
Prostate cancer exosomes as modulators of the tumor microenvironment


Abstract
Researchers are currently trying to understand why some men with prostate cancer go on to develop aggressive disease whilst others maintain slow growing tumors. Although endogenous genetic anomalies within the tumor cell are important, the prevailing view is that the tissue microenvironment as a whole is the determinant factor. Many studies have focussed on the role of soluble factors in modulating the nature of the tumor microenvironment. There is however a growing interest in the role of extracellular vesicles, including exosomes, as regulators of disease progression. A variety of resident cells, as well as infiltrating cells, all contribute to a heterogeneous population of exosomes within the tumor microenvironment. Studies focussing on the role of exosomes in prostate cancer are however relatively rare. In this review, evidence from various cancers, including prostate, is used to present numerous potential roles of exosomes in prostate cancer. Whilst further validation of some functions may remain necessary it is clear that exosomes play a major role in intercellular communication between various cell types within the tumor microenvironment and are necessary for driving disease progression.
Keywords
Introduction
Prostate cancer is the most common form of cancer to affect men in the UK. Current survival rates suggest that of those men who develop the disease approximately eighty four percent will survive for 10 or more years. For some men, however, the disease is far more aggressive. Ongoing studies are in place to try to understand the mechanisms responsible for this difference between slow growing, indolent tumors, and the aggressive disease. Many of these studies have focussed on the role of soluble growth factors as modulators of the tumor microenvironment thereby supporting aggressive metastatic forms of the disease. There is, however, a growing precedent to explore the role of extracellular vesicles (EV) in this process.
All cells are capable of secreting vesicles into the extracellular space. Vesicle secretion becomes elevated when cells are subjected to cellular stress[1,2], which can also result in altered molecular cargo within the vesicle[3]. This is particularly relevant in cancer where stress can come from the hypoxic environment, nutrient deficiency, altered extracellular matrix, and other environmental factors. Such vesicles are often regarded as one of two broad subtypes, microvesicles and exosomes. Microvesicles are large, tending to be greater than 200 nm in diameter, dense, and are formed from outward budding of the plasma membrane. Exosomes are much smaller, typically 30 to 150 nm in diameter, float at a characteristic density of 1.1 to 1.2 g/mL[4] and originate within multivesicular endosomes[4]. The secretion of small, exosome-like, vesicles has also been reported from the plasma membrane[5]. It remains a challenge to accurately define vesicle subtypes based on size alone. To aid researchers, the International Society for Extracellular Vesicles has released a position paper detailing the minimal experimental requirements for defining EV[6]. Although, the challenge of defining EV subtypes remains, and is further compounded by overlap in EV composition[7], hence the term EV is often used. The majority of EV present within both cell conditioned media or biological fluids tend to be small[8], suggesting a predominant exosome-like population. The biological significance of any one EV subtype compared to another, however, remains unknown.
The role of EV in cancer has been the studied intensively over recent years[9]. Relatively few of these studies have focused on the potential role of EV, and more specifically exosomes, in prostate cancer. In this current article, we review past studies into the role of exosomes, in diverse malignancies, to identify their potential functions in disease processes of relevance to prostate cancer.
Exosome-mediated angiogenesis
Angiogenesis, or the formation of new blood vessels from pre-existing vasculature, is a vital component in numerous physiological and pathological responses. A variety of angiogenic signals are required to drive endothelial maturation and subsequent re-organisation with vascular smooth muscle cells and pericytes to form a functional vessel network[10], thereby allowing nutrient and waste product exchange[11,12]. In cancer, multiple modulators of vascular remodelling contribute to tumor growth and progression[13]. Once a tumor lesion forms it will become hypoxic and nutrient deprived. The secretion of growth factors activates normal surrounding quiescent cells, to initiate a cascade of events that become quickly dysregulated. This involves an “angiogenic” switch, regulated by both anti- and pro-angiogenic cytokines, examples of which include endothelial growth factor, fibroblast growth factor (FGF), hepatocyte growth factor (HGF), platelet-derived growth factor (PDGF) and vascular endothelial growth factor (VEGF)[14-16]. These responses may initially provide the tumor with more nutrients and oxygen, however, the structural organization of the vessel network is poor, and the continuously remodeled tumor vasculature is disorganized and leaky[17]. This causes irregular blood flow and provides invasive tumors with access to the circulatory system.
Pro-angiogenic activity of exosome-associated proteins
Cancer cell-derived EV have been shown in several studies to promote angiogenesis. In the case of prostate cancer it is well established that c-Src tyrosine kinase, insulin-like growth factor 1 receptor (IGF-1R) and focal adhesion kinase (FAK) play important roles in tumor growth and disease progression[18]. Src-family kinases are normally expressed in prostatic epithelium and reported to transform normal cells when constitutively active and up-regulated during disease initiation and progression[19]. Cross-talk between Src and IGF-1R has previously been shown to promote angiogenesis[20]. It has been reported that Src, IGF-1R and FAK are enriched in prostate cancer exosomes[21]. Src and c-Src are also present in plasma exosomes derived from prostate tumor bearing mice; suggesting that Src-enriched exosomes can promote angiogenesis in vivo. Src is known to stimulate transcription of VEGF and modulate angiogenesis[22] whilst IGF-1R has been demonstrated to induce VEGF-C expression and stimulate angiogenesis[23]. These observations suggest that prostate cancer exosomes enriched with c-Src, IGF-1R and FAK may be able to stimulate angiogenic activity within the tumor microenvironment.
Prostate cancer EV are also likely to be capable of delivering growth factors with known pro-angiogenic function. For instance, EV from aggressive prostate cancer cells have been shown to contain urokinase-type plasminogen activator (uPA)[24], known to be involved in activation of the protease plasminogen which is responsible for vascular remodeling[25]. Addition of uPA positive vesicles to less aggressive prostate cancer cells stimulated cell migration and invasiveness[24]. Although this study did not investigate the impact of uPA positive vesicles on the ability of treated cells to drive angiogenesis, it is conceivable that prostate cancer derived EV can support endothelial tubule formation via delivery of pro-angiogenic growth factors. Additional pro-angiogenic factors have been identified on EV from a variety of different cancer cell types and are summarized in Table 1. Further studies are required to ascertain whether these factors are present on prostate cancer EV.
EV-associated pro-angiogenic proteins
| Protein | Pro-angiogenic function | Cancer cell of EV origin | Reference |
|---|---|---|---|
| Angiogenin | Translocates to the nucleus of recipient cells and enhances RNA transcription, stimulating expression of pro-angiogenic proteins | Multiple myeloma | [131] |
| EGFR | Induces VEGF expression in recipient cells through Akt signaling | Lung, glioma | [132,133] |
| FAK | Interactions between FAK, IGF-1R and Src result in various downstream signaling events and modulation of angiogenesis | Prostate | [21] |
| FGF2 | Promotes proliferation and differentiation of endothelial cells | Multiple myeloma | [131] |
| IGF-1R | Interactions between FAK, IGF-1R and Src result in various downstream signaling events and modulation of angiogenesis | Prostate | [21] |
| MMP-2, MMP-9 | Degradation of extracellular matrix components | Ovarian | [134] |
| Src | Activation of FAK, and subsequent formation of focal adhesions between endothelial cells | Prostate, myeloid leukemia | [21,135] |
| Tspan8 | Induces uPA, VEGFR and vWF in recipient endothelial cells | Pancreatic | [134,135] |
| uPA | Activation of plasminogen leading to vascular remodeling | Prostate | [24] |
| VEGF | Rearranges the cytoskeleton through the FAK/paxillin pathway, induces capillary formation via RhoA/ROCK signaling and controls vascular permeability through PLCγ | Multiple myeloma, ovarian | [131,134] |
Delivery of pro-angiogenic RNAs by exosomes
Whilst direct evidence of RNA delivery by prostate cancer EV is currently lacking, EV from several cancer types are known to be enriched with mRNA transcripts related to pro-angiogenic function that can then be translated by recipient cells[26,27]. Similar studies have shown an enhanced proliferative impact on endothelial cells[28,29] and enhanced tubule formation within 3D cell cultures[28]. The transfer of exosomal miRNA, such as miRNA-92a and miR-17-92, may also play a role in this process[30] and miR-17-92 may play a role in this process[30]. Furthermore, transmittance of the miR-17-92 cluster from EV to endothelial cells has been shown to attenuate endothelial expression of integrin αv resulting in enhanced endothelial cell migration and tube formation[30]. Numerous studies highlight a role of cancer exosomes in delivery of RNAs to endothelial cells, thereby promoting angiogenesis, and it is therefore likely that prostate cancer exosomes share this functionality.
