
A versatile method for enumeration and characterization of circulating tumor cells from patients with breast cancer

Abstract
Aim: To establish a standardized protocol for the isolation and enumeration of circulating tumor cells (CTCs) from peripheral blood of patients with metastatic breast cancer.
Methods: The protocol used tumor cells spiked in a lymphoid cell line with detection by flow cytometry and quantitative reverse transcription polymerase chain reaction (QRT-PCR). Cells of the human mammary cancer subtypes were spiked into Jurkat cells, which served as the lymphocyte designate in numbers from 10 to 500 per 105 Jurkat cells. This mixed population was probed for CD45, EpCAM, and pancytokeratin acquired from flow cytometry and characterized by microscopy. QRT-PCR was done for CK-19, MUC-1, EpCAM, and GAPDH. Validation was attained with blood samples from 22 patients with metastatic breast cancer and 20 healthy individuals.
Results: Flow cytometry could detect 1 breast cancer cell per 100,000 Jurkat cells, with similar detection levels in the breast cancer subtypes. Samples from patients with breast cancer showed a range of CTCs from 1-85 per 10 mL of blood. Quantitation of expression for EpCAM, CK-19, Muc-1, and Her2neu confirmed the presence of CTCs in 76% of samples.
Conclusion: Density gradient and immunomagnetic enrichment accomplished isolation of CTCs and quantitation was achieved using flow cytometry. Combined QRT-PCR and imaging further validated these findings, rendering a robust methodology.
Keywords
Introduction
Circulating tumor cells (CTCs) in peripheral blood has emerged as an important surrogate marker for prognosis of cancer.[1-4] Various studies have demonstrated the presence of CTCs in peripheral blood of patients and their association with tumor progression and metastatic development.[5-7] Reports have also shown that a change in CTCs number predicts response to therapy and can evaluate residual disease.[8-11] Hence, to establish CTC number and molecular characteristics, a necessary requirement is a feasible and suitable approach.
There are several technologies which have been developed and are available for CTC enrichment and detection as cited in literature.[12,13] The current standard method that has been widely used in large multicenter clinical trials world-wide and which continues to be preferred is the FDA-approved quantitative, semi-automated, Cell Search system,[14,15] which assesses 7.5 mL of blood. It is based on the epithelial cell adhesion molecule (EpCAM/CD326) conjugated immunomagnetic enrichment followed by microscopic imaging using positive immunostaining of cytokeratin (CK), negative immunostaining of leucocyte common antigen CD45, and DNA staining with DAPI.
The literature shows that almost 60% of studies on detection and enumeration of CTCs have preferred methods using reverse transcription polymerase chain reaction (RT-PCR) because of its sensitivity and low cost as compared to using the Cell Search technology. One such RT-PCR kit-based technology is the Adnagen assay.[16,17] This entails enrichment on the basis of epithelial and tumour-specific antigens, isolation of RNA, conversion to cDNA and subsequent amplification of tumour-associated genes and a control gene for normalization. Amplified cDNA samples are then analyzed on a chip system (Bioanalyzer) and simultaneously compared with positive controls of each gene provided in the kit.
Strategies using cell-imaging, quantitative RT-PCR (QRT-PCR)[18] and flow cytometric detection[19-21] for enumeration and characterization of CTC shave also been employed.
The purpose of this study was to devise a feasible, relatively low-cost methodology which would enumerate CTCs as well as validate their assessment. The protocol for enumeration was developed using tumor cells from cell lines of breast cancer subtypes spiked in a lymphoid cell line and its quantitation by flow cytometry. Cells of human mammary cancer subtypes were spiked into a Jurkat cell line which served as the lymphocyte designate in numbers from 10 to 500 per 105 Jurkat cells. This mixed population was then probed for CD45, EpCAM, and pancytokeratin acquired on a flowcytometer and quantitated. Similar spiking experiments were done for QRT-PCR and expression of CK-19, MUC-1, EpCAM, and GAPDH. The methods described by others[22,23] have been adapted for quantitation of CK 19, EpCAM, MUC-1, and Her2neu. Data pertaining to these methods have been generated and any one of these methods can be used. Our method will overcome the differences in expression of the selected genes across clinical samples, estimate copies/mL in blood samples and enable direct comparison of samples across time points of collection. Thus, besides characterization, a quantitative measure of gene expression translating to numbers of CTCs can be obtained.
This methodology was substantiated with blood samples obtained from patients and healthy normal individuals. Blood from 22 patients with metastatic breast cancer and 20 healthy individuals was separated on a Ficoll gradient to obtain the peripheral blood mononuclear cells (PBMCs) fraction, positively enriched for EpCAM, quantitatedby flow cytometry and validated with QRT-PCR for the presence of CTCs.
Methods
Materials
DMEM, RPMI 1640, Fetal Calf Serum (FCS), Trizol, High capacity cDNA kit, TaqMan assays for CK-19, EpCAM, MUC1, Her2neuand GAPDH, 2 X TMM, and PBS were procured from Invitrogen Life Technologies (Carlsbad, CA, USA). RNeasy plus micro kit was from Qiagen, GmBh, D-40724, Hilden, Germany. FcR blocking reagent, CD326 (EpCAM) Microbeads, MACS BSA Stock Solution, rinsing buffer, and washing buffer were purchased from Miltenyi Biotec GmBH, Germany. Ficoll-Paque PLUS was procured from GE Healthcare, Bio-Sciences AB, Uppsala, Sweden. EDTA vacutainer tubes were from Greiner Bio-One, Austria. All other chemicals were obtained locally and were of analytical grade.
Antibodies
CD326 (EpCAM) antibody conjugated to APC (clone HEA-125), pan-Cytokeratin antibody conjugated to FITC (clone CK3-6H5), CD45 antibody conjugated to PerCP (clone 30F11.1), mouse IgG1 isotype control antibodies (clone-IS5-21F5) conjugated to FITC, APC, and PerCP were purchased from Miltenyi Biotec GmBH, Bergisch-Gladbach, Germany.
Buffers
Separation buffer composition: phosphate-buffered saline (PBS; pH 7.2), 5% bovine serum albumin (BSA), 2 mmol/L EDTA prepared by diluting MACS BSA Stock Solution with auto MACS™ rinsing solution (1:20); FACS buffer contains 0.02% NaN3, 1% FCS in PBS (pH 7.4); Saponin buffer for permeabilization contained 0.1% saponin in FACS buffer.
Cell lines
Human mammary carcinoma cell lines: MCF-7, T-47D, ZR75-1, BT-474, and MDA-MB-468 and the lymphoid cell line, Jurkat, were used for cell spiking and standardization of the flow cytometry assay, and qPCR. MCF-7, BT-474, and MDA MB-468 were maintained in DMEM containing 10% FCS; T-47-D and ZR75-1 in RPMI-1640 with L-glutamine (2 mmol/L) and 10% FCS; Jurkat in DMEM containing 10% FCS and antibiotic mixture containing gentamycin, streptomycin and anti-fungal, Forcan. Cell line ZR75-1 was purchased from the National Centre for Cell Science, Pune, India. All other cell lines were available in the institute or laboratory and procured for ongoing studies.
