
The synergy of Helicobacter pylori and lipid metabolic disorders in induction of Th17-related cytokines in human gastric cancer


Abstract
Aim: To study the impact of Helicobacter pylori (H. pylori) and lipid metabolic disorder on the expression of Th17-related cytokines in gastric cancer (GC). Methods: GC specimens were randomly collected from 42 patients, of whom 15 had H. pylori infection and 27 were without. Tumor RNA was extracted for reverse transcription quantitative polymerase chain reaction quantification of gene expression. Results: The mRNA levels of interleukin (IL)-6 and leptin, which are known to regulate Th17 differentiation, were upregulated by 20 and 6 folds, respectively, in H. pylori-infected compared to uninfected patients. IL-17A and granulocyte-macrophage colony-stimulating factor, two cytokines produced by Th17 cells, were 5- and 6-fold higher in tumors with H. pylori infection, respectively. Consistently, RORγt, a transcription factor regulating Th17 differentiation, was increased 6-fold in H. pylori-positive vs. negative tumors. Further elevation of RORγt was seen in advanced H. pylori-associated tumors. In addition, H. pylori infection was also associated with enhanced expression of CXCL1 (5 folds), chemotactic factor capable of driving bone marrow-derived immature myeloid cells. Interestingly, we observed that H. pylori-associated increase of IL-17A was enhanced in the group with higher plasma triglycerides. Conclusion: The findings demonstrate a cross-talk and synergistic role of H. pylori infection and abnormal lipid metabolism in GC development, at least partly via cooperative induction of Th17 differentiation and activation.
Keywords
Introduction
Gastric cancer (GC) is currently the third leading cause of cancer-related death worldwide due to the highly metastatic property and poor prognosis.[1,2] The overall 5-year survival rate of GC patients is only between 15 to 35%.[3] Epidemiological studies show that persistent Helicobacter pylori (H. pylori) infection accounts for approximate 75% of confound risk factors for GC.[4-6] Understanding the underlying mechanism of GC development associated with H. pylori infection will be important for developing novel therapeutic methods.
H. pylori, a gram-negative spiral-shaped pathogenic bacterium, specifically colonizes and induces damage to the gastric epithelium leading to chronic gastritis, ulcers and even cancer.[2,7,8] Considerable studies have demonstrated that a mixed response of Th1 and Th17 cells plays a critical role in H. pylori-induced inflammatory gastric diseases and cancer.[9,10] The phenotypes of T helper subsets are determined by the local cytokine milieu and their lineage-specific transcription factors.[11-13]H. pylori elicits Th1 response to produce interferon-γ and tumor necrosis factor-α causing chronic gastritis and ulcers.[9,13] Th17 cells are also frequently recruited by H. pylori to the gastric mucosa, and are characterized by expression of interleukin (IL)-17A/F, granulocyte-macrophage colony-stimulating factor (GM-CSF), IL-21, IL-22 and IL-23, and the transcription factor of RORγt.[11,14] While activation of Th17 cells contributes to bacterial eradication,[15] Th17-mediated immune-response can be detrimental to gastric epithelium during gastritis.[9,14] Th17 cells can be further activated in tumor microenvironment due to involvement of IL-6 and transforming growth factor-β.[16,17] Although activation of Th17 cells might have antitumor activity by facilitating the recruitment of other effector immune cells,[18] Th17-derived IL-17A favors angiogenesis and tumor growth through inducing IL-6 that in turn activates STAT3 signaling to promote tumor survival and angiogenesis.[19,20]
It has been reported that obesity and diabetes can worsen the process of Helicobactor-associated GC.[19,21] However, the cross-talk between Helicobactor infection and metabolic disorders in the gastric carcinogenesis remains not completely understood. We recently demonstrated that high fat diet (HFD) and obesity could strongly enhance H. felis-induced GC in mice.[19] We observed that H. felis infection potently stimulates stomach Th17 recruitment and development, and enhanced mobilization of bone-marrow derived IMCs via CXCL1 expression. Interestingly, the local Th17-associated gastric inflammation results in increased IL-17A in blood and causes adipose inflammation in HFD-fed obese mice. In turn, fat-derived IL-6 and leptin can promote gastric Th17 expansion, thus forming a positive loop in Th17 activation. These findings suggest that Th17 and IL-17A play a critical role in the synergy of Helicobacter infection and metabolic abnormalities in accelerating GC progression. In present study, we used clinical GC specimens to document Th17-related cytokines and explore the roles of H. pylori infection and lipid metabolic disorders in GC development. Our results suggest that dysregulated lipid metabolism may synergize with H. pylori to promote GC development.