Hypoxic tumor-derived exosomes enhance angiogenesis
As a tumor grows diffusion distances from the existing vascular supply increase, resulting in hypoxia. Sustained growth of the tumor mass often requires new blood vessels to provide rapidly proliferating tumor cells with an adequate supply of metabolites and oxygen. Under hypoxic conditions the cellular secretome becomes altered and a proportion of these changes may reside within the exosome fraction. Exosomes derived from solid tumors, which have been cultured in hypoxic conditions, become enriched with hypoxia-regulated mRNAs and proteins such as Caveolin 1, IL-8, matrix metalloproteinase (MMP) and PDGF, and are capable of promoting angiogenesis[31]. Similarly, under hypoxic conditions, the secretion of exosomes from breast cancer[32] or leukemic cells[33] demonstrate elevated levels of exosomal miR-210, with the capacity to enhance HUVEC tube formation compared to exosomes from normoxic conditions. Although EV from hypoxic prostate cancer cells are yet to be investigated, based on this evidence, it is highly likely that the cargo of prostate cancer exosomes is also influenced by hypoxic conditions. The impact of hypoxia-derived vesicles on angiogenesis and subsequent development of prostatic tumors remains unknown.
Indirect exosome-mediated angiogenesis
In addition to direct modulation of angiogenesis within the tumor microenvironment, exosomes have the potential to regulate angiogenesis indirectly through interactions with various non-endothelial cell types. Prostate cancer exosomes, expressing transforming growth factor beta (TGFβ), can activate fibroblasts resulting in elevated secretion of multiple pro-angiogenic factors including VEGF, HGF, FGF-2, and uPA[34,35]. Furthermore, prostate cancer exosomes were shown to induce pro-angiogenic function within primary prostate stromal cells and were shown to facilitate in vivo tumor growth[35]. Other studies have reported cancer-associated myofibroblasts can secrete pro-angiogenic growth factors and promote angiogenesis at the primary tumor site[36-39]. Also, HGF and stromal cell-derived factor-1 derived from these myofibroblasts can indirectly enhance angiogenesis by inducing the secretion of angiogenic factors from tumor cells[40,41]. Collectively, these studies demonstrate that exosomes derived from solid tumors, including prostate cancer, drive activation of fibroblasts to a pro-angiogenic phenotype.
The ability of prostate cancer exosomes to trigger secretion of pro-angiogenic factors extends to bone marrow-derived mesenchymal stem cells (BM-MSCs), which can also gain exosome-induced pro-angiogenic function[42]. Exosome-activated MSCs were shown to secrete elevated levels of HGF, VEGF and MMPs, and support the formation of endothelial vessel-like structures. Exosomes from metastatic melanomas have also been shown to interact with bone marrow progenitor cells via the tyrosine kinase MET[43], which induced vascular leakiness at pre-metastatic sites and reprogrammed bone marrow progenitors towards a pro-vasculogenic phenotype. This “reprogramming” of the bone marrow progenitors resulted in significantly increased tumor vascular density in vivo.
A wide range of studies have demonstrated various roles of cancer exosomes in promoting angiogenesis either through direct or indirect interaction with endothelial cells. Prostate cancer exosomes are likely to be dynamic in response to hypoxia and may act as a means to deliver a variety of factors capable of supporting the formation of tumor-associated vasculature in vivo.
Exosome-driven tumor-stroma interactions
Stromal cells surrounding a tumor can undergo a desmoplastic response, characterized by aberrant cell growth and morphological transformation of the stroma, resulting in a more aggressive tumor microenvironment[44]. A key feature of this tumor reactive stroma is the presence of cells with a myofibroblast-like phenotype[45]. Myofibroblasts are contractile cells, characterized by the formation of α smooth muscle actin (αSMA) stress fibers[46], loss of the spindle phenotype and formation of a hyaluronic acid pericellular coat[47]. During wound healing myofibroblasts are present to aid wound closure. In various cancers, however, a chronic wound response can occur resulting in sustained presence of myofibroblasts within the tumor microenvironment[48].
Cancer associated myofibroblasts display an altered phenotype compared to wound associated myofibroblasts[49] and have been termed activated fibroblasts, tumor associated fibroblasts and cancer associated fibroblasts. There is conflicting evidence as to whether myofibroblasts promote or suppress tumorigenesis. Rhim et al.[50] observed that removal of myofibroblasts from the stroma of pancreatic ductal adenocarcinoma (PDAC) in vivo results in more aggressive tumors and reduced mouse survival rates. However, the prevailing view is that stroma rich in myofibroblasts has an increased ability to drive tumor growth, angiogenesis, metastasis and treatment resistance[45,51,52].
Activation and modulation of stromal cells by exosomes
Fibroblast differentiation is known to be induced by TGFβ1 via SMAD dependent and independent signaling pathways[53-55]. It has been established that exosomes secreted by prostate cancer cells express latent TGFβ1[56], tethered to the exosome surface via proteoglycans and capable of activating SMAD3 dependent signaling[34]. The authors demonstrate that prostate cancer derived exosomes, with greater than 6 pg TGFβ1/µg exosome, can induce fibroblast differentiation[34]. Differentiation could be sustained for at least 2 weeks in the absence of further exosome treatment, indicating the resulting myofibroblast-like phenotype is self-maintaining. In contrast, EV originating from MDA-MB231 breast cancer cells and u87 glioblastoma cells could only induce transient fibroblast differentiation[57], potentially suggesting differences between EV from distinct tissue types.
A subsequent study by Webber et al.[35] identified that exosomal TGFβ1 induces a more aggressive, pro-angiogenic myofibroblast phenotype compared to the soluble form of the growth factor. These results were replicated in primary stromal cells from normal prostate tissue, resulting in a myofibroblast-like phenotype that matched that found within disease-associated stromal tissue. Furthermore, pre-treating normal stroma with prostate cancer derived exosomes prior to administration enhanced tumor growth in mice. In contrast, pre-treatment with soluble TGFβ1 led to tumor control. Consistent with this report, a separate study showed that metastatic rat prostate tumor EV are capable of activating primary rat prostate fibroblasts, leading to upregulation of αSMA, HGF and VEGFA[58]. Exosomes therefore appear to play a crucial role in communication between prostate cancer cells and the surrounding stroma, with exosome-associated TGFβ1 essential for inducing fibroblast differentiation towards a disease supporting phenotype.
The exact origin of disease-associated myofibroblasts remains unclear, and it has been shown that other cells are capable of myofibroblastic differentiation. MSCs, a multipotent cell type capable of generating many different types of connective tissue, can also differentiate into myofibroblasts in response to secreted factors from tumors[40]. MSCs make up 1.1% of cells within the prostate cancer stroma[59] and exhibit similar tumor promoting effects to cancer associated stroma[40,60,61].
Exosomes secreted by breast[62], ovarian[63] and gastric[64] cancer cells induce TGFβ1-dependent differentiation of adipose or cord blood derived MSCs to myofibroblasts. In addition, chronic lymphocytic leukemia exosomes have been shown to enhance tumor growth in vivo by inducing differentiation of BM-MSCs[65]. Adipose derived MSCs, meanwhile, differentiate in response to EV from metastatic breast cancer in 2D and 3D culture. This process was shown to require TGFβ dependent MAPK signaling involving phosphorylation of ERK1/2 and JNK1/2[66].
Chowdhury et al.[42] demonstrated that prostate cancer exosomes can also drive BM-MSC differentiation, resulting in myofibroblasts with increased VEGFA, HGF and MMP secretion, capable of enhancing cancer cell growth. Exosome differentiated BM-MSCs drove prostate cancer cell invasion in a 3D spheroid model and stimulated endothelial cell migration, proliferation and angiogenic potential. As previously observed, exosomal TGFβ1 treatment resulted in myofibroblasts with an enhanced pro-tumorigenic phenotype compared to soluble TGFβ1. Interestingly, exosomes also modulated BM-MSC derived myofibroblast expression of ITGB6 and ITGB8, encoding for components of integrins αvβ6 and αvβ8, which are involved in converting latent TGFβ1 to the active form[67,68]. This may therefore explain how latent TGFβ1 delivered by exosomes becomes functionally active. The predominant population of myofibroblast precursors remains unclear. Regardless of the precursor cell, it is evident that prostate cancer exosomes can trigger differentiation to a stromal phenotype with disease promoting properties.
Delivery of TGFβ1 is not the only mechanism by which exosomes can stimulate pro-tumorigenic phenotypes in stromal cells. EV transfer of mRNA, miRNA and membrane proteins have all been implicated. For instance, acute myeloid leukemia cell exosomes promote proliferation and migration of bone marrow stromal cells via transfer of IGF-IR mRNA[69]. Similar results have been shown in solid cancers whereby exosomal miRNAs regulate stromal cell behavior. Metastatic breast cancer cells, for example, were shown to enhance vascular permeability, and promote tumor metastasis, via the suppression of the tight junction protein ZO-1 by exosome delivered miR-105[70]. Gastric cancer exosomes stimulate primary mouse liver myofibroblasts and hepatic pericytes by exosome mediated delivery of the membrane protein epidermal growth factor receptor (EGFR)[71]. After insertion into the stromal cell membrane, where it co-localizes with E-cadherin, EGFR activates HGF secretion by potentially suppressing upstream miRNAs such as miR-26a/b. The subsequent increase in HGF secretion promotes gastric cancer cell proliferation, migration and invasion.