Patients and controls
A total of 22 breast cancer patients with metastatic disease were included in this study. In addition, blood samples from 20 healthy volunteers were accrued. After collection, blood samples were immediately processed for isolation and detection of CTCs.
Blood collection and PBMCs isolation
Peripheral blood (25 mL) was drawn by phlebotomy in EDTA vacutainer tubes. The initial 5 mL was routed for other routine tests to avoid contamination with cells from the skin and blood vessels. Tumor cells were isolated along with the PBMCs on a Ficoll Hypaque gradient. Blood diluted with PBS was layered over Ficoll Hypaque and centrifuged at 400 g for 30 min at 25 °C. The interphase was collected, washed twice, with separation buffer, spun at 400 g for 15 min at 4 °C to obtain a cell pellet, which was suspended in separation buffer and the total yield and viability was assessed.
Immunomagnetic enrichment and CTCs isolation
CTCs were enriched from the total cell pellet. Depending on the total cell count, initially 50-100 µL of FcR blocking reagent (Miltenyi Biotec) was added to block Fc receptors and eliminate non-specific binding. Subsequently, 50-100 µL of micro beads conjugated to monoclonal antibody for EpCAM (Miltenyi Biotec) was added and this mixture was incubated for 30 min at 4 °C. Subsequently, cells were washed and re-suspended in 700 µL of separation buffer. This labeled cell suspension was then acquired on autoMACS Pro® Separator (Miltenyi Biotec) with a double-positive selection. The positive fraction constituted cells that expressed EpCAM and thus was an enriched fraction of EpCAM-expressing cells. The negative fraction constituted lymphocytes, cells that do not express EpCAM and flow through the column. The positive and negative fractions were divided into two aliquots: one for multi-parameter flow cytometry, the other for quantitative RT-PCR.
Cell spiking experiments
Human mammary cancer cell lines representing the molecular subtypes were grown as adherent cultures and harvested using sterile trypsin-EDTA solution at 80% confluency. The Harvested cells were collected, washed, and re-suspended in a fixed volume of culture medium. An average of 3 cell counts and viability were determined by the trypan blue dye exclusion method. This cell suspension was used for serial dilution in cell spiking experiments and cells were spiked into the Jurkat cell line, which served as the WBC designate of the PBMC fraction. Jurkat cell number was selected on the basis of the average cell count obtained in the positive fraction following immunomagnetic enrichment of 20 mL of peripheral blood, which is approximately 1 × 105 cells counted with a hemocytometer. Human mammary tumor cells were spiked into 1 × 105 Jurkat cells at levels ranging from 0.5% to 0.001%. These cell suspensions were labeled with antibodies as described below, acquired on the flowcytometer and the percentage expected to the percentage recovered was plotted as shown below.[24] For each sub-type, experiments were repeated at least thrice.
Antibody labeling and acquisition on flow cytometer
Serially diluted samples of tumor cells spiked into Jurkat cells were surface-stained with monoclonal antibodies that target epithelial cell antigens EpCAM (CD326) and CD45 or the corresponding isotype control antibody by incubating the cells in 50 µL of FACS buffer for 30 min in the dark at room temperature. Cells were then washed with FACS buffer and fixed with 1% paraformaldehyde for 15 min at 4 °C before permeabilization for intracellular staining. To permeabilize cells, the pellet was resuspended in 0.1% saponin buffer. Cells were subsequently stained with anti-pan CK antibody or the relevant isotype control and incubated for 45 min in the dark at room temperature. After staining, cells were washed with saponin buffer, resuspended in 300 µL of FACS buffer, and immediately acquired on a FACS AriaTM flowcytometer (Becton Dickinson, USA), which is equipped with a 488 nm blue laser (for excitation of FITC and PerCP) and a 633 nm red laser (for excitation of APC) and the following filters: FITC: 530 nm band pass; PerCP: 670 nm long pass; APC: 660 nm band pass. Setup and automatic compensation were performed using cells stained for each marker. PMT voltages used for recording fluorescence signals were as follows: FITC = 429, PerCP = 599, APC = 396. The absolute number of spiked tumor cells was estimated by acquiring a minimum of 10,000 events in the analyzed sample. Data were analyzed with BD FACSDivaTM software v6.1.3 (BD Biosciences).
RNA isolation and cDNA synthesis
A Trizol/RNeasy hybrid RNA extraction protocol was used for isolation of total RNA under RNase-free conditions. The aqueous phase obtained was passed through a gDNA eliminator column to remove genomic DNA contamination. Total RNA isolated from each sample was suspended in RNAse-free water. Concentration and purity were determined on Nanodrop ND-1000 and stored at -80 °C until further use. cDNA synthesis was carried out with high capacity reverse transcriptase kit (Applied Biosystems) according to manufacturer’s instructions.
Preparation of qRT-PCR calibrators
QRT-PCR calibrators were prepared adapting the Aerts et al.[22] method with selection of the average cell number obtained in the positive fraction post-immunomagnetic enrichment from patient samples. Serial dilutions corresponding to five log steps of cell number by diluting RNAzol lysates of MCF-7 in the WBC cell line (Jurkat) with ratios of 1:1 to 1:104 of CK19+ and EpCAM+ cells per CK19- and EpCAM- cells were made. Dilution of lysates was performed to avoid the need for permanent maintenance of cell cultures for preparation of cell-cell dilutions. RNA extraction and cDNA synthesis were performed as described. Each sample was measured in triplicate.
We also prepared qRT-PCR calibrators by adapting the Strati et al.[23] method as a qRT-PCR requires analysis of samples across time frames. Individual PCR amplicons corresponding to gene-targets CK-19, EpCAM, MUC-1, and Her2Neu that could serve as quantification calibrators were generated. For this purpose, total RNA was extracted from MCF-7 and BT474 cells, DNase-treated, and quantified. cDNA was synthesized from 1 µg of DNase-treated RNA using a high capacity cDNA kit according to manufacturer’s instructions. PCR for each target gene was carried out with TaqMan primers on a Bio-Rad Peltier Thermal cycler to ensure production of calibrators from the same amplicon that would be synthesized during the qRT-PCR of clinical samples. PCR amplicons were separated on a 3% Agarose gel, excised under UV, purified using QIAquickGel Extraction Kit (Qiagen), and quantified on Nanodrop. Copy number was calculated from the concentrations using Avogadro constant and molecular weight of each amplicon number of bases of the PCR product multiplied by the mean molecular weight of a pair of nucleic acids, which is 660. Serial dilutions of these stock amplicons were made in TE buffer (1 × 106 copies to 1 × 100 copies) and used as quantification calibrators throughout the study. For quantification of a gene transcript, a TaqMan quantitative PCR assay was done for the gene transcript for every dilution in duplicate and an external calibration curve was obtained by plotting the concentration as copy number vs. the corresponding threshold cycle.[25] Each of these calibration curves were repeated at least thrice.
Quantitative PCR
TaqMan assays were done for cytokeratin-19 (CK19), epithelial cell adhesion molecule (EpCAM), Mucin1 (MUC1), Her2-neu, and Glyceraldehyde-3-phosphate dehydrogenase (GAPDH). Reactions for all the above genes were performed with 10 µL volume with cDNA corresponding to 20 ng. All the samples were analyzed in duplicates and the average value of the two was used as the quantitative value. A non-template control, also in duplicate, was used for all genes.
