing RPMI-1640 medium (500 µL) was added to the lower chambers. After incubating for 24 h at 37 °C, filter inserts were removed from the wells, the cells that had invaded were stained with PI, and fluorescence images were taken. The number of invaded cells was determined using Analytical Imaging Station Software Package (Imaging Research, ON, Canada). The migration assay was performed accordingly but with 8-µm pore polycarbonate filters, which were not coated with Matrigel.
Western blotting
UP-LN1 P, A, and F cells lysates were prepared using ReadyPrep Protein Extraction Kit (Bio-Rad, Hercules, CA, USA) according to the instructions provided. Total cell lysates (50 µg) were separated electrophoretically by a 10% polyacrylamide SDS-PAGE gel and transferred to a polyvinylidene fluoride membrane using the BioRad Mini Protean transfer system. The blots were then blocked with 5% skim milk in PBST for 1 h and probed with primary antibodies overnight at 4 °C. All primary antibodies were purchased from cell signaling unless otherwise specified. The membranes were sequentially detected with an appropriate peroxidase-conjugated secondary antibody incubated at room temperature for 1 h. Blots were washed 3 times with PBS. Signals were then detected using the enhanced chemiluminescence detection system and the BioSpectrum Imaging System (UVP, Upland, CA, USA).
In vivo evaluation of WA-mediated anti-UP-LN1 F cell effects
All animal studies were performed strictly under the animal experimentation protocols approved by Taipei Medical University. UP-LN1 F cells were first modified to express dual reporter system, FUW-Luc-mCherry-Puro (a generous gift from Dr. Andrew Kung, Lurie family Imaging Center, Dana-Farber Cancer Institute, MA) according to an established protocol.[14] Imaging-ready UP-LN1 F cells were harvested and subcutaneously injected into the left flank for NOD/SCID mice (3 × 105 cells/mouse; 5 mice/group). Tumor-bearing mice were then subdivided into control and WA-treated groups (10 mg/kg intraperitoneal injection [i.p.], 3 times a week). For intravenous (i.v.) tumor injection, 5.5 × 105 cells/mouse were injected, followed by WA i.p injection as described for subcutaneous (s.c.) tumor injected animals. For either the s.c. or i.v.-tumor injection group, WA treatment was initiated 2 weeks after tumor injection into the animals. Tumor burden was then non-invasively assessed based on bioluminescence intensity for 6 weeks using IVIS200 system (Caliper Life Sciences Inc., Hopkinton, MA, USA). Tumor autopsies were obtained at the end of the experimental period by humanely sacrificing the animals for pathological and immunohistological analyses. All experiments were conducted in accordance with the Guide for the Care and Use of Laboratory Animals of the National Health Research Institutes of Taiwan and following the Institutional Animal Care and Use Committee protocol authorized by Taipei Medical University.
Histology and immunohistochemical staining
Tumor tissues were fixed in 10% formalin and embedded in paraffin. Serial sections of the embedded specimens were deparaffinized and then rehydrated in a gradient fashion and stained with hematoxylin and eosin. For immunohistochemical staining, the deparaffinized slides were subjected to antigen retrieval and probed with anti-CXCR4 (1:100), anti-caspase-3 (1:200), anti-PARP (1:100) antibodies, or isotype IgG control. Slides were washed and incubated with biotinylated link universal antiserum, followed by horseradish peroxidase-streptavidin conjugate (LSAB 1 kit). The slides were rinsed, and the color was developed using 3,3-diaminobenzidine hydrochloride as a chromogen. Finally, sections were rinsed in distilled water, counterstained with Mayer’s hematoxylin, and mounted with DPX mounting medium for evaluation. Pictures were captured with a Photometrics CoolSnap CF color camera (Nikon, Lewisville, TX, USA).
Statistical analysis
Each experiment was performed in triplicate. The results were expressed as means ± standard deviation. The significant difference between control and experimental groups was analyzed using t-test (*P < 0.05; **P < 0.01).
Results
F subset of UP-LN1 cells are enriched with cancer stem cells
We utilized the SP method to compare and analyze the percentage of F cells in the UP-LN1 cell line. In the absence and presence of verapamil, the percentage of SP cells in each group was calculated. Results of SP analysis from 1 of 3 independent experiments showed that parental (P) UP-LN1 cells contained an intermediate of 2.93% SP cells, A cells contained the least among the three groups at 1.07%, and F cells contained the highest of 4.20% [Figure 1a, upper frame]. The results show Hoechst 33342 dye exclusion was verapamil-sensitive; they suggest that F cells contained the highest proportion of CSCs in UP-LN1, and A cells contained the least. Quantitative results based on the 3 experiments reveal the statistical differences between F versus A and between F versus P cells in terms of the percentage of SP cells as follows: F > A with P < 0.01, and F > P with P < 0.05 [Figure 1a, lower frame]. To reinforce our SP data, an embryonic stem cell specific fluorescent dye CDy1[22] was used to stain UP-LN1 cells. The red fluorescent signal was strongly associated with F cells as compared to A cells [arrowheads, Figure 1b]. Notably, red fluorescence was significantly stronger in F cell aggregates [arrowheads, Figure 1b]. To add support to F cells identity as potential CSCs, we examined the expression of stemness gene signatures such as Nanog, Oct4, Sox2, and c-Myc in the UP-LN1 cell line. F cells exhibited the highest expression level of these stemness genes followed by P and A cells [Figure 1c], establishing F cells as the major subpopulation containing the CSCs and their CSC niche of the UP-LN1 cell line.