Methods
Clinical specimens
Forty-two GC specimens were randomly collected from Fujian Provincial Cancer Hospital in China. H. pylori infection was clinically diagnosed and confirmed with the expression of CagA, VacA or both.[22,23] The patients were also divided into high and normal lipid groups with a diagnostic cut-off of 1.7 mmol/L of plasma triglyceride (TG).[24] All tumors were histological diagnosed according to the World Health Organization classification. The pathological TNM stage and clinical stages were also recorded.[25]
Extraction of RNA and quantitative real-time PCR
Total RNA was extracted using Triazol kit (Invitrogen Company, USA) with slight modifications of protocol. The RNA was reverse transcribed using Hifair™ III 1st Strand cDNA Synthesis Kit (Yesen Company, China). The cDNAs were then used in quantitative polymerase chain reaction (qPCR) quantitative analysis of IL-6, leptin, IL-17A, GM-CSF, CXCL1 and RORγt mRNA expression levels in ABI 7500 system (Applied Biosystems, Foster, CA) by using Hieff™ qPCR SYBR® Green Master Mix (Yesen Company, China). Their relative levels were normalized to β-actin expression. Specific primers used in this study were listed in Table 1.
The primers used in RT-qPCR
| Target gene | Primer sequence (5’ to 3’) |
|---|---|
| IL-6 | AGACAGCCACTCACCTCTTC |
| TTTCACCAGGCAAGTCTCCT | |
| IL-17A | AATCTCCACCGCAATGAGGA |
| ACCAGTATCTTCTCCAGCCG | |
| CXCL1 | TCACAGTGTGTGGTCAACAT |
| AGCCCCTTTGTTCTAAGCCA | |
| GM-CSF | ATTCTACAAGCCCAGCCCAG |
| CCCTCCTTGGCTGAACAGAG | |
| CagA | GAAGCAATCAATCAAGAACC |
| GACTCCCCATTAACACAGAA | |
| VacA | CGGTATCAATCTGTCCAATC |
| AATTCACAAATCTTCCCAAA | |
| β-actin | GCGTGACATTAACCACAAGC |
| CCACGTCACACTTCATGATGG |
Statistical analysis
Data analysis was conducted by using Graph pad 6.0 Software. After log transformation, normal distribution was analyzed. Comparison between the two groups was done using t-test and Spearman analysis of correlation was performed between the groups. The contingency were analyzed by using Chi-square testing.
Results
The effects of metabolic milieu on H. pylori-induced IL-17A expression
Since obesity was suggested to play an important role in H. felis-induced GC by stimulating Th17 response in mice,[19] we determined the effects of H. pylori infection and lipid metabolic disorders on Th17-related cytokines in human GC. Our results showed that IL-17A was 5-fold higher in H. pylori-infected than uninfected patients [Figure 1A], while approximately 1.8-fold increase in IL-17A levels was seen in patients with high TG (> 1.7 mmol/L) comparing to those with normal TG [Figure 1B]. However, abnormal TG could not alone induce IL-17A expression as evidenced in H. pylori-negative patients [Figure 1C]. H. pylori could increase IL-17A expression by 3.2 folds in patients with normal plasma TG, but could further increase IL-17A expression (5.5 folds) in the milieu of high TG content [Figure 1C]. However, the contingency analysis showed that there was no significant correlation between H. pylori infection and metabolic factors including plasma glucose, cholesterol, low density lipoprotein, high density lipoprotein and TG [Table 2]. Taken together, the data demonstrated that H. pylori infection and lipid metabolic disorders could synergistically increase IL-17A expression.