Statistical analysis
For all groups studied, values were represented as mean ± SD. A simple linear regression model was used to assess the fit for the recovery of expected cells.
Results
Flow cytometry for CTCs analysis
A tricolor setup was configured to detect the CD45 PerCP signal in 670 nm long pass, EpCAM-APC signal in 660 nm bandpass, and Cytokeratin-FITC signal in 530 nm band pass filter. Events that fell within the region P2, i.e. EpCAM, and CK dual-positive were counted as meeting criteria for human tumor cells [Figure 1A and B]. Thresholds for specific EpCAM APC and CK FITC signals were determined using the sample stained with isotype control antibodies. The same gating strategy was then applied for detecting EpCAM+CK+CD45- cells in the clinical sample stained with the specific antibodies.
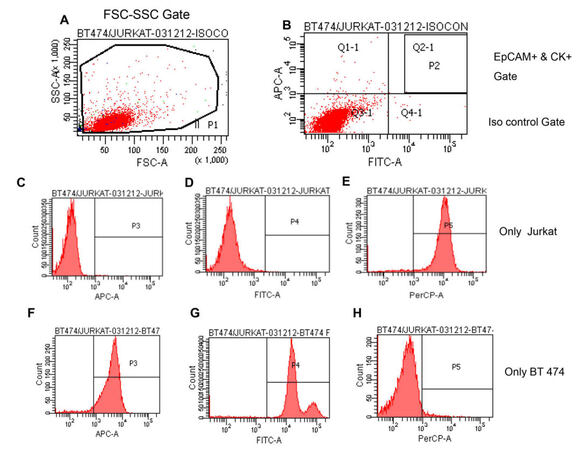
Figure 1. EpCAM+CK+CD45- tumor cells detected by flow cytometry by first gating out cell debris and cell clumps in forward/side scatter plot (A); threshold for specific EpCAM (APC) and CK (FITC) signals determined using sample stained with isotype control antibodies. Gates set to have no positive events above these thresholds in control sample (B); representative histograms shown of individual criteria gated on (C-E) Jurkat cells and (F-H) BT-474 human tumor cells
Specificity (ability to differentiate between epithelial tumour cellsand WBC designate)
EpCAM-APC (clone HEA-125) and Pan-Cytokeratin-FITC (clone CK3-6H5) Abs were found to be specific for human mammary cancer cells, with no non-specific binding to Jurkat cells [Figure 1C-E]. Similarly, human CD45 PerCP antibody (clone 30F11.1) was found to be highly specific for leukocytes and did not stain human mammary tumour cells [Figure 1F-H]. To determine applicability of the method to various human mammary cancer cell lines, the ability to detect MCF-7, T-47D, ZR-75-1, BT-474, and MDA-MB-468 spiked in Jurkat cells was tested. In all cases, tumor cells exhibited similar staining to EpCAM and CK antibodies and could be clearly differentiated from Jurkat cells.
Sensitivity (ability to detect lowest number of epithelial tumor cells)
The assay of serial dilutions (0.001-0.5%) of human breast tumor cells in Jurkat cells established that the lower detection limit for sensitivity of the method was 0.001% or 10-5, corresponding to 1 human cell per 100,000 Jurkat cells. Recovery and linearity were highly reproducible across separate experiments [Figure 2] and the number of tumor cell events could be positively correlated.Linear correlation and regression analysis showed R2 > 0.98 for ZR-75-1, BT-474, and MDA-MB-468 cell lines, whereas in the case of MCF-7 and T-47D, R2 was 0.9465 and 0.9589, respectively [Supplementary Figure 1]. The percentage of tumor cells recovered was not significantly different from the percentage of tumor cells expected, based on serial dilutions.
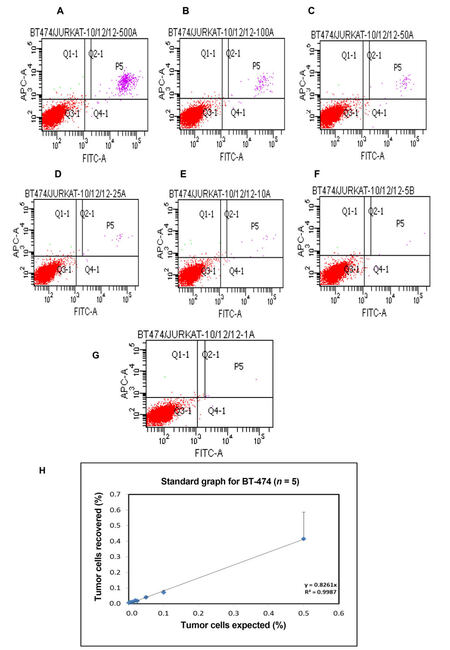
Figure 2. Enumeration of circulating tumor cell in spiking experiments. Representative BT-474 cells shown for (A) 0.5%, (B) 0.1%, (C) 0.05%, (D) 0.025%, (E) 0.01%, (F) 0.005%, (G) 0.001%; (H) Correlation and regression analysis of recovered versus expected number of positive tumor events at different dilutions
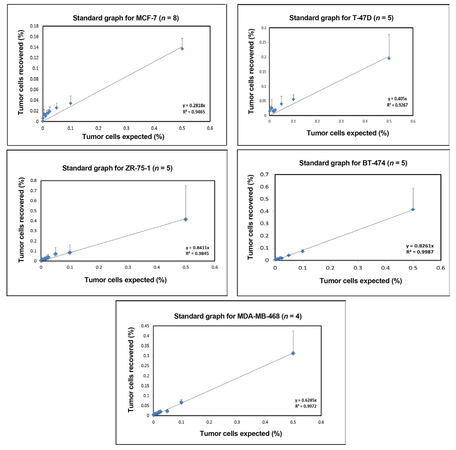
Supplementary Figure 1. Correlation and regression analysis of recovered versus expected number of positive tumour events at different dilutions
Recovery
Recovery of the flow cytometry protocol was determined by spiking 1-500 tumor cells in 1 × 105 Jurkat cells. The gating strategy for detection of EpCAM+CK+CD45- cells is as shown in Figure 1A and B. Recovery of spiked cells was highly linear and revealed a mean recovery of 75% of spiked tumor cells (range: 43-100%), as shown in Table 1.
