
Effects of Gas1 on gliomas: a review on current preclinical studies


Abstract
Glioblastoma multiforme (GBM) is the most common and lethal brain tumor. Its prognosis remains very poor, despite the use of combined treatments such as surgical resection, radiation and chemotherapy. The major limitations for the treatment of GBM are its high invasiveness, tumor recurrence and resistance to treatments. Therefore, gene therapy appears as a relevant strategy for its treatment. Thus, we have investigated the use of growth-arrest-specific 1 (Gas1) for the treatment of GBM. Gas1 is a tumor suppressor protein that inhibits glioma growth by inducing arrest and apoptosis of tumor cells. Moreover, we have shown that a soluble form of Gas1 acting in both autocrine and paracrine manners is also effective inhibiting tumor growth in animal models, indicating its potential as an adjuvant for the treatment of GBM.
Keywords
Introduction
Gliomas are the most frequent and aggressive tumors of the central nervous system (CNS) and current treatments have not improved their prognosis. In children and adolescents, tumors of the CNS are the most common and lethal;[1] and glioblastoma multiforme (GBM) is the most frequent malignant primary brain tumor.[2-5] Gliomas are the main neuroepithelial tumors of the CNS that originate from mature or precursor ectodermal-derived glial cells. The World Health Organization (WHO) has classified gliomas on 4 grades from I to IV (GI-GIV) according to the histological dedifferentiation and the expression of the KI-67 protein, which indicates the rate of proliferation. WHO-GI gliomas are considered as benign tumors, since the malignant features are only present on low-grade (WHO-GII) and high-grade (WHO-GIII and -GIV) gliomas; in this respect, WHO classification correlates with the prognosis of the patient, regardless of multimodal therapeutic treatments;[6,7] the 5-year life span rates after diagnosis are 50%, 30% and 5% for WHO-GII, -GIII and -GIV glioma patients, respectively.[8]
Tumors of glial origin are considered gliomas and are divided into: astrocytoma, oligodendroglioma, ependymoma, mixed gliomas and not otherwise specified.[2,3] On the other hand, the Cancer Genome Atlas (TCGA) designed a sub-classification of GBM based on their molecular signature [Table 1], which comprises: classical, mesenchymal, proneural and neural tumors.[8,9] Classical GBM are tumors that present high expression of the epidermal growth factor receptor (EGFR) and absence of tumor suppressor proteins such as p16 and p14. In mesenchymal GBM, the phosphatase and tensin homolog (PTEN) gene is mutated and loss of its activity leads to activation of the serine-threonine protein kinase (AKT) survival pathway. Additionally, this subtype expresses chitinase 3-like-1 and MET transcripts characteristics of mesenchymal cells and shows low expression of the transcript for the tumor suppressor protein neurofibromin 1. On the other hand, proneural GBM expresses oligodendrocyte transcripts NK2 homeobox 2 and oligodendrocyte transcription factor (OLIG2), as well as high levels of platelet-derived growth factor (PDGF) receptor, alpha polypeptide, and mutations of the dehydrogenase 1 and/or P53 genes. The neural GBM is the most dedifferentiated subtype because it expresses neuronal markers such as: negative regulatory factor (NEFK), γ-aminobutyric acid A receptor A, synaptotagmin 1 and the symporter K+: Cl- (SLC12A5).[8,9] The variability of the molecular signature of GBM suggests that the characterization of gliomas must be analyzed to devise specific treatments.
Subclassification of GBM based on their molecular signature[9]
| GBM subclassification | Genes altered | Signal pathway | Status | Physiological activity |
|---|---|---|---|---|
| Classical | CDKN2A (p14/p16) | RB | Homozygote deletion | Cell cycle: G1/S transition |
| EGFR | EGF/TNF-α | Overexpression and mutations | Cell Cycle | |
| NESTIN | Overexpression | Neural stemness | ||
| NOTCH3, JAG1, LENG | NOTCH | |||
| SMO, GAS1, GLI2 | SHH | |||
| Mesenchymal | NF1 | AKT | Deletion and/or mutation | Survival and proliferation pathways |
| PTEN | Mutation | |||
| TRADD, RELB, NFRSF1A | NF-κβ/TNF superfamily | Overexpression | Cell stress response | |
| CH3IL1, MET | Expression | Mesenchymal transtion | ||
| Proneural | PDGFRA | PDGF | Overpression and mutation | Cell cycle and angiogenesis, |
| IDH1 | Mutation | cytoplasmic NADPH production | ||
| PIK3CA/PIK3R1 | AKT | Mutation | Survival and proliferation pathways | |
| P53 | P53 | Mutation | Cell cycle: G1/S transition | |
| CDKN1A (p21) | Low expression | |||
| DCX, DLL3, ASCL1, TCF4 | SOX | Overexpression | CNS cell fate determination | |
| NKX2-2 | ||||
| OLIG2 | ||||
| Neural | NEFL | Expression | Neuronal markers | |
| GABRA1 | GABA | |||
| SYT1 | ||||
| SLC12A5 |
Clinical management of gliomas
Advanced anaplasia in high-grade gliomas difficults the complete surgical resection of the tumor; as a consequence, tumor recurrences are unavoidable. On the other hand, postoperative radiotherapy has been the standard treatment for GBM. However, the survival after radiation is low and overall survival remains poor.[10-12]
Concomitant and adjuvant chemotherapy for high grade gliomas include alkylating agents that damage DNA, such as: carmustine, procarbazine, lomustine, vincristine and temozolamide (TMZ). Recently, it has been reported that both bevacizumab and cediranib prevent angiogenesis by inhibiting the vascular endothelial growth factor (VEGF) signaling pathway.[11-13] Moreover, TMZ is the drug of choice for the treatment of high-grade glioma. TMZ alkylates guanine, inducing the methylation of gene promoters and leading to apoptosis, when the mismatch repair system is intact.[14,15] This drug is well tolerated by most patients, furthermore it has a favorable safety profile that is associated with only mild side-effects compared with nitrosoureas.[10,16] The addition of chemotherapy to standard postoperative radiotherapy improves in 2.5 months the median survival relative to postoperative radiotherapy alone.[10-12] To enhance the effect of TMZ it has been proposed the use of lipid-based nanoparticles, which cross the blood brain barrier more efficiently causing an increment of brain levels of TMZ and reducing the adverse effects in other organs such as the heart and kidneys.[16-18] Despite the above GBMs that express high levels of O6-methylguanine DNA methyltransferase (MGMT) protein are resistant to TMZ chemotherapy.[19-22] Small molecule inhibitors of MGMT exist, but their use in combination with TMZ is limited due to toxicity to peripheral organs.[23] Furthermore, the mutation in the mutS homolog (MSH) 6 mismatch repair gene facilitates resistance to TMZ and recurrence of GBM.[14] Until now, the surgical approach is still the most effective measure to treat gliomas, followed by radiotherapy and chemotherapy; however the clinical prognosis of the patients remains very poor. Therefore, new strategies and therapeutic agents should be investigated, based on the molecular characteristics of gliomas.
