
Therapeutic strategies for targeting cancer stem cells

Abstract
The therapeutic limitations of conventional chemotherapeutic drugs present a challenge for cancer therapy; these shortcomings are largely attributed to the ability of cancer cells to repopulate and metastasize after initial therapies. Compelling evidence suggests that cancer stem cells (CSCs) have a crucial impact in current shortcomings of cancer therapy because they are largely responsible for tumor initiation, relapse, metastasis, and chemo-resistance. Thus, a better understanding of the properties and mechanisms underlying CSC resistance to treatments is necessary to improve patient outcomes and survival rates. In this review, the authors characterize and compare different CSC-specific biomarkers that are present in various types of tumors. We further discuss multiple targeting approaches currently in preclinical or clinical testing that show great potential for targeting CSCs. This review discusses numerous strategies to eliminate CSCs by targeting surface biomarkers, regulating CSC-associated oncogenes and signaling pathways, inhibiting drug-efflux pumps involved in drug resistance, modulating the tumor microenvironment and immune system, and applying drug combination therapy using nanomedicine.
Keywords
Introduction
Cancer stem cells (CSCs) are a small subset of cancer cells with the ability to self-renew and initiate tumor growth. They were first discovered in acute myeloid leukemia (AML) in the late 1990s.[1] Since then, CSCs have been discovered in many solid tumors.[2-6] Within the last two decades, CSCs have become a subject of intense research as a potential target for cancer therapeutics.
The discovery of CSCs led to a major shift in cancer modeling. Previously, cancers were thought to be made up of equipotent malignant cells which either renewed or differentiated stochastically, giving rise to a heterogeneous tumor. In contrast, the CSC model suggests that a hierarchy exists among tumor cells, with CSCs at the top, producing the bulk of the tumor cells while maintaining their own renewal.[5] A third model, clonal evolution, states that heterogeneity comes from genetic or epigenetic changes that arise during cancer progression. The CSC and clonal evolution models are not mutually exclusive, as CSCs can also evolve over time, generating different clonal subpopulations within the tumor.[6]
CSCs share a number of properties with normal stem cells (SCs). Both typically make up a small percentage of the total number of cells in a tissue, they are largely quiescent, and, most notably, they are multipotent and can self-renew indefinitely. Many pathways vital to SC function, such as Wnt, Hedgehog, Notch,[7] and PI3K/Akt,[8] are dysregulated in CSCs, potentially contributing to neoplastic transformation. For example, cases of multiple myeloma have displayed abnormal signaling in response to elevated levels of Hedgehog ligand secreted by tumor stromal cells,[9] and upregulated Notch4 signaling has been implicated in drug-resistant breast CSC activity.[10] Like SCs, CSCs are able to repair damaged DNA more quickly and overexpress drug-efflux pumps such as ATP-binding cassette (ABC) transporters. In a glioblastoma model, aberrant Akt signaling contributed to overactivation of the ABC transporter ABCG2 in CSCs, leading to increased drug expulsion and rendering them resistant to mitoxantrone.[11]
CSCs may also contribute to metastasis. During normal wound healing, cells are able to migrate to the wound site through the epithelial to mesenchymal transition (EMT) process. CSCs may also undergo EMT when migrating from the primary tumor site. Another theory hypothesizes that the CSC microenvironment -- including the surrounding vasculature -- facilitates metastasis.[12] While the exact mechanisms have not been discovered, there are many reports of CSC-driven metastasis. In fact, numerous studies have used breast CSC-rich cell lines such as MDA-MB-231 to first produce primary tumors and then seed lung metastases.[13,14]
Studies of CSC-targeted therapy depend on the isolation and enrichment of CSCs. They can be identified, isolated, and characterized by several methodologies, including flow cytometric analysis of CSC-specific cell surface markers, detection of side-population (SP) phenotypes by Hoechst 33342 dye exclusion, changes in aldehyde dehydrogenase (ALDH) enzymatic activities using an aldeflour assay, ability to grow as suspension spheres in serum-free medium, SC-related gene expression, and auto-fluorescence.[6,15-17] There are no widely accepted techniques solely developed to isolate CSCs, necessitating the use of combination markers and methods rather than single strategies.
Surface marker-based assays have become the mostly commonly used method.[18]Table 1 summarizes the list of cell surface phenotypes of CSCs in different tumors. The detection can be performed with specific antibodies in flow cytometry, competitive ELISA, or magnetic beads.[19] Dick and coworkers showed the first evidence of the presence of CSCs in human AML by the flow cytometric display of the CD34+CD38- surface marker phenotype.[20] A breast CSC subpopulation was identified and isolated by the combination of CD44 and CD24 markers.[2]
Cancer stem cell surface markers in human cancers
| Tumor types | Surface marker on the CSCs | References |
|---|---|---|
| Breast | CD44+/CD24-, CD133+, EpCAM+ | [2,17,18] |
| Colon | CD133+, EpCAM+, CD44+ | [17,18,36] |
| Glioma (brain) | CD133+, CD15+, CD49f+, CD90+ | [3,17,18] |
| Leukemia (AML) | CD34+/CD38-, CD123+ | [1,17,18] |
| Lung | ABCG2, CD133+ | [16-18] |
| Melanoma | ABCB5, CD133+, CD20+, CD271+ | [18] |
| Ovarian | CD44+, CD117+, CD133+ | [39] |
Functional CSC properties like intracellular ALDH enzymatic activities and ABC transporter efflux activities of vital DNA dyes such as Hoechst 33342 have been used for CSC isolation.[21,22] Increased aldehyde dehydrogenase isoform 1 (ALDH1) activity has been used to identify and analyze different types of CSCs. Furthermore, CSCs have a distinct efflux mechanism, stemming from their high expression of ABC transporter proteins.[15] These cells, referred to as the “side population” (SP), are able to actively transport fluorescent dyes such as Hoechst 33342 out of the cells. Flow cytometric SP analysis has been performed with numerous cancer cell lines and the SP has shown enriched CSC activities.[21]
A subpopulation of CSCs exhibit intrinsic autofluorescence and were shown to be exclusively linked to a functional CSC phenotype in different epithelial tumors. These autofluorescent cells had CSC characteristics such as high self-renewal, long-term tumorigenic capacity, invasiveness, and chemoresistance. These cells have intrinsic autofluorescence with excitation wavelengths at 488 nm and emission at about 520 nm. This new marker has been proven to be a more reliable and accurate way to identify and characterize CSCs.[16,23]
Another important functional property of CSCs, as well as normal stem SCs, is the ability to produce sphere-forming colonies from a single cell in serum-free medium or in soft agar medium, as differentiated cells cannot survive and proliferate in this environment.[24] Thus, several studies have used the sphere formation assay as an efficient method for isolating, enriching and maintaining CSCs from various primary tumors. Generally, these CSC-driven spheres are greater in both number and size as compared to ones generated from non-CSCs.[18,25] These spheres clearly demonstrated stem-like properties and expressed characteristics of CSCs.[16]
Here, we will focus on cancer therapeutics which can target CSCs. The development of various strategies that can act effectively against CSCs has been categorized into six groups, as shown in Figure 1.