Recovery of spiked cancer cells as determined by flow cytometry analysis (%)
| MCF-7 | T47 D | ZR 75-1 | BT 474 | MDA MB 468 | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Tumor Cells Expected | Mean tumor cells recovered | Average recovery | Mean tumor cells recovered | Average recovery | Mean tumor cells recovered | Average recovery | Mean tumor cells recovered | Average recovery | Mean tumor cells recovered | Average recovery |
| 0.001 | 0.00575 | > 100 | 0.006 | > 100 | 0.0078 | > 100 | 0.006 | > 100 | 0.00575 | > 100 |
| 0.005 | 0.0065 | > 100 | 0.015 | > 100 | 0.016 | > 100 | 0.008 | > 100 | 0.00825 | > 100 |
| 0.01 | 0.00975 | 97.5 | 0.01433 | > 100 | 0.0178 | > 100 | 0.01 | 100 | 0.01025 | ≈ 100 |
| 0.015 | 0.01475 | 98.3 | 0.015 | 100 | 0.0262 | > 100 | 0.014 | 93 | 0.01225 | 81.6 |
| 0.02 | 0.01575 | 78.75 | 0.014 | 70 | 0.0328 | > 100 | 0.0198 | 99 | 0.01825 | 91.25 |
| 0.025 | 0.01875 | 75 | 0.019 | 76 | 0.0402 | > 100 | 0.0173 | 69 | 0.0205 | 82 |
| 0.05 | 0.02571 | 51.4 | 0.049 | 99 | 0.0726 | > 100 | 0.04 | 80 | 0.023 | 46 |
| 0.1 | 0.034125 | 34.13 | 0.0596 | 60 | 0.085 | 85 | 0.072 | 72 | 0.06725 | 67.25 |
| 0.5 | 0.137 | 27.4 | 0.216 | 43 | 0.415 | 83 | 0.415 | 83 | 0.3135 | 62.7 |
Analysis of CTCs in patients
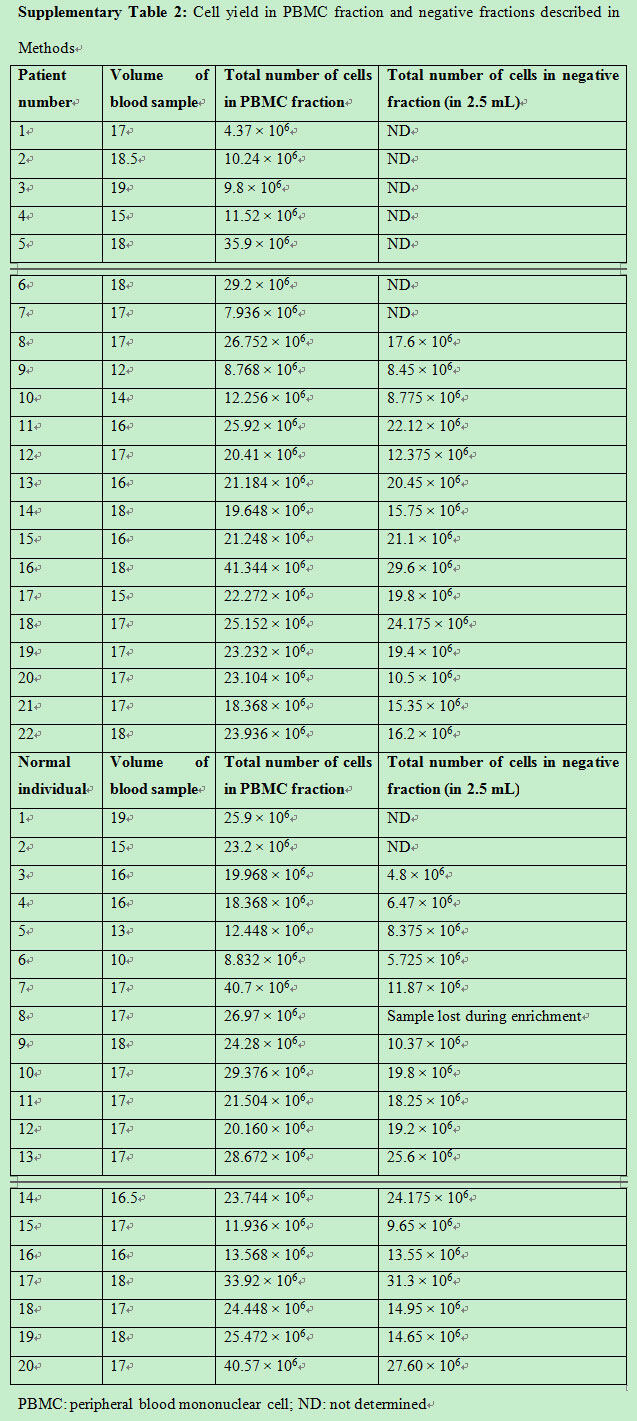
To validate the use of this technique in a clinical setting, 17 patient samples and 13 normal controls were assessed. As described in Methods, the PBMC fraction was separated from peripheral blood sample with an average PBMC cell count of 20.2 × 106 ± 9 × 106 cells for patient samples and 23.7 × 106 ± 8.6 × 106 cells in normal controls [Supplementary Figure 2]. CTCs were enriched from the PBMC fraction using immunomagnetic double-positive selection for EpCAM. The number of cells in the positive fraction represented cells that were enriched for EpCAM and, as the number was very low, the hemocytometer count would not be an appropriate representation. CTCs would be detected in this fraction. Average cell number obtained in the negative fraction (as described in Methods) for the patient sample was 17.3 × 106 ± 5.7 × 106 cells and in the normal control was 15.6 × 106 ± 8.03 × 106[Supplementary Figure 2 and Supplementary Table 2]. The pre-enrichment fraction, positive and negative fractions, and controls were acquired and analyzed by flow cytometry. CTCs were selected on the basis of cell size and presence of EpCAM and CK. Figure 3 and Table 2 show the number of CTCs identified in patient samples and normal controls. Double-positive cells (CK+, EpCAM+) were CTCs obtained from the positive-enriched fraction from patient blood samples and normal controls. The positive enriched fraction was split equally to be analyzed by flow cytometry and quantitative RT-PCR. Patient blood samples showed a range of 1-85 per 10 mL of blood, with an average of 23.35 ± 22.85, and for normal controls the range was 0-14 per 10 mL of blood, with an average of 5 ± 4.