Exosomes are not the only EV subgroup shown to alter the prostate stroma phenotype. Prostate cancer cells also secrete large oncosomes, EV between 100-400 nm in diameter[72], which have sustained AKT1 activity[73,74]. A recent study by Minciacchi et al.[75] reported that internalization of large oncosomes by prostate fibroblasts resulted in the induction of a αSMA-positive myofibroblast phenotype. Interestingly, induction of other myofibroblast markers, such as MMP1, thrombospondin-1 (TSP-1) and TGFβ1 did not occur, potentially suggesting that oncosomes induce a distinct myofibroblast-like phenotype. Analysis of transcription factor DNA binding in treated prostate fibroblasts highlighted that MYC binding was essential for this induction of a myofibroblast-like phenotype. The mechanism by which large oncosomes stimulate MYC-DNA binding has not yet been elucidated, however, as MYC has not been found to be present inside the EV it appears MYC is activated rather than delivered. This study also explored the impact of large oncosomes in vivo and found that prostate fibroblasts pre-treated with oncosomes facilitated enhanced tumor growth. These findings are similar to the earlier results obtained with exosomes and lend support to the critical role of diverse vesicle subtypes in tumor-stroma communication in prostate cancer.
Secretion of stroma-derived exosomes
Stromal cells activated by cancer cell secreted EV can initiate a positive feedback mechanism via release of stromal cell EV which promote tumorigenesis after internalization by tumor cells. A study by Josson et al.[76] highlighted this cyclical system in prostate cancer. Activated prostate fibroblasts were shown to release miR-409 containing EV, which are taken up by prostate cancer cells. Upon EV internalization miR-409 downregulates the tumor suppressors Ras suppressor 1 and stromal antigen 2, promoting cancer cell tumorigenesis and stimulating EMT and stemness in epithelial cells. This effect can also be observed in other tissues. For example, activated PDAC fibroblasts secrete ANXA6 positive EV containing the ANXA6/LRP1/TSP1 complex. Uptake of these EV by PDAC cells was shown to enhance tumorigenesis by stimulating cancer cell migration and driving tumor growth in vivo[77]. Activation of the Wnt-planar cell polarity (PCP) pathway, and subsequent stimulation of cell motility and metastasis, can also be induced by stromal cell EV. Luga et al.[78] determined that CD81+ vesicles secreted from activated fibroblasts are capable of activating the Wnt-PCP pathway in breast cancer cells via transfer of Wnt11.
Stromal cells can also confer chemoresistance on surrounding tumor cells via EV communication. Activated fibroblasts resistant to the chemotherapy drug Gemcitabine (GEM) release exosomes containing miR-146a and mRNA for its upstream transcription factor Snail[79]. Incubation of PDAC cells with exosomes from GEM treated fibroblasts results in increased levels of Snail mRNA and miR-146a in the cancer cells, leading to cell proliferation and chemoresistance. Similar findings have been observed in colorectal[80] and breast cancers[81], with the latter study identifying activation of antiviral signaling pathways through stimulation of the pattern recognition receptor RIG-I by exosomal RNA. RIG-I activates STAT1 dependent signaling which cooperates with NOTCH3 to mediate NOTCH target gene transcription, supporting maintenance of therapy resistant tumor initiating cells.
Activation of stromal cells by cancer cell-derived exosomes results in a pro-proliferative and pro-angiogenic stromal phenotype. In turn, EV and exosomes from activated stromal cells may then drive surrounding cancer cells towards a more aggressive, chemoresistant, phenotype. This suggests a network of reciprocal communication based on EV exists to exacerbate disease.
Exosome modulation of myeloid cells
There have been numerous studies demonstrating immunological control by EV, as reviewed previously[9]. Despite such studies, there is a surprising paucity of information relating to prostate cancer exosomes and their influence on myeloid cells. This topic is highly relevant, however, as the presence of CD14+ macrophages and chronic inflammation within the microenvironment is a key risk factor in prostate cancer[82].
Exosome-mediated antigen presentation
Some of the early discoveries of exosome function have centered on their potential as immune-activating factors[4], where professional antigen presenting cells derived from monocyte precursors were able to secrete exosomes carrying MHC-peptide complexes that were functional in T cell stimulation[83]. Antigen presenting cells (APC), educated with cancer antigens in the form of protein or peptide fragments, therefore produce nanovesicles as APC-surrogates to disseminate the activation of T cells. Isolated APC-exosomes can also be manipulated directly, by pulsing with antigenic peptides of desired specificity, and this scheme has been proposed as a cancer vaccine strategy[84]. APC can, however, also receive a complex set of antigenic information in the form of exosomes secreted by tumor cells[85,86], providing not only tumor-associated antigens but importantly additional information such as cellular stress signals (e.g. heat shock proteins[87]), or even encapsulated RNA[88], to modulate APC-phenotype and control subsequent functions. Some researchers argue that cancer cell-derived exosomes may be an advantageous form of antigen delivery to APCs in vivo[89]. There are, however, conflicting examples where the interaction of cancer-exosomes with myeloid cells may lead to disease exacerbating effects.
Amongst the earliest examples are reports detailing the skewing of dendritic cell differentiation away from a competent antigen presentation phenotype, and towards TGFβ producing myeloid cells capable of negatively regulating T cell responses[90,91]. More recent reports also point to this phenomenon, where monocytes stimulated with cancer cell-derived EV become alternatively-activated/M2-type macrophages, expressing elevated levels of VEGF, IL6, Cox2, and arginase-1 amongst many other tumor-supportive factors[92,93]. Similar modulation of myeloid cells are seen using pancreatic cancer exosomes, giving a suppressive CD14+HLA-DRlow/neg phenotype akin to those elevated within the circulation of patients[94]. Similarly, myeloma-derived EV present within the bone marrow microenvironment can activate myeloid-derived suppressor cells (MDSC) and promote progression[95]. In acute myeloid leukemia, vesicles may play a role in modulating normal myeloporesis and select for cells destined for suppressor-like differentiation[96,97]. It currently remains unclear as to whether this latter phenomenon is also true of solid cancers.
Mechanisms of myeloid activation by EV
Whilst there remains much to be learned about how EV exert such influences on myeloid cells, evidence points to delivery of EV-associated ligands to trigger signaling cascades mediated through toll-like[98,99] or other receptors[100,101]. Moreover, there is a likely additional effect of EV-encapsulated RNAs which may also be delivered to myeloid cells. In one elegant experimental system, cancer cells were engineered to express Cre-recombinase. Cre mRNA was detectable in various EV sub-fractions secreted by these cells, with the predominant EV type appearing to be exosome-like. Transplantation of these cells into mice with a Cre-reporter background led to recombination events at the tumor site, as indicated by β-galactosidase expression following receipt of vesicular Cre mRNA. These Cre-recombined cells were 90% CD45+ leukocytes, principally of a Gr1+CD11b+ MDSC phenotype[102]. The MDSC which had taken up vesicular RNA exhibited more potent suppressive functions compared to their counterparts that had not. The study highlights the in vivo transfer of vesicle-encapsulated RNA to myeloid cells within the tumor microenvironment, resulting in enhanced immune-suppressive function of MDSC.
The influence of EV may, however, not be limited to the local environment. In a highly metastatic breast cancer model EV were again taken up principally by CD45+ bone marrow-derived cells present at distant sites of the lung and liver. These myeloid cells were implicated thereafter in aiding the colonization of these organs by metastasizing cells. Part of this effect may also be due to localized natural killer and T cell suppressive effects attenuating anti-cancer immunity in the premetastatic organs[103]. Dissemination of EV may be more limited in some other cancer types, like glioma, where influences on myeloid phenotypes are not always found in the periphery[104]. In one study, attenuating TLR2-dependent interaction between cancer exosome and MSDS was an effective strategy for limiting MDSC numbers and activation in vivo, and in fact potentiated the effect of chemotherapeutics that would otherwise lead to heightened release of MDSC-activating vesicles. Preventing vesicle effects on MDSC may be a worthwhile therapeutic approach to consider[99].
If lessons are to be gained from these studies of other diverse cancer types, it is indeed likely that EV of prostate cancer origin may also exert local and possibly distant influences on the myeloid cell components of tissues, and profoundly impact the course of the disease. New studies are, however, required in order to examine this further.
Exosome driven metastasis and multidrug resistance
A key step in the progression of various cancers is the invasion of cancer cells into surrounding tissues and subsequent metastasis from the primary tumor site. The 5-year survival rates of patients with prostate cancer drop dramatically following metastasis from the primary tumor. The primary site of metastasis of prostate cancer is the bone; such metastasis remains incurable. An increased concentration of circulating microvesicles has been reported in in vivo models of metastatic prostate cancer[74] and studies by Peinado et al.[43] have demonstrated a role of exosomes in the support of tumor metastasis to the bone.