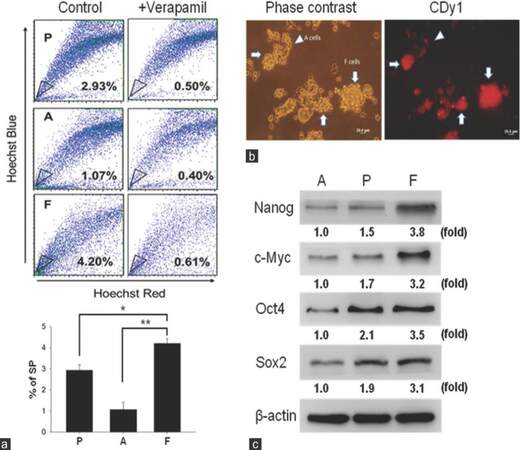
Figure 1. Characterization of cancer stem-like properties of UP-LN1 cells. (a) The side-population method was employed to compare and analyze the percentage of potential stem cell-like cells in UP-LN1 cells. With the absence and presence of verapamil, the percentage of side-population cells was calculated. In the upper frame, based on representative results of one experiment, parental (P) contained an intermediate of 2.93% side-population cells, adherent (A) cells contained the least among the three groups at 1.07%, and floating (F) cells contained the highest percentage at 4.20%. The quantitative results shown in the lower frame reveal that the percentage of side-population in F cells is significantly higher than that in A and P cells with P < 0.01 and P < 0.05, respectively; (b) to support our side-population data, an embryonic stem cell-specific fluorescent dye CDy1 was used to stain UP-LN1 cells. The red fluorescent signal was strongly associated with F cells as compared to A cells (arrowheads). Notably, red fluorescence was signifi cantly stronger in F-cell aggregates (arrows); (c) when examined by Western blot analysis, F cells were found to express a significantly higher level of stemness genes (including Nanog, c-Myc, Oct4, and Sox2) than P and A cells. Note that the relative densities in the expression of each stemness gene among F, P, and A cells are also shown by fold difference in this figure
WA reduces SP and cell aggregates in UP-LN1 cells
We next sought to examine the potential CSC inhibitory effect of WA. Our cytofluorometric data demonstrated that WA reduced the percentage of SP cells in UP-LN1 in a dose-dependent manner [Figure 2a]. The ability of WA to affect UP-LN1 viability was then tested on F and A cells. The viability of SP in A cells was affected least among the three groups. WA preferentially targeted F cells in a dose-dependent fashion [Figure 2b]. Since F cells spontaneously formed grape-like cell aggregates, they are a close representation of the so-called tumor spheres or CSCs reported. WA treatment also prevented the formation of F-cell aggregates [Figure 2c]. At 10 µmol/L, WA reduced F-cell aggregates by approximately 80%.
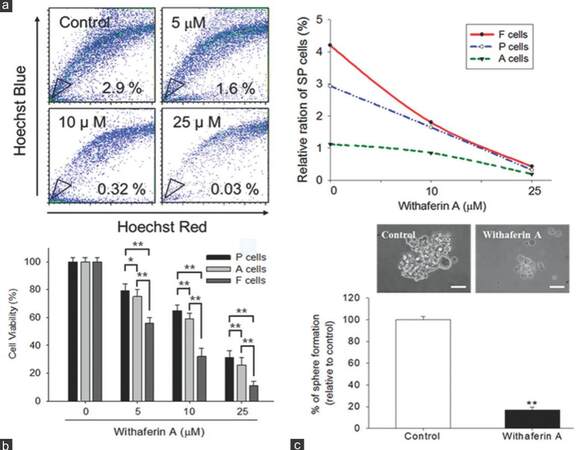
Figure 2. Withaferin A reduced side-population and cell aggregates in UP-LN1 cells. (a) Withaferin A reduced the percentage of side-population cells in UP-LN1 in a dose-dependent manner as indicated by our cytofluorometric analysis. The reduction of side-population cells in different populations of UP-LN1 cells was plotted against the concentration of withaferin A (right panel); (b) sulforhodamine B viability assay indicated that withaferin A targeted floating (F) cells most effectively and also in a dose-dependent manner as compared with adherent (A) or parental (P) cells. The difference between F and A cells is signifi cant (P < 0.01), and so is the difference between F and P cells (P < 0.01) at each of the three withaferin A dose levels (5, 10, 25 μmol/L) tested; (c) microscopic analysis of F cells under the influence of withaferin A demonstrated a significant reduction in the formation of F-cell aggregates or spheres. At 10 μmol/L, withaferin A reduced F-cell aggregates by approximately 80%
WA preferentially induces apoptosis in F cells
Since WA has been shown to induce apoptosis in cervical cancer cells,[16] we wished to determine if it exerted a similar function in UP-LN1 cells. Using Annexin V as an apoptotic indicator, we demonstrated that WA promoted apoptosis in P, F, and A cells in a dose-dependent manner [Figure 3a-c, respectively]. When analyzed quantitatively, WA appeared to preferentially target F cells and triggered apoptosis to a higher extent in F cells than in P and A cells [Figure 3b]. This observation corroborates the preference of WA in suppressing the formation of F-cell aggregates in the aforementioned section. In addition, Western blot analysis of cell lysates obtained from WA-treated F cells indicated an increased expression in some pro-apoptotic molecules including caspase-3, -8, -9 and PARP at higher concentrations [≥ 5 µmol/L, Figure 3c]. The remaining pro-apoptotic molecule, Fas receptor, and anti-apoptotic molecules such as Bcl-2 and survivin were clearly down-regulated in a dose-dependent manner.