Molecular approach against multi-resistant gliomas
Resistance to various treatments and the recurrence of tumors has been attributed to the presence of a subpopulation of cells in gliomas with properties of stem cells, known as glioma-initiating cells (GICs). These cells express neural stem cell markers such as NESTIN, OLIG2, sex determining region Y-box 2 (SOX2) and fucosyltransferase 4.[20,24,25] The classification of GICs is based on the expression of prominin-1 (CD133); the CD133+ GICs are more invasive than those that do not express the antigen, and constitute 3-29% of the glioma mass.[26,27] Also, GICs from secondary cultures conserve the characteristics of the original tumor, even after several passages.[28] Self-renewal, aggressiveness and stemness of GICs are associated with the expression of Cyclin E and proteins of the family of inhibitor of DNA-binding/differentiation proteins, increased activity of several signaling pathways including: transforming growth factor (TGF)-β; protein kinase A and jagged-NOTCH;[26,29] as well as the activation of the receptors for PDGF, epidermal growth factor (EGF) and fibroblast growth factor (FGF).[26] Surprisingly, CD133+ GICs also contribute to malignant transformation of the adjacent normal glial cells through paracrine activity of PDGF-α and stimulate angiogenesis by activation of the NOTCH signaling pathway on endothelial tissue.[26,28,30]
It is noteworthy that CD133+ GICs are predisposed to become resistant to chemo- and radiotherapy with respect to non-GICs cells, implicating the activation of extracellular signal-regulated kinases 1 and 2 (ERK1/2).[20,31,32] In this context, the use of classical therapies against gliomas, facilitate the selection of multi-resistant GICs, leading to the formation of more aggressive tumors, resistant to chemo- and radiotherapy.[33] Moreover, in vitro and in vivo studies have shown that with the adequate stimulus, GICs differentiate to either neuronal or astrocytic cells, however many of these responses are deregulated in gliomas.[31]
Currently, the analysis of protein expression and of the transcriptome, including micro-RNAs (miRNAs), has helped to uncover molecular markers involved in the susceptibility or resistance to treatments. Indeed, differential expression patterns of miRNAs have been reported in high-grade gliomas.[34] Even now, the association of miRNAs with the methylation of the MGMT promoter is still controversial since miRNAs are not considered as direct epigenetic regulators.[21,35] However, overexpression of miR-222, -145 and -132 is related with TMZ resistance coupled with MGMT promoter methylation.[35] Additionally, miR-181b and -181c are downregulated in patients that responded to radiotherapy and concomitant TMZ.[36] However, sensibility to chemotherapy is not only dependent of the presence of MGMT.[37,38]
Screening of molecular changes after radiotherapy showed overexpression of miR-1, -125a, -144, -150, -151-5p, -221/22, -425 and -1285.[39-42] The ectopic expression of miR-1, -125a, -150 and -425 increases cell survival and confers radioresistance through the induction of the cell-cycle.[39] On the other hand, TGF-α and -β, and the EGFR also contribute to radioresistance in classical-GBM.[43] Thus, inhibiting TGF-β has been proposed as a treatment since it induces radiosensitivity in gliomas through decreasing the expression of miR-1 and -125a.[23,39,44] Additionally, miR-221/222 downregulate PTEN, leading to activation of proteins that promote cell proliferation or prevent cell death, such as: AKT, B-cell lymphoma 2 (Bcl-2), Cyclin-D, matrix metallopeptidase 2 and 9,[41,45,46] Interestingly, the phosphorylation of AKT is the main mechanism for developing radioresistance, regardless of the activity of its negative regulator PTEN.[40,41] In summary, molecular analysis could help to reconsider whether conventional treatments are suitable, or therapeutic modifications should be adapted to the requirements of the patient.
Growth-arrest-specific 1
The balance between proliferation or growth arrest is regulated by several extrinsic and intrinsic factors. Cells can exit the cell cycle and enter in the non-proliferative phase, known as the G0 phase. Particularly, in the this phase six genes named growth-arrest-specific (Gas) genes are expressed, from 1 to 6.[47] The gas1 transcript is the most abundant in NIH3T3 cells arrested in the G0 phase by deprivation of serum or high cell density.[47-49]Gas1 induces growth arrest by inhibiting DNA synthesis in NIH3T3 cells when it is ectopically expressed.[49]
Transcriptional and translational regulation
Human and mouse gas1 genes are located in the long arms of chromosome 9 (9q21.3-q22)[50,51] and chromosome 13[52] respectively, with 77.04% of homology between them. Gas1 is an intronless gene, suggesting that it probably originated from a retrotransposon.[53]
There are few studies about the regulation of the gas1 gene, however it has been reported that Menin and Myb-like (coded by the men1 and dmp1 genes respectively), induce the transcriptional repression of gas1.[54,55] Also, c-Myc and Src repress gas1 transcription, since they facilitate re-entering to the cell cycle.[56,57] c-Myc protein requires the Myc-Box2 domain to be present on the N-terminus to repress the transcription of gas1. Furthermore, the basic Helix-loop-Helix leucine zipper domain located at the C-terminus of c-Myc is also necessary to induce the transcriptional repression of gas1, perhaps together with an accessory protein not yet identified.[56,58] Both c-Myc and Src are key components for the proliferation, growth, and survival of glioma cells. The expression of c-Myc closely correlates both with cellular dedifferentiation and the grade of malignancy,[59-61] since its activity induces the transcripcion of cyclin D1 and repression of the p21WAF1/CIP1 cyclin-dependent kinase inhibitor.[62] Interestingly, the histone chaperone, Facilitate Chromatin Transcription protein complex (FACT) increases the transcription of Myc, a recent report showed that the downregulation or inhibition of FACT decreased the formation of metastasis and delayed tumor growth, it also proved to be an excellent cytotoxic adjuvant.[63] Thus, these data suggest that the inhibition both of FACT and Myc, could increase the anti-tumoral effect of Gas1. Moreover, transgenic mice that express Src under the transcriptional control of the glial fibrillary acidic protein develop hypervascularized glioblastomas with morphological and molecular characteristics of human GBM.[64,65]
On the other hand, estrogens like estradiol, induce their biological effects through binding to intra-cellular hormone-specific estrogen receptors (ERα and ERβ), and this binding produces a conformational change in the receptors, causing the activation of their transcriptional domains. Specifically, estradiol reduced the levels of gas1 mRNA, however it is not yet known whether the gas1 promoter has an estrogen response element.[66]
Little is known about the transcription factors that up-regulate gas1. For example the transcription factor Tbox5 increases the activity of the mouse gas1 promoter.[67] Moreover, microarray experiments indicate that gas1 could be a retinoic acid responsive gene.[68,69] Since both retinoic receptors and Gas1 are expressed during embryonic development,[70-74] we insinuate that retinoic acid may induce the expression of Gas1 to promote exit from the cell cycle and initiate the differentiation process.
Four miRNAs have been reported to interact with the human gas1 transcript: miR-34a-5p, -148a-3p, -130b-5p and -183-5p.[75,76] Only miR-34a, derived from the 5’ arm of the pre-miRNA sequence (miR-34a-5p), has been shown to downregulate the translation of Gas1 when the miRNA interacts with nucleotides located at position 812-832 from the 3’ untranslated region of gas1, preventing the activity of Gas1 on the phosphatidylinositol 3-kinase (PI3K)-AKT dependent cell survival pathway.[77,78] In fact, the repression of gas1 by miR-34a-5p promotes cell survival and proliferation, preventing apoptosis by reducing the cleavage of Caspase-3 on papillary thyroid carcinoma cell cultures.[77]
Gas1 protein structure and expression
The nucleotide sequence of the gas1 of both the human and mouse genes reveals an open reading frame of 345 and 384 amino acids, respectively.[49] The proteins encoded by these genes undergo post-translational modifications in the endoplasmic reticulum consisting of an N-linked glycosylation, signal peptide cleavage and addition of a glycosylphosphatidylinositol (GPI) group at the C-terminal. The mature form of the Gas1 protein has a molecular mass of about 37 kDa and is anchored to the outer cell membrane by a GPI molecule.[49,51,79] The region of Gas1 from amino acid 182 to amino acid 234 is essential to induce growth arrest whereas neither the GPI nor the C-terminal domain are necessary for this function.[80]
We previously showed that Gas1 possesses significant structural homology with the glial cell-derived neurotrophic factor (GDNF) family of receptors (GFRαs). Gas1 has two domains, called D-N and D-C, which have high similarity to the D2 and D3 domains of the GFRαs. These domains have cysteines that participate in the formation of five disulfide bridges.[81,82] It is noteworthy to mention that Gas1 binds to RET in either the presence or the absence of GDNF.[82] Based on the above information we and other research groups showed that Gas1 inhibits the signaling pathway induced by GDNF, an aspect that we will discuss later.
Interestingly, it has been reported that a soluble form of Gas1 inhibits the proliferation of mesangial cells. Disintegrin and metalloproteinase (ADAM) 10 and 17 are responsible of cleaving the Gas1 GPI anchor.[83,84] In glioma cells, ADAM17 increases the shedding of soluble VEGF and activates the EGFR-PI3K-AKT pathway, contributing to invasiveness, and angiogenesis.[85] For its part, ADAM10 promotes glioma cell migration by cleaving the adhesion molecule N-cadherin from the cell surface.[86] On the other hand, we constructed a lentiviral vector that produce a soluble and secretable form of Gas1 (tGas1), lacking the GPI consensus sequence. tGas1 induces cell arrest and apoptosis of GBM cells and inhibit glioma tumor growth in vivo.[87,88] This soluble form of Gas1 acts in both autocrine and paracrine manners. However, previous data suggests that the full form of Gas1 (with GPI) can have paracrine effects, since the GPI anchor of Gas1 could be cleaved by ADAM 10 and 17 in gliomas.