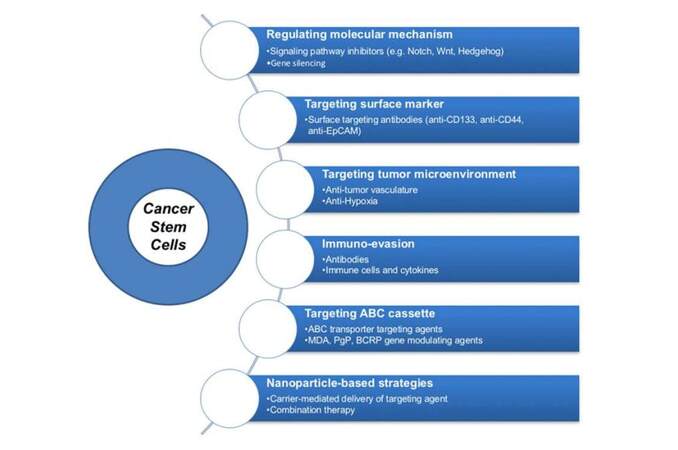
Figure 1. Novel therapeutic strategies for targeting cancer stem cells
Regulating CSC signaling pathways
Many signaling pathways are deregulated in CSCs and are potential targets in anti-CSC therapies. Overactivation of the Notch pathway has been implicated particularly in breast CSCs, possibly by influencing the EMT and contributing to the invasiveness of the CSCs.[26] One group investigated the effects of the bioactive compound psoralidin on Notch signaling in a breast cancer model. The plant-derived drug inhibited Notch signaling in both bulk tumor and CSCs, resulting in decreased mammosphere formation, upregulation of pro-apoptotic proteins, and inhibition of CSC proliferation.[27] Other studies have demonstrated that inhibiting Notch signaling resensitizes breast cancer cells to doxorubicin and docetaxel.[28]
The Hedgehog signaling pathway may also contribute to CSC formation. Hedgehog signaling controls cell fate and proliferation in embryonic SCs, but dysregulation of the pathway has been associated with CSC generation. Cyclopamine was the first Hedgehog antagonist to be identified and its effects have been studied in many cancers.[29] Cyclopamine depleted CSCs and induced tumor regression in a chronic myeloid leukemia model,[30] decreased tumor growth rate in a medulloblastoma model,[31] and inhibited proliferation of pancreatic CSCs.[32]
Hedgehog abnormalities are linked to aberrant Wnt pathway activity. It is believed that Wnt plays a role in maintaining the self-renewal capabilities of CSCs. CD44, a CSC marker, is an important target for Wnt signaling, and CD44 knockdown resulted in decreased tumor formation in an intestinal cancer model.[33] Another group tested several small molecule antagonists of Wnt and reported reduced mammosphere formation in vitro and halted tumor growth in vivo in a breast cancer model.[34]
Silencing oncogenes
RNAi is biological processes in which small interfering RNAs (siRNA) cause complementary target mRNA to be degraded, thereby silencing the gene. While there are many RNAi-based strategies which target bulk tumor cells, fewer studies have shown CSC-specific RNAi. One group used lentiviral short hairpin RNA (shRNA) to silence the human papillomavirus gene E6 in CSC-enriched cervical cancers. They discovered that after shRNA exposure, CSC growth and sphere formation were dramatically inhibited. E6 is upregulated in cervical CSCs, and E6 silencing also led to decreased CSC self-renewal through TGF-β modulation.[35] Another study also used shRNA to silence HMGA1, an oncogene overexpressed in CD133+ colon CSCs. HMGA1 knockdown restored normal SC properties to CSCs, including quiescence, increased asymmetric division, and decreased self-renewing division. HMGA1 silencing was also linked to increased p53 expression.[36] Both E6 and HMGA1 may be viable targets for anti-CSC therapy.
The application of RNAi in the clinic has been hampered by the inability to deliver high enough doses to the tumor site. One group has used targeted siRNAs to downregulate CSC oncogenes in vivo. They used PEGylated EpCAM aptamers to guide siRNA to EpCAM-overexpressing breast CSCs. The siRNA accumulated at the tumor site and resulted in an 80% knockdown of the survivin gene, which inhibits apoptosis and promotes chemoresistance in CSCs. When combined with doxorubicin, the aptamer-siRNA chimera improved survival rates of tumor-bearing mice,[37] demonstrating the effectiveness of anti-CSC RNAi in vivo as well as in vitro.
Targeting CSC surface markers
One potential CSC therapeutics approach is targeting CSC surface markers. One of the most established and commonly used CSC biomarkers is CD44, which is a cell-surface extracellular matrix receptor.[6] Many studies represented CD44 antibody therapy as the major anti-CSC approach. The first of these studies showed that H90 anti-CD44 therapy successfully eradicated AML.[38]
CD133 is a transmembrane glycoprotein and is another well-known CSC marker in several tumors such as glioblastoma, hepatocellular and colon cancers. CD133+ CSCs have shown resistance to chemotherapy and radiotherapy due to their slower cell cycle, lower proliferation, higher expression of DNA repair and anti-apoptotic genes.[39,40] In a study by Carter et al.,[38] the AC133 antibody was conjugated to a potent cytotoxic drug, monomethyl auristatin, using a protease cleavable linker. This antibody drug conjugate was efficiently internalized, co-localized with the lysosome and showed high effectiveness against hepatocellular cancer cells.