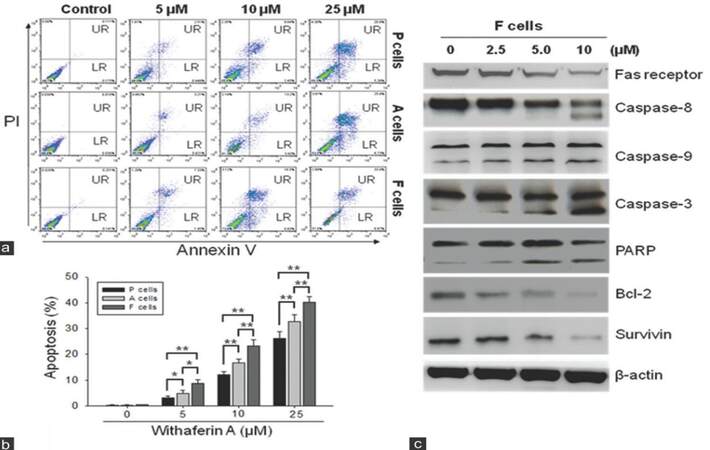
Figure 3. Withaferin A induced apoptosis in floating (F) cells. (a) Using cytofluorometric technique and Annexin V as an apoptotic indicator, we demonstrated that withaferin A promoted apoptosis in parental (P), F and adherent (A) cells in a dose-dependent manner; (b) quantitative representation of withaferin A-induced apoptosis. Withaferin A appeared to trigger apoptosis to a higher extent in F cells than in P cells (P < 0.01) or in A cells (P < 0.01); (c) immunoblots of total cell lysates obtained from withaferin A-treated F cells showed an increased expression of pro-apoptotic molecules such as caspase-3, -8, -9, and poly ADP-ribose polymerase at higher concentrations of withaferin A (5-10 μmol/L), except Fas receptor. On the other hand, anti-apoptotic molecules, survivin, and Bcl-2 were clearly down-regulated when the two higher concentrations of withaferin A were used
WA treatment suppresses two major metastasis signaling pathways (STAT3 and CXCR4) in F cells
The presence of IFN-γ in tumor microenvironment or NK/LAK culture conditioned medium has been reported to promote the metastatic ability of cancer cells through the modulation of CXCR4 expression in cancer cells.[9,23] We wished to determine if WA treatment could overcome metastatic potential induced by IFN-γ in the CSC-like F-cell population. It was observed that the addition of IFN-γ increased the invasive ability in all 3 cell populations of the UP-LN1 cell line, with F cells at the highest efficiency.[9] In the presence of WA, IFN-γ-induced invasion was significantly suppressed, particularly in the F-cell population [Figure 4a]. We subsequently examined the 2 major signaling axes involved in cellular trafficking, namely CXCR4 and STAT3.[24,25] Using cytofluorometric analysis, we demonstrated, as shown previously,[9] that the surface expression of CXCR4 was elevated in the presence of IFN-γ at concentrations as low as 10 U/mL, in terms of increased percentage of CXCR4-positive cells. Notably, each positive cell bore a relatively constant number of CXCR4 receptor sites, as revealed by a constant value of mean fluorescence intensity. The addition of a low concentration of WA (2.5 µmol/L) reduced the percentage of CXCR4-positive cells [Figure 4b], and with the addition of this agent at a higher concentration (5 µmol/L), CXCR4-positive cells could hardly be detected.
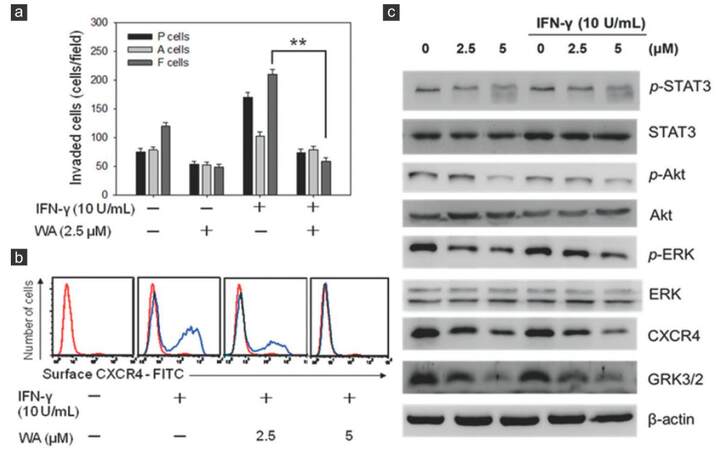
Figure 4. Withaferin A suppressed metastatic potential via modulating signaling pathways participating in invasive tumor activity. (a) Withaferin A treatment was able to suppress interferon-γ-induced invasive ability in both parental (P) and to floating (F) cells; (b) withaferin A treatment also suppressed the expression of CXCR4 expression even under the stimulation of interferon-γ. The difference between F cells treated with 2.5 μmol/L withaferin A and F cells without withaferin A treatment is significant (P < 0.01) regardless of whether or not interferon-γ (10 U/mL) was used to stimulate F cells; (c) Western blot analysis of withaferin A-mediated suppression in invasive ability in F cells. Several major signaling pathways including Akt, ERK, CXCR4, GRK3/2 and STAT3, all of which are known to participate in cell mobility, appeared to be down-regulated by withaferin A treatment in a dose-dependent manner
Next, we examined STAT3 and several key signaling pathways involved in cancer metastasis using Western blot analysis. WA treatment suppressed the expression of Akt/ ERK, CXCR4, STAT3, and GRK3/2 in F cells with or without the induction of mCSCs by IFN-γ treatment [Figure 4c]. A note of explanation is needed regarding the appearance of CXCR4 bands in the absence of IFN-γ added, which is contradictory to the results obtained cytofluorometrically. This was most likely due to the fact that in Western blotting, both surface and cytoplasmic CXCR4 molecules were detected, whereas in the cytofluorometric results, only the surface CXCR4 molecules were seen. Moreover, the inhibitory effect on the phosphorylation of STAT3 was readily noted at the highest concentration of WA (5 μmol/L) used. Collectively, we concluded that WA blocked the formation of IFN-γ-mediated induction of mCSCs through the inhibition of both STAT3 and CXCR4 pathways.