Exosome-mediated regulation of MMPs
Cancer cell invasion, and disease progression, has been linked to an altered expression of MMPs, key regulators of the extracellular matrix. Fibronectin-mediated binding of exosomes to myeloma cells has been shown to activate p38 and ERK signaling, resulting in elevate expression of DKK1 and MMP-9 and subsequent myeloma progression[105]. More recently, it has been shown that prostate cancer exosomes can regulate MMP-9 expression within osteoclast precursor cells and impair osteoclastic differentiation[106]. Collectively these studies suggest a role of prostate cancer exosomes in the modulation of the bone environment, and subsequent preparation of the metastatic site.
Proteomic analyses have revealed that both cell surface-anchored and soluble matrix metalloproteinases are present in EV isolated from either cell conditioned media or from biofluids[107]. Such vesicular-associated MMPs have been shown to be proteolytically active, and may play a variety of functional roles including direct interaction or cleavage of extracellular matrix proteins or removal of membrane-anchored receptors from target cells[108]. This is supported by further evidence from Hakulinen et al.[109] demonstrating that cancer exosomes can express functionally active MMP-14.
It has long been recognized that platelets can play a role in tumor progression by promoting angiogenesis, resulting in leaky capillaries, and therefore facilitating tumor metastasis. The mechanism of their action has however remained unclear until relatively recently. In a study by Janowska-Wieczorek et al.[110], it was shown that platelet-derived EV, and exosomes released from α-granules, can contribute to metastatic spread via phosphorylation of mitogen activated protein kinase p42/44 and serine/threonine kinase as well as the expression of membrane type 1-MMP (MT1-MMP). The authors also showed that platelet-derived EV are capable of inducing MMP-9 mRNA expression. This study demonstrates that platelet-derived EV can simultaneously activate MT1-MMPs and induce de novo expression of MMPs within cancer cells.
Collectively, these studies suggests that exosomes may be capable of direct contribution to matrix remodeling both within the tumor microenvironment and potentially at distant sites away from the primary tumor.
Exosome-regulated metabolism and drug resistance
Altered cell metabolism is a hallmark of cancer, with many cancer cells demonstrating an increase of aerobic glycolysis. This results in subsequent lowering of pH, leading to increased tumor invasion, proliferation, migration and drug resistance[111,112]. There is growing interest in the role of exosomes, and other EV, as modulators of cancer cell metabolism. It has been reported that pH of the tumor microenvironment is a key factor in regulating both the release and uptake of exosomes by cancer cells[113], suggesting a positive-feedback mechanism resulting in elevated secretion of EV from the tumor microenvironment.
Several studies have demonstrated a link between altered cell metabolism and the development of multidrug resistance in multiple cancer types[114-116], including prostate[117]. Prostate cancer progression is a complex process. In early stage disease the cancer remains androgen sensitive and can be treated with androgen-deprivation therapy. Over time, however, the cancer cells become androgen insensitive. Chemotherapeutic agents, such as docetaxel, can be used to treat androgen-independent disease[118]. By this stage, however, disease relapse is extremely likely and the development of multidrug resistant cancers results in impaired treatment. Several factors have been linked to multidrug resistance[119] including the overexpression of transporter proteins such as P-glycoprotein[120], a well characterized ATP-binding cassette transporter that is involved in the transportation of various substances across the plasma membrane.
Drug-resistant prostate cancer cell lines can transfer drug resistance to non-resistant cells via uptake of exosomes[121], and other EV[122], shed from drug-resistant cells. The initiation of drug-resistance is triggered by vesicular-mediated metabolic alteration of drug-sensitive cells towards a drug-resistant phenotype, with an increase in glycolysis and glycolytic capacity[123]. Such changes in metabolic profile may also be reflected in cargo of EV secreted from the cancer, and may represent a source of biomarkers useful for both diagnosis and monitoring prognosis of disease[124].
In addition, cancer-associated fibroblasts can also regulate metabolic processes within neighboring cancer cells[125]. It was recently shown that cancer-associated fibroblast-derived exosomes can reprogram prostate cancer cell metabolism by downregulating mitochondrial function[126]. Specifically, fibroblast-derived exosomes were shown to inhibit mitochondrial oxidative phosphorylation, resulting in an increase in glycolysis. This may be in part due to delivery of metabolite cargo consisting of lactate, acetate, amino acids, tricarboxylic acid cycle intermediates and lipids from fibroblast-exosomes[126]. Activated stromal cells therefore appear capable of inducing the Warburg effect[127,128], an increased rate of glycolysis followed by lactic acid fermentation, in surrounding cancer cells through EV mediated processes. Despite further studies being required to clarify the effects of metabolic change on cancer progression, stromal cell EV appear to contribute to cancer proliferation and survival in environments low in oxygen and nutrients.
Conclusion
As studies into the role of exosomes in prostate cancer continue, we are likely to learn of further ways in which exosomes regulate disease progression. Whilst studies specifically on prostate cancer/stroma-derived exosomes may appear limited in number there is a great wealth of knowledge on the role of exosomes within other solid cancers that remain useful in informing us of the potential role of exosomes in prostate cancer [Figure 1].
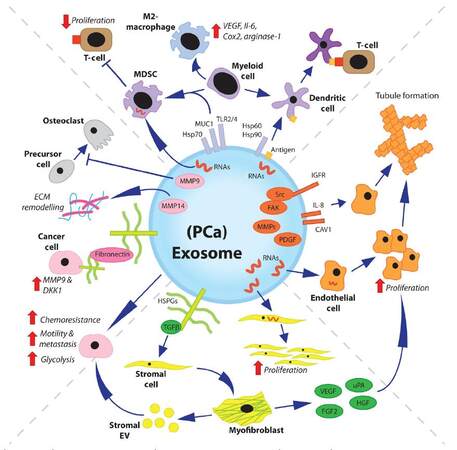
Figure 1. Overview of multiple roles of exosomes in prostate cancer. The previously described roles of prostate cancer exosomes are varied. Many other potential roles demonstrated for exosomes, and/or EV, from other cancer types may also be applicable to prostate cancer exosomes. Cancer exosomes can modulate the immune system. They can transmit tumor antigens to DC, or direct differentiation of myeloid cells towards MDSC/anti-inflammatory (M2) macrophage phenotypes. Exosome-mediated delivery of RNAs can induce endothelial cell proliferation, and exosome-associated proteins can induce endothelial tubule formation. Exosomal-TGFβ can induce differentiation of stromal fibroblasts or MSC towards a pro-angiogenic and tumor supporting myofibroblast-like phenotype. Stromal cell-derived EV can transfer chemoresistance to cancer cells and modulate both cancer cell metastasis and metabolism. Disease progression is further enhanced by cancer exosomes, which have been shown to drive extracellular matrix remodeling and impair osteoclastic differentiation. EV: extracellular vesicles; DC: dendritic cells; MDSC: myeloid-derived suppressor cell; MSC: mesenchymal stem cell; VEGF: vascular endothelial growth factor; MMP: matrix metalloproteinase; FAK: focal adhesion kinase; IGFR: insulin-like growth factor receptor; Src: proto-oncogene tyrosine-protein kinase Src; FGF2: fibroblast growth factor 2; uPA: urokinase-type plasminogen activator; HGF: hepatocyte growth factor, PDGF: platelet-derived growth factor; TGFβ: transforming growth factor beta
Prostate cancer exosomes have been shown to regulate angiogenesis, which may occur through exosome-mediated delivery of growth factors or RNAs. Prostate cancer exosomes have also been shown to further regulate the tumor microenvironment through activation of stromal cells to a disease-supporting myofibroblast-like phenotype and may be capable of modulating myeloid cells, thereby regulating immune and inflammatory responses within the tumor microenvironment. There is sufficient evidence to suggest that exosomes are capable of regulating cancer cell metabolism and tumor metastasis, and are capable of transferring drug resistance from one cell to another. Such exosome-mediated effects, may impact tumor progression through direct or indirect mechanisms. Furthermore, it is not just cancer cell-derived exosomes, but also exosomes from other cell types within the tumor microenvironment, which may facilitate cancer progression. Whilst we may currently only be scratching the surface in terms of the possible roles for exosomes in prostate cancer, it is clear that exosomes are present and actively contribute to the disease process.