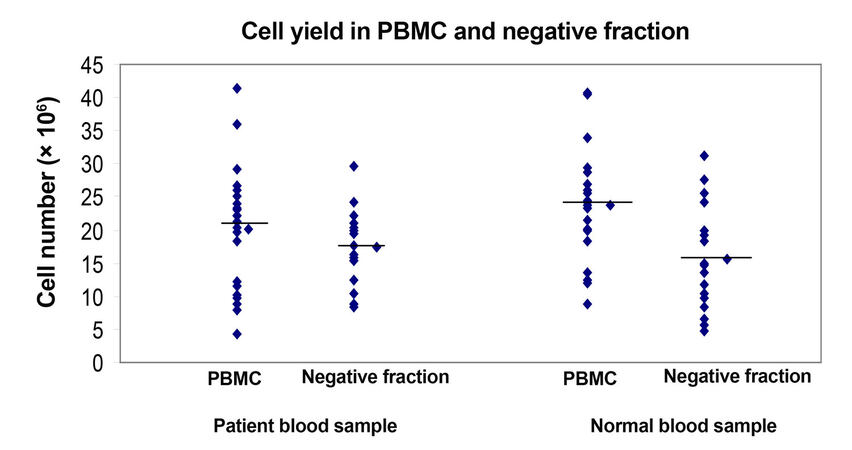
Supplementary Figure 2. Total cell yield in PBMCs fraction obtained from patients and normal individuals and cell number in negative fraction after positive enrichment selection of EpCAM positive cells as assessed by trypan blue staining. PBMC: peripheral blood mononuclear cell
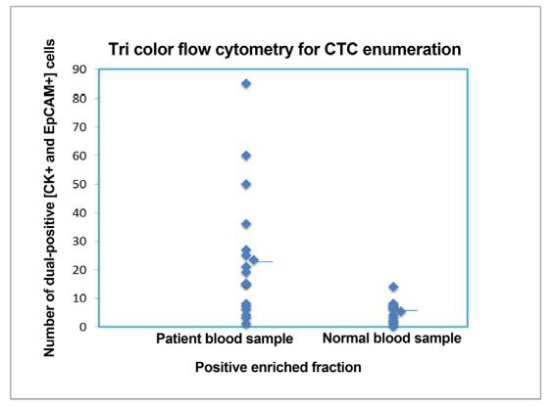
Figure 3. Tri color flow cytometry for CTC enumeration. CK+/EpCAM+ dual-positive cells represent CTCs seen in positive enriched fraction. The number of dual-positive cells in patient and normal healthy individuals is shown. CTC: circulating tumor cell
Tricolor flow cytometry for enumeration of CTC done as described in Methods (complements Figure 3)
| Patient number | Number of dual positive cells [EpCAM+ CK+] CTCs |
|---|---|
| 1 | 25 |
| 2 | 36 |
| 3 | 85 |
| 4 | 60 |
| 5 | 15 |
| 6 | 15 |
| 7 | 27 |
| 8 | 21 |
| 9 | 50 |
| 11 | 7 |
| 13 | 1 |
| 14 | 3 |
| 15 | 4 |
| 16 | 8 |
| 17 | 15 |
| 20 | 19 |
| 22 | 6 |
| For 5 patients only RT-PCR no flow | |
| Normal individual number | Number of dual positive cells [EpCAM + CK] |
| 1 | 8 |
| 2 | 4 |
| 3 | 14 |
| 4 | 7 |
| 5 | 7 |
| 6 | 3 |
| 7 | 8 |
| 9 | 0 |
| 10 | 1 |
| 17 | 2 |
| 18 | 6 |
| 19 | 7 |
| 20 | 2 |
| For 6 normal individuals only RT-PCR done | |
| Flow cytometry not done | |
| One sample lost during enrichment |
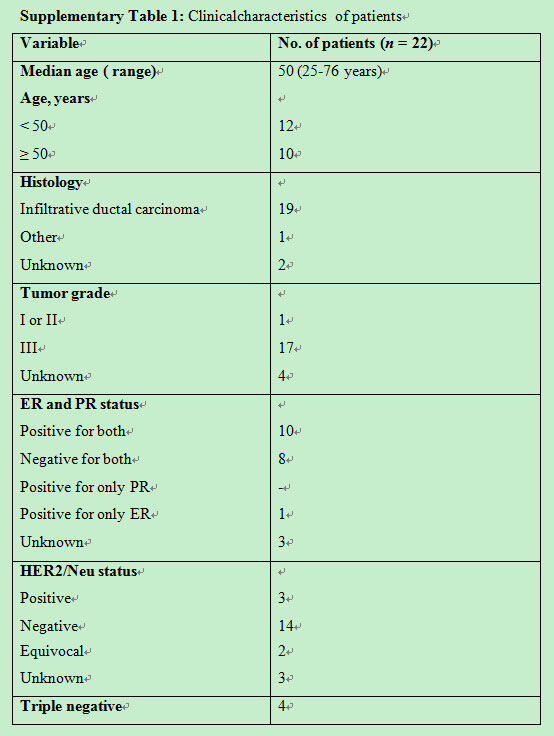
Clinical characteristics of patients are as described in Supplementary Table 1. Hormone receptor status showed that 10 (45%) were ER-positive, 6 (27%) ER-negative an equal number of 9 (41%) were PR-positive and PR-negative, 3 (14%) were Her2-positive, and 13 (60%) were Her2-negative.
Clinicalcharacteristics of patients
 |
Cell yield in PBMC fraction and negative fractions described in Methods
 |
Quantitative PCR
Quantification using cell line dilution series
Adapting the Aerts et al.[22] method, a standard curve of ∆Ct values was generated from the dilution series, as described in Methods, to determine the number of circulating tumor cells in a clinical blood sample by interpolation from this standard curve. This curve displayed a linear relationship between ∆Ct values and the logarithm of marker-positive. As seen in Figure 4 and Supplementary Figure 3A and B, standard curves were generated for CK-19, EpCAM, and MUC1 using breast cancer cell lines T47D, MCF-7, and ZR-75-1. All Ct values were normalized to the values obtained from Taqman PCR for GAPDH as a “house-keeping” gene to overcome differences of efficiency of cDNA synthesis byformulae ∆Ct = Ct (CK-19/EpCAM/ MUC1)-Ct (GAPDH). Table 2 shows the cell numbers obtained by this method in patients with EpCAM, CK-19, and MUC-1.
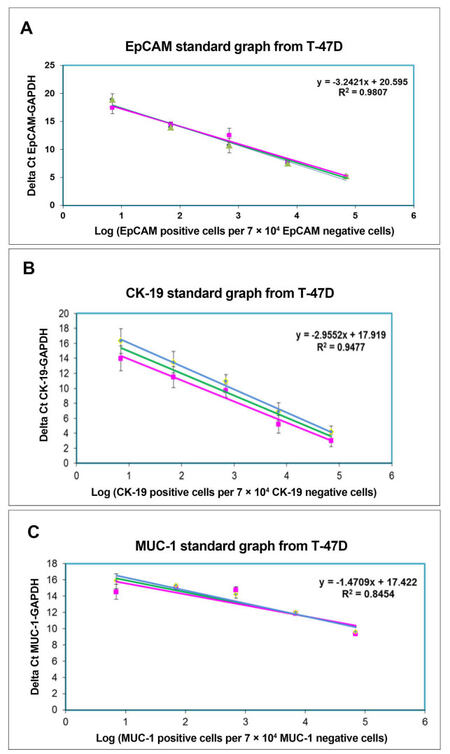
Figure 4. Standard curve for Aerts et al.[22] method for T47-D cell line. (A) Log number of EpCAM-positive cells versus Delta Ct (EpCAM-GAPDH); (B) log number of CK-19-positive cells versus Delta Ct (CK-19-GAPDH); (C) log number of MUC-1-positive cells versus Delta Ct (MUC-1-GAPDH)
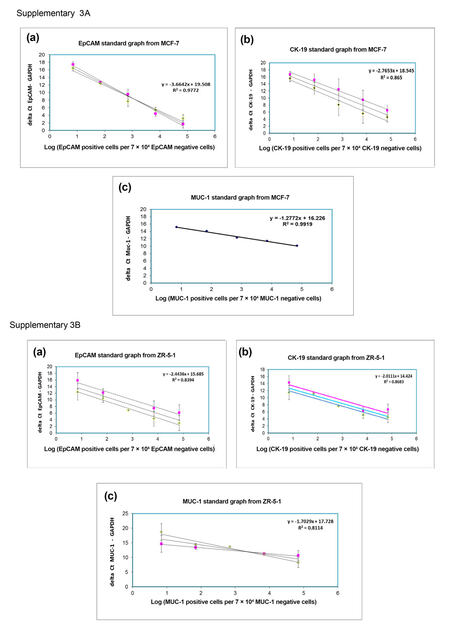
Supplementary Figure 3. (A) Standard curve for Aerts et al.[22] method for MCF-7 cell line. (a) Log number of EpCAM-positive cells versus Delta Ct (EpCAM-GAPDH); (b) log number of CK-19-positive cells versus Delta Ct (CK-19-GAPDH); (c) log number of MUC-1-positive cells versus Delta Ct (MUC-1-GAPDH); (B) standard curve for Aerts et al.[22] method for ZR-75-1 cell line. (a) Log number of EpCAM-positive cells versus Delta Ct (EpCAM-GAPDH); (b) log number of CK-19-positive cells versus Delta Ct (CK-19-GAPDH); (c) log number of MUC-1-positive cells versus Delta Ct (MUC-1-GAPDH)
Quantification using concentration (copy number) of the marker transcript
Based on the Strati et al.[23] method, we developed a CTC gene expression qRT-PCR assay by using quantification calibrators for four gene transcripts (CK-19, EpCAM, MUC1, Her2) containing a known number of copies, prepared as described in Methods. These were synthesized as described and aliquoted to eliminate experimental variation. The basis for adopting this method was to enable comparisons of CTC analysis as copy number in clinical samples collected and analyzed at different time points, and also to allow for the possibility of a low detection limit such as 1 copy. Figure 5 shows the calibration curve for the four selected transcripts in the range from 106 copy number to 1 copy, with a linearity observed from 106 copies to 10 copies with correlation coefficient close to 1 (0.99), indicating a precise log-linear relationship.