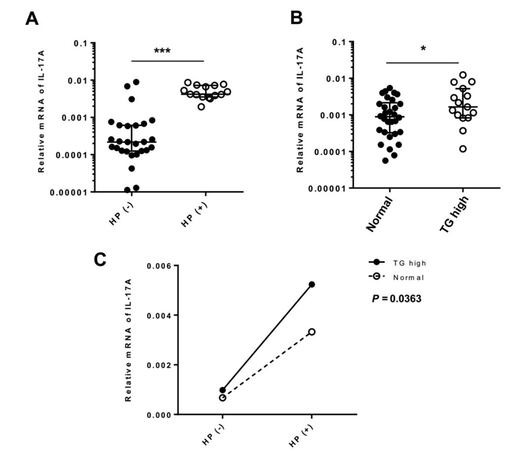
Figure 1. IL-17A expression in H. pylori-associated GC was enhanced in abnormal lipid milieu. (A) H. pylori induced IL-17A expression. RNA was extracted from GC specimens. The levels of IL-17A were quantitated with RT-qPCR and compared between HP (+) and HP (-) groups; (B) the effect of high TG content on H. pylori-induced IL-17A expression. The GC patients were divided into groups with high TG (> 1.7 mmol/L) and normal TG (≤ 1.7 mmol/L). IL-17A levels were compared between these two groups; (C) the synergistic effects of H. pylori and aberrant lipid metabolism on IL-17A induction. Regression analysis was employed. ***P < 0.001, HP (+) vs. HP (-); *P < 0.05, high TG vs. normal TG. H. pylori: Helicobacter pylori; GC: gastric cancer; IL: interleukin; RT-qPCR: reverse transcription quantitative polymerase chain reaction; TG: triglyceride
Altered glycolipid metabolic factors and H. pylori infection
| HP (+) | HP (-) | P value | |
|---|---|---|---|
| GLU | |||
| High | 6 | 14 | 0.53 |
| Normal | 9 | 13 | |
| CHO | |||
| High | 7 | 10 | 0.74 |
| Normal | 8 | 17 | |
| LDL | |||
| High | 10 | 18 | 0.74 |
| Normal | 5 | 7 | |
| HDL | |||
| Low | 1 | 3 | 1 |
| Normal | 14 | 22 | |
| TG | |||
| High | 7 | 8 | 0.32 |
| Normal | 8 | 19 | |
H. pylori infection contributes to Th17 differentiation and response
IL-6 and leptin have been reported to promote Th17 differentiation and play roles in tumor progression.[19,26] Consistently, our results showed that the levels of IL-6 and leptin expression were increased 20 and 6 folds, respectively, in H. pylori-infected vs. uninfected tumors [Figure 2A and B]. This data suggested that H. pylori infection promoted the expression of factors that regulate Th17 differentiation.
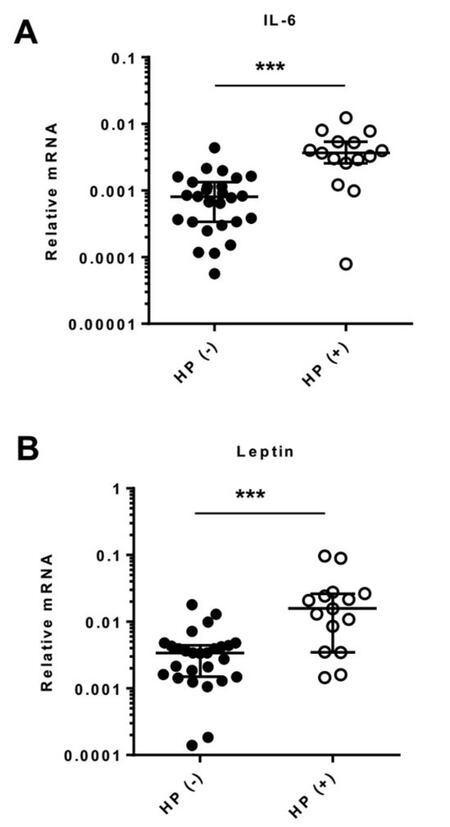
Figure 2. H. pylori induced IL-6 and leptin expression. (A) The levels of IL-6 expression was quantitated by RT-qPCR and compared between HP (+) and HP (-) GCs; (B) leptin was similarly assayed to determine the effects of H. pylori infection. ***P < 0.001, HP (+) vs. HP (-). H. pylori: Helicobacter pylori; GC: gastric cancer; IL: interleukin; RT-qPCR: reverse transcription quantitative polymerase chain reaction
GM-CSF is an additional cytokine released by Th17 cells, and importantly, has been strongly linked to pathogenicity of Th17 cells in other disease states.[27] Thus, we next examined whether GM-CSF was associated with H. pylori infection in human HC. Indeed, higher levels of gastric GM-CSF were seen in H. pylori-positive vs. negative tumors [Figure 3A]. Further, we observed that H. pylori-induced GM-CSF expression was closely associated with enhanced IL-17A expression [Figure 3B]. Our results suggest that H. pylori infection may activate Th17 responses as evidenced by the induced expression of IL-17A and GM-CSF.