It remains unclear why some men with prostate cancer have slow growing, indolent, tumors whilst others develop aggressive late stage disease that is resistant to treatment. There is therefore a growing demand for improved assays capable of predicting those men who are likely to develop aggressive disease. Due to the elevated secretion of exosomes from neoplastic cells, their altered cargo, and their presence within numerous biological fluids, there is substantial interest in the use of exosomes as biomarkers for both diagnostic and prognostic monitoring of disease. Methodologies for isolation of exosomes from biofluids, such as urine and plasma[129], already exist and early testing of exosomes as potential biomarkers of prostate cancer appear promising[130]. Further studies are however required to validate the clinical utility of such assays and to fully understand the relationship between EV, biomarkers, and disease outcome.
Declarations
Authors’ contributionsConceived the study: A. Clayton, J.P. Webber
Performed literature searches and prepared the manuscript: A.P. Shephard, V. Yeung, A. Clayton, J.P. Webber
Revised the manuscript: J.P. Webber
Financial support and sponsorshipThe authors are supported by funding from Prostate Cancer UK (awarded to JPW and AS), Cancer Research Wales (awarded to JPW and AC), Life Science Research Network Wales (an initiative funded through the Welsh Government’s Ser Cymru programme; awarded to AC, JPW, and VY), Tenovus Cancer Care (awarded to AC), Cardiff University (awarded to AC) and Welsh Crucible (awarded to JPW).
Conflicts of interestThere are no conflicts of interest.
Patient consentNot applicable.
Ethics approvalNot applicable.
Copyright© The Author(s) 2017.
REFERENCES
1. Park JE, Tan HS, Datta A, Lai RC, Zhang H, Meng W, Lim SK, Sze SK. Hypoxic tumor cell modulates its microenvironment to enhance angiogenic and metastatic potential by secretion of proteins and exosomes. Mol Cell Proteomics 2010;9:1085-99.
2. Clayton A, Turkes A, Navabi H, Mason MD, Tabi Z. Induction of heat shock proteins in B-cell exosomes. J Cell Sci 2005;118:3631-8.
3. de Jong OG, Verhaar MC, Chen Y, Vader P, Gremmels H, Posthuma G, Schiffelers RM, Gucek M, van Balkom BW. Cellular stress conditions are reflected in the protein and RNA content of endothelial cell-derived exosomes. J Extracell Vesicles 2012;1:18396.
4. Raposo G, Nijman HW, Stoorvogel W, Liejendekker R, Harding CV, Melief CJ, Geuze HJ. B lymphocytes secrete antigen-presenting vesicles. J Exp Med 1996;183:1161-72.
5. Booth AM, Fang Y, Fallon JK, Yang JM, Hildreth JE, Gould SJ. Exosomes and HIV Gag bud from endosome-like domains of the T cell plasma membrane. J Cell Biol 2006;172:923-35.
6. Lötvall J, Hill AF, Hochberg F, Buzás EI, Di Vizio D, Gardiner C, Gho YS, Kurochkin IV, Mathivanan S, Quesenberry P, Sahoo S, Tahara H, Wauben MH, Witwer KW, Théry C. Minimal experimental requirements for definition of extracellular vesicles and their functions: a position statement from the International Society for Extracellular Vesicles. J Extracell Vesicles 2014;3:26913.
7. Kowal J, Arras G, Colombo M, Jouve M, Morath JP, Primdal-Bengtson B, Dingli F, Loew D, Tkach M, Théry C. Proteomic comparison defines novel markers to characterize heterogeneous populations of extracellular vesicle subtypes. Proc Natl Acad Sci U S A 2016;113:E968-77.
9. Webber J, Yeung V, Clayton A. Extracellular vesicles as modulators of the cancer microenvironment. Semin Cell Dev Biol 2015;40:27-34.
11. McDonald DM, Baluk P. Significance of blood vessel leakiness in cancer. Cancer Res 2002;62:5381-5.
12. Nagy JA, Chang SH, Shih SC, Dvorak AM, Dvorak HF. Heterogeneity of the tumor vasculature. Semin Thromb Hemost 2010;36:321-31.
13. Adams RH, Alitalo K. Molecular regulation of angiogenesis and lymphangiogenesis. Nat Rev Mol Cell Biol 2007;8:464-78.
15. Boccaccio C, Andò M, Tamagnone L, Bardelli A, Michieli P, Battistini C, Comoglio PM. Induction of epithelial tubules by growth factor HGF depends on the STAT pathway. Nature 1998;391:285-8.
16. Dunn IF, Heese O, Black PM. Growth factors in glioma angiogenesis: FGFs, PDGF, EGF, and TGFs. J Neurooncol 2000;50:121-37.
17. Hoeben A, Landuyt B, Highley MS, Wildiers H, Van Oosterom AT, De Bruijn EA. Vascular endothelial growth factor and angiogenesis. Pharmacol Rev 2004;56:549-80.
18. Chang YM, Kung HJ, Evans CP. Nonreceptor tyrosine kinases in prostate cancer. Neoplasia 2007;9:90-100.
19. Tatarov O, Mitchell TJ, Seywright M, Leung HY, Brunton VG, Edwards J. SRC family kinase activity is up-regulated in hormone-refractory prostate cancer. Clin Cancer Res 2009;15:3540-9.
20. Min HY, Yun HJ, Lee JS, Lee HJ, Cho J, Jang HJ, Park SH, Liu D, Oh SH, Lee JJ, Wistuba II, Lee HY. Targeting the insulin-like growth factor receptor and Src signaling network for the treatment of non-small cell lung cancer. Mol Cancer 2015;14:113.
21. DeRita RM, Zerlanko B, Singh A, Lu H, Iozzo RV, Benovic JL, Languino LR. c-Src, insulin-like growth factor I receptor, G-protein-coupled receptor kinases and focal adhesion kinase are enriched into prostate cancer cell exosomes. J Cell Biochem 2017;118:66-73.
22. Marx M, Warren SL, Madri JA. pp60(c-src) modulates microvascular endothelial phenotype and in vitro angiogenesis. Exp Mol Pathol 2001;70:201-13.
23. Lopez T, Hanahan D. Elevated levels of IGF-1 receptor convey invasive and metastatic capability in a mouse model of pancreatic islet tumorigenesis. Cancer Cell 2002;1:339-53.
24. Angelucci A, D'Ascenzo S, Festuccia C, Gravina G, Bologna M, Dolo V, Pavan A. Vesicle-associated urokinase plasminogen activator promotes invasion in prostate cancer cell lines. Clin Exp Metastasis 2000;18:163-70.
25. Drew AF, Tucker HL, Kombrinck KW, Simon DI, Bugge TH, Degen JL. Plasminogen is a critical determinant of vascular remodeling in mice. Circ Res 2000;87:133-9.
26. Skog J, Wurdinger T, van Rijn S, Meijer DH, Gainche L, Sena-Esteves M, Curry WT Jr, Carter BS, Krichevsky AM, Breakefield XO. Glioblastoma microvesicles transport RNA and proteins that promote tumour growth and provide diagnostic biomarkers. Nat Cell Biol 2008;10:1470-6.
27. Thompson CA, Purushothaman A, Ramani VC, Vlodavsky I, Sanderson RD. Heparanase regulates secretion, composition, and function of tumor cell-derived exosomes. J Biol Chem 2013;288:10093-9.
28. Hong BS, Cho JH, Kim H, Choi EJ, Rho S, Kim J, Kim JH, Choi DS, Kim YK, Hwang D, Gho YS. Colorectal cancer cell-derived microvesicles are enriched in cell cycle-related mRNAs that promote proliferation of endothelial cells. BMC Genomics 2009;10:556.
29. Huang Z, Feng Y. Exosomes derived from hypoxic colorectal cancer cells promotes angiogenesis through Wnt4 induced beta-catenin signaling in endothelial cells. Oncol Res 2017;25:651-61.
30. Umezu T, Ohyashiki K, Kuroda M, Ohyashiki JH. Leukemia cell to endothelial cell communication via exosomal miRNAs. Oncogene 2013;32:2747-55.
31. Kucharzewska P, Christianson H, Welch J, Svensson K, Fredlund E, Ringnér M, Mörgelin M, Bourseau-Guilmain E, Bengzon J, Belting M. Exosomes reflect the hypoxic status of glioma cells and mediate hypoxia-dependent activation of vascular cells during tumor development. Proc Natl Acad Sci U S A 2013;110:7312-7.
32. King HW, Michael MZ, Gleadle JM. Hypoxic enhancement of exosome release by breast cancer cells. BMC Cancer 2012;12:421.
33. Tadokoro H, Umezu T, Ohyashiki K, Hirano T, Ohyashiki JH. Exosomes derived from hypoxic leukemia cells enhance tube formation in endothelial cells. J Biol Chem 2013;288:34343-51.
34. Webber J, Steadman R, Mason MD, Tabi Z, Clayton A. Cancer exosomes trigger fibroblast to myofibroblast differentiation. Cancer Res 2010;70:9621-30.
35. Webber JP, Spary LK, Sanders AJ, Chowdhury R, Jiang WG, Steadman R, Wymant J, Jones AT, Kynaston H, Mason MD, Tabi Z, Clayton A. Differentiation of tumour-promoting stromal myofibroblasts by cancer exosomes. Oncogene 2015;34:290-302.