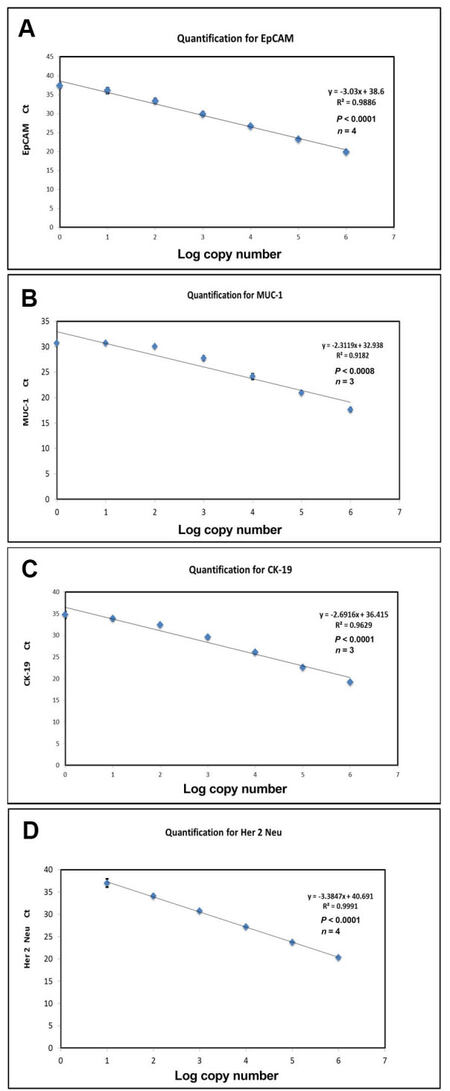
Figure 5. Standard curve with Strati et al.[23] method done as described in the text. (A) Log number of EpCAM copies versus EpCAM Ct values; (B) log number of CK19 copies versus CK19 Ct values; (C) log number of Muc-1 copies versus Muc-1 Ct values; (D) log number of Her-2 copies versus Her-2 Ct values
CTCs in patients and controls
Quantitative PCR was done with 17 patient samples and 15 controls. These were probed for at least three genes: EpCAM, CK-19, GAPDH. Her2 and MUC 1 were assessed in a few samples subject to the IHC report and availability of cDNA [Table 2]. All patients as well as control samples showed Ct values lower than 30 for GAPDH. From the 17 patient samples, 14 (82%) gave Ct values below 36. Of the 14, 9 (64%) were found to be positive for two marker genes, 10 (71.4%) for EpCAM, 9 (64%) for CK19, and 6 (43%) for Muc-1. There were two samples that were positive for all the four markers, with a high copy number recorded for Her2. Her2 expression levels were comparable to protein levels obtained by IHC, which was reported as 3+. In the 15 control samples, 4 recorded Ct values that could be converted to copies/mL and these values were below 1.
Imaging CTCs
In blood samples from patients, where CTCs were higher, captured cells were subjected to morphological characterization and examined for presence of a nucleus. Figure 6 shows representative images of two CTCs characterized as EpCAM and CK positive, with a well-defined nucleus and were negative for the leucocyte antigen, CD45. Imaging further confirmed the presence of CTCs in samples.
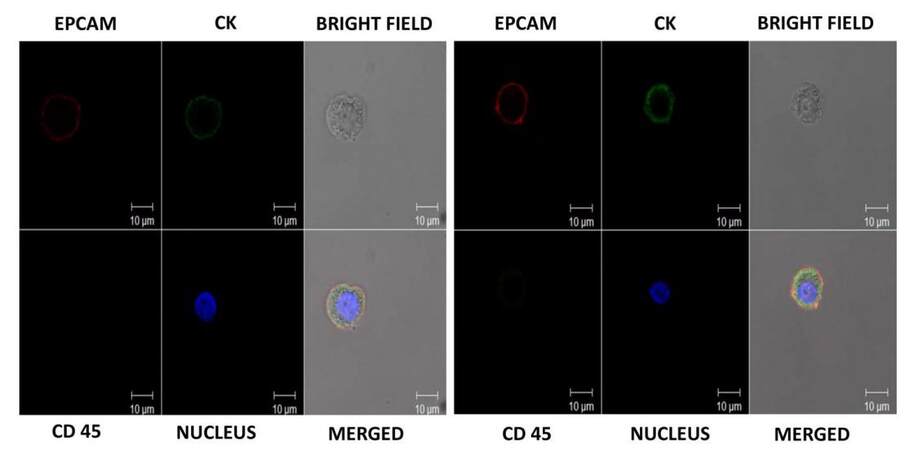
Figure 6. Images of captured CTCs from blood sample of a patient with breast cancer. CTCs are positive for EpCAM and CK with the presence of a well-defined nucleus and negative for the common leucocyte antigen CD45. CTCs: circulating tumor cells; CK: cytokeratin
Discussion
We propose a workable method for the isolation and enumeration of CTCs wherein a two-tier protocol of cell isolation with an initial separation based on density gradient centrifugation, followed by EpCAM immunomagnetic-positive double enrichment has been described and adopted. The enriched fraction of tumour cells is further divided, one analyzed for enumeration of CTCs using flow cytometry based on large size of tumor cells, with the presence of CK, EpCAM, and the absence of CD45. Dissimilar expression levels of EpCAM could compromise the detection of CTCs,[26,27] hence, the initial standardization for flow and QRT-PCR analysis was done with cell spiking of cancer cells from different breast cancer subtypes to assess possible differences. Tumor cells from the different subtypes exhibited similar staining to EpCAM and CK antibodies and could be clearly distinguished from Jurkat cells. As described for serial dilutions [Figure 2] recovery and linearity showed reproducibility and were highly consistent across independent experiments. A positive correlation was observed between recovered tumor events and expected tumor events. Based on the serial dilution assay, the percentage of tumor cells recovered was not significantly different from the percentage of tumor cells expected.