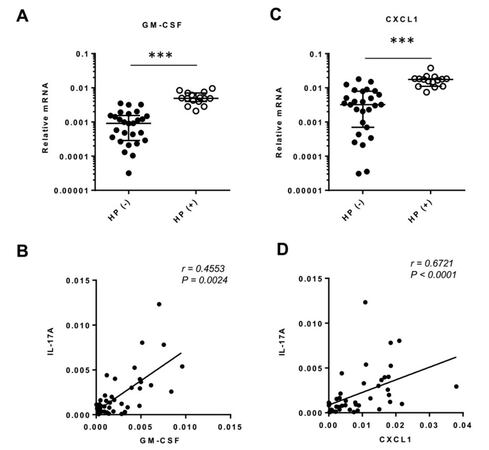
Figure 3. H. pylori induced GM-CSF and CXCL1 expression. (A and C) The quantitative assays of GM-CSF (A) and CXCL1 (C) were performed by RT-qPCR. ***P < 0.001, HP (+) vs. HP (-); (B and D) the correlation of GM-CSF (B) and CXCL1 (D) with IL-17A expression was analyzed. H. pylori: Helicobacter pylori; RT-qPCR: reverse transcription quantitative polymerase chain reaction; IL: interleukin; CXCL1: chemokine (C-X-C motif) ligand 1; GM-CSF: granulocyte-macrophage colony-stimulating factor
CXCL1 has been demonstrated to be secreted by inflamed stomach and adipose associated with H. pylori infection,[19] acting as a potent mobilizer of bone marrow-derived IMCs. Consistently, our present results showed that the expression of CXCL1 was significantly increased in H. pylori-positive vs. negative patients [Figure 3C], and a close correlation of CXCL1 with IL-17A expression was established [Figure 3D].
H. pylori induces tumor progression and RORγt expression
In agreement with previous reports,[28,29] we observed that H. pylori infection was related to tumor progression, as H. pylori-associated tumors were usually more aggressive than the tumors from uninfected individuals. Tumors larger than 4 cm were seen in 67% of H. pylori-infected patients but only in 44% of uninfected patients [Figure 4A]. Consistently, approximately 80% H. pylori-associated tumors advanced to T3/T4 stages, whereas 69% of tumors of uninfected individuals did so [Figure 4B].
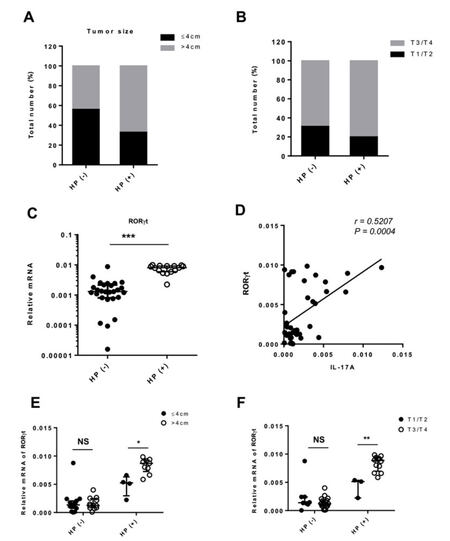
Figure 4. Roles of H. pylori and RORγt in tumor progression. (A and B) The association of H. pylori infection with tumor sizes (A) and stages (B); (C) RORγt was enhanced by H. pylori. RORγt was assayed via RT-qPCR; (D) the correlation of RORγt with IL-17A; (E and F) the expression levels of RORγt were compared in H. pylori-related and unrelated tumors. The levels of RORγt were also compared in tumors with different sizes and different stages. HP (+) vs. HP (-): ***P < 0.001; ≤ 4 cm vs. > 4 cm: *P < 0.05; T1/T2 vs. T3/T4: **P < 0.01. H. pylori: Helicobacter pylori; RT-qPCR: reverse transcription quantitative polymerase chain reaction; IL: interleukin
RORγt is the most important transcription factor for the differentiation and activation of Th17 cells.[30] We thus analyzed RORγt expression in H. pylori-associated GC and explored its potential role in tumor progression. We found that an overall 6-fold increase of RORγt in H. pylori-infected vs. uninfected GC [Figure 4C], and that H. pylori-associated expression of RORγt and IL-17A were positively correlated [Figure 4D]. In the absence of H. pylori infection, the levels of RORγt were not different between early and advanced tumors [Figure 4E and F]. In contrast, RORγt expression was further enhanced in H. pylori-associated tumor progression, with higher expression in larger tumors (> 4 cm) [Figure 4E] and those with more metastatic capability (T3/T4 stages) [Figure 4F].