36. Xin X, Yang S, Ingle G, Zlot C, Rangell L, Kowalski J, Schwall R, Ferrara N, Gerritsen ME. Hepatocyte growth factor enhances vascular endothelial growth factor-induced angiogenesis in vitro and in vivo. Am J Pathol 2001;158:1111-20.
37. Zhang YW, Su Y, Volpert OV, Vande Woude GF. Hepatocyte growth factor/scatter factor mediates angiogenesis through positive VEGF and negative thrombospondin 1 regulation. Proc Natl Acad Sci U S A 2003;100:12718-23.
38. Guo X, Oshima H, Kitmura T, Taketo MM, Oshima M. Stromal fibroblasts activated by tumor cells promote angiogenesis in mouse gastric cancer. J Biol Chem 2008;283:19864-71.
39. Noma K, Smalley KS, Lioni M, Naomoto Y, Tanaka N, El-Deiry W, King AJ, Nakagawa H, Herlyn M. The essential role of fibroblasts in esophageal squamous cell carcinoma-induced angiogenesis. Gastroenterology 2008;134:1981-93.
40. Mishra PJ, Humeniuk R, Medina DJ, Alexe G, Mesirov JP, Ganesan S, Glod JW, Banerjee D. Carcinoma-associated fibroblast-like differentiation of human mesenchymal stem cells. Cancer Res 2008;68:4331-9.
41. Lee KH, Kim JR. Hepatocyte growth factor induced up-regulations of VEGF through Egr-1 in hepatocellular carcinoma cells. Clin Exp Metastasis 2009;26:685-92.
42. Chowdhury R, Webber JP, Gurney M, Mason MD, Tabi Z, Clayton A. Cancer exosomes trigger mesenchymal stem cell differentiation into pro-angiogenic and pro-invasive myofibroblasts. Oncotarget 2015;6:715-31.
43. Peinado H, Aleckovic M, Lavotshkin S, Matei I, Costa-Silva B, Moreno-Bueno G, Hergueta-Redondo M, Williams C, García-Santos G, Ghajar C, Nitadori-Hoshino A, Hoffman C, Badal K, Garcia BA, Callahan MK, Yuan J, Martins VR, Skog J, Kaplan RN, Brady MS, Wolchok JD, Chapman PB, Kang Y, Bromberg J, Lyden D. Melanoma exosomes educate bone marrow progenitor cells toward a pro-metastatic phenotype through MET. Nat Med 2012;18:883-91.
44. Quail DF, Joyce JA. Microenvironmental regulation of tumor progression and metastasis. Nat Med 2013;19:1423-37.
45. Tuxhorn JA, Ayala GE, Smith MJ, Smith VC, Dang TD, Rowley DR. Reactive stroma in human prostate cancer: induction of myofibroblast phenotype and extracellular matrix remodeling. Clin Cancer Res 2002;8:2912-23.
46. Hinz B, Celetta G, Tomasek JJ, Gabbiani G, Chaponnier C. Alpha-smooth muscle actin expression upregulates fibroblast contractile activity. Mol Biol Cell 2001;12:2730-41.
47. Webber J, Meran S, Steadman R, Phillips A. Hyaluronan orchestrates transforming growth factor-beta1-dependent maintenance of myofibroblast phenotype. J Biol Chem 2009;284:9083-92.
48. Dvorak HF. Tumors: wounds that do not heal. Similarities between tumor stroma generation and wound healing. N Engl J Med 1986;315:1650-9.
50. Rhim AD, Oberstein PE, Thomas DH, Mirek ET, Palermo CF, Sastra SA, Dekleva EN, Saunders T, Becerra CP, Tattersall IW, Westphalen CB, Kitajewski J, Fernandez-Barrena MG, Fernandez-Zapico ME, Iacobuzio-Donahue C, Olive KP, Stanger BZ. Stromal elements act to restrain, rather than support, pancreatic ductal adenocarcinoma. Cancer Cell 2014;25:735-47.
51. Olumi AF, Grossfeld GD, Hayward SW, Carroll PR, Tlsty TD, Cunha GR. Carcinoma-associated fibroblasts direct tumor progression of initiated human prostatic epithelium. Cancer Res 1999;59:5002-11.
52. Orimo A, Gupta PB, Sgroi DC, Arenzana-Seisdedos F, Delaunay T, Naeem R, Carey VJ, Richardson AL, Weinberg RA. Stromal fibroblasts present in invasive human breast carcinomas promote tumor growth and angiogenesis through elevated SDF-1/CXCL12 secretion. Cell 2005;121:335-48.
53. Tomasek JJ, Gabbiani G, Hinz B, Chaponnier C, Brown RA. Myofibroblasts and mechano-regulation of connective tissue remodelling. Nat Rev Mol Cell Biol 2002;3:349-63.
54. Calvo F, Ege N, Grande-Garcia A, Hooper S, Jenkins RP, Chaudhry SI, Harrington K, Williamson P, Moeendarbary E, Charras G, Sahai E. Mechanotransduction and YAP-dependent matrix remodelling is required for the generation and maintenance of cancer-associated fibroblasts. Nat Cell Biol 2013;15:637-46.
55. Shimoda M, Principe S, Jackson HW, Luga V, Fang H, Molyneux SD, Shao YW, Aiken A, Waterhouse PD, Karamboulas C, Hess FM, Ohtsuka T, Okada Y, Ailles L, Ludwig A, Wrana JL, Kislinger T, Khokha R. Loss of the Timp gene family is sufficient for the acquisition of the CAF-like cell state. Nat Cell Biol 2014;16:889-901.
56. Verona EV, Elkahloun AG, Yang J, Bandyopadhyay A, Yeh IT, Sun LZ. Transforming growth factor-beta signaling in prostate stromal cells supports prostate carcinoma growth by up-regulating stromal genes related to tissue remodeling. Cancer Res 2007;67:5737-46.
57. Antonyak MA, Li B, Boroughs LK, Johnson JL, Druso JE, Bryant KL, Holowka DA, Cerione RA. Cancer cell-derived microvesicles induce transformation by transferring tissue transglutaminase and fibronectin to recipient cells. Proc Natl Acad Sci U S A 2011;108:4852-7.
58. Halin Bergstrom S, Hagglof C, Thysell E, Bergh A, Wikstrom P, Lundholm M. Extracellular vesicles from metastatic rat prostate tumors prime the normal prostate tissue to facilitate tumor growth. Sci Rep 2016;6:31805.
59. Brennen WN, Chen S, Denmeade SR, Isaacs JT. Quantification of mesenchymal stem cells (MSCs) at sites of human prostate cancer. Oncotarget 2013;4:106-17.
60. Karnoub AE, Dash AB, Vo AP, Sullivan A, Brooks MW, Bell GW, Richardson AL, Polyak K, Tubo R, Weinberg RA. Mesenchymal stem cells within tumour stroma promote breast cancer metastasis. Nature 2007;449:557-63.
61. Jung Y, Kim JK, Shiozawa Y, Wang J, Mishra A, Joseph J, Berry JE, McGee S, Lee E, Sun H, Wang J, Jin T, Zhang H, Dai J, Krebsbach PH, Keller ET, Pienta KJ, Taichman RS. Recruitment of mesenchymal stem cells into prostate tumours promotes metastasis. Nat Commun 2013;4:1795.
62. Cho JA, Park H, Lim EH, Lee KW. Exosomes from breast cancer cells can convert adipose tissue-derived mesenchymal stem cells into myofibroblast-like cells. Int J Oncol 2012;40:130-8.
63. Cho JA, Park H, Lim EH, Kim KH, Choi JS, Lee JH, Shin JW, Lee KW. Exosomes from ovarian cancer cells induce adipose tissue-derived mesenchymal stem cells to acquire the physical and functional characteristics of tumor-supporting myofibroblasts. Gynecol Oncol 2011;123:379-86.
64. Gu J, Qian H, Shen L, Zhang X, Zhu W, Huang L, Yan Y, Mao F, Zhao C, Shi Y, Xu W. Gastric cancer exosomes trigger differentiation of umbilical cord derived mesenchymal stem cells to carcinoma-associated fibroblasts through TGF-beta/Smad pathway. PLoS One 2012;7:e52465.
65. Paggetti J, Haderk F, Seiffert M, Janji B, Distler U, Ammerlaan W, Kim YJ, Adam J, Lichter P, Solary E, Berchem G, Moussay E. Exosomes released by chronic lymphocytic leukemia cells induce the transition of stromal cells into cancer-associated fibroblasts. Blood 2015;126:1106-17.
66. Song YH, Warncke C, Choi SJ, Choi S, Chiou AE, Ling L, Liu HY, Daniel S, Antonyak MA, Cerione RA, Fischbach C. Breast cancer-derived extracellular vesicles stimulate myofibroblast differentiation and pro-angiogenic behavior of adipose stem cells. Matrix Biol 2017;60-61:190-205.