Gas1 is expressed during the early stages of development in the primitive streak, somites, heart, limb, otic vesicle, kidney, lung, muscle, gonads, brain and placenta.[51,70,72,73] Its expression is fundamental during embryonic development since Gas1 knockout (K.O.) mice die immediately after birth.[89-91] The K.O. mice develop several defects including decreased cell proliferation in cerebellum, morphological alterations in the gastrointestinal tract and microform holoprosencephaly associated with multiple craniofacial defects.[72,89-93] The defects in Gas1-/- mice, are associated with the loss of the signaling induced by Sonic hedgehog (Shh). Interestingly, some patients with holoproscencephaly present mutations in the Gas1 gene with or without additional mutations on the Shh gene.[93,94] During development, Gas1 has both negative and positive effects on cell proliferation, for example: in the limbs, Gas1 promotes the death of the interdigital tissue;[95] whereas it promotes proliferation of granular cell progenitors in the cerebellum.[96]
Previous studies using in situ hybridization showed the expression of the gas1 gene in the brain of adult mice (http://www.brain-map.org/;www.genepaint.org;www.stjudebgem.org). Additionally, we reported that Gas1 is mainly found in neurons and in a more restricted manner in glial cells in different regions of the CNS of adult mice.[97] Furthermore, the expression of Gas1 decreases when neural stem cells are differentiated to a glial phenotype.[98] However, the role of Gas1 in glial cells is unknown in the adult CNS. In hippocampal neurons, Gas1 induces cell death after excitotoxic insults, inhibiting the signaling induced by GDNF.[99,100] Nevertheless, during cerebellar development Gas1 induces the proliferation of cerebellar granule neuron progenitors in a Shh-dependent manner.[71,96]
Gas1 promotes shh signaling
Shh is a secreted and diffusible morphogen implicated in the development of tissues and organs, including the CNS. The receptor for Shh is Patched (Ptc) which constitutively inhibits Smoothened [Smo; Figure 1]. The binding of Shh-Ptc produces the disinhibition of Smo and allows its signaling.[101] Downstream, Gli1, 2 and 3 proteins activate the transcription of genes such as N-myc, cyclin D and bcl-2 which promote cell proliferation. On the other hand, there are evidences of the interaction between Gas1 with Shh, and Indian hedgehog.[70] This interaction was originally interpreted as antagonistic, however recently it has been shown that Gas1 promotes Shh signaling during the development of the neural tube and cerebellum [Figure 1].[89,96,102-104] Ptc and Gli are highly expressed in gliomas and are considered oncogenes.[105,106] Therefore this would suggest that Gas1 could enhance the effect of Shh, inducing the proliferation of glioma and neuroblastoma cells, however we showed that Gas1 inhibits cell proliferation of glioma cells even in the presence of the Shh molecular machinery,[107,108] which suggest that in tumors Gas1 inhibits the GDNF signaling pathway.
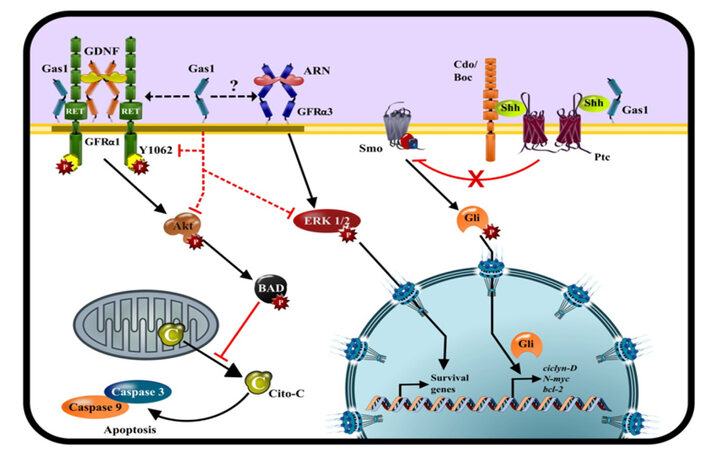
Figure 1. Graphic description of the interaction and effects of Gas1 with different intracellular pathways. Black arrows indicate positive regulation or activation; red cut-ending lines indicate negative regulation. Black dotted arrows show the interaction of Gas1 with GFLs signaling pathway, whereas red dotted lines indicate the disruption of mechanisms of phosphorylation caused by Gas1
Gas1 inhibits the signaling induced by GDNF and artemin
The GDNF family of ligands (GFLs), GDNF, neurturin (NRTN), artemin (ARTN) and persephin (PSPN), belong to a distant branch of the TGF-β superfamily.[109] GFLs play a pivotal role in the differentiation and maintenance of both the central and the peripheral nervous system. The cellular responses to GFLs are mediated by a multicomponent receptor complex composed by GPI anchored co-receptors (GFRα1-4) and as ligand binding component the Ret receptor which is a tyrosine kinase. The co-receptors provide specificity for the binding of the ligand to the receptor complex; GDNF preferentially binds to GFRα1, NRTN to GFRα2, ARTN to GFRα3 and PSPN to GFRα4. Although there are promiscuity of the ligand-receptor interactions.[109]
The binding of GDNF to GFRα-1 induces the recruitment of two RET proteins on lipid rafts.[109] Like other receptor tyrosine kinases, RET can activate various signaling pathways including ERK, PI3K/AKT, the p38 mitogen activated protein kinase and the c-Jun N-terminal kinase (JNK) pathways.[109,110] AKT is constitutively expressed in GBM cells and its activation induces uncontrolled growth, resistance to apoptosis, and enhanced tumor invasiveness,[111] by inactivating pro-apoptotic proteins as BAD and procaspase-9[Figure 1], as well as the transcription factor forkhead box O (FOXO).[111] Thus, the inhibition of AKT is an important therapeutic target for the treatment of gliomas. The activation of AKT is regulated by PI3K, a member of the intracellular lipid kinase family, which catalyzes the generation of phosphatidylinositol-3,4,5-triphosphate (PIP3) from phosphatidylinositol-4,5-triphosphate (PIP2).[112] PIP3 recruits AKT to the plasma membrane where it is phosphorylated in Thr308 by phosphoinositide dependent kinase 1 and in Ser473 by PDK2, which results in the full activation of AKT.[113] On the other hand, the activation of the PI3K/AKT signaling pathway is reduced when PIP3 is dephosphorylated and converted to PIP2 by the activity of PTEN.[114] In neuroblastoma and glioma cells, Gas1 blocks cell cycle progression, inhibits proliferation and induces cell death by inhibiting the GDNF/AKT pathway.[107,108,115,116] We showed that Gas1 prevents the phosphorylation of Ret Tyr1062 and reduces the activation of AKT[Figure 1]. This leads to the translocation of BAD to the mitochondria and the release of cytochrome-C to the cytosol which in turn induces the activation of Caspases 9 and 3.[107,108,115-118] Recently Wang and et al.[100] demonstrated that Gas1 promotes excitotoxicity in dopaminergic neurons by inhibiting the GDNF signaling pathway.
AKT phosphorylates, activates, or inhibits a number of proteins that regulate several processes related with cell survival.[114,119] First, AKT has anti-apoptotic effects through the phosphorylation and inhibition of pro-apoptotic proteins, such as BAD, MDM2 and members of the FOX family. Second, AKT promotes the progression of the cell cycle by blocking the degradation of cyclin D and inactivating the inhibitors of the cell cycle p21 and p27. Finally, AKT activates the mammalian target of rapamycin (mTOR) kinase by inhibiting a complex formed by the tumor suppressor proteins tuberous sclerosis 1 and 2. In turn mTOR increases protein synthesis and cell proliferation.[114,119]
Additionality, we found that Gas1 inhibits cell growth through a RET-independent mechanism. Gas1 decreases the viability of MDA-MB-231 human breast cancer cells, interfering with the interaction between ARTN and GFRα3, leading to a decrement of the activation of ERK1/2.[120] In turn, the activation of the ERK pathway is triggered by a wide variety of receptor tyrosine kinases activated by growth factors and cytokines. ERK1/2 is activated by the small G protein Ras-Raf family members (Raf-1, A-Raf, B-Raf) followed by MEK1/2. ERK1/2 controls either cell survival or apoptosis by regulating the activity of anti- and pro-apoptotic transcription factors.[121] The phosphorylations of ERK 1/2 promote cell survival by enhancing the transcription and activity of the anti-apoptotic molecules Bcl-2, myeloid cell leukemia 1 and B-cell lymphoma-extra large.[122] Alternatively, ERK1/2 downregulate the expression and inhibit the activity of the pro-apoptotic protein Bcl2-interacting mediator. Moreover, under conditions of oxidative stress, ERK has pro-apoptotic effects;[123] however this process it is not well understood yet.