Time course study of inhibition of IFN-γ-enhanced CXCR4 expression in F cells by WA
Vimentin is known to affect the mobility and invasiveness of cancer cells.[26] Increasing evidence also indicates that the expression of vimentin is closely associated with CSCs and EMT positive circulating tumor cells.[27,28] In addition, we have shown that IFN-γ induces surface CXCR4 expression on the F but not A subset of the UP-LN1 cell line, while the same treatment decreases cytoplasmic expression of CXCL12 in the F, but not the A, subset.[9] No changes were found in the expression of CXCR4 and CXCL12 in A cells.[9] These findings prompted us also to look into the possible correlation between vimentin, CXCL12, and CXCR4 expression by F cells pretreated with 10 U/mL IFN-γ for 48 h, followed by incubation with 5 µmol/L WA for indicated time periods in vitro. In Figure 4c, we showed that WA could exhibit a direct inhibitory effect on IFN-γ-mediated enhancement of surface CXCR4 expression in F cells in Western blot analysis. The expression of both CXCL12 and vimentin was inhibited in a similar manner as early as 12 h after WA treatment, although the extent of inhibition for CXCL12 was not as obvious as that for CXCR4 or vimentin [Figure 5a].
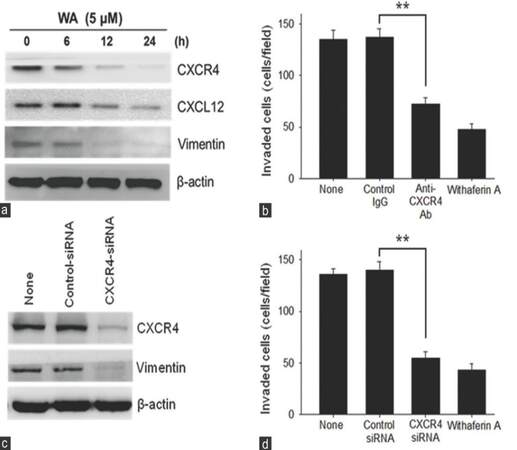
Figure 5. Inhibition of the CXCL12/CXCR4 axis expression by withaferin A in F cells which were pretreated with interferon-γ (10 U/mL for 24 h). (a) Time-dependent inhibition of CXCR4, CXCL12, and vimentin expression by withaferin A. Interferon-γ pretreated floating (F) cells were treated with 5 μmol/L withaferin A for various lengths of time as indicated, and the expression of each protein was measured by Western blot analysis; (b) suppression of cellular invasion by interferon-γ pretreated F cells by neutralizing anti-CXCR4 antibody. Interferon-γ pretreated F cells were incubated with 5 μg/mL control IgG, 5 μg/mL anti-CXCR4 antibody, or 5 μmol/L withaferin A for 24 h. Asterisks denote a statistically significant difference between anti-CXCR4 antibody treatment and control IgG treatment (P < 0.01); (c) knockdown of CXCR4 expression by CXCR4-siRNA. Interferon-γ pretreated F cells were transfected with control-siRNA or CXCR4-siRNA, and the expression of CXCR4 and vimentin were measured by Western blot analysis; (d) suppression of cellular invasion by interferon-γ pretreated F cells by treatment with CXCR4-siRNA. Interferon-γ pretreated F cells were transfected with control-siRNA or CXCR4-siRNA. Interferon-γ pretreated F cells were then harvested, and incubated in a chamber for 24 h. Asterisks denote a statistically significant (P < 0.01) difference between CXCR4-siRNA treatment and control-siRNA treatment
Effect of inhibition of CXCR4 expression on in vitro invasion of IFN-γ-treated F cells
IFN-γ-induced surface CXCR4 expression on F cells was blocked by anti-CXCR4 mAb, and cell invasion was examined by in vitro assay. IFN-γ-treated F cell invasion was clearly much reduced when compared to the control IgG group or the untreated group, each with P < 0.01 [Figure 5b]. Similarly, when the expression of CXCR4 was knocked down by CXCR4 siRNA treatment, the invasion of IFN-γ-treated F cells was again significantly reduced as compared with either the control siRNA group or the untreated group, each with P < 0.01 [Figure 5c and d]. Taken together, we herein clearly demonstrated that the extent of attenuation patterns of IFN-γ-induced CXCR4 expression in F cells following WA treatment was similar to that following blocking by anti-CXCR4 [Figure 5b] or that following knockdown of CXCR4 by CXCR4 siRNA [Figure 5c and d]. The observed attenuation of CXCR4 expression by F cells seemed to be accompanied by a decrease in vimentin and CXCL12 expression. The similarity in the response profile between vimentin and CXCR4 expression is striking.
In vivo validation of WA-mediated suppression of tumor growth and metastasis
Finally, we wished to validate the anti-cancer effects using tumor-bearing mouse model. F cells expressing dual luciferase reporter (enhanced green fluorescent protein and firefly luciferase, L2G) were subcutaneously injected into NOD/SCID mice for in vivo validation of WA-mediated anti-cancer effects. Tumor burden and spreading were monitored using the bioluminescent imaging technique. Tumor burden was significantly larger in the control group than in the WA-treated group [left panel, Figure 6a], reflected by the change in bioluminescent intensity. Importantly, anterior spreading of tumor cells was evident in the control animals starting from week 3, while WA-treated animals exhibited suppressed and restricted bioluminescent signal at the primary lesion site [left panel, Figure 6a]. Tumor burden was quantitatively measured as fold changes in bioluminescent intensity and plotted against time [right panel, Figure 6a]. In another model, when F cells were intravenously injected, they appeared to localize to the abdominal region of the animals, and WA treatment appeared to prevent the spreading of the F cells [Figure 6b]. Subcutaneous tumor biopsies were obtained for immunohistochemical analysis. Immunostaining of CXCR4 was significantly less intense in the tumor sections from WA-treated mice than in control samples [Figure 6c]. In contrast, caspase-3, -8, -9 and PARP staining was markedly higher in the WA-treated xenografted tumor samples. These in vivo observations, while preliminary, corroborated our in vitro data (induction of apoptosis), suggesting that WA could indeed suppress metastatic propensity and induce apoptosis.