67. Aluwihare P, Mu Z, Zhao Z, Yu D, Weinreb PH, Horan GS, Violette SM, Munger JS. Mice that lack activity of alphavbeta6- and alphavbeta8-integrins reproduce the abnormalities of Tgfb1- and Tgfb3-null mice. J Cell Sci 2009;122:227-32.
68. Minagawa S, Lou J, Seed RI, Cormier A, Wu S, Cheng Y, Murray L, Tsui P, Connor J, Herbst R, Govaerts C, Barker T, Cambier S, Yanagisawa H, Goodsell A, Hashimoto M, Brand OJ, Cheng R, Ma R, McKnelly KJ, Wen W, Hill A, Jablons D, Wolters P, Kitamura H, Araya J, Barczak AJ, Erle DJ, Reichardt LF, Marks JD, Baron JL, Nishimura SL. Selective targeting of TGF-beta activation to treat fibroinflammatory airway disease. Sci Transl Med 2014;6:241ra79.
69. Huan J, Hornick NI, Shurtleff MJ, Skinner AM, Goloviznina NA, Roberts CT Jr, Kurre P. RNA trafficking by acute myelogenous leukemia exosomes. Cancer Res 2013;73:918-29.
70. Zhou W, Fong MY, Min Y, Somlo G, Liu L, Palomares MR, Yu Y, Chow A, O'Connor ST, Chin AR, Yen Y, Wang Y, Marcusson EG, Chu P, Wu J, Wu X, Li AX, Li Z, Gao H, Ren X, Boldin MP, Lin PC, Wang SE. Cancer-secreted miR-105 destroys vascular endothelial barriers to promote metastasis. Cancer Cell 2014;25:501-15.
71. Zhang H, Deng T, Liu R, Bai M, Zhou L, Wang X, Li S, Wang X, Yang H, Li J, Ning T, Huang D, Li H, Zhang L, Ying G, Ba Y. Exosome-delivered EGFR regulates liver microenvironment to promote gastric cancer liver metastasis. Nat Commun 2017;8:15016.
72. Meehan B, Rak J, Di Vizio D. Oncosomes - large and small: what are they, where they came from? J Extracell Vesicles 2016;5:33109.
73. Di Vizio D, Kim J, Hager MH, Morello M, Yang W, Lafargue CJ, True LD, Rubin MA, Adam RM, Beroukhim R, Demichelis F, Freeman MR. Oncosome formation in prostate cancer: association with a region of frequent chromosomal deletion in metastatic disease. Cancer Res 2009;69:5601-9.
74. Di Vizio D, Morello M, Dudley AC, Schow PW, Adam RM, Morley S, Mulholland D, Rotinen M, Hager MH, Insabato L, Moses MA, Demichelis F, Lisanti MP, Wu H, Klagsbrun M, Bhowmick NA, Rubin MA, D'Souza-Schorey C, Freeman MR. Large oncosomes in human prostate cancer tissues and in the circulation of mice with metastatic disease. Am J Pathol 2012;181:1573-84.
75. Minciacchi VR, Spinelli C, Reis-Sobreiro M, Cavallini L, You S, Zandian M, Li X, Mishra R, Chiarugi P, Adam RM, Posadas EM, Viglietto G, Freeman MR, Cocucci E, Bhowmick NA, Di Vizio D. MYC mediates large oncosome-induced fibroblast reprogramming in prostate cancer. Cancer Res 2017;77:2306-17.
76. Josson S, Gururajan M, Sung SY, Hu P, Shao C, Zhau HE, Liu C, Lichterman J, Duan P, Li Q, Rogatko A, Posadas EM, Haga CL, Chung LW. Stromal fibroblast-derived miR-409 promotes epithelial-to-mesenchymal transition and prostate tumorigenesis. Oncogene 2015;34:2690-9.
77. Leca J, Martinez S, Lac S, Nigri J, Secq V, Rubis M, Bressy C, Sergé A, Lavaut MN, Dusetti N, Loncle C, Roques J, Pietrasz D, Bousquet C, Garcia S, Granjeaud S, Ouaissi M, Bachet JB, Brun C, Iovanna JL, Zimmermann P, Vasseur S, Tomasini R. Cancer-associated fibroblast-derived annexin A6+ extracellular vesicles support pancreatic cancer aggressiveness. J Clin Invest 2016;126:4140-56.
78. Luga V, Zhang L, Viloria-Petit AM, Ogunjimi AA, Inanlou MR, Chiu E, Buchanan M, Hosein AN, Basik M, Wrana JL. Exosomes mediate stromal mobilization of autocrine Wnt-PCP signaling in breast cancer cell migration. Cell 2012;151:1542-56.
79. Richards KE, Zeleniak AE, Fishel ML, Wu J, Littlepage LE, Hill R. Cancer-associated fibroblast exosomes regulate survival and proliferation of pancreatic cancer cells. Oncogene 2017;36:1770-8.
80. Hu Y, Yan C, Mu L, Huang K, Li X, Tao D, Wu Y, Qin J. Fibroblast-derived exosomes contribute to chemoresistance through priming cancer stem cells in colorectal cancer. PLoS One 2015;10:e0125625.
81. Boelens MC, Wu TJ, Nabet BY, Xu B, Qiu Y, Yoon T, Azzam DJ, Twyman-Saint Victor C, Wiemann BZ, Ishwaran H, Ter Brugge PJ, Jonkers J, Slingerland J, Minn AJ. Exosome transfer from stromal to breast cancer cells regulates therapy resistance pathways. Cell 2014;159:499-513.
82. MacLennan GT, Eisenberg R, Fleshman RL, Taylor JM, Fu P, Resnick MI, Gupta S. The influence of chronic inflammation in prostatic carcinogenesis: a 5-year followup study. J Urol 2006;176:1012-6.
83. Zitvogel L, Regnault A, Lozier A, Wolfers J, Filament C, Tenza D, Ricciardi-Castagnoli P, Raposo G, Amigorena S. Eradication of established murine tumours using a novel cell-free vaccine: dendritic cell-derived exosomes. Nat Med 1998;4:594-600.
84. Andre F, Chaput N, Schartz NEC, Flament C, Aubert N, Bernard J, Lemonnier F, Raposo G, Escudier B, Hsu DH, Tursz T, Amigorena S, Angevin E, Zitvogel L. Exosomes as potent cell-free peptide-based vaccine. I. Dendritic cell-derived exosomes transfer functional MHC class I/peptide complexes to dendritic cells. J Immunol 2004;172:2126-36.
85. Wolfers J, Lozier A, Raposo G, Regnault A, Théry C, Masurier C, Flament C, Pouzieux S, Faure F, Tursz T, Angevin E, Amigorena S, Zitvogel L. Tumor-derived exosomes are a source of shared tumor rejection antigens for CTL cross-priming. Nat Med 2001;7:297-303.
86. Andre F, Schartz NE, Movassagh M, Flament C, Pautier P, Morice P, Pomel C, Lhomme C, Escudier B, Le Chevalier T, Tursz T, Amigorena S, Raposo G, Angevin E, Zitvogel L. Malignant effusions and immunogenic tumour-derived exosomes. Lancet 2002;360:295-305.
87. Chen W, Wang J, Shao C, Liu S, Yu Y, Wang Q, Cao X. Efficient induction of antitumor T cell immunity by exosomes derived from heat-shocked lymphoma cells. Eur J Immunol 2006;36:1598-607.
88. Pegtel DM, Cosmopoulos K, Thorley-Lawson DA, van Eijndhoven MA, Hopmans ES, Lindenberg JL, de Gruijl TD, Würdinger T, Middeldorp JM. Functional delivery of viral miRNAs via exosomes. Proc Natl Acad Sci U S A 2010;107:6328-33.
89. Zeelenberg IS, Ostrowski M, Krumeich S, Bobrie A, Jancic C, Boissonnas A, Delcayre A, Le Pecq JB, Combadière B, Amigorena S, Théry C. Targeting tumor antigens to secreted membrane vesicles in vivo induces efficient antitumor immune responses. Cancer Res 2008;68:1228-35.
90. Valenti R, Huber V, Filipazzi P, Pilla L, Sovena G, Villa A, Corbelli A, Fais S, Parmiani G, Rivoltini L. Human tumor-released microvesicles promote the differentiation of myeloid cells with transforming growth factor-beta-mediated suppressive activity on T lymphocytes. Cancer Res 2006;66:9290-8.
91. Yu S, Liu C, Su K, Wang J, Liu Y, Zhang L, Li C, Cong Y, Kimberly R, Grizzle WE, Falkson C, Zhang HG. Tumor exosomes inhibit differentiation of bone marrow dendritic cells. J Immunol 2007;178:6867-75.