Potential therapeutic effect of GAS1 for the treatment of gliomas
Several studies suggest that GAS1 is a tumor suppressor and that its downregulation facilitates the uncontrolled growth of several types of cancer cells.[107,108,115-118,124,125] It is worth noting that the downregulation of GAS1 is associated with the progression of thyroid and prostate cancer and with a poor prognosis of survival.[126,127] Additionally the loss of GAS1 increases the metastasis of breast, prostate and gastric cancers.[124,128] On the other hand, GAS1 has been proposed as a molecular marker for prostate cancer.[129] The mechanisms that regulate the expression of GAS1 in gliomas have not been identified yet; however these tumors express several transcription factors that negatively regulate GAS1 such as c-Myc and v-Src.[56,57]
We previously showed that GAS1 induces apoptosis and inhibits cell growth in glioma cell lines and human glioma primary cultures of low and high grade.[87,88,107,116-118] Furthermore, we demonstrated that GAS1 decreased the proliferation and induced apoptosis through the inhibition of AKT as well as the induction of apoptosis mediated by caspase 3, independently of the activity of p53 in C6 glioma cells and U373 human astrocytoma cells.[87,88,116-118] Interestingly, GAS1 produces death of glioma cells even in the presence of the molecular machinery of Shh,[92] suggesting that it acts through the GDNF pathway [Figure 2].
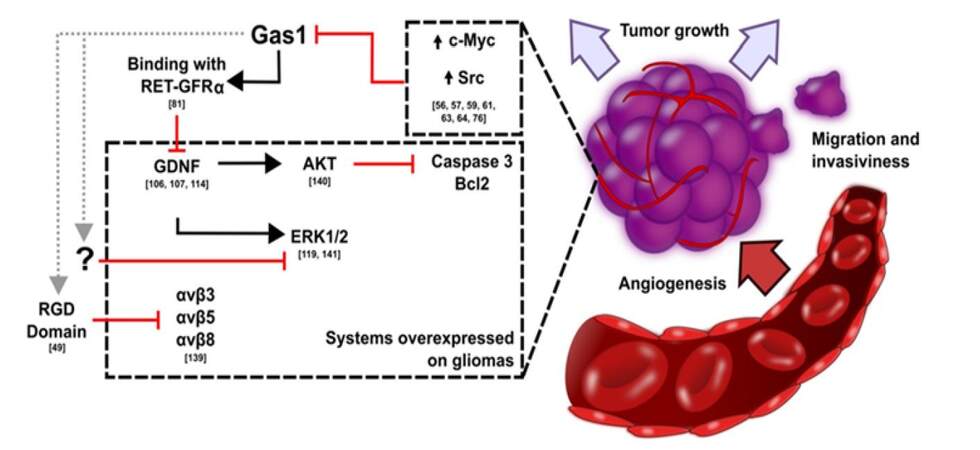
Figure 2. Pathways activated in gliomas and corresponding targets of Gas1
Gas1 binds to Ret in a manner independent of the presence of GDNF.[82] On the other hand, the expression and activity of GDNF and its receptor GFRα1 are increased by their soluble forms in gliomas.[130-133] Based on the above, we developed a lentiviral vector in which the expression of tGAS1 is inducible.[87,88] This soluble form of GAS1 acts both in autocrine and paracrine manners in GBM cells and inhibits glioma tumor growth in vivo. Subsequent to this study, we used neural stem cells as a vehicle to deliver tGAS1 into intracranial gliomas, since they have innate tropism towards tumors. We found that tGAS1 decreased tumor growth and increased the overall health and survival of nude mice implanted with GBM.[88]
There is evidence indicating that GAS1 is a metastasis suppressor in mouse 67NR breast cancer cells and B16-F0 melanoma cells.[128] Extracranial metastasis is a rare manifestation of GBM, this is probably due to the shortened survival of patients, that will not allow glioblastoma cells generate metastasis.[134] On the other hand, gliomas overexpress ERK1/2 and GDNF, molecules that promote migration and invasiveness of gliomas into the brain parenchyma.[104] There is evidence that GAS1 inhibits the migration of breast cancer cells by blocking ERK in a RET-independent manner.[120] Moreover, GAS1 decreased tumor vascularization in a breast cancer model.[120] All these suggest that GAS1 can be an important molecule to counter the migration of glioma cells. Additionally, GAS1 has a RGD domain which is essential for the binding and blockade of some integrins that promote the migration and invasiveness of gliomas.[49,135] It has been found that RGD-integrin antagonists can inhibit cell adhesion and angiogenesis.[135] On the other hand, it has been reported that integrin α5β1 (in the absence of attachment to fibronectin) decreased the proliferation of HT29 colon carcinoma cells by inducing the transcription of GAS1.[136] Until now, however, there is no evidence of a relationship between the RGD domain of GAS1 and cell migration [Figure 2].
The recurrence of gliomas that occurs after surgical resection, is attributed to the presence of GIC´s. Alternatively the activation of ERK is involved with the maintenance of the expression of MGMT and resistance to TMZ of GBM-GICs.[32] As previously mentioned GAS1 inhibits the activation of ERK1/2, thus it may promote the elimination of the GIC´s population [Figure 2]. It is relevant that the overexpression of GAS1 in human adenocarcinoma cells (A549) increases their sensibility to cisplatin, which inhibits proliferation and induces cell cycle arrest and apoptosis.[137,138] Also, the downregulation of GAS1 promotes resistance to epirubicin in human gastric cancer by regulating drug efflux and apoptosis.[139] On the other hand, it was reported that GAS1 could be an important biomarker for the prognosis of gastric cancer patients, since it was found that reduced or negative GAS1 expression is associated with shorter survival time and worse patient prognosis.[124] In conclusion, current data suggest that GAS1 is a potential adjuvant for the treatment of gliomas and other tumors. The use of GAS1 with current treatments may improve their efficacy.
Financial support and sponsorship
This work was partially supported by CONACYT grant # 239516 (JS).
Conflicts of interest
There are no conflicts of interest.
REFERENCES
1. Ostrom QT, de Blank PM, Kruchko C, Petersen CM, Liao P, Finlay JL, Stearns DS, Wolff JE, Wolinsky Y, Letterio JJ, Barnholtz-Sloan JS. Alex's Lemonade Stand Foundation Infant and Childhood Primary Brain and Central Nervous System Tumors Diagnosed in the United States in 2007-2011. Neuro Oncol 2015;16 Suppl 10:x1-36.
2. Ostrom QT, Gittleman H, Farah P, Ondracek A, Chen Y, Wolinsky Y, Stroup NE, Kruchko C, Barnholtz-Sloan JS. CBTRUS statistical report: Primary brain and central nervous system tumors diagnosed in the United States in 2006-2010. Neuro Oncol 2013;15 Suppl 2:ii1-56.
3. Ostrom QT, Gittleman H, Liao P, Rouse C, Chen Y, Dowling J, Wolinsky Y, Kruchko C, Barnholtz-Sloan J. CBTRUS statistical report: primary brain and central nervous system tumors diagnosed in the United States in 2007-2011. Neuro Oncol 2014;16 Suppl 4:iv1-63.
4. Larjavaara S, Mantyla R, Salminen T, Haapasalo H, Raitanen J, Jaaskelainen J, Auvinen A. Incidence of gliomas by anatomic location. Neuro Oncol 2007;9:319-25.
5. Zada G, Bond AE, Wang YP, Giannotta SL, Deapen D. Incidence trends in the anatomic location of primary malignant brain tumors in the United States: 1992-2006. World Neurosurg 2012;77:518-24.
6. Louis DN, Ohgaki H, Wiestler OD, Cavenee WK, Burger PC, Jouvet A, Scheithauer BW, Kleihues P. The 2007 WHO classification of tumours of the central nervous system. Acta Neuropathol 2007;114:97-109.
7. Mischel PS, Cloughesy TF, Nelson SF. DNA-microarray analysis of brain cancer: molecular classification for therapy. Nat Rev Neurosci 2004;5:782-92.
9. Verhaak RG, Hoadley KA, Purdom E, Wang V, Qi Y, Wilkerson MD, Miller CR, Ding L, Golub T, Mesirov JP, Alexe G, Lawrence M, O'Kelly M, Tamayo P, Weir BA, Gabriel S, Winckler W, Gupta S, Jakkula L, Feiler HS, Hodgson JG, James CD, Sarkaria JN, Brennan C, Kahn A, Spellman PT, Wilson RK, Speed TP, Gray JW, Meyerson M, Getz G, Perou CM, Hayes DN. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 2010;17:98-110.
10. Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJ, Belanger K, Brandes AA, Marosi C, Bogdahn U, Curschmann J, Janzer RC, Ludwin SK, Gorlia T, Allgeier A, Lacombe D, Cairncross JG, Eisenhauer E, Mirimanoff RO. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med 2005;352:987-96.