EpCAM has been discovered as a CSC marker in solid tumors and is correlated with all the characteristics of CSCs. EpCAM+/CD44+/CD24- population in breast cancer had a significantly higher frequency of tumor-initiating cells. Moreover, ovarian cancer cells with high EpCAM expression were involved in EMT, leading to metastasis.[2,41] Humanized EpCAM antibodies have been successful in both preclinical and early clinical studies, showing potent anti-tumor activity.[38,41]
Inhibiting ABC transporters
CSC chemoresistance is due in large part to the overexpression of drug efflux pumps such as ABC transporters. Several pharmacological agents have demonstrated inhibitory or neutralizing effects on these transporters. There are three generations of inhibitors of one of the main ABC transporters, P-glycoprotein (P-gp). However, none have been approved for clinical use due to a lack of specificity and adverse side effects. Recently, a more specific P-gp inhibitor, vardenafil, has shown promise in mitigating the effects of P-gp overexpression. Vardenafil appeared to directly block P-gp-mediated drug efflux and resulted in increased intracellular concentration and cytotoxicity of paclitaxel and vincristine.[42]
RNAi has also been used to silence ABC transporter genes. siRNA targeting P-gp reversed drug resistance in a doxorubicin-resistant breast cancer model. Doxorubicin-resistant cell lines are enriched with CSCs upon prolonged doxorubicin exposure.[43] Exposing the resistant, CSC-enriched cells to P-gp siRNA resulted in downregulation of P-gp gene expression and led to increased intracellular accumulation of doxorubicin and a 4-fold resensitization.[44]
Nanotechnology can be used alone or in combination with drugs or RNAi of ABC transporters. Triblock copolymers by themselves have been shown to resensitize P-gp-overexpressing tumors to chemotherapeutic drugs; one group incorporated such a copolymer into polylactic acid micelles and reported overcoming multidrug resistance (MDR) in a paclitaxel-resistant breast cancer cell line.[45] One ABC transporter inhibitor, ritonavir, was conjugated with copolymer nanoparticles to increase uptake into tumor cells and enhance the cytotoxic effect of doxorubicin in drug resistant murine leukemic cells.[46] Another study suppressed P-gp using siRNA-loaded dextran polymeric nanoparticles in conjunction with doxorubicin treatment.[47]
Enhancing immune responses
It is hypothesized that CSCs are able to evade cancer immunosurveillance due to phenotypic and functional properties that allow them to survive in immunocompetent hosts. Antitumor immune cells are detectable and relevant to disease prognosis. Tumor associated antigens (TAA) are encoded by lineage specific genes and are often present or overexpressed on tumor cells.[48] In patients with metastatic melanoma, circulating CD8+ T cells targeting the TAA MART-1 were detected, although they were functionally unresponsive.[49,50] These TAA-specific T cells may be rendered anergic in vivo, and it is also plausible that CSCs downregulate their expression of human leukocyte antigen class 1 molecules or TAAs as another means of immunoevasion[49,50] Consequently, immunotherapy has become one of the most promising treatments for patients with metastatic cancer. Examples of strategies developed to enhance the host immune system are nonspecific immunomodulation to activate the host’s immune response, and adoptive cell transfer of ex vivo expanded lymphocytes, such as T cells and natural killer (NK) cells.[48,51]
Nonspecific immunomodulation includes treatment of patients with metastatic cancer using FDA-approved cytokines such as IFNα and IL-2.[48,52] Administration of high doses of IL-2 into experimental animals was reported to reduce lung and liver metastases, and further investigation was conducted in human patients with metastatic melanoma, which demonstrated 15-20% objective clinical response.[53,54] Several researches have attempted to explain the role of IL-2 in immunomodulation, and proposed that IL-2 induces expansion of T cells with major histocompatibility complex (MHC) -specific recognition of TAA to eliminate target cells.[54,55] One disadvantage of this nonspecific antitumor immune activation is it also upregulates the CD4+CD25hi Foxp3+ regulatory T cell (Treg) population, which impedes general antitumor T cell function and contributes to tumor immunoevasion.[52,56]
Another promising strategy in targeting cancers in vivo is adoptive transfer of chimeric antigen receptor (CAR) engineered T cells, which can specifically target any TAAs or cancer stromal antigens with high binding affinity. Preclinical models have been developed as a proof of concept that CARs could also be used to target CSCs.[57] Deng et al.,[58] for instance, demonstrated that CAR T cell therapy could inhibit tumor growth of highly metastatic prostate cancer that expresses low levels of EpCAM. Other CSC-targeting adoptive T cell therapies include CAR T cells which bind to a CSC-specific N-glycosylation-dependent epitope of CD133,[59] high-molecular weight melanoma associated antigen or chondroitin sulfate proteoglycan 4-specific CAR T cells that were reported to specifically eliminate melanoma with a CSC phenotype,[60,61] and epidermal growth factor receptor variant III (EGFRvIII) specific CAR T cells which target glioma SCs.[62]
Cells in the tumor microenvironment was also found to express several negative immune regulators such as programmed cell death 1(PD-1) and its ligand (PD-L1), cytotoxic T lymphocyte associated 4 (CTLA-4), and transforming growth factor β (TGF-β).[63] Engagement of CTLA-4 attenuates activation of downstream inflammatory cytokines, which contribute to T cell antitumor immunity, such as IL-2 and IFN-γ.[64,65] In Tregs, the engagement of CTLA-4 is required for immune suppression.[66] Antibodies blocking CTLA-4 engagement were developed and tested for their cancer therapeutic potential.[67] Wu et al.[68] showed that CTLA-4 monoclonal antibody (mAb) was able to inhibit early stages of tumor growth in a murine mesothelioma model and improved tumor infiltration of CD8+ and CD4+ T cells. In human patients with advanced melanoma, the CTLA-4 mAbs ipilimumab and tremelimumab prolonged T cell activation. However, only ipilimumab demonstrated improved survival in phase III study of patients with previously treated melanoma and gained FDA approval for treatment of metastatic melanoma in 2011.[69] Future applications of mAb CTLA-4 will most likely come in the form of combination therapy to modulate the host immune system in a more effective synergistic fashion.[67,68,70,71]
The second T cell regulatory pathway is the PD1/PD-L1 axis, which inhibits lymphocyte activation. PD-L1 or B7 homolog 1 (B7-H1) is expressed in many tumors including melanoma and cancers of the lung, colon, ovarian, liver and breast.[63,72] PD1/PD-L1 binding triggers apoptosis of B and T cells in the tumor microenvironment. In tumors with upregulated PD-L1 expression, there is decreased T cell infiltration, activation, and expansion, effectively shielding CSCs against the host’s immune response.[63,72,73] Most recently, the new immunotherapy drug which has been approved by FDA in May 2016.Tecentriq is a monoclonal antibody that targets the PD-1/PD-L1 pathway by directly binding with a PD-L1 protein expressed on tumor cells and tumor-infiltrating immune cells. This immune checkpoint inhibitor will help the body’s immune system fight against cancer cells.[74]
In addition to previously reported suppressor molecules, CD200 (OX-2) is another immunosuppressive factor that may have an important role in CSC’s immunoevasion.[75] CD200 is co-expressed with CSC markers such as CD133+ glioblastoma, colon and melanoma CSCs, CD44+/CD24- in breast CSCs and CD44+ prostate CSCs.[76,77] Upregulation of CD200 negatively correlates with the levels of Th1 cytokines required for effective T cell activation, such as IL-2 and IFNγ.[78-80] Shifting of Th1 to Th2 cytokine production is observed in the progression of many cancer types and is a characteristic of the tumor microenvironment, especially in carcinomas with poor prognosis.[78]
Targeting the tumor microenvironment
The tumor microenvironment of CSCs has three major characteristics: (1) chronic inflammation and secretion of inflammatory cytokines,[81] (2) hypoxia,[82] and (3) perivascular niches that regulate the capacity of proliferation and differentiation.[83] Inflammatory cytokines such as IL-1β, IL-6 and IL-8 activate the Stat3/NF-κB pathways in tumor and stromal cells to further secrete cytokines in a positive feedback loop that prompts CSC self-renewal, angiogenesis, and metastasis.[81,84] Moreover, the CSC population along with other cells which coevolved in the tumor microenvironment are near blood vessels that form a niche characterized by severe hypoxia and increased angiogenesis.[82,83] These aspects of the tumor microenvironment have been explored as possible pharmaceutical targets of CSCs.