The flow cytometry protocol was validated with blood samples obtained from patients with metastatic breast cancer. Eighteen of these patients were with tumor grade II-III and 17 were diagnosed with verified metastasis. Clinical characteristics are as described in Supplementary Table 1. Median age was 50 years (range: 25-76 years). All, except two, were diagnosed as invasive ductal carcinoma, the most common type of breast cancer. Hormone receptor status showed 10 (45%) were ER-positive, 6 (27%), ER-negative, equal number 9 (41%) PR-positive and PR-negative, 3 (14%) Her2-positive, and 13 (60%) Her2-negative. CTCs showed a range of 1-85 per 10 mL of blood, with an average of 23.35 ± 22.85. Twenty healthy women volunteers were also included in this validation, with values ranging from 0-14 per 10 mL of blood, with an average of 5 ± 4. A cut-off of 10 and above has been selected, based on these results.
For clinical samples where CTCs were higher, captured cells were subjected to morphological characterization and examined for presence of a nucleus. Figure 6 shows representative images of two CTCs characterized as EpCAM and CK positive, with a well-defined nucleus and negative for the leucocyte antigen, CD45. Immunophenotyping and imaging further confirmed the presence of CTCs in samples.
Determination by quantitative RT-PCR for CK 19, EpCAM, and MUC-1 in the enriched fraction was also a specific approach which was applied in validating CTCs numbers obtained by flow cytometry. Further, to overcome representation of results as expression in relative fold and to show absolute quantitation, the Strati et al.[23] and Aerts et al.[22] methods were adapted. These methods can overcome differences in expression of the selected genes across samples, estimate the quantities as copies/mL in blood samples, and enable direct comparison of samples across time points of collection.
Sixty-four percent of patient samples showed concordance with flow and RT-PCR evaluation of EpCAM and CK. The lower number was because all samples were not analyzed by both methods and five for each group were analyzed by only one method.
We have developed a protocol that is technically feasible and economically viable in the laboratory settings for the study of CTCs. In the literature presence of CTCs has been correlated with poor prognosis as well as progression-free survival, and CTCs have been identified as indicators of treatment efficacy in different tumours.[6,28-30] Their role in the metastatic process is endorsed by several studies, yet their clinical use for tumour staging, disease monitoring, and choice of treatment seems a distant reality. Never the less, monitoring CTCs has the potential to gauge the extent of disease and serve as a liquid biopsy. Attempts to formulate economically viable and feasible protocols become an essential requisite.
Financial support and sponsorship
None.
Conflicts of interest
There are no conflicts of interest.
Patient consent
The patient consent was obtained for all patients participating in this study.
Ethics approval
Blood samples collected were from an approved study and all the human subjects participating in the study have given the required informed consent.
REFERENCES
1. Botteri E, Sandri MT, Bagnardi V, Munzone E, Zorzino L, Rotmensz N, Casadio C, Cassatella MC, Esposito A, Curigliano G, Salvatici M, Verri E, Adamoli L, Goldhirsch A, Nolè F. Modeling the relationship between circulating tumour cells number and prognosis of metastatic breast cancer. Breast Cancer Res Treat 2010;122:211-7.
2. Smerage JB, Budd GT, Doyle GV, Brown M, Paoletti C, Muniz M, Miller MC, Repollet MI, Chianese DA, Connelly MC, Terstappen LW, Hayes DF. Monitoring apoptosis and Bcl-2 on circulating tumor cells in patients with metastatic breast cancer. Mol Oncol 2013;7:680-92.
3. Lucci A, Hall CS, Lodhi AK, Bhattacharyya A, Anderson AE, Xiao L, Bedrosian I, Kuerer HM, Krishnamurthy S. Circulating tumour cells in non-metastatic breast cancer: a prospective study. Lancet Oncol 2012;13:688-95.
4. Bidard FC, Belin L, Delaloge S, Lerebours F, Ngo C, Reyal F, Alran S, Giacchetti S, Marty M, Lebofsky R, Pierga JY. Time-dependent prognostic impact of circulating tumor cells detection in non-metastatic breast cancer: 70-month analysis of the REMAGUS02 study. Int J Breast Cancer 2013;2013:130470.
5. Bidard FC, Peeters DJ, Fehm T, Nolé F, Gisbert-Criado R, Mavroudis D, Grisanti S, Generali D, Garcia-Saenz JA, Stebbing J, Caldas C, Gazzaniga P, Manso L, Zamarchi R, de Lascoiti AF, De Mattos-Arruda L, Ignatiadis M, Lebofsky R, van Laere SJ, Meier-Stiegen F, Sandri MT, Vidal-Martinez J, Politaki E, Consoli F, Bottini A, Diaz-Rubio E, Krell J, Dawson SJ, Raimondi C, Rutten A, Janni W, Munzone E, Carañana V, Agelaki S, Almici C, Dirix L, Solomayer EF, Zorzino L, Johannes H, Reis-Filho JS, Pantel K, Pierga JY, Michiels S. Clinical validity of circulating tumour cells in patients with metastatic breast cancer: a pooled analysis of individual patient data. Lancet Oncol 2014;15:406-14.
6. Rack B, Schindlbeck C, Jückstock J, Andergassen U, Hepp P, Zwingers T, Friedl TW, Lorenz R, Tesch H, Fasching PA, Fehm T, Schneeweiss A, Lichtenegger W, Beckmann MW, Friese K, Pantel K, Janni W; SUCCESS Study Group. Circulating tumor cells predict survival in early average-to-high risk breast cancer patients. J Natl Cancer Inst 2014;106.
7. Giuliano M, Giordano A, Jackson S, De Giorgi U, Mego M, Cohen EN, Gao H, Anfossi S, Handy BC, Ueno NT, Alvarez RH, De Placido S, Valero V, Hortobagyi GN, Reuben JM, Cristofanilli M. Circulating tumor cells as early predictors of metastatic spread in breast cancer patients with limited metastatic dissemination. Breast Cancer Res 2014;16:440.
8. Lorente D, Olmos D, Mateo J, Bianchini D, Seed G, Fleisher M, Danila DC, Flohr P, Crespo M, Figueiredo I, Miranda S, Baeten K, Molina A, Kheoh T, McCormack R, Terstappen LW, Scher HI, de Bono JS. Decline in circulating tumor cell count and treatment outcome in advanced prostate cancer. Eur Urol 2016;70:985-92.
9. Čapoun O, Mikulová V, Jančíková M, Honová H, Kološtová K, Sobotka R, Michael P, Zima T, Hanuš T, Soukup V. Prognosis of castration-resistant prostate cancer patients-use of the AdnaTest(R) system for detection of circulating tumor cells. Anticancer Res 2016;36:2019-26.
10. Agelaki S, Kalykaki A, Markomanolaki H, Papadaki MA, Kallergi G, Hatzidaki D, Kalbakis K, Mavroudis D, Georgoulias V. Efficacy of lapatinib in therapy-resistant HER2-positive circulating tumor cells in metastatic breast cancer. PLoS One 2015;10:e0123683.