11. Chao ST, Suh JH. When should radiotherapy for low-grade glioma be given--immediately after surgery or at the time of progression? Nat Clin Pract Oncol 2006;3:136-7.
12. Chamberlain MC. Temozolomide: therapeutic limitations in the treatment of adult high-grade gliomas. Expert Rev Neurother 2010;10:1537-44.
14. Cahill DP, Levine KK, Betensky RA, Codd PJ, Romany CA, Reavie LB, Batchelor TT, Futreal PA, Stratton MR, Curry WT, Iafrate AJ, Louis DN. Loss of the mismatch repair protein MSH6 in human glioblastomas is associated with tumor progression during temozolomide treatment. Clin Cancer Res 2007;13:2038-45.
15. Chao TF, Xiong HH, Liu W, Chen Y, Zhang JX. MiR-21 mediates the radiation resistance of glioblastoma cells by regulating PDCD4 and hMSH2. J Huazhong Univ Sci Technolog Med Sci 2013;33:525-9.
16. Gao J, Wang Z, Liu H, Wang L, Huang G. Liposome encapsulated of temozolomide for the treatment of glioma tumor: preparation, characterization and evaluation. Drug Discov Ther 2015;9:205-12.
17. Song S, Mao G, Du J, Zhu X. Novel RGD containing, temozolomide-loading nanostructured lipid carriers for glioblastoma multiforme chemotherapy. Drug Deliv 2015;27:1-5.
18. Kim SS, Rait A, Kim E, DeMarco J, Pirollo KF, Chang EH. Encapsulation of temozolomide in a tumor-targeting nanocomplex enhances anti-cancer efficacy and reduces toxicity in a mouse model of glioblastoma. Cancer Lett 2015;369:250-8.
19. Grossman R, Burger P, Soudry E, Tyler B, Chaichana KL, Weingart J, Olivi A, Gallia GL, Sidransky D, Quinones-Hinojosa A, Ye X, Brem H. MGMT inactivation and clinical response in newly diagnosed GBM patients treated with Gliadel. J Clin Neurosci 2015;22:1938-42.
20. Natsume A, Kinjo S, Yuki K, Kato T, Ohno M, Motomura K, Iwami K, Wakabayashi T. Glioma-initiating cells and molecular pathology: implications for therapy. Brain Tumor Pathol 2011;28:1-12.
21. Lakomy R, Sana J, Hankeova S, Fadrus P, Kren L, Lzicarova E, Svoboda M, Dolezelova H, Smrcka M, Vyzula R, Michalek J, Hajduch M, Slaby O. MiR-195, miR-196b, miR-181c, miR-21 expression levels and O-6-methylguanine-DNA methyltransferase methylation status are associated with clinical outcome in glioblastoma patients. Cancer Sci 2011;102:2186-90.
22. Network TC. Corrigendum: Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature 2013;494:506.
23. Sabharwal A, Middleton MR. Exploiting the role of O6-methylguanine-DNA-methyltransferase (MGMT) in cancer therapy. Curr Opin Pharmacol 2006;6:355-63.
24. Kahlert UD, Bender NO, Maciaczyk D, Bogiel T, Bar EE, Eberhart CG, Nikkhah G, Maciaczyk J. CD133/CD15 defines distinct cell subpopulations with differential in vitro clonogenic activity and stem cell-related gene expression profile in in vitro propagated glioblastoma multiforme-derived cell line with a PNET-like component. Folia Neuropathol 2012;50:357-68.
25. Trepant AL, Bouchart C, Rorive S, Sauvage S, Decaestecker C, Demetter P, Salmon I. Identification of OLIG2 as the most specific glioblastoma stem cell marker starting from comparative analysis of data from similar DNA chip microarray platforms. Tumour Biol 2015;36:1943-53.
26. Jeon HM, Kim SH, Jin X, Park JB, Joshi K, Nakano I, Kim H. Crosstalk between glioma-initiating cells and endothelial cells drives tumor progression. Cancer Res 2014;74:4482-92.
27. Ilkanizadeh S, Lau J, Huang M, Foster DJ, Wong R, Frantz A, Wang S, Weiss WA, Persson AI. Glial progenitors as targets for transformation in glioma. Adv Cancer Res 2014;121:1-65.
28. Singh SK, Clarke ID, Terasaki M, Bonn VE, Hawkins C, Squire J, Dirks PB. Identification of a cancer stem cell in human brain tumors. Cancer Res 2003;63:5821-8.
29. Wang H, Sun T, Hu J, Zhang R, Rao Y, Wang S, Chen R, McLendon RE, Friedman AH, Keir ST, Bigner DD, Li QJ, Wang XF. miR-33a promotes glioma-initiating cell self-renewal via PKA and NOTCH pathways. J Clin Invest 2014;124:4489-502.
30. Chen Y, Wang Z, Dai X, Fei X, Shen Y, Zhang M, Wang A, Li X, Huang Q, Dong J. Glioma initiating cells contribute to malignant transformation of host glial cells during tumor tissue remodeling via PDGF signaling. Cancer Lett 2015;365:174-81.
31. Aldaz B, Sagardoy A, Nogueira L, Guruceaga E, Grande L, Huse JT, Aznar MA, Diez-Valle R, Tejada-Solis S, Alonso MM, Fernandez-Luna JL, Martinez-Climent JA, Malumbres R. Involvement of miRNAs in the differentiation of human glioblastoma multiforme stem-like cells. PLoS One 2013;8:e77098.
32. Sato A, Sunayama J, Matsuda K, Seino S, Suzuki K, Watanabe E, Tachibana K, Tomiyama A, Kayama T, Kitanaka C. MEK-ERK signaling dictates DNA-repair gene MGMT expression and temozolomide resistance of stem-like glioblastoma cells via the MDM2-p53 axis. Stem Cells 2011;29:1942-51.
33. Bao S, Wu Q, McLendon RE, Hao Y, Shi Q, Hjelmeland AB, Dewhirst MW, Bigner DD, Rich JN. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature 2006;444:756-60.
34. Rao SA, Santosh V, Somasundaram K. Genome-wide expression profiling identifies deregulated miRNAs in malignant astrocytoma. Mod Pathol 2010;23:1404-17.
35. Cheng W, Ren X, Cai J, Zhang C, Li M, Wang K, Liu Y, Han S, Wu A. A five-miRNA signature with prognostic and predictive value for MGMT promoter-methylated glioblastoma patients. Oncotarget 2015;6:29285-95.
36. Slaby O, Lakomy R, Fadrus P, Hrstka R, Kren L, Lzicarova E, Smrcka M, Svoboda M, Dolezalova H, Novakova J, Valik D, Vyzula R, Michalek J. MicroRNA-181 family predicts response to concomitant chemoradiotherapy with temozolomide in glioblastoma patients. Neoplasma 2010;57:264-9.
37. Ujifuku K, Mitsutake N, Takakura S, Matsuse M, Saenko V, Suzuki K, Hayashi K, Matsuo T, Kamada K, Nagata I, Yamashita S. miR-195, miR-455-3p and miR-10a(*) are implicated in acquired temozolomide resistance in glioblastoma multiforme cells. Cancer Lett 2010;296:241-8.
38. Boccard SG, Marand SV, Geraci S, Pycroft L, Berger FR, Pelletier LA. Inhibition of DNA-repair genes Ercc1 and Mgmt enhances temozolomide efficacy in gliomas treatment: a pre-clinical study. Oncotarget 2015;6:29456-68.
39. Moskwa P, Zinn PO, Choi YE, Shukla SA, Fendler W, Chen CC, Lu J, Golub TR, Hjelmeland A, Chowdhury D. A functional screen identifies miRs that induce radioresistance in glioblastomas. Mol Cancer Res 2014;12:1767-78.
40. Li W, Guo F, Wang P, Hong S, Zhang C. miR-221/222 confers radioresistance in glioblastoma cells through activating Akt independent of PTEN status. Curr Mol Med 2014;14:185-95.
41. Chun-Zhi Z, Lei H, An-Ling Z, Yan-Chao F, Xiao Y, Guang-Xiu W, Zhi-Fan J, Pei-Yu P, Qing-Yu Z, Chun-Sheng K. MicroRNA-221 and microRNA-222 regulate gastric carcinoma cell proliferation and radioresistance by targeting PTEN. BMC Cancer 2010;10:367.
42. Yu L, Yang Y, Hou J, Zhai C, Song Y, Zhang Z, Qiu L, Jia X. MicroRNA-144 affects radiotherapy sensitivity by promoting proliferation, migration and invasion of breast cancer cells. Oncol Rep 2015;34:1845-52.