Discussion
It has been long recognized that unresolved inflammation induced by H. pylori will favor gastric carcinogenesis. Eradication of H. pylori has been shown to be beneficial in preventing GC development.[31,32] Obesity and diabetes have become a great problem in modern societies, which profoundly increase the frequencies of malignant neoplasms, including GC.[21,33,34] Although H. pylori infection and metabolic disorders can independently promote tumor progression, there are considerable evidences showing that they can also exert a synergistic effect on tumorigenesis.[19] However, the molecular mechanisms behind this synergy remain elusive. We previously reported that H. felis-induced GC in obese mice can be influenced by the gastric homing and activation of Th17 cells, which trigger a series of inflammatory responses in both stomach and adipose tissues through releasing IL-17A.[19] Our current results further the concept that chronic H. pylori infection and aberrant lipid metabolism can interact to activate Th17 responses and facilitate GC.
We demonstrate that H. pylori infection is associated with striking elevation of IL-17A content in GC [Figure 1A]. The expression of IL-17A is also increased in the patients with abnormal high plasma levels of TG, though to a lesser extent compared to that of H. pylori infection [Figure 1B]. We consistently observe a significant synergy between H. pylori infection and abnormal lipid metabolism in producing IL-17A [Figure 1C]. CD4+ T-cells are widely observed in Helicobacter-associated GC, and of these, IL-17A is predominantly produced by the Th17 subset.[35] However, future studies are needed to understand whether IL-17A is also produced from other sources, such as CD8+ T-cells and/or innate lymphoid cells (ILCs). Nevertheless, given the observation that other Th17-related cytokines, such as IL-6 and GM-CSF, are also increased, our results suggest that H. pylori infection and altered TG metabolism cooperate in enhancing the Th17 response.
As a pro-inflammatory subset of T cells, it is possible that Th17 cells can also activate antitumor immunity.[19,37] Myeloid-derived suppressor cells (MDSCs) are known to home to the site of tumors and facilitate their avoidance of cytotoxic T cells. Thus, the production of CXCL1 and GM-CSF may be critical members of the cytokine milieu, as these have been reported to promote the recruitment and function of MDSCs, respectively.[38] In agreement with previous observations, we found that both GM-CSF and CXCL1 were greatly increased in H. pylori positive tumors [Figure 3A and C] and their upregulation was coincident with elevated IL-17A [Figure 3B and D]. In addition, it has been shown that inflamed adipose and stomach tissues induced by H. felis/HFD can enhance IL-6 and leptin production to stimulate Th17 differentiation and stabilization.[19,39] In present study, we note that significant amount of IL-6 and leptin are seen in H. pylori-associated GC tissues, suggesting that tumor microenvironment may be sufficient foster Th17 development.
We finally demonstrate that RORγt, a transcriptional activator of IL-17A,[40,41] is extensively induced in H. pylori-infected patients [Figure 4C], and the expression is further enhanced in advanced tumors [Figure 4E and F]. RORγt has been widely investigated in Th17 cells but seldom in malignant diseases. Thus, future studies should include analysis of the important association of RORγt and H. pylori infection.
In summary, we demonstrate that H. pylori infection and abnormal lipid metabolism can exert a synergistic role in Th17 activation and response to promote GC development. These observations are important to understand GC pathogenesis and can be of therapeutic significance.
Declarations
AcknowledgmentsWe are grateful to all the patients who have contributed to the data of this work.