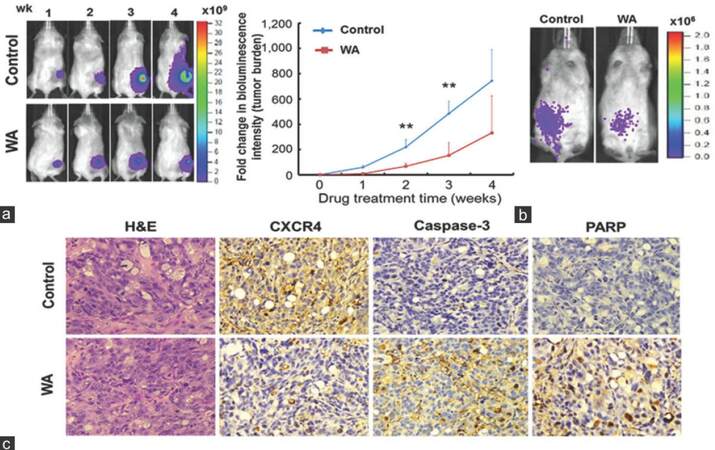
Figure 6. In vivo validation of withaferin A-mediated anti-cancer effects. (a) Representative bioluminescence images of control and withaferin A-treatment mice subcutaneously inoculated with floating (F) cells. Withaferin A treatment was initiated 2 weeks post-tumor implantation to allow tumor establishment. Note, inoculated F cells were found to start disseminating anteriorly during week 3-4 as compared to localized signal found in withaferin A-treated mice (left panels). The bioluminescent intensity was significantly lower in withaferin A-treated group than in the control group. The data were quantitatively represented by the fold change in bioluminescence intensity over time (right panel); (b) systematic injection of F cells mimicking metastatic model. Representative images of F cell-inoculated mice (via tail vein injection) demonstrated that withaferin A treatment given 2 weeks after intervenous tumor injection not only suppressed tumor growth but also controlled the spread of F cells; (c) immunohistochemical analysis of tumor samples harvested from both control and withaferin A-treated animals (from subcutaneous tumor samples). Representative sections of the tumor samples were stained with CXCR4, caspase-3, and poly ADP-ribose polymerase antibodies. Samples treated with withaferin A demonstrated decreased CXCR4 immunostaining, while increased caspase-3 and poly ADP-ribose polymerase immunostaining as compared with the control samples (×200)
Discussion
CSCs, which are a small subpopulation of tumor cells, are characterized by their tumor-initiating/self-renewal capacities and the ability to generate bulk populations of non-tumorigenic progenies through differentiation. CSCs have been identified in many human malignancies, and their abundance in clinical specimens has been correlated with disease progression.[3] Importantly, clinical cancer progression driven by CSCs may contribute to the failure of both conventional and targeted therapies.[4] CSC is targeted by a novel fluorescent dye, CDy1, which has specific affinity for pluripotent stem cells.[22] Suspended F cells spontaneously formed tumor aggregates or spheres under normal culture conditions and gave rise to A cells with a greater differentiated phenotype, which have been shown to be more sensitive to the conventional therapeutic modalities, such as chemotherapy and/or radiotherapy. The dynamic phenotypic transition between F and A cells closely resembles CSC physiology, thereby representing an ideal in vitro model. Therapeutic approaches represent translational strategies, which could improve the clinical outcome for patients with malignancies that are currently refractory to conventional treatments.[4,5] However, developing such an agent or strategy has been hindered by the lack of an experimental CSC cell model, which could be maintained with relative ease and replicate most of the clinical CSC characteristics.
Previously, we identified the UP-LN1 cell line which was characterized by a co-existence of 2 unique cell populations.[8,9] The CD44high/CD24low F cells attracted our attention particularly due to their phenotypic and cellular resemblance of CSCs in the form of tumor spheres. Using SP methods, we demonstrated that F cell population was enriched with CSCs with clearly elevated levels of stemness markers including Nanog, c-Myc, Oct4 and Sox2 noted, as compared with the CD44low/CD24high A-cell population [Figure 1a].
Having established this CSC cell model,[9] we intended to examine WA as a potential anti-CSC and anti-mCSC agent. WA has been indicated for its anti-cancer effects by modulating multiple molecular pathways, predominantly through the induction of intracellular oxidative stress.[12,13,17,29] WA dose-dependently reduced the percentage of SP cells in A, F, and parental UP-LN1 cells [Figure 2a]. Interestingly, F cells appeared to be more sensitive to WA treatment than A and parental cells [Figure 2b]. In addition, we demonstrated that WA promoted apoptosis in F cells in a dose-dependent manner as evidenced by both cytofluorometric [Figure 3a and b] and Western blot [Figure 3c] analyses via up-regulating caspases-3, -8 and -9, as well as PARP, which are collectively a family of proteins involved in a number of cellular processes, such as DNA repair and programmed cell death. It has been suggested that CSCs were more sensitive to ROS-induced apoptosis.[26,27] F cells were found to contain a lower intracellular glutathione (GSH) level (data not shown), which could partially explain why WA eliminated F cells more efficiently. On the other hand, A cells are in general more sensitive to killing by chemotherapeutic drugs and NK/LAK cells.[8,9]
In addition to promotion of apoptosis in F cells, WA appeared to be potent in suppressing the metastatic potential of F cells. As we observed in our previous study,[9] we have also demonstrated here that the migratory/invasive ability of UP-LN1 cells could be stimulated by low levels of IFN-γ via an increase in only the percentage of F cells expressing CXCR4, while the number of CXCR4 density on a per-cell basis remained relatively constant [Figure 4b]. This study showed that WA suppressed IFN-γ-induced invasiveness in the UP-LN1 parental, A and F cells in a dose-dependent manner [Figure 4a], with the greatest effect on F cells, the intermediate effect on parental cells, and least effect on A cells. The WA-mediated effect was found to be through down-regulation of CXCR4 and vimentin expression [Figure 4b]. This finding suggests that the increase in the number of CXCR4-positive F cells, also known as mCSCs, played a pivotal role in lymph node metastasis of some types of GI cancer, including UP-LN1.[8,9,30] This observation was further supported by a recent study where the positive CXCR4 expression was shown to be significantly associated with lymph node metastases (P = 0.028) and higher stages III/ IV (P = 0.047) in gastric cancer.