43. Schmidt-Ullrich RK, Valerie KC, Chan W, McWilliams D. Altered expression of epidermal growth factor receptor and estrogen receptor in MCF-7 cells after single and repeated radiation exposures. Int J Radiat Oncol Biol Phys 1994;29:813-9.
44. Maachani UB, Tandle A, Shankavaram U, Kramp T, Camphausen K. Modulation of miR-21 signaling by MPS1 in human glioblastoma. Oncotarget 2015. Epub ahead of print.
45. Zhang C, Zhang J, Hao J, Shi Z, Wang Y, Han L, Yu S, You Y, Jiang T, Wang J, Liu M, Pu P, Kang C. High level of miR-221/222 confers increased cell invasion and poor prognosis in glioma. J Transl Med 2012;10:119.
46. Zhang Z, Cui BZ, Wu LH, Xu QL, Wang Z, Yang B. The inhibition effect of expressions of miR-221 and miR-222 on glioma and corresponding mechanism. Bratisl Lek Listy 2014;115:685-91.
47. Schneider C, King RM, Philipson L. Genes specifically expressed at growth arrest of mammalian cells. Cell 1988;54:787-93.
48. Ciccarelli C, Philipson L, Sorrentino V. Regulation of expression of growth arrest-specific genes in mouse fibroblasts. Mol Cell Biol 1990;10:1525-9.
49. Del Sal G, Ruaro ME, Philipson L, Schneider C. The growth arrest-specific gene, gas1, is involved in growth suppression. Cell 1992;70:595-607.
50. Evdokiou A, Webb GC, Peters GB, Dobrovic A, O'Keefe DS, Forbes IJ, Cowled PA. Localization of the human growth arrest-specific gene (GAS1) to chromosome bands 9q21.3-q22, a region frequently deleted in myeloid malignancies. Genomics 1993;18:731-3.
51. Del Sal G, Collavin L, Ruaro ME, Edomi P, Saccone S, Valle GD, Schneider C. Structure, function, and chromosome mapping of the growth-suppressing human homologue of the murine gas1 gene. Proc Natl Acad Sci U S A 1994;91:1848-52.
52. Colombo MP, Martinotti A, Howard TA, Schneider C, D'Eustachio P, Seldin MF. Localization of growth arrest-specific genes on mouse chromosomes 1, 7, 8, 11, 13, and 16. Mamm Genome 1992;2:130-4.
53. Hatinen T, Holm L, Airaksinen MS. Loss of neurturin in frog--comparative genomics study of GDNF family ligand-receptor pairs. Mol Cell Neurosci 2007;34:155-67.
54. Mallakin A, Sugiyama T, Kai F, Taneja P, Kendig RD, Frazier DP, Maglic D, Matise LA, Willingham MC, Inoue K. The Arf-inducing transcription factor Dmp1 encodes a transcriptional activator of amphiregulin, thrombospondin-1, JunB and Egr1. Int J Cancer 2010;126:1403-16.
55. Gurung B, Feng Z, Iwamoto DV, Thiel A, Jin G, Fan CM, Ng JM, Curran T, Hua X. Menin epigenetically represses Hedgehog signaling in MEN1 tumor syndrome. Cancer Res 2013;73:2650-8.
56. Lee TC, Li L, Philipson L, Ziff EB. Myc represses transcription of the growth arrest gene gas1. Proc Natl Acad Sci U S A 1997;94:12886-91.
57. Grossi M, La Rocca SA, Pierluigi G, Vannucchi S, Ruaro EM, Schneider C, Tato F. Role of Gas1 down-regulation in mitogenic stimulation of quiescent NIH3T3 cells by v-Src. Oncogene 1998;17:1629-38.
58. Gartel AL, Shchors K. Mechanisms of c-myc-mediated transcriptional repression of growth arrest genes. Exp Cell Res 2003;283:17-21.
59. Cappellen D, Schlange T, Bauer M, Maurer F, Hynes NE. Novel c-MYC target genes mediate differential effects on cell proliferation and migration. EMBO reports 2006;8:70-6.
60. Lassman AB, Dai C, Fuller GN, Vickers AJ, Holland EC. Overexpression of c-MYC promotes an undifferentiated phenotype in cultured astrocytes and allows elevated Ras and Akt signaling to induce gliomas from GFAP-expressing cells in mice. Neuron Glia Biology 2004;1:157-63.
61. Herms JW, von Loewenich FD, Behnke J, Markakis E, Kretzschmar HA. c-myc oncogene family expression in glioblastoma and survival. Surg Neurol 1999;51:536-42.
62. Wang J, Wang H, Li Z, Wu Q, Lathia JD, McLendon RE, Hjelmeland AB, Rich JN. c-Myc is required for maintenance of glioma cancer stem cells. PLoS One 2008;3:e3769.
63. Carter DR, Murray J, Cheung BB, Gamble L, Koach J, Tsang J, Sutton S, Kalla H, Syed S, Gifford AJ, Issaeva N, Biktasova A, Atmadibrata B, Sun Y, Sokolowski N, Ling D, Kim PY, Webber H, Clark A, Ruhle M, Liu B, Oberthuer A, Fischer M, Byrne J, Saletta F, Thwe le M, Purmal A, Haderski G, Burkhart C, Speleman F, De Preter K, Beckers A, Ziegler DS, Liu T, Gurova KV, Gudkov AV, Norris MD, Haber M, Marshall GM. Therapeutic targeting of the MYC signal by inhibition of histone chaperone FACT in neuroblastoma. Sci Transl Med 2015;7:312ra176.
64. Theurillat J-P, Hainfellner J, Maddalena A, Weissenberger J, Aguzzi A. Early Induction of Angiogenetic Signals in Gliomas of GFAP-v-src Transgenic Mice. The American Journal of Pathology 1999;154:581-90.
65. Weissenberger J, Steinbach JP, Malin G, Spada S, Rulicke T, Aguzzi A. Development and malignant progression of astrocytomas in GFAP-v-src transgenic mice. Oncogene 1997;14:2005-13.
66. Ferrero M, Cairo G. Estrogen-regulated expression of a growth arrest specific gene (gas-1) in rat uterus. Cell Biol Int 1993;17:857-62.
67. Xie L, Hoffmann AD, Burnicka-Turek O, Friedland-Little JM, Zhang K, Moskowitz IP. Tbx5-hedgehog molecular networks are essential in the second heart field for atrial septation. Dev Cell 2012;23:280-91.
68. Eifert C, Sangster-Guity N, Yu LM, Chittur SV, Perez AV, Tine JA, McCormick PJ. Global gene expression profiles associated with retinoic acid-induced differentiation of embryonal carcinoma cells. Mol Reprod Dev 2006;73:796-824.
69. Wang L, Mear JP, Kuan CY, Colbert MC. Retinoic acid induces CDK inhibitors and growth arrest specific (Gas) genes in neural crest cells. Dev Growth Differ 2005;47:119-30.
70. Lee CS, Buttitta L, Fan CM. Evidence that the WNT-inducible growth arrest-specific gene 1 encodes an antagonist of sonic hedgehog signaling in the somite. Proceedings of the National Academy of Sciences 2001;98:11347-52.
71. Liu Y, May NR, Fan C-M. Growth Arrest Specific Gene 1 Is a Positive Growth Regulator for the Cerebellum. Developmental Biology 2001;236:30-45.
72. Lee CS, Fan CM. Embryonic expression patterns of the mouse and chick Gas1 genes. Mech Dev 2001;101:293-7.
73. Lee KK, Leung AK, Tang MK, Cai DQ, Schneider C, Brancolini C, Chow PH. Functions of the growth arrest specific 1 gene in the development of the mouse embryo. Dev Biol 2001;234:188-203.
74. Dolle P, Ruberte E, Leroy P, Morriss-Kay G, Chambon P. Retinoic acid receptors and cellular retinoid binding proteins. I. A systematic study of their differential pattern of transcription during mouse organogenesis. Development 1990;110:1133-51.
75. Hafner M, Landthaler M, Burger L, Khorshid M, Hausser J, Berninger P, Rothballer A, Ascano M Jr, Jungkamp AC, Munschauer M, Ulrich A, Wardle GS, Dewell S, Zavolan M, Tuschl T. Transcriptome-wide identification of RNA-binding protein and microRNA target sites by PAR-CLIP. Cell 2010;141:129-41.
76. Hsu SD, Tseng YT, Shrestha S, Lin YL, Khaleel A, Chou CH, Chu CF, Huang HY, Lin CM, Ho SY, Jian TY, Lin FM, Chang TH, Weng SL, Liao KW, Liao IE, Liu CC, Huang HD. miRTarBase update 2014: an information resource for experimentally validated miRNA-target interactions. Nucleic Acids Res 2014;42:D78-85.