11. Grande E, Capdevila J, Castellano D, Teulé A, Durán I, Fuster J, Sevilla I, Escudero P, Sastre J, García-Donas J, Casanovas O, Earl J, Ortega L, Apellaniz-Ruiz M, Rodriguez-Antona C, Alonso-Gordoa T, Díez JJ, Carrato A, García-Carbonero R. Pazopanib in pretreated advanced neuroendocrine tumors: a phase II, open-label trial of the spanish task force group for neuroendocrine tumors (GETNE). Ann Oncol 2015;26:1987-93.
12. Alix-Panabieres C, Pantel K. Challenges in circulating tumour cell research. Nat Rev Cancer 2014;14:623-31.
13. Harouaka R, Kang Z, Zheng SY, Cao L. Circulating tumor cells: advances in isolation and analysis, and challenges for clinical applications. Pharmacol Ther 2014;141:209-21.
14. Kagan M, Howard D, Bendele T, Rao C, Terstappen LWMM. Circulating tumor cells as cancer markers, a sample preparation and analysis system. In: Diamandis EP, Fritsche HA, Lilja H, Chan DW, Schwartz M, editors. Tumor markers: physiology, pathobiology, technology, and clinical applications. Washington (DC): AACC Press; 2002. pp. 495-8.
15. Riethdorf S, Fritsche H, Müller V, Rau T, Schindlbeck C, Rack B, Janni W, Coith C, Beck K, Jänicke F, Jackson S, Gornet T, Cristofanilli M, Pantel K. Detection of circulating tumor cells in peripheral blood of patients with metastatic breast cancer: a validation study of the cellsearch system. Clin Cancer Res 2007;13:920-8.
16. Lankiewicz S, Rivero BG, Bocher O. Quantitative real-time RT-PCR of disseminated tumor cells in combination with immunomagnetic cell enrichment. Mol Biotechnol 2006;34:15-27.
17. Zieglschmid V, Hollmann C, Gutierrez B, Albert W, Strothoff D, Gross E, Böcher O. Combination of immunomagnetic enrichment with multiplex RT-PCR analysis for the detection of disseminated tumor cells. Anticancer Res 2005;25:1803-10.
18. Blassl C, Kuhlmann JD, Webers A, Wimberger P, Fehm T, Neubauer H. Gene expression profiling of single circulating tumor cells in ovarian cancer-establishment of a multi-marker gene panel. Mol Oncol 2016;10:1030-42.
19. Magbanua MJ, Park JW. Isolation of circulating tumor cells by immunomagnetic enrichment and fluorescence-activated cell sorting (IE/FACS) for molecular profiling. Methods 2013;64:114-8.
20. Takao M, Takeda K. Enumeration, characterization, and collection of intact circulating tumor cells by cross contamination-free flow cytometry. Cytometry A 2011;79:107-17.
21. Watanabe M, Uehara Y, Yamashita N, Fujimura Y, Nishio K, Sawada T, Takeda K, Koizumi F, Koh Y. Multicolor detection of rare tumor cells in blood using a novel flow cytometry-based system. Cytometry A 2014;85:206-13.
22. Aerts J, Wynendaele W, Paridaens R, Christiaens MR, van den Bogaert W, van Oosterom AT, Vandekerckhove F. A real-time quantitative reverse transcriptase polymerase chain reaction (RT-PCR) to detect breast carcinoma cells in peripheral blood. Ann Oncol 2001;12:39-46.
23. Strati A, Markou A, Parisi C, Politaki E, Mavroudis D, Georgoulias V, Lianidou E. Gene expression profile of circulating tumor cells in breast cancer by RT-qPCR. BMC Cancer 2011;11:422.
24. Hu Y, Fan L, Zheng J, Cui R, Liu W, He Y, Li X, Huang S. Detection of circulating tumor cells in breast cancer patients utilizing multiparameter flow cytometry and assessment of the prognosis of patients in different CTCs levels. Cytometry A 2010;77:213-9.
25. Greenfield LJ, Proctor MC, Saluja A. Clinical results of greenfield filter use in patients with cancer. Cardiovasc Surg 1997;5:145-9.
26. Osta WA, Chen Y, Mikhitarian K, Mitas M, Salem M, Hannun YA, Cole DJ, Gillanders WE. EpCAM is overexpressed in breast cancer and is a potential target for breast cancer gene therapy. Cancer Res 2004;64:5818-24.
27. Alberti S, Ambrogi F, Boracchi P, Fornili M, Querzoli P, Pedriali M, La Sorda R, Lattanzio R, Tripaldi R, Piantelli M, Biganzoli E, Coradini D. Cytoplasmic Trop-1/Ep-CAM overexpression is associated with a favorable outcome in node-positive breast cancer. Jpn J Clin Oncol 2012;42:1128-37.
28. Cohen SJ, Punt CJ, Iannotti N, Saidman BH, Sabbath KD, Gabrail NY, Picus J, Morse M, Mitchell E, Miller MC, Doyle GV, Tissing H, Terstappen LW, Meropol NJ. Relationship of circulating tumor cells to tumor response, progression-free survival, and overall survival in patients with metastatic colorectal cancer. J Clin Oncol 2008;26:3213-21.
29. Cristofanilli M, Budd GT, Ellis MJ, Stopeck A, Matera J, Miller MC, Reuben JM, Doyle GV, Allard WJ, Terstappen LW, Hayes DF. Circulating tumor cells, disease progression, and survival in metastatic breast cancer. N Engl J Med 2004;351:781-91.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Warawdekar UM, Parmar V, Prabhu A, Kulkarni A, Chaudhari M, Badwe RA. A versatile method for enumeration and characterization of circulating tumor cells from patients with breast cancer. J Cancer Metastasis Treat 2017;3:23-33. http://dx.doi.org/10.20517/2394-4722.2016.66
AMA Style
Warawdekar UM, Parmar V, Prabhu A, Kulkarni A, Chaudhari M, Badwe RA. A versatile method for enumeration and characterization of circulating tumor cells from patients with breast cancer. Journal of Cancer Metastasis and Treatment. 2017; 3: 23-33. http://dx.doi.org/10.20517/2394-4722.2016.66
Chicago/Turabian Style
Warawdekar, Ujjwala M., Vani Parmar, Aruna Prabhu, Abhay Kulkarni, Meenal Chaudhari, Rajendra A. Badwe. 2017. "A versatile method for enumeration and characterization of circulating tumor cells from patients with breast cancer" Journal of Cancer Metastasis and Treatment. 3: 23-33. http://dx.doi.org/10.20517/2394-4722.2016.66
ACS Style
Warawdekar, UM.; Parmar V.; Prabhu A.; Kulkarni A.; Chaudhari M.; Badwe RA. A versatile method for enumeration and characterization of circulating tumor cells from patients with breast cancer. J. Cancer. Metastasis. Treat. 2017, 3, 23-33. http://dx.doi.org/10.20517/2394-4722.2016.66
About This Article
Special Issue
Copyright

Data & Comments
Data


 Cite This Article 6 clicks
Cite This Article 6 clicks

 Like This Article 18
likes
Like This Article 18
likes






Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.