Authors’ contributionsConceived and designed the study: J.Z. Zeng, W. Han, A.S.T. Wong, R.E. Ericksen
Performed the study: H. Wang, G. Chen, J. Liu, M. Yang, Z.X. Wu
Prepared the manuscript: J.Z. Zeng
Revised the manuscript: W. Han, R.E. Ericksen
Financial support and sponsorshipThis work is supported by grants from the 10th Singapore-China Joint Research Program (S2014GR0448), Natural Science Foundation of China (NSFC) (81673467, 31471273, 21303145, 31340029, 30971445, 31501145), Research Grants Council (RGC) of Hong Kong Joint Research Scheme (NSFC/RGC) (31461163002/N_HKU740/14 and 30931160431/N_HKU 735/09), Marine Drug Research Project of South Marine Center (14GYY023NF23), Fujian Province’s 100-Talent Program of Innovation of Science and Technology (201510117), and the Fundamental Research Funds for the Central Universities (No. 2013121037).
Conflicts of interestThere are no conflicts of interest.
Patient consentInformed consents were signed by patients.
Ethics approvalOur experimental protocols were approved by The Hospital Ethics Committee and The Ethics Committee of Xiamen University, China.
REFERENCES
1. Khatoon J, Rai RP, Prasad KN. Role of Helicobacter pylori in gastric cancer: updates. World J Gastrointest Oncol 2016;8:147-58.
2. Amieva M, Peek RM, Jr. Pathobiology of helicobacter pylori-induced gastric cancer. Gastroenterology 2016;150:64-78.
3. O'Connor A, O'Morain CA, Ford AC. Population screening and treatment of Helicobacter pylori infection. Nat Rev Gastroenterol Hepatol 2017;14:230-40.
4. de Martel C, Forman D, Plummer M. Gastric cancer: epidemiology and risk factors. Gastroenterol Clin North Am 2013;42:219-40.
5. Lee YC, Chiang TH, Chou CK, Tu YK, Liao WC, Wu MS, Graham DY. Association between helicobacter pylori eradication and gastric cancer incidence: a systematic review and meta-analysis. Gastroenterology 2016;150:1113-24 e5.
6. Ford AC, Axon AT. Epidemiology of Helicobacter pylori infection and public health implications. Helicobacter 2010;15 Suppl 1:1-6.
7. Wang F, Meng W, Wang B, Qiao L. Helicobacter pylori-induced gastric inflammation and gastric cancer. Cancer Lett 2014;345:196-202.
8. Bornschein J, Kandulski A, Selgrad M, Malfertheiner P. From gastric inflammation to gastric cancer. Dig Dis 2010;28:609-14.
9. Hitzler I, Kohler E, Engler DB, Yazgan AS, Muller A. The role of Th cell subsets in the control of Helicobacter infections and in T cell-driven gastric immunopathology. Front Immunol 2012;3:142.
10. Ren Z, Pang G, Clancy R, Li LC, Lee CS, Batey R, Borody T, Dunkley M. Shift of the gastric T-cell response in gastric carcinoma. J Gastroenterol Hepatol 2001;16:142-8.
11. Maruyama T, Kono K, Mizukami Y, Kawaguchi Y, Mimura K, Watanabe M, Izawa S, Fujii H. Distribution of Th17 cells and FoxP3(+) regulatory T cells in tumor-infiltrating lymphocytes, tumor-draining lymph nodes and peripheral blood lymphocytes in patients with gastric cancer. Cancer Sci 2010;101:1947-54.
12. Li B, Chen L, Sun H, Yang W, Hu J, He Y, Wei S, Zhao Z, Zhang J, Li H, Zou Q, Wu C. Immunodominant epitope-specific Th1 but not Th17 responses mediate protection against Helicobacter pylori infection following UreB vaccination of BALB/c mice. Sci Rep 2015;5:14793.
13. Gray BM, Fontaine CA, Poe SA, Eaton KA. Complex T cell interactions contribute to Helicobacter pylori gastritis in mice. Infect Immun 2013;81:740-52.
14. Serelli-Lee V, Ling KL, Ho C, Yeong LH, Lim GK, Ho B, Wong SB. Persistent Helicobacter pylori specific Th17 responses in patients with past H. pylori infection are associated with elevated gastric mucosal IL-1beta. PLoS One 2012;7:e39199.