[27] In addition, several other major metastasis-associated pathways such as Akt/ ERK, GRK3/2, and STAT3 could also be attenuated by the addition of WA [Figure 4c]. Interestingly, GRK3/2 expression, in particular, has been shown to form complexes with CXCR4 and/or FAK in human primary monocyte-macrophages and to play a part in their trafficking, upon the stimulus by inflammatory cytokines such as IL-4 and IL-13.[30] Equally important, it has been demonstrated in small cell lung cancer where the activation of CXCR4/CXCL12 axis leads to the activation of the JAK/STAT3 pathway.[31-34] STAT3 has been found and deposit and proliferate at the new site constitutively activated as a result of carcinogenesis in different cancer types, and aberrant STAT3 signaling has been implicated as an important process in malignant transformation[35-37] and induction of angiogenesis.[37] Activation of CXCR4 and STAT3 has been linked to tumor progression in different cell types, such as hematopoietic progenitor cells[38] and small cell lung carcinoma lines[39] with high mobility. Interestingly, STAT3 activation was shown to allow a crosstalk between tumor cells and dendritic cells which forms an immunosuppressive microenvironment favorable for tumor survival and perpetuation.[40] Thus, the observation that WA treatment negatively regulated the CXCR4/CXCL12 chemotactic axis and molecules involved in IFN-γ-stimulated F cells, leading to making more CXCR4-positive mCSCs in cell numbers but not in increasing the number of CXCR4 sites on a per-cell basis, provides support for using WA as a potent anti-metastasis agent. The extent of the inhibitory effect of WA on invasiveness of IFN-γ-treated F cells was as great as that achieved by IFN-γ-mediated induction of CXCR4 in F cells attenuated by neutralizing anti-CXCR4 antibody [Figure 5b], or by knocking down the CXCR4 expression using siRNA [Figure 5c]. It should be pointed out that IFN-γ is not the only agent or means known to induce mCSCs,[9] since other agents/methods such as HGF[41] and hypoxia conditions[42] have also been reported to be able to do the same. Interestingly, the fluctuations of vimentin and CXCR4 expression in the experiments stated above were very similar, suggesting the importance of these 2 molecules in the appearance of EMT phenotype during the process of cancer migration and invasion.[26,27] Interestingly, a recent study by Bargagna-Mohan et al. indicated that WA acted as a tumor inhibitor, as well as the antiangiogenic agent through targeting the intermediate filament protein vimentin.[18]
Based on our own current findings and those by others,[13,15,17,19] the apoptotic process triggered by WA works through multiple mechanisms. They involved the mitochondrial pathway and associated Bcl-2 down-regulation, caspase-8, -9 and -3 activations, DNA fragmentation, and the inhibition of both CXCR4/CXCL12 and STAT3/IL-6 pathways. To our knowledge, WA inhibition of IFN-γ-induced mCSCs via CXCR4/CXCL12 axis is reported herein for the first time. In addition, WA exerts the anti-angiogenic effect by targeting and binding vimentin, and WA cytotoxicity requires early ROS production and GSH depletion, and the inhibition of ROS increase resulting in complete suppression of a series of cellular events.[18] Collectively, these results strongly support the therapeutic potential of WA against different types of human solid tumors including GI malignancies.
Finally, we evaluated the anti-human cancer effects of WA in a xenograft mouse model. Using bioluminescent imaging technique,[14] we demonstrated that WA not only significantly suppressed the proliferation of F cells but also dissemination [Figure 6a]. A similar observation was made when F cells were injected intravenously [Figure 6b] that WA suppressed the dissemination of F cells around the abdominal region of the animals. Investigations on a larger panel of cancers or tumor cell lines similarly exhibiting the features of A and F subsets are warranted to confirm our current conclusion.
It is now considered that the CSC phenotype is more fluid than previously envisioned and is strongly modulated by the tumor microenvironment. This concept has been referred to as the dynamic CSC model.[42,43] Our data imply that UP-LN1 cells represent a micro-niche where the dynamic transitions between adherent cells (A cells, with the more differentiated status) and floating cells (F cells, with the undifferentiated CSC phenotype) occur, and may closely replicate the pathophysiological characteristics of CSCs in vivo. Accordingly, we believe that this unique cell line is valuable for the further study of the biology of CSCs and mCSCs and drug screening. Using our unique UP-LN1 cell model, we have provided experimental evidence that WA is a potent CSC- and an mCSC-targeting agent which preferentially promotes apoptosis in F cells and suppresses tumorigenic (CSCs) and metastatic (mCSCs) activities. It achieves the latter through the metastasis-associated signaling pathways, notably of CXCR4/CXCL12 and STAT3/IL-6. Therefore, we propose that WA is considered for clinical trials in patients with a subset of GI-cancer like UP-LN1 exhibiting grape-like tumor spheres.
Acknowledgments
The authors thank Dr. Alexander TH Wu and Dr. Chi-Tai Yeh for their interest and constructive advice in the experimental designs and technical aspects of this project and Zhi-Hua Chen and Guei-Yun Cheng for their skillful technical support.
Financial support and sponsorship
This study was supported by grants from the National Science Council of Taiwan (NSC99-2314-B-038-032, NSC100-2314-B-038-016 to SK Liao) and Chang Gung Medical Research Fund (CMRPD170043, CMRPG3A0811 to ST Pang and SK Liao).
Conflicts of interest
There are no conflicts of interest.
REFERENCES
1. Spano D, Zollo M. Tumor microenvironment: a main actor in the metastasis process. Clin Exp Metastasis 2012;29:381-95.
2. Thiery JP. Epithelial-mesenchymal transitions in development and pathologies. Curr Opin Cell Biol 2003;15:740-6.
3. Reya T, Morrison SJ, Clarke MF, Weissman IL. Stem cells, cancer, and cancer stem cells. Nature 2001;414:105-11.
4. Eyler CE, Rich JN. Survival of the fittest: cancer stem cells in therapeutic resistance and angiogenesis. J Clin Oncol 2008;26:2839-45.