Recent studies have demonstrated decreased tumor growth after blocking IL-6 and/or IL-8 cytokine signaling.[85,86] One of the pharmaceutical molecules tested was repertaxin, a non-competitive inhibitor of IL-8 and CXCR1 signaling, which decreased tumor size and increased efficacy of chemotherapy.[87] However, the effects of blocking single cytokines is limited as both IL-6 and IL-8 are critical for xenograft tumor growth and the combined expression of these genes correlates with poor prognosis in patients with breast cancer. Therefore, co-inhibition of both IL-6 and IL-8 was suggested to be a more advantageous method to induce substantial effects on tumor growth.[88]
Tumor hypoxia is another intriguing method for attacking CSC niches. Hypoxia activates the hypoxia inducible factor (HIF) pathway and upregulates HIF-1α, which mediates multiple biological effects of hypoxia in tissues and increases resistance against chemotherapy and radiation.[89] Several small molecule inhibitors of the HIF pathway have been pursued in clinical trials, although only a few of them were successful. Bortezomib (Velcade®, PS-341) was approved by the FDA in 2003 for use against multiple myeloma, followed by Temsirolimus (Torisel®, CCI-779), approved in 2007 for use against renal cell carcinoma. The majority of the other drugs -- including Perifosine, 2-methoxyestradiol, Echinomycin, Geldanamycin -- were terminated in either phase I or II trials when they failed to show significant advantages.[89]
Lastly, targeting tumor vasculature is another way to disrupt the CSC niche. Several agents blocking the activity of vascular endothelial growth factor, which drives the migration of endothelial cells and stimulates angiogenesis, are being tested in initial phases of clinical therapy with moderate success. These include Bevacizumab (Avastin®), Cediranib (AZD2171), Sunitinib and Vandetanib.[82,90-93]
Nanomedicine in combination therapy
Frequently after treatment, surviving CSCs induce new tumor formation and metastases in which cancer reappears in an even more aggressive form. With this phenomenon in mind, an increasing number of CSC-targeted therapeutic agents have been developed over the past several years such as salinomycin, curcumin, thioridazine hydrochloride, sulforaphane, miR-34a, and miR-130b.[94-97] Despite their therapeutic potential in targeting CSCs, their clinical application has been hindered by their hydrophobicity, poor specificity and poor pharmacokinetics (PK) profiles.[98-100]
Recent developments in nanoparticle delivery systems have provided new strategies to efficiently deliver therapeutics that can overcome the challenges posed by CSCs and improve therapeutic efficacy of CSC-targeting agents by controlling release kinetics, prolonging circulation time and improving bio-distribution. In a study by Zhou et al.,[98] they used HPMA polymeric nanoparticles to deliver a Hedgehog pathway inhibitor that efficiently eliminated CD133+ cells within prostate tumors. Mamada and coworkers designed mesoporous silica nanoparticles to deliver a potent inhibitor of the Notch signaling pathway. Their nanoparticle drug treatments efficiently targeted CSC populations in the tumor. Furthermore, in the study done by Wei and colleagues, salinomycin was conjugated to a hyaluronic acid-based nanogel to target CD44+ drug resistant cells which enhanced the therapeutic efficacy of salinomycin.[97,98]
Another advantage of using nanoparticles is the additional capability to modify their surfaces with targeting agents such as mAbs and peptides. High target selectivity and internalization can be achieved by surface modification of nanoparticles with targeting moieties. As previously discussed, CSCs are characterized by certain surface markers; this allows specific targeting of CSCs as a therapeutic strategy for drug delivery. Swaminathan et al.[101] demonstrated that their targeted nanoparticles induced a significant tumor volume reduction compared to untreated control and non-targeted groups in an in vivo MDA-MB 231 xenograft tumor model by developing paclitaxel-loaded polymeric PLGA nanoparticles conjugated with CD133 mAb. In another study done by Dou et al.,[94] myeloma CSCs were treated with silver nanoparticles decorated with anti-ABCG2 antibodies on the surface along with vincristine. Despite these advances in the laboratory, targeted nanoparticle approaches in the CSC field are still in the early preclinical development stage due to limitations such as potential systemic toxicity, unwanted side effects, and poor extravasation and exposure to their targets.[97,98,102]
It has been shown that using a CSC-targeted inhibitor alone is not very effective in reducing the tumor bulk due to the fact that these inhibitors are not highly cytotoxic as compared with conventional chemotherapeutics. Thereby, dual targeting nanoparticles loaded with CSC inhibitors and conventional cytotoxic agents can improve clinical outcomes by effectively eradicating both CSCs and bulk tumor cells at the same time. When compared with the free drugs, the nanoparticle formulated drugs were significantly more effective and less toxic both in vitro and in vivo.[103-105]
Conclusion and future perspectives
It is clear now that conventional chemotherapy is not enough to overcome the abilities of CSCs to self-renew and metastasize. A combination of surface markers and their functional properties have been used to identify and isolate CSCs. Despite this progress, there is still a lack of reliable and accurate CSC markers. This must be overcome in order to develop therapeutic strategies with higher specificity and fewer side effects.
Using either small molecule inhibitors or RNAi to target CSC-associated oncogenes and signaling pathways have resulted in decreased functionality and numbers of CSCs and tumor regression in several pre-clinical models. CSCs develop resistance to conventional chemotherapeutics, but targeting ABC transporters resensitizes CSCs to those same drugs. Several studies have shown greater CSC targeting effects by employing antibodies against CSC-specific biomarkers. Anti-CSC approaches such as CD44 and EpCAM antibodies could selectively induce differentiation and inhibit proliferation.
Modulating the immune system and tumor microenvironment as a means of targeting CSCs has shown encouraging results. However, the efficacy of immunotherapy alone may be inadequate to produce clinical results. Therefore, combination therapy with conventional modalities as well as with immunomodulatory agents may be of future interest to enhance therapeutic effects.
As discussed above, nanocarriers enhanced the delivery and cytotoxic activity of CSC-inhibitors. Several studies introduced active targeting strategies of nanoparticle surfaces to increase their specificity and cellular uptake by CSCs. Lastly, researchers have been focusing on nanoparticle-mediated drug combinatorial therapy. One important advantage of nanocarriers is their capability to incorporate multiple therapeutic agents in one carrier system, allowing co-delivery of cytotoxic drugs and CSC inhibitors to simultaneously target both bulk tumor and CSCs. Patient cures will rely on the ablation of the entire tumor. Ultimately, nanoparticle mediated combination therapy may prove to be the most successful in eradicating whole tumors.
The CSC field is relatively new, and CSC-targeting therapeutics is in their early stages. While many advances have been made in CSC research, many of these studies have been performed in vitro only, and none are past the early clinical stages. Important factors such as effective dosages and side effects must be elucidated before employing cancer treatment plans that target both differentiated tumor cells and CSCs. There is need to improve the existing methods to precisely isolate, identify and target CSCs. As mentioned, increasing amount of nanomedicine have been evaluated about their application potentials in CSC therapy, but only a small amount of them can be approved to translate to clinical treatment. With the fact that every cancer acts differently in different patients, the development of personalized combinational therapies may serve as a key to successful treatments. Furthermore, it is important to realize that the combination of nanomedicine and immunotherapy may present a novel direction which shows great potential in personalized cancer therapy.