77. Ma Y, Qin H, Cui Y. MiR-34a targets GAS1 to promote cell proliferation and inhibit apoptosis in papillary thyroid carcinoma via PI3K/Akt/Bad pathway. Biochemical and Biophysical Research Communications 2013;441:958-63.
78. Zhang L, He S, Guo S, Xie W, Xin R, Yu H, Yang F, Qiu J, Zhang D, Zhou S, Zhang K. Down-regulation of miR-34a alleviates mesangial proliferation in vitro and glomerular hypertrophy in early diabetic nephropathy mice by targeting GAS1. Journal of Diabetes and its Complications 2014;28:259-64.
79. Stebel M, Vatta P, Ruaro ME, Del Sal G, Parton RG, Schneider C. The growth suppressing gas1 product is a GPI-linked protein. FEBS Lett 2000;481:152-8.
80. Ruaro ME, Stebel M, Vatta P, Marzinotto S, Schneider C. Analysis of the domain requirement in Gas1 growth suppressing activity. FEBS Lett 2000;481:159-63.
81. Schueler-Furman O, Glick E, Segovia J, Linial M. Is GAS1 a co-receptor for the GDNF family of ligands? Trends in Pharmacological Sciences 2006;27:72-7.
82. Cabrera JR, Sanchez-Pulido L, Rojas AM, Valencia A, Manes S, Naranjo JR, Mellstrom B. Gas1 Is Related to the Glial Cell-derived Neurotrophic Factor Family Receptors and Regulates Ret Signaling. Journal of Biological Chemistry 2006;281:14330-9.
83. van Roeyen CRC, Zok S, Pruessmeyer J, Boor P, Nagayama Y, Fleckenstein S, Cohen CD, Eitner F, Gröne H-J, Ostendorf T, Ludwig A, Floege J. Growth arrest-specific protein 1 is a novel endogenous inhibitor of glomerular cell activation and proliferation. Kidney International 2012;83:251-63.
84. Zhang L, He S, Guo S, Xie W, Xin R, Yu H, Yang F, Qiu J, Zhang D, Zhou S, Zhang K. Down-regulation of miR-34a alleviates mesangial proliferation in vitro and glomerular hypertrophy in early diabetic nephropathy mice by targeting GAS1. J Diabetes Complications 2014;28:259-64.
85. Zheng X, Jiang F, Katakowski M, Lu Y, Chopp M. ADAM17 promotes glioma cell malignant phenotype. Mol Carcinog 2012;51:150-64.
86. Kohutek ZA, diPierro CG, Redpath GT, Hussaini IM. ADAM-10-mediated N-cadherin cleavage is protein kinase C-alpha dependent and promotes glioblastoma cell migration. J Neurosci 2009;29:4605-15.
87. López-Ornelas A, Mejía-Castillo T, Vergara P, Segovia J. Lentiviral transfer of an inducible transgene expressing a soluble form of Gas1 causes glioma cell arrest, apoptosis and inhibits tumor growth. Cancer Gene Therapy 2010;18:87-99.
88. López-Ornelas A, Vergara P, Segovia J. Neural stem cells producing an inducible and soluble form of Gas1 target and inhibit intracranial glioma growth. Cytotherapy 2014;16:1011-23.
89. Seppala M, Depew MJ, Martinelli DC, Fan C-M, Sharpe PT, Cobourne MT. Gas1 is a modifier for holoprosencephaly and genetically interacts with sonic hedgehog. Journal of Clinical Investigation 2007;117:1575-84.
90. Biau S, Jin S, Fan CM. Gastrointestinal defects of the Gas1 mutant involve dysregulated Hedgehog and Ret signaling. Biology Open 2012;2:144-55.
91. Seppala M, Xavier GM, Fan CM, Cobourne MT. Boc modifies the spectrum of holoprosencephaly in the absence of Gas1 function. Biology Open 2014;3:728-40.
92. Martinelli DC, Fan CM. A Sonic Hedgehog Missense Mutation Associated with Holoprosencephaly Causes Defective Binding to GAS1. Journal of Biological Chemistry 2009;284:19169-72.
93. Ribeiro LA, Quiezi RG, Nascimento A, Bertolacini CP, Richieri-Costa A. Holoprosencephaly and holoprosencephaly-like phenotype and GAS1 DNA sequence changes: Report of four Brazilian patients. American Journal of Medical Genetics Part A 2010;152A:1688-94.
94. Pineda-Alvarez DE, Roessler E, Hu P, Srivastava K, Solomon BD, Siple CE, Fan CM, Muenke M. Missense substitutions in the GAS1 protein present in holoprosencephaly patients reduce the affinity for its ligand, SHH. Hum Genet 2012;131:301-10.
95. Liu Y, Liu C, Yamada Y, Fan CM. Growth arrest specific gene 1 acts as a region-specific mediator of the Fgf10/Fgf8 regulatory loop in the limb. Development 2002;129:5289-300.
96. Izzi L, Lévesque M, Morin S, Laniel D, Wilkes Brian C, Mille F, Krauss Robert S, McMahon Andrew P, Allen Benjamin L, Charron F. Boc and Gas1 Each Form Distinct Shh Receptor Complexes with Ptch1 and Are Required for Shh-Mediated Cell Proliferation. Developmental Cell 2011;20:788-801.
97. Zarco N, Bautista E, Cuellar M, Vergara P, Flores-Rodriguez P, Aguilar-Roblero R, Segovia J. Growth Arrest Specific 1 (GAS1) Is Abundantly Expressed in the Adult Mouse Central Nervous System. Journal of Histochemistry & Cytochemistry 2013;61:731-48.
98. Obayashi S, Tabunoki H, Kim SU, Satoh J-i. Gene Expression Profiling of Human Neural Progenitor Cells Following the Serum-Induced Astrocyte Differentiation. Cellular and Molecular Neurobiology 2009;29:423-38.
99. Mellstrom B. Gas1 Is Induced during and Participates in Excitotoxic Neuronal Death. Molecular and Cellular Neuroscience 2002;19:417-29.
100. Wang K, Zhu X, Zhang K, Zhou F, Zhu L. Gas1 Knockdown Increases the Neuroprotective Effect of Glial Cell-Derived Neurotrophic Factor Against Glutamate-Induced Cell Injury in Human SH-SY5Y Neuroblastoma Cells. Cellular and Molecular Neurobiology 2015; Epub ahead of print.
101. Varjosalo M, Taipale J. Hedgehog: functions and mechanisms. Genes & Development 2008;22:2454-72.
102. Allen BL, Tenzen T, McMahon AP. The Hedgehog-binding proteins Gas1 and Cdo cooperate to positively regulate Shh signaling during mouse development. Genes & Development 2007;21:1244-57.
103. Martinelli DC, Fan CM. Gas1 extends the range of Hedgehog action by facilitating its signaling. Genes & Development 2007;21:1231-43.
104. Tenzen T, Allen BL, Cole F, Kang JS, Krauss RS, McMahon AP. The Cell Surface Membrane Proteins Cdo and Boc Are Components and Targets of the Hedgehog Signaling Pathway and Feedback Network in Mice. Developmental Cell 2006;10:647-56.
105. Jeng KS, Sheen IS, Jeng WJ, Lin CC, Lin CK, Su JC, Yu MC, Fang HY. High Expression of Patched Homolog-1 Messenger RNA and Glioma-Associated Oncogene-1 Messenger RNA of Sonic Hedgehog Signaling Pathway Indicates a Risk of Postresection Recurrence of Hepatocellular Carcinoma. Annals of Surgical Oncology 2012;20:464-73.
106. Shahi MH, Lorente A, Castresana JS. Hedgehog signalling in medulloblastoma, glioblastoma and neuroblastoma. Oncol Rep 2008;19:681-8.
107. Domínguez-Monzón G, Benítez JA, Vergara P, Lorenzana R, Segovia J. Gas1 inhibits cell proliferation and induces apoptosis of human primary gliomas in the absence of Shh. International Journal of Developmental Neuroscience 2009;27:305-13.
108. Zarco N, González-Ramírez R, González RO, Segovia J. GAS1 induces cell death through an intrinsic apoptotic pathway. Apoptosis 2012;17:627-35.
109. Airaksinen MS, Saarma M. The Gdnf Family: Signalling, Biological Functions and Therapeutic Value. Nature Reviews Neuroscience 2002;3:383-94.