7. Croker AK, Allan A. Cancer stem cells: implications for the progression and treatment of metastatic disease. J Cell Mol Med 2008;12:374-90.
8. Chang CJ, Weng WH, Chou AS, Chuang CK, Porwit-McDonalds A, Pang ST, Larsson C, Liao SK. Immunophenotyic and molecular cytogenetic features of the cell line UP-LN1 established from a lymph node metastasis of a poorly-differentiated carcinoma. Anticancer Res 2005;25:683-92.
9. Chen HC, Chou AS, Liu YC, Hsieh CH, Kang CC, Pang ST, Yeh CT, Liu HP, Liao SK. Induction of metastatic cancer stem cells from the NK/LAK-resistant floating, but not adherent, subset of the UP-LN1 carcinoma cell line by IFN-gamma. Lab Invest 2011;91:1502-13.
10. Wu MS, Lia CH, Ruppertb JG, Chang CC. Cytokeratin 8-MHC class I interactions: a potential novel immune escape phenotype by a lymph node metastatic carcinoma cell line. Biochem Biophys Res Commun 2013;441:618-23.
11. Chaffer CL, Brueckmann I, Scheel C, Kaestli AJ, Wiggins PA, Rodrigues LO, Brooks M, Reinhardt F, Su Y, Polyak K, Arendt LM, Kuperwasser C, Bierie B, Weinberg RA. Normal and neoplastic nonstem cells can spontaneously convert to a stem-like state. Proc Natl Acad Sci U S A 2011;108:7950-5.
12. Choi MJ, Park EJ, Min KJ, Park JW, Kwon TK. Endoplasmic reticulum stress mediates withaferin A-induced apoptosis in human renal carcinoma cells. Toxicol In Vitro 2011;25:692-8.
13. Hahm ER, Moura MB, Kelley EE, Van Houten B, Shiva S, Singh SV. Withaferin A-induced apoptosis in human breast cancer cells is mediated by reactive oxygen species. PLoS One 2011;6:e23354.
14. Tai CJ, Wu AT, Chou JF, Jan HJ, Hsu CH, Lin CT, Chiu WH, Wu CW, Lee HM, Deng WP. The investigation of mitogen-activated protein kinase phosphatase-1 as a potential pharmacological target in non-small cell lung carcinoma, assisted by non-invasive molecular imaging. BMC Cancer 2010;10:95.
15. Mayola E, Gallerne C, Esposti DD, Martel C, Pervaiz S, Larue L, Debuire B, Lemoine A, Brenner C, Lemaire C. Withaferin A induces apoptosis in human melanoma cells through generation of reactive oxygen species and down-regulation of Bcl-2. Apoptosis 2011;16:1014-27.
16. Munagala R, Kausar H, Munjal C, Gupta RC. Withaferin A induces p53-dependent apoptosis by repression of HPV oncogenes and upregulation of tumor suppressor proteins in human cervical cancer cells. Carcinogenesis 2011;32:1697-705.
17. Stan SD, Hahm ER, Warin R, Singh SV. Withaferin A causes FOXO3a- and Bim-dependent apoptosis and inhibits growth of human breast cancer cells in vivo. Cancer Res 2008;68:7661-9.
18. Bargagna-Mohan P, Hamza A, Kim YE, Khuan Abby Ho Y, Mor-Vaknin N, Wendschlag N, Liu J, Evans RM, Markovitz DM, Zhan CG, Kim KB, Mohan R. The tumor inhibitor and antiangiogenic agent withaferin A targets the intermediate filament protein vimentin. Chem Biol 2007;14:623-34.
19. Kakar SS, Ratajczak MZ, Powell KS, Moghadamfalahis M, Miller DM. Withaferin A alone and in combination with cisplatin suppresses growth and metastasis of ovarian cancer by targeting putative cancer stem cells. PLoS One 2014;9:e10756.
20. Patrawala L, Calhoun T, Schneider-Broussard R, Zhou J, Claypool K, Tang DG. Side population is enriched in tumorigenic, stem-like cancer cells, whereas ABCG2+ and ABCG2- cancer cells are similarly tumorigenic. Cancer Res 2005;65:6207-19.
21. Yang Y, Fan Y, Qi Y, Liu D, Wu K, Wen F, Zhao S. Side population cells separated from A549 lung cancer cell line possess cancer stem cell-like properties and inhibition of autophagy potentiates the cytotoxic effect of cisplatin. Oncol Rep 2015;34:929-35.
22. Kang NY, Yun SW, Ha HH, Park SJ, Chang YT. Embryonic and induced pluripotent stem cell staining and sorting with the live-cell fluorescence imaging probe CDy1. Nat Protoc 2011;6:1044-52.
23. Zhang I, Yeger H, Das B, Irwin MS, Baruchel S. Tissue microenvironment modulates CXCR4 expression and tumor metastasis in neuroblastoma. Neoplasia 2007;9:36-46.
24. Zhou J, Wulfkuhle J, Zhang H, Gu P, Yang Y, Deng J, Margolick JB, Liotta LA, Petricoin E 3rd, Zhang Y. Activation of the PTEN/mTOR/STAT3 pathway in breast cancer stem-like cells is required for viability and maintenance. Proc Natl Acad Sci U S A 2007;104:16158-63.
25. Li HD, Huang C, Huang KJ, Wu WD, Jiang T, Cao J, Feng ZZ, Qiu ZJ. STAT3 knockdown reduces pancreatic cancer cell invasiveness and matrix metalloproteinase-7 expression in nude mice. PLoS One 2011;6:e25941.
26. Zao Y, Yan Q, Long X, Chen X, Wang Y. Vimentin affects mobility and invasiveness of prostate cancer cells. Cancer Biochem Funct 2008;26:571-7.