Financial support and sponsorship
This work was supported by National Institutes of Health grants (R01AI068978, R01CA170820 and P01CA132681), a translational acceleration grant from the Joint Center for Translational Medicine, the National Cancer Institute (P30CA014089), and a grant from the Ming Hsieh Institute for Research on Engineering Medicine for Cancer.
Conflicts of interests
There are no conflicts of interest.
REFERENCES
1. Bonnet D, Dick JE. J. Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat Med 1997;3:730-7.
2. Al-Hajj M, Wicha M, Benito-Hernandez A, Morrison S, Clarke M. Prospective identification of tumorigenic breast cancer cells. PNAS 2003;100:3983-8.
3. Singh SK, Clarke ID, Terasaki M, Bonn VE, Hawkins C, Squire J, Dirks PB. Identification of a cancer stem cell in human brain tumors. Cancer Res 2003;63:5821-8.
4. Kim CF, Jackson EL, Woolfenden AE, Lawrence S, Babar I, Vogel S, Crowley D, Bronson RT, Jacks T. Identification of bronchioalveolar stem cells in normal lung and lung cancer. Cell 2005;121:823-35.
6. Chen K, Huang YH, Chen JL. Understanding and targeting cancer stem cells: therapeutic implications and challenges. Acta Pharmacol Sin 2013;34:732-40.
7. Wang J, Sullenger BA, Rich JN. Notch signaling in cancer stem cells. Adv Exp Med Biol 2012;727:174-85.
8. Hambardzumyan D1, Becher OJ, Rosenblum MK, Pandolfi PP, Manova-Todorova K, Holland EC. PI3K pathway regulates survival of cancer stem cells residing in the perivascular niche following radiation in medulloblastoma in vivo. Genes Dev 2008;22:436-48.
9. Campbell V, Copland M. Hedgehog signaling in cancer stem cells: a focus on hematological cancers. Stem Cells Cloning 2015;8:27-38.
10. Simões BM, O'Brien CS, Eyre R, Silva A, Yu L, Sarmiento-Castro A, Alférez DG, Spence K, Santiago-Gómez A, Chemi F, Acar A, Gandhi A, Howell A, Brennan K, Rydén L, Catalano S, Andó S, Gee J, Ucar A, Sims AH, Marangoni E, Farnie G, Landberg G, Howell SJ, Clarke RB. Anti-estrogen resistance in human breast tumors is driven by JAG1-NOTCH4-dependent cancer stem cell activity. Cell Rep 2015;12:1968-77.
11. Bleau AM, Hambardzumyan D, Ozawa T, Fomchenko EI, Huse JT, Brennan CW, Holland EC. PTEN/PI3K/Akt pathway regulates the side population phenotype and ABCG2 activity in glioma tumor stem-like cells. Cell Stem Cell 2009;4:226-35.
13. Price JE, Polyzos A, Zhang RD, Daniels LM. Tumorigenicity and metastasis of human breast carcinoma cell lines in nude mice. Cancer Res 1990;50:717-21.
14. Minn AJ, Gupta GP, Siegel PM, Bos PD, Shu W, Giri DD, Viale A, Olshen AB, Gerald WL, Massagué J. Genes that mediate breast cancer metastasis to lung. Nature 2005;436:518-24.
15. Allahverdiyev AM, Bagirova M, Oztel ON, Yaman S, Abamor ES, Koc RC, Ates SC, Elcicek S, Baydar SY. Aldehyde Dehydrogenase: Cancer and Stem Cells. In: Canuto RA, editor. Dehydrogenases. InTech: Rijeka, Croatia; 2012. pp. 3-28.
16. Duan JJ, Qiu W, Xu SL, Wang B, Ye XZ, Ping YF, Zhang X, Bian XW, Yu SC. Strategies for isolating and enriching cancer stem cells: well begun is half done. Stem Cells Dev 2013;22:2221-39.
17. Sheridan C, Kishimoto H, Fuchs RK, Mehrotra S, Bhat-Nakshatri P, Turner CH, Goulet R Jr, Badve S, Nakshatri H. CD44+/CD24- breast cancer cells exhibit enhanced invasive properties: an early step necessary for metastasis. Breast Cancer Res 2006;8:R59.
18. Klonisch T, Wiechec E, Hombach-Klonisch S, Ande SR, Wesselborg S, Schulze-Osthoff K, Los M. Cancer stem cell markers in common cancers - therapeutic implications. Trends Mol Med 2008;10:450-60.
19. Ali HR, Dawson SJ, Blows FM, Provenzano E, Pharoah PD, Caldas C. Cancer stem cell markers in breast cancer: pathological, clinical and prognostic significance. Breast Cancer Res 2011;6:R118.
20. Tirino V, Desiderio V, Paino F, De Rosa A, Papaccio F, La Noce M, Laino L, De Francesco F, Papaccio G. Cancer stem cells in solid tumors: an overview and new approaches for their isolation and characterization. FASEB J 2013;1:13-24.
21. Greve B, Kelsch R, Spaniol K, Eich HT, Götte M. Flow cytometry in cancer stem cell analysis and separation. Cytometry A 2012;4:284-93.
22. Komuro H, Saihara R, Shinya M, Takita J, Kaneko S, Kaneko M, Hayashi Y. Identification of side population cells (stem-like cell population) in pediatric solid tumor cell lines. J Pediatr Surg 2007;42:2040-5.
23. Miranda-Lorenzo I, Dorado J, Lonardo E, Alcala S, Serrano AG, Clausell-Tormos J, Cioffi M, Megias D, Zagorac S, Balic A, Hidalgo M, Erkan M, Kleeff J, Scarpa A, Sainz B Jr, Heeschen C. Intracellular autofluorescence: a biomarker for epithelial cancer stem cells. Nat Methods 2014;11:1161-9.
24. Cao L, Zhou Y, Zhai B, Liao J, Xu W, Zhang R, Li J, Zhang Y, Chen L, Qian H, Wu M, Yin Z. Sphere-forming cell subpopulations with cancer stem cell properties in human hepatoma cell lines. BMC Gastroenterol 2011;11:71.
25. Wang K, Wu X, Wang J, Huang J. Cancer stem cell theory: therapeutic implications for nanomedicine. Int J Nanomedicine 2013;8:899-908.
26. Smalley M, Piggott L, Clarkson R. Breast cancer stem cells: obstacles to therapy. Cancer Lett 2013;338:57-62.
27. Suman S, Das TP, Damodaran C. Silencing NOTCH signaling causes growth arrest in both breast cancer stem cells and breast cancer cells. Br J Cancer 2013;109:2587-96.