110. Coulpier M, Anders J, Ibanez CF. Coordinated activation of autophosphorylation sites in the RET receptor tyrosine kinase: importance of tyrosine 1062 for GDNF mediated neuronal differentiation and survival. J Biol Chem 2002;277:1991-9.
111. Altomare DA, Testa JR. Perturbations of the AKT signaling pathway in human cancer. Oncogene 2005;24:7455-64.
112. Katso R, Okkenhaug K, Ahmadi K, White S, Timms J, Waterfield MD. Cellular function of phosphoinositide 3-kinases: implications for development, homeostasis, and cancer. Annu Rev Cell Dev Biol 2001;17:615-75.
113. Alessi DR, James SR, Downes CP, Holmes AB, Gaffney PR, Reese CB, Cohen P. Characterization of a 3-phosphoinositide-dependent protein kinase which phosphorylates and activates protein kinase Balpha. Curr Biol 1997;7:261-9.
114. Carracedo A, Pandolfi PP. The PTEN-PI3K pathway: of feedbacks and cross-talks. Oncogene 2008;27:5527-41.
115. López-Ramírez MA, Domínguez-Monzón G, Vergara P, Segovia J. Gas1 reduces Ret tyrosine 1062 phosphorylation and alters GDNF-mediated intracellular signaling. International Journal of Developmental Neuroscience 2008;26:497-503.
116. Benítez JA, Arregui L, Vergara P, Segovia J. Targeted-simultaneous expression of Gas1 and p53 using a bicistronic adenoviral vector in gliomas. Cancer Gene Therapy 2007;14:836-46.
117. Zamorano A, Lamas M, Vergara P, Naranjo JR, Segovia J. Transcriptionally mediated gene targeting of gas1 to glioma cells elicits growth arrest and apoptosis. J Neurosci Res 2003;71:256-63.
118. Zamorano A, Mellström B, Vergara P, Naranjo JR, Segovia J. Glial-specific retrovirally mediated gas1 gene expression induces glioma cell apoptosis and inhibits tumor growth in vivo. Neurobiology of Disease 2004;15:483-91.
119. Testa JR, Bellacosa A. AKT plays a central role in tumorigenesis. Proc Natl Acad Sci U S A 2001;98:10983-5.
120. Jiménez A, López-Ornelas A, Estudillo E, González-Mariscal L, González RO, Segovia J. A soluble form of GAS1 inhibits tumor growth and angiogenesis in a triple negative breast cancer model. Experimental Cell Research 2014;327:307-17.
122. Boucher MJ, Morisset J, Vachon PH, Reed JC, Laine J, Rivard N. MEK/ERK signaling pathway regulates the expression of Bcl-2, Bcl-X(L), and Mcl-1 and promotes survival of human pancreatic cancer cells. J Cell Biochem 2000;79:355-69.
123. Cagnol S, Chambard JC. ERK and cell death: mechanisms of ERK-induced cell death--apoptosis, autophagy and senescence. FEBS J 2010;277:2-21.
124. Wang H, Zhou X, Zhang Y, Zhu H, Zhao L, Fan L, Wang Y, Gang Y, Wu K, Liu Z, Fan D. Growth arrest-specific gene 1 is downregulated and inhibits tumor growth in gastric cancer. FEBS J 2012;279:3652-64.
125. Sacilotto N, Castillo J, Riffo-Campos AL, Flores JM, Hibbitt O, Wade-Martins R, Lopez C, Rodrigo MI, Franco L, Lopez-Rodas G. Growth Arrest Specific 1 (Gas1) Gene Overexpression in Liver Reduces the In Vivo Progression of Murine Hepatocellular Carcinoma and Partially Restores Gene Expression Levels. PLoS One 2015;10:e0132477.
126. Huang Y, Prasad M, Lemon WJ, Hampel H, Wright FA, Kornacker K, LiVolsi V, Frankel W, Kloos RT, Eng C, Pellegata NS, de la Chapelle A. Gene expression in papillary thyroid carcinoma reveals highly consistent profiles. Proc Natl Acad Sci U S A 2001;98:15044-9.
127. Bettuzzi S, Davalli P, Astancolle S, Carani C, Madeo B, Tampieri A, Corti A. Tumor progression is accompanied by significant changes in the levels of expression of polyamine metabolism regulatory genes and clusterin (sulfated glycoprotein 2) in human prostate cancer specimens. Cancer Res 2000;60:28-34.
128. Gobeil S, Zhu X, Doillon CJ, Green MR. A genome-wide shRNA screen identifies GAS1 as a novel melanoma metastasis suppressor gene. Genes & Development 2008;22:2932-40.
129. Rizzi F, Belloni L, Crafa P, Lazzaretti M, Remondini D, Ferretti S, Cortellini P, Corti A, Bettuzzi S. A novel gene signature for molecular diagnosis of human prostate cancer by RT-qPCR. PLoS One 2008;3:e3617.
130. Wiesenhofer B, Stockhammer G, Kostron H, Maier H, Hinterhuber H, Humpel C. Glial cell line-derived neurotrophic factor (GDNF) and its receptor (GFR-alpha 1) are strongly expressed in human gliomas. Acta Neuropathol 2000;99:131-7.
131. Song H, Moon A. Glial cell-derived neurotrophic factor (GDNF) promotes low-grade Hs683 glioma cell migration through JNK, ERK-1/2 and p38 MAPK signaling pathways. Neurosci Res 2006;56:29-38.
132. Ng WH, Wan GQ, Peng ZN, Too HP. Glial cell-line derived neurotrophic factor (GDNF) family of ligands confer chemoresistance in a ligand-specific fashion in malignant gliomas. Journal of Clinical Neuroscience 2009;16:427-36.
133. Ku MC, Wolf SA, Respondek D, Matyash V, Pohlmann A, Waiczies S, Waiczies H, Niendorf T, Synowitz M, Glass R, Kettenmann H. GDNF mediates glioblastoma-induced microglia attraction but not astrogliosis. Acta Neuropathol 2013;125:609-20.
134. Widjaja A, Mix H, Golkel C, Flemming P, Egensperger R, Holstein A, Rademaker J, Becker H, Hundt M, Wagner S, Manns MP. Uncommon metastasis of a glioblastoma multiforme in liver and spleen. Digestion 2000;61:219-22.
135. Russo MA, Paolillo M, Sanchez-Hernandez Y, Curti D, Ciusani E, Serra M, Colombo L, Schinelli S. A small-molecule RGD-integrin antagonist inhibits cell adhesion, cell migration and induces anoikis in glioblastoma cells. Int J Oncol 2013;42:83-92.
136. Varner JA, Emerson DA, Juliano RL. Integrin alpha 5 beta 1 expression negatively regulates cell growth: reversal by attachment to fibronectin. Mol Biol Cell 1995;6:725-40.
137. Zhang Y, Wang X, Han L, Zhou Y, Sun S. Green tea polyphenol EGCG reverse cisplatin resistance of A549/DDP cell line through candidate genes demethylation. Biomedicine & Pharmacotherapy 2015;69:285-90.
138. Zhang YW, Zheng Y, Wang JZ, Lu XX, Wang Z, Chen LB, Guan XX, Tong JD. Integrated analysis of DNA methylation and mRNA expression profiling reveals candidate genes associated with cisplatin resistance in non-small cell lung cancer. Epigenetics 2014;9:896-909.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Segovia J, Bautista E, Lara-Lozano M. Effects of Gas1 on gliomas: a review on current preclinical studies. J Cancer Metastasis Treat 2016;2:101-11. http://dx.doi.org/10.20517/2394-4722.2015.76
AMA Style
Segovia J, Bautista E, Lara-Lozano M. Effects of Gas1 on gliomas: a review on current preclinical studies. Journal of Cancer Metastasis and Treatment. 2016; 2: 101-11. http://dx.doi.org/10.20517/2394-4722.2015.76
Chicago/Turabian Style
Segovia, Jose, Elizabeth Bautista, Manuel Lara-Lozano. 2016. "Effects of Gas1 on gliomas: a review on current preclinical studies" Journal of Cancer Metastasis and Treatment. 2: 101-11. http://dx.doi.org/10.20517/2394-4722.2015.76
ACS Style
Segovia, J.; Bautista E.; Lara-Lozano M. Effects of Gas1 on gliomas: a review on current preclinical studies. J. Cancer. Metastasis. Treat. 2016, 2, 101-11. http://dx.doi.org/10.20517/2394-4722.2015.76
About This Article
Special Issue
Copyright

Data & Comments
Data


 Cite This Article 10 clicks
Cite This Article 10 clicks

 Like This Article 31
likes
Like This Article 31
likes






Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.