27. Mitra A. Cell surface vimentin (CSV) redefines CTCs to monitor cancer progression in patients. J Cancer Prev Curr Res 2014;1:00014.
28. Velasco-Vlaquez MA, Popov VM, Lisanti MP, Pestell RG. The role of breast cancer stem cells in metastasis and therapeutic implications. Am J Pathol 2011;179:2-11.
29. Grover A, Shandilya A, Punetha A, Bisaria VS, Sundar D. Inhibition of the NEMO/IKKß association complex formation, a novel mechanism associated with the NF-κB activation suppression by Withania somnifera's key metabolite withaferin A. BMC Genomics 2010;11 Suppl 4:S25.
30. Zhao BC, Wang ZJ, Mao WZ, Ma HC, Han JG, Zhao B, Xu HM. CXCR4/SDF-1 axis is involved in lymph node metastasis of gastric carcinoma. World J Gastroenterol 2011;17:2389-929.
31. Lee HJ, Kim SW, Kim HY, Li S, Yun HJ, Song KS, Kim S, Jo DY. Chemokine receptor CXCR4 expression, function, and clinical implications in gastric cancer. Int J Oncol 2009;34:473-80.
32. Wang J, Guan E, Roderiquez G, Calvert V, Alvarez R, Norcross MA. Role of tyrosine phosphorylation in ligand-independent sequestration of CXCR4 in human primary monocytes-macrophages. J Biol Chem 2001;276:49236-43.
33. Vila-Coro AJ, Rodríguez-Frade JM, Martín De Ana A, Moreno-Ortíz MC, Martínez AC, Mellado M. The chemokine SDF-1alpha triggers CXCR4 receptor dimerization and activates the JAK/STAT pathway. FASEB J 1999;13:1699-710.
34. Ahr B, Denizot M, Robert-Hebmann V, Brelot A, Biard-Piechaczyk M. Identification of the cytoplasmic domains of CXCR4 involved in Jak2 and STAT3 phosphorylation. J Biol Chem 2005;280:6692-700.
35. Bromberg JF, Darnell JE Jr. Potential roles of Stat1 and Stat3 in cellular transformation. Cold Spring Harb Symp Quant Biol 1999;64:425-8.
36. Bromberg JE, Wrzeszczynska MH, Devgan G, Zhao Y, Pestell Y, Albanese C, Darnell JE Jr. Stat3 as an oncogene. Cell 1999;89:295-303.
37. Niu G, Wright KL, Huang M, Song L, Haura E, Turkson J, Zhang S, Wang T, Sinibaldi D, Coppola D, Heller R, Ellis LM, Karras J, Bromberg J, Pardoll D, Jove R, Yu H. Constitutive Stat3 activity up-regulates VEGF expression and tumor angiogenesis. Oncogene 2002;21:2000-8.
38. Zhang XW, Audet J, Piret JM, Li YX. Cell cycle distribution of primitive haematopoietic cells stimulated in vitro and in vivo. Cell Prolif 2001;34:321-30.
39. Pfeiffer M, Hartmann TN, Leick M, Catusse J, Schmitt-Graeff A, Burger M. Alternative implication of CXCR4 in JAK2/STAT3 activation in small cell lung cancer. Br J Cancer 2009;100:1949-56.
40. Maroni P, Bendinelli P, Matteucci E, Desiderio MA. HGF induces CXCR4 and CXCL12-mediated tumor invasion through Ets1 and NF-kB. Carcinogenesis 2007;28:267-79.
41. Struchmann K, Mertz K, Steu S, Storz M, Staller P, Krek W, Schraml P, Moch H. pVHL co-ordinately regulates CXCR4/ CCXCL12 and MMP2/MMP9 expression in human clear cell renal cell acrcinoma. J Pathol 2008;214:464-71.
42. Yu H, Kortylewski M, Pardoll D. Crosstalk between cancer and immune cells: role of STAT3 in the tumour microenvironment. Nat Rev Immunol 2007;7:41-51.
43. Vermeulen L, de Sousa e Melo F, Richel DJ, Medema JP. The developing cancer stem-cell model: clinical challenges and opportunities. Lancet Oncol 2012;13:e83-9.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Ting LL, Chou AB, Hsieh CH, Hsiung SC, Pang ST, Liao SK. Withaferin A targeting both cancer stem cells and metastatic cancer stem cells in the UP-LN1 carcinoma cell model. J Cancer Metastasis Treat 2016;2:29-40. http://dx.doi.org/10.4103/2394-4722.172008
AMA Style
Ting LL, Chou AB, Hsieh CH, Hsiung SC, Pang ST, Liao SK. Withaferin A targeting both cancer stem cells and metastatic cancer stem cells in the UP-LN1 carcinoma cell model. Journal of Cancer Metastasis and Treatment. 2016; 2: 29-40. http://dx.doi.org/10.4103/2394-4722.172008
Chicago/Turabian Style
Ting, Lai-Lei, Andy Shau-Bin Chou, Chin-Hsuan Hsieh, Shih-Chieh Hsiung, See-Tong Pang, Shuen-Kuei Liao. 2016. "Withaferin A targeting both cancer stem cells and metastatic cancer stem cells in the UP-LN1 carcinoma cell model" Journal of Cancer Metastasis and Treatment. 2: 29-40. http://dx.doi.org/10.4103/2394-4722.172008
ACS Style
Ting, L.L.; Chou A.B.; Hsieh C.H.; Hsiung S.C.; Pang S.T.; Liao S.K. Withaferin A targeting both cancer stem cells and metastatic cancer stem cells in the UP-LN1 carcinoma cell model. J. Cancer. Metastasis. Treat. 2016, 2, 29-40. http://dx.doi.org/10.4103/2394-4722.172008
About This Article
Copyright

Author Biographies

Data & Comments
Data


 Cite This Article 9 clicks
Cite This Article 9 clicks

 Like This Article 0
likes
Like This Article 0
likes






Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.