15. Dixon BR, Radin JN, Piazuelo MB, Contreras DC, Algood HM. IL-17a and IL-22 induce expression of antimicrobials in gastrointestinal epithelial cells and may contribute to epithelial cell defense against Helicobacter pylori. PLoS One 2016;11:e0148514.
16. Kryczek I, Banerjee M, Cheng P, Vatan L, Szeliga W, Wei S, Huang E, Finlayson E, Simeone D, Welling TH, Chang A, Coukos G, Liu R, Zou W. Phenotype, distribution, generation, and functional and clinical relevance of Th17 cells in the human tumor environments. Blood 2009;114:1141-9.
17. Pinchuk IV, Morris KT, Nofchissey RA, Earley RB, Wu JY, Ma TY, Beswick EJ. Stromal cells induce Th17 during Helicobacter pylori infection and in the gastric tumor microenvironment. PLoS One 2013;8:e53798.
18. Li J, Yue L, Wang H, Liu C, Liu H, Tao J, Qi W, Wang Y, Zhang W, Fu R, Shao Z. Th17 cells exhibit antitumor effects in MDS possibly through augmenting functions of CD8+ T cells. J Immunol Res 2016;2016:9404705.
19. Ericksen RE, Rose S, Westphalen CB, Shibata W, Muthupalani S, Tailor Y, Friedman RA, Han W, Fox JG, Ferrante AW Jr, Wang TC. Obesity accelerates Helicobacter felis-induced gastric carcinogenesis by enhancing immature myeloid cell trafficking and TH17 response. Gut 2014;63:385-94.
20. Kennedy CL, Najdovska M, Jones GW, McLeod L, Hughes NR, Allison C, Ooi CH, Tan P, Ferrero RL, Jones SA, Dev A, Sievert W, Bhathal PS, Jenkins BJ. The molecular pathogenesis of STAT3-driven gastric tumourigenesis in mice is independent of IL-17. J Pathol 2011;225:255-64.
21. Li Q, Zhang J, Zhou Y, Qiao L. Obesity and gastric cancer. Front Biosci (Landmark Ed) 2012;17:2383-90.
22. Monteiro L, Oleastro M, Lehours P, Megraud F. Diagnosis of Helicobacter pylori infection. Helicobacter 2009;14 Suppl 1:8-14.
23. Camorlinga-Ponce M, Torres J, Perez-Perez G, Leal-Herrera Y, Gonzalez-Ortiz B, Madrazo de la Garza A, Gomez A, Munoz O. Validation of a serologic test for the diagnosis of Helicobacter pylori infection and the immune response to urease and CagA in children. Am J Gastroenterol 1998;93:1264-70.
24. Connelly PW, Petrasovits A, Stachenko S, MacLean DR, Little JA, Chockalingam A. Prevalence of high plasma triglyceride combined with low HDL-C levels and its association with smoking, hypertension, obesity, diabetes, sedentariness and LDL-C levels in the Canadian population. Canadian Heart Health Surveys Research Group. Can J Cardiol 1999;15:428-33.
25. Sobin LH, Gospodarowicz MK, Wittekind C. TNM classification of malignant tumours. 7th ed. Hoboken, NJ: Wiley-Blackwell; 2011.
26. Reis BS, Lee K, Fanok MH, Mascaraque C, Amoury M, Cohn LB, Rogoz A, Dallner OS, Moraes-Vieira PM, Domingos AI, Mucida D. Leptin receptor signaling in T cells is required for Th17 differentiation. J Immunol 2015;194:5253-60.
28. Uemura N, Okamoto S, Yamamoto S, Matsumura N, Yamaguchi S, Yamakido M, Taniyama K, Sasaki N, Schlemper RJ. Helicobacter pylori infection and the development of gastric cancer. N Engl J Med 2001;345:784-9.
29. Liu N, Zhou N, Chai N, Liu X, Jiang H, Wu Q, Li Q. Helicobacter pylori promotes angiogenesis depending on Wnt/beta-catenin-mediated vascular endothelial growth factor via the cyclooxygenase-2 pathway in gastric cancer. BMC Cancer 2016;16:321.
30. Solt LA, Kumar N, Nuhant P, Wang Y, Lauer JL, Liu J, Istrate MA, Kamenecka TM, Roush WR, Vidovic D, Schurer SC, Xu J, Wagoner G, Drew PD, Griffin PR, Burris TP. Suppression of TH17 differentiation and autoimmunity by a synthetic ROR ligand. Nature 2011;472:491-4.