28. Zang S, Chen F, Dai J, Guo D, Tse W, Qu X, Ma D, Ji C. RNAi-mediated knockdown of Notch-1 leads to cell growth inhibition and enhanced chemosensitivity in human breast cancer. Oncol Rep 2010:23893-9.
29. Gupta S, Takebe N, Lorusso P. Targeting the Hedgehog pathway in cancer. Ther Adv Med Oncol 2010;2:237-50.
30. Zhao C, Chen A, Jamieson CH, Fereshteh M, Abrahamsson A, Blum J, Kwon HY, Kim J, Chute JP, Rizzieri D, Munchhof M, VanArsdale T, Beachy PA, Reya T. Hedgehog signalling is essential for maintenance of cancer stem cells in myeloid leukaemia. Nature 2009;458:776-9.
31. Berman DM, Karhadkar SS, Hallahan AR, Pritchard JI, Eberhart CG, Watkins DN, Chen JK, Cooper MK, Taipale J, Olson JM, Beachy PA. Medulloblastoma growth inhibition by hedgehog pathway blockade. Science 2002;297:1559-61.
32. Thayer SP, di Magliano MP, Heiser PW, Nielsen CM, Roberts DJ, Lauwers GY, Qi YP, Gysin S, Fernández-del Castillo C, Yajnik V, Antoniu B, McMahon M, Warshaw AL, Hebrok M. Hedgehog is an early and late mediator of pancreatic cancer tumorigenesis. Nature 2003;425:851-6.
33. Zeilstra J, Joosten SP, Dokter M, Verwiel E, Spaargaren M, Pals ST. Deletion of the WNT target and cancer stem cell marker CD44 in Apc(Min/+) mice attenuates intestinal tumorigenesis. Cancer Res 2008;68:3655-61.
34. Hallett RM, Kondratyev MK, Giacomelli AO, Nixon AM, Girgis-Gabardo A, Ilieva D, Hassell JA. Small molecule antagonists of the Wnt/β-catenin signaling pathway target breast tumor-initiating cells in a Her2/Neu mouse model of breast cancer. PLoS One 2012;7:e33976.
35. Gu W, Yeo E, McMillan N, Yu C. Silencing oncogene expression in cervical cancer stem-like cells inhibits their cell growth and self-renewal ability. Cancer Gene Ther 2011;18:897-905.
36. Puca F, Colamaio M, Federico A, Gemei M, Tosti N, Bastos AU, Del Vecchio L, Pece S, Battista S, Fusco A. HMGA1 silencing restores normal stem cell characteristics in colon cancer stem cells by increasing p53 levels. Oncotarget 2014;5:3234-45.
37. Wang T, Gantier MP, Xiang D, Bean AG, Bruce M, Zhou SF, Khasraw M, Ward A, Wang L, Wei MQ, AlShamaileh H, Chen L, She X, Lin J, Kong L, Shigdar S, Duan W. EpCAM aptamer-mediated survivin silencing sensitized cancer stem cells to doxorubicin in a breast cancer model. Theranostics 2015;5:1456-72.
38. Deonarain MP, Kousparou CA, Epenetos AA. Antibodies targeting cancer stem cells: a new paradigm in immunotherapy? MAbs 2009;1:12-25.
39. Zinzi L, Contino M, Cantore M, Capparelli E, Leopoldo M, Colabufo NA. ABC transporters in CSCs membranes as a novel target for treating tumor relapse. Front Pharmacol 2014;5:163.
40. Morrison R, Schleicher SM, Sun Y, Niermann KJ, Kim S, Spratt DE, Chung CH, Lu B. Targeting the mechanisms of resistance to chemotherapy and radiotherapy with the cancer stem cell hypothesis. J Oncol 2011;2011:941876.
41. Vaz AP, Ponnusamy MP, Batra SK. Cancer stem cells and therapeutic targets: an emerging field for cancer treatment. Drug Deliv Transl Res 2013;3:113-20.
42. Ding PR, Tiwari AK, Ohnuma S, Lee JW, An X, Dai CL, Lu QS, Singh S, Yang DH, Talele TT, Ambudkar SV, Chen ZS. The phosphodiesterase-5 inhibitor vardenafil is a potent inhibitor of ABCB1/P-glycoprotein transporter. PLoS One 2011;6:e19329.
43. Calcagno AM, Salcido CD, Gillet JP, Wu CP, Fostel JM, Mumau MD, Gottesman MM, Varticovski L, Ambudkar SV. Prolonged drug selection of breast cancer cells and enrichment of cancer stem cell characteristics. J Natl Cancer Inst 2010;102:1637-52.
44. Dönmez Y, Gündüz U. Reversal of multidrug resistance by small interfering RNA (siRNA) in doxorubicin-resistant MCF-7 breast cancer cells. Biomed Pharmacother 2011;65:85-9.
45. Li X, Li P, Zhang Y, Zhou Y, Chen X, Huang Y, Liu Y. Novel Mixed Polymeric Micelles for Enhancing Delivery of Anticancer Drug and Overcoming Multidrug Resistance in Tumor Cell Lines Simultaneously. Pharm Res 2010;27:1498-511.
46. Šubr V, Koziolová E, Sivák L, Říhová B, Kovář M, Ulbrich K. Polymer inhibitors of ABC transporter overcoming multidrug resistance: Synthesis, characterization and in vitro evaluation. J Control Release 2015;213:e107-8.
47. Falasca M, Linton KJ. Investigaional ABC transporter inhibitors. Expert Opin Investig Drugs 2012;21:657-66.
48. Schatton T, Frank MH. Antitumor immunity and cancer stem cells. Ann N Y Acad Sci 2009;1176:154-69.
49. Lee PP, Yee C, Savage PA, Fong L, Brockstedt D, Weber JS, Johnson D, Swetter S, Thompson J, Greenberg PD, Roederer M, Davis MM. Characterization of circulating T cells specific for tumor-associated antigens in melanoma patients. Nat Med 1999;5:677-85.
50. Khong HT, Wang QJ, Rosenberg SA. Identification of multiple antigens recognized by tumor-infiltrating lymphocytes from a single patient: tumor escape by antigen loss and loss of MHC expression. J Immunother 2004;27:184-90.
51. Ames E, Canter RJ, Grossenbacher SK, Mac S, Chen M, Smith RC, Hagino T, Perez-Cunningham J, Sckisel GD, Urayama S, Monjazeb AM, Fragoso RC, Sayers TJ, Murphy WJ. NK Cells Preferentially Target Tumor Cells with a Cancer Stem Cell Phenotype. J Immunol 2015;195:4010-9.
52. Ahmadzadeh M, Rosenberg SA. IL-2 administration increases CD4+ CD25(hi) Foxp3+ regulatory T cells in cancer patients. Blood 2006;107:2409-14.
53. Rosenberg SA, Yang JC, Topalian SL, Schwartzentruber DJ, Weber JS, Parkinson DR, Seipp CA, Einhorn JH, White DE. Treatment of 283 consecutive patients with metastatic melanoma or renal cell cancer using high-dose bolus interleukin 2. JAMA 1994;271:907-13.
54. Rosenberg SA, Mulé JJ, Spiess PJ, Reichert CM, Schwarz SL. Regression of established pulmonary metastases and subcutaneous tumor mediated by the systemic administration of high-dose recombinant interleukin 2. J Exp Med 1985;161:1169-88.
55. Itoh K, Platsoucas CD, Balch CM. Autologous tumor-specific cytotoxic T lymphocytes in the infiltrate of human metastatic melanomas. Activation by interleukin 2 and autologous tumor cells, and involvement of the T cell receptor. J Exp Med 1988;168:1419-41.
56. Powell DJ Jr, de Vries CR, Allen T, Ahmadzadeh M, Rosenberg SA. Inability to mediate prolonged reduction of regulatory T Cells after transfer of autologous CD25-depleted PBMC and interleukin-2 after lymphodepleting chemotherapy. J Immunother 2007;30:438-47.
57. Canter RJ, Grossenbacher SK, Ames E, Murphy WJ. Immune targeting of cancer stem cells in gastrointestinal oncology. J Gastrointest Oncol 2016;7 Suppl 1:S1-10.
58. Deng Z, Wu Y, Ma W, Zhang S, Zhang YQ. Adoptive T-cell therapy of prostate cancer targeting the cancer stem cell antigen EpCAM. BMC Immunol 2015;16:1.
59. Zhu X, Prasad S, Gaedicke S, Hettich M, Firat E, Niedermann G. Patient-derived glioblastoma stem cells are killed by CD133-specific CAR T cells but induce the T cell aging marker CD57. Oncotarget 2015;6:171-84.
60. Schmidt P, Kopecky C, Hombach A, Zigrino P, Mauch C, Abken H. Eradication of melanomas by targeted elimination of a minor subset of tumor cells. Proc Natl Acad Sci U S A 2011;108:2474-9.
61. Beard RE, Zheng Z, Lagisetty KH, Burns WR, Tran E, Hewitt SM, Abate-Daga D, Rosati SF, Fine HA, Ferrone S, Rosenberg SA, Morgan RA. Multiple chimeric antigen receptors successfully target chondroitin sulfate proteoglycan 4 in several different cancer histologies and cancer stem cells. J Immunother Cancer 2014;2:25.
62. Morgan RA, Johnson LA, Davis JL, Zheng Z, Woolard KD, Reap EA, Feldman SA, Chinnasamy N, Kuan CT, Song H, Zhang W, Fine HA, Rosenberg SA. Recognition of glioma stem cells by genetically modified T cells targeting EGFRvIII and development of adoptive cell therapy for glioma. Hum Gene Ther 2012;23:1043-53.
63. Dong H, Strome SE, Salomao DR, Tamura H, Hirano F, Flies DB, Roche PC, Lu J, Zhu G, Tamada K, Lennon VA, Celis E, Chen L. Tumor-associated B7-H1 promotes T-cell apoptosis: A potential mechanism of immune evasion. Nat Med 2002;8:793-800.
64. Riley JL, Mao M, Kobayashi S, Biery M, Burchard J, Cavet G, Gregson BP, June CH, Linsley PS. Modulation of TCR-induced transcriptional profiles by ligation of CD28, ICOS, and CTLA-4 receptors. Proc Natl Acad Sci U S A 2002;99:11790-5.
65. Rabinovich GA, Gabrilovich D, Sotomayor EM. Immunosuppressive strategies that are mediated by tumor cells. Annu Rev Immunol 2007;25:267-96.
66. Wing K, Onishi Y, Prieto-Martin P, Yamaguchi T, Miyara M, Fehervari Z, Nomura T, Sakaguchi S. CTLA-4 control over Foxp3+ regulatory T cell function. Science 2008;322:271-5.
67. Tarhini AA, Iqbal F. CTLA-4 blockade: therapeutic potential in cancer treatments. Onco Targets Ther 2010;3:15-25.
68. Hu Y, Fu L. Targeting cancer stem cells: a new therapy to cure cancer patients. Am J Cancer Res 2012;2:340-56.
69. Hodi FS, O'Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, Gonzalez R, Robert C, Schadendorf D, Hassel JC, Akerley W, van den Eertwegh AJ, Lutzky J, Lorigan P, Vaubel JM, Linette GP, Hogg D, Ottensmeier CH, Lebbé C, Peschel C, Quirt I, Clark JI, Wolchok JD, Weber JS, Tian J, Yellin MJ, Nichol GM, Hoos A, Urba WJ. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med 2010;363:711-23.
70. Fong L, Small EJ. Anti-cytotoxic T-lymphocyte antigen-4 antibody: the first in an emerging class of immunomodulatory antibodies for cancer treatment. J Clin Oncol 2008;26:5275-83.
71. Langer LF, Clay TM, Morse MA. Update on anti-CTLA-4 antibodies in clinical trials. Expert Opin Biol Ther 2007;7:1245-56.
72. Hirano F, Kaneko K, Tamura H, Dong H, Wang S, Ichikawa M, Rietz C, Flies DB, Lau JS, Zhu G, Tamada K, Chen L. Blockade of B7-H1 and PD-1 by monoclonal antibodies potentiates cancer therapeutic immunity. Cancer Res 2005;65:1089-96.
73. Chemnitz JM, Parry RV, Nichols KE, June CH, Riley JL. SHP-1 and SHP-2 associate with immunoreceptor tyrosine-based switch motif of programmed death 1 upon primary human T cell stimulation, but only receptor ligation prevents T cell activation. J Immunol 2004;173:945-54.
74. FDA approves new, targeted treatment for bladder cancer: Tecentriq is the first PD-L1 inhibitor approved by the FDA. Food and Drug Administration, FDA News Release 2016.
75. Kawasaki BT, Farrar WL. Cancer Stem Cells, CD200 and Immunoevasion. Trends Immunol 2008;29:464-8.
76. Kawasaki BT, Mistree T, Hurt EM, Kalathur M, Farrar WL. Co-expression of the toleragenic glycoprotein, CD200, with markers for cancer stem cells. Biochem Biophys Res Commun 2007;364:778-82.
77. Jung YS, Vermeer PD, Vermeer DW, Lee SJ, Goh AR, Ahn HJ, Lee JH. CD200: association with cancer stem cell features and response to chemoradiation in head and neck squamous cell carcinoma. Head Neck 2015;37:327-35.
78. McWhirter JR, Kretz-Rommel A, Saven A, Maruyama T, Potter KN, Mockridge CI, Ravey EP, Qin F, Bowdish KS. Antibodies selected from combinatorial libraries block a tumor antigen that plays a key role in immunomodulation. Proc Natl Acad Sci U S A 2006;103:1041-6.
79. Gorczynski RM, Chen Z, Hu J, Kai Y, Lei J. Evidence of a role for CD200 in regulation of immune rejection of leukaemic tumour cells in C57BL/6 mice. Clin Exp Immunol 2001;126:220-9.