31. Ohba R, Iijima K. Pathogenesis and risk factors for gastric cancer after Helicobacter pylori eradication. World J Gastrointest Oncol 2016;8:663-72.
32. Graham DY. Helicobacter pylori update: gastric cancer, reliable therapy, and possible benefits. Gastroenterology 2015;148:719-31 e3.
33. Tseng CH, Tseng FH. Diabetes and gastric cancer: the potential links. World J Gastroenterol 2014;20:1701-11.
34. Yoon JM, Son KY, Eom CS, Durrance D, Park SM. Pre-existing diabetes mellitus increases the risk of gastric cancer: a meta-analysis. World J Gastroenterol 2013;19:936-45.
35. Mills KH. Induction, function and regulation of IL-17-producing T cells. Eur J Immunol 2008;38:2636-49.
36. Kim DH, Sandoval D, Reed JA, Matter EK, Tolod EG, Woods SC, Seeley RJ. The role of GM-CSF in adipose tissue inflammation. Am J Physiol Endocrinol Metab 2008;295:E1038-46.
37. Wen L, Gong P, Liang C, Shou D, Liu B, Chen Y, Bao C, Chen L, Liu X, Liang T, Gong W. Interplay between myeloid-derived suppressor cells (MDSCs) and Th17 cells: foe or friend? Oncotarget 2016;7:35490-6.
38. Zhang T, Tseng C, Zhang Y, Sirin O, Corn PG, Li-Ning-Tapia EM, Troncoso P, Davis J, Pettaway C, Ward J, Frazier ML, Logothetis C, Kolonin MG. CXCL1 mediates obesity-associated adipose stromal cell trafficking and function in the tumour microenvironment. Nat Commun 2016;7:11674.
39. Yu Y, Liu Y, Shi FD, Zou H, Matarese G, La Cava A. Cutting edge: Leptin-induced RORgammat expression in CD4+ T cells promotes Th17 responses in systemic lupus erythematosus. J Immunol 2013;190:3054-8.
40. Valmori D, Raffin C, Raimbaud I, Ayyoub M. Human RORgammat+ TH17 cells preferentially differentiate from naive FOXP3+Treg in the presence of lineage-specific polarizing factors. Proc Natl Acad Sci U S A 2010;107:19402-7.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Liu J, Wang H, Chen G, Yang M, Wu ZX, Ericksen RE, Wong AST, Han W, Zeng JZ. The synergy of Helicobacter pylori and lipid metabolic disorders in induction of Th17-related cytokines in human gastric cancer. J Cancer Metastasis Treat 2017;3:169-76. http://dx.doi.org/10.20517/2394-4722.2017.46
AMA Style
Liu J, Wang H, Chen G, Yang M, Wu ZX, Ericksen RE, Wong AST, Han W, Zeng JZ. The synergy of Helicobacter pylori and lipid metabolic disorders in induction of Th17-related cytokines in human gastric cancer. Journal of Cancer Metastasis and Treatment. 2017; 3: 169-76. http://dx.doi.org/10.20517/2394-4722.2017.46
Chicago/Turabian Style
Liu, Jie, Han Wang, Gang Chen, Mo Yang, Zhi-Xian Wu, Russell Erick Ericksen, Alice Sze Tsai Wong, Weiping Han, Jin-Zhang Zeng. 2017. "The synergy of Helicobacter pylori and lipid metabolic disorders in induction of Th17-related cytokines in human gastric cancer" Journal of Cancer Metastasis and Treatment. 3: 169-76. http://dx.doi.org/10.20517/2394-4722.2017.46
ACS Style
Liu, J.; Wang H.; Chen G.; Yang M.; Wu Z.X.; Ericksen RE.; Wong AST.; Han W.; Zeng J.Z. The synergy of Helicobacter pylori and lipid metabolic disorders in induction of Th17-related cytokines in human gastric cancer. J. Cancer. Metastasis. Treat. 2017, 3, 169-76. http://dx.doi.org/10.20517/2394-4722.2017.46
About This Article
Copyright

Data & Comments
Data


 Cite This Article 2 clicks
Cite This Article 2 clicks

 Like This Article 8
likes
Like This Article 8
likes






Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.