80. Siva A, Xin H, Qin F, Oltean D, Bowdish KS, Kretz-Rommel A. Immune modulation by melanoma and ovarian tumor cells through expression of the immunosuppressive molecule CD200. Cancer Immunol Immunother 2008;57:987-96.
81. Korkaya H, Liu S, Wicha MS. Regulation of cancer stem cells by cytokine networks: attacking cancer's inflammatory roots. Clin Cancer Res 2011;17:6125-9.
82. Persano L, Rampazzo E, Basso G, Viola G. Glioblastoma cancer stem cells: role of the microenvironment and therapeutic targeting. Biochem Pharmacol 2013;85:612-22.
83. Charles N, Holland EC. The perivascular niche microenvironment in brain tumor progression. Cell Cycle 2010;9:3012-21.
84. Scheller J, Rose-John S. Interleukin-6 and its receptor: from bench to bedside. Med Microbiol Immunol 2006;195:173-83.
85. Trikha M, Corringham R, Klein B, Rossi JF. Targeted anti-interleukin-6 monoclonal antibody therapy for cancer: a review of the rationale and clinical evidence. Clin Cancer Res 2003;9:4653-65.
86. Guo Y, Xu F, Lu T, Duan Z, Zhang Z. Interleukin-6 signaling pathway in targeted therapy for cancer. Cancer Treat Rev 2012;38:904-10.
87. Ginestier C, Liu S, Diebel ME, Korkaya H, Luo M, Brown M, Wicinski J, Cabaud O, Charafe-Jauffret E, Birnbaum D, Guan JL, Dontu G, Wicha MS. CXCR1 blockade selectively targets human breast cancer stem cells in vitro and in xenografts. J Clin Invest 2010;120:485-97.
88. Hartman ZC, Poage GM, den Hollander P, Tsimelzon A, Hill J, Panupinthu N, Zhang Y, Mazumdar A, Hilsenbeck SG, Mills GB, Brown PH. Growth of triple-negative breast cancer cells relies upon coordinate autocrine expression of the proinflammatory cytokines IL-6 and IL-8. Cancer Res 2013;73:3470-80.
89. Burroughs SK, Kaluz S, Wang D, Wang K, Van Meir EG, Wang B. Hypoxia inducible factor pathway inhibitors as anticancer therapeutics. Future Med Chem 2013;5:553-72.
90. Batchelor TT, Duda DG, di Tomaso E, Ancukiewicz M, Plotkin SR, Gerstner E, Eichler AF, Drappatz J, Hochberg FH, Benner T, Louis DN, Cohen KS, Chea H, Exarhopoulos A, Loeffler JS, Moses MA, Ivy P, Sorensen AG, Wen PY, Jain RK. Phase II study of cediranib, an oral pan-vascular endothelial growth factor receptor tyrosine kinase inhibitor, in patients with recurrent glioblastoma. J Clin Oncol 2010;28:2817-23.
91. Pan E, Yu D, Yue B, Potthast L, Chowdhary S, Smith P, Chamberlain M. A prospective phase II single-institution trial of sunitinib for recurrent malignant glioma. J Neurooncol 2012;110:111-8.
92. Brossa A, Grange C, Mancuso L, Annaratone L, Satolli MA, Mazzone M, Camussi G, Bussolati B. Sunitinib but not VEGF blockade inhibits cancer stem cell endothelial differentiation. Oncotarget 2015;6:11295-309.
94. Krishnamurthy S, Ke X, Yang YY. Delivery of therapeutics using nanocarriers for targeting cancer cells and cancer stem cells. Nanomedicine (Lond) 2015;10:143-60.
95. Gupta PB, Onder TT, Jiang G, Tao K, Kuperwasser C, Weinberg RA, Lander ES. Identification of selective inhibitors of cancer stem cells by high-throughput screening. Cell 2009;138:645-59.
96. Malhi S, Gu X. Nanocarrier-mediated drugs targeting cancer stem cells: an emerging delivery approach. Expert Opin Drug Deliv 2015;12:1177-201.
97. Shen S, Xia JX, Wang J. Nanomedicine-mediated cancer stem cell therapy. Biomaterials 2016;74:1-18.
98. Zhao Y, Alakhova DY, Kabanov AV. Can nanomedicines kill cancer stem cells? Adv Drug Deliv Rev 2013;65:1763-83.
99. Bertrand N, Wu J, Xu X, Kamaly N, Farokhzad OC. Cancer nanotechnology: the impact of passive and active targeting in the era of modern cancer biology. Adv Drug Deliv Rev 2014;66:2-25.
100. Liu Y, Rohrs J, Wang P. Advances and challenges in the use of nanoparticles to optimize PK/PD interactions of combined anti-cancer therapies. Curr Drug Metab 2014;15:818-28.
101. Swaminathan SK, Roger E, Toti U, Niu L, Ohlfest JR, Panyam J. CD133-targeted paclitaxel delivery inhibits local tumor recurrence in a mouse model of breast cancer. J Control Release 2013;171:280-7.
102. Vinogradov S, Wei X. Cancer stem cells and drug resistance: the potential of nanomedicine. Nanomedicine (Lond) 2012;7:597-615.
103. Xiao H, Li W, Qi R, Yan L, Wang R, Liu S, Zheng Y, Xie Z, Huang Y, Jing X. Co-delivery of daunomycin and oxaliplatin by biodegradable polymers for safer and more efficacious combination therapy. J Control Release 2012;163:304-14.
104. Kim YJ, Liu Y, Li S, Rohrs J, Zhang R, Zhang X, Wang P. Co-Eradication of Breast Cancer Cells and Cancer Stem Cells by Cross-Linked Multilamellar Liposomes Enhances Tumor Treatment. Mol Pharm 2015;12:2811-22.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Kim YJ, Siegler EL, Siriwon N, Wang P. Therapeutic strategies for targeting cancer stem cells. J Cancer Metastasis Treat 2016;2:233-42. http://dx.doi.org/10.20517/2394-4722.2016.26
AMA Style
Kim YJ, Siegler EL, Siriwon N, Wang P. Therapeutic strategies for targeting cancer stem cells. Journal of Cancer Metastasis and Treatment. 2016; 2: 233-42. http://dx.doi.org/10.20517/2394-4722.2016.26
Chicago/Turabian Style
Kim, Yu Jeong, Elizabeth L. Siegler, Natnaree Siriwon, Pin Wang. 2016. "Therapeutic strategies for targeting cancer stem cells" Journal of Cancer Metastasis and Treatment. 2: 233-42. http://dx.doi.org/10.20517/2394-4722.2016.26
ACS Style
Kim, YJ.; Siegler EL.; Siriwon N.; Wang P. Therapeutic strategies for targeting cancer stem cells. J. Cancer. Metastasis. Treat. 2016, 2, 233-42. http://dx.doi.org/10.20517/2394-4722.2016.26
About This Article
Special Issue
Copyright

Author Biographies

Data & Comments
Data


 Cite This Article 25 clicks
Cite This Article 25 clicks

 Like This Article 0
likes
Like This Article 0
likes






Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.