92. de Vrij J, Maas SL, Kwappenberg KM, Schnoor R, Kleijn A, Dekker L, Luider TM, de Witte LD, Litjens M, van Strien ME, Hol EM, Kroonen J, Robe PA, Lamfers ML, Schilham MW, Broekman ML. Glioblastoma-derived extracellular vesicles modify the phenotype of monocytic cells. Int J Cancer 2015;137:1630-42.
93. Xiang X, Poliakov A, Liu C, Liu Y, Deng ZB, Wang J, Cheng Z, Shah SV, Wang GJ, Zhang L, Grizzle WE, Mobley J, Zhang HG. Induction of myeloid-derived suppressor cells by tumor exosomes. Int J Cancer 2009;124:2621-33.
94. Javeed N, Gustafson MP, Dutta SK, Lin Y, Bamlet WR, Oberg AL, Petersen GM, Chari ST, Dietz AB, Mukhopadhyay D. Immunosuppressive CD14+HLA-DRlo/neg monocytes are elevated in pancreatic cancer and "primed" by tumor-derived exosomes. Oncoimmunology 2016;6:e1252013.
95. Wang J, De Veirman K, De Beule N, Maes K, De Bruyne E, Van Valckenborgh E, Vanderkerken K, Menu E. The bone marrow microenvironment enhances multiple myeloma progression by exosome-mediated activation of myeloid-derived suppressor cells. Oncotarget 2015;6:43992-4004.
96. Pyzer AR, Stroopinsky D, Rajabi H, Washington A, Tagde A, Coll M, Fung J, Bryant MP, Cole L, Palmer K, Somaiya P, Karp Leaf R, Nahas M, Apel A, Jain S, McMasters M, Mendez L, Levine J, Joyce R, Arnason J, Pandolfi PP, Kufe D, Rosenblatt J, Avigan D. MUC1-mediated induction of myeloid-derived suppressor cells in patients with acute myeloid leukemia. Blood 2017;129:1791-801.
97. Umansky V, Blattner C, Fleming V, Hu X, Gebhardt C, Altevogt P, Utikal J. Myeloid-derived suppressor cells and tumor escape from immune surveillance. Semin Immunopathol 2017;39:295-305.
98. Bretz NP, Ridinger J, Rupp AK, Rimbach K, Keller S, Rupp C, Marmé F, Umansky L, Umansky V, Eigenbrod T, Sammar M, Altevogt P. Body fluid exosomes promote secretion of inflammatory cytokines in monocytic cells via Toll-like receptor signaling. J Biol Chem 2013;288:36691-702.
99. Gobbo J, Marcion G, Cordonnier M, Dias AM, Pernet N, Hammann A, Richaud S, Mjahed H, Isambert N, Clausse V, Rébé C, Bertaut A, Goussot V, Lirussi F, Ghiringhelli F, de Thonel A, Fumoleau P, Seigneuric R, Garrido C. Restoring anticancer immune response by targeting tumor-derived exosomes with a HSP70 peptide aptamer. J Natl Cancer Inst 2015;108:djv330.
100. Wu L, Zhang X, Zhang B, Shi H, Yuan X, Sun Y, Pan Z, Qian H, Xu W. Exosomes derived from gastric cancer cells activate NF-kappaB pathway in macrophages to promote cancer progression. Tumour Biol 2016;37:12169-80.
101. Song X, Ding Y, Liu G, Yang X, Zhao R, Zhang Y, Zhao X, Anderson GJ, Nie G. Cancer cell-derived exosomes induce mitogen-activated protein kinase-dependent monocyte survival by transport of functional receptor tyrosine kinases. J Biol Chem 2016;291:8453-64.
102. Ridder K, Sevko A, Heide J, Dams M, Rupp AK, Macas J, Starmann J, Tjwa M, Plate KH, Sültmann H, Altevogt P, Umansky V, Momma S. Extracellular vesicle-mediated transfer of functional RNA in the tumor microenvironment. Oncoimmunology 2015;4:e1008371.
103. Wen SW, Sceneay J, Lima LG, Wong CS, Becker M, Krumeich S, Lobb RJ, Castillo V, Wong KN, Ellis S, Parker BS, Möller A. The biodistribution and immune suppressive effects of breast cancer-derived exosomes. Cancer Res 2016;76:6816-27.
104. Iorgulescu JB, Ivan ME, Safaee M, Parsa AT. The limited capacity of malignant glioma-derived exosomes to suppress peripheral immune effectors. J Neuroimmunol 2016;290:103-8.
105. Purushothaman A, Bandari SK, Liu J, Mobley JA, Brown EE, Sanderson RD. Fibronectin on the surface of myeloma cell-derived exosomes mediates exosome-cell interactions. J Biol Chem 2016;291:1652-63.
106. Karlsson T, Lundholm M, Widmark A, Persson E. Tumor cell-derived exosomes from the prostate cancer cell line TRAMP-C1 impair osteoclast formation and differentiation. PloS One 2016;11:e0166284.
108. Yanez-Mo M, Siljander PR, Andreu Z, Zavec AB, Borras FE, Buzas EI, Buzas K, Casal E, Cappello F, Carvalho J, Colás E, Cordeiro-da Silva A, Fais S, Falcon-Perez JM, Ghobrial IM, Giebel B, Gimona M, Graner M, Gursel I, Gursel M, Heegaard NH, Hendrix A, Kierulf P, Kokubun K, Kosanovic M, Kralj-Iglic V, Krämer-Albers EM, Laitinen S, Lässer C, Lener T, Ligeti E, Linē A, Lipps G, Llorente A, Lötvall J, Manček-Keber M, Marcilla A, Mittelbrunn M, Nazarenko I, Nolte-'t Hoen EN, Nyman TA, O'Driscoll L, Olivan M, Oliveira C, Pállinger É, Del Portillo HA, Reventós J, Rigau M, Rohde E, Sammar M, Sánchez-Madrid F, Santarém N, Schallmoser K, Ostenfeld MS, Stoorvogel W, Stukelj R, Van der Grein SG, Vasconcelos MH, Wauben MH, De Wever O. Biological properties of extracellular vesicles and their physiological functions. J Extracell Vesicles 2015;4:27066.
109. Hakulinen J, Sankkila L, Sugiyama N, Lehti K, Keski-Oja J. Secretion of active membrane type 1 matrix metalloproteinase (MMP-14) into extracellular space in microvesicular exosomes. J Cell Biochem 2008;105:1211-8.
110. Janowska-Wieczorek A, Wysoczynski M, Kijowski J, Marquez-Curtis L, Machalinski B, Ratajczak J, Ratajczak MZ. Microvesicles derived from activated platelets induce metastasis and angiogenesis in lung cancer. Int J Cancer 2005;113:752-60.
111. Gatenby RA, Gawlinski ET, Gmitro AF, Kaylor B, Gillies RJ. Acid-mediated tumor invasion: a multidisciplinary study. Cancer Res 2006;66:5216-23.
112. Salimian Rizi B, Caneba C, Nowicka A, Nabiyar AW, Liu X, Chen K, Klopp A, Nagrath D. Nitric oxide mediates metabolic coupling of omentum-derived adipose stroma to ovarian and endometrial cancer cells. Cancer Res 2015;75:456-71.
113. Parolini I, Federici C, Raggi C, Lugini L, Palleschi S, De Milito A, Coscia C, Iessi E, Logozzi M, Molinari A, Colone M, Tatti M, Sargiacomo M, Fais S. Microenvironmental pH is a key factor for exosome traffic in tumor cells. J Biol Chem 2009;284:34211-22.
114. Semenza GL. HIF-1 mediates metabolic responses to intratumoral hypoxia and oncogenic mutations. J Clin Invest 2013;123:3664-71.
115. Zub KA, Sousa MM, Sarno A, Sharma A, Demirovic A, Rao S, Young C, Aas PA, Ericsson I, Sundan A, Jensen ON, Slupphaug G. Modulation of cell metabolic pathways and oxidative stress signaling contribute to acquired melphalan resistance in multiple myeloma cells. PloS One 2015;10:e0119857.
116. Koczula KM, Ludwig C, Hayden R, Cronin L, Pratt G, Parry H, Tennant D, Drayson M, Bunce CM, Khanim FL, Günther UL. Metabolic plasticity in CLL: adaptation to the hypoxic niche. Leukemia 2016;30:65-73.
117. Wartenberg M, Richter M, Datchev A, Günther S, Milosevic N, Bekhite MM, Figulla HR, Aran JM, Pétriz J, Sauer H. Glycolytic pyruvate regulates P-Glycoprotein expression in multicellular tumor spheroids via modulation of the intracellular redox state. J Cell Biochem 2010;109:434-46.
118. Shelley M, Harrison C, Coles B, Staffurth J, Wilt TJ, Mason MD. Chemotherapy for hormone-refractory prostate cancer. Cochrane Database Syst Rev 2006;(4):CD005247.
119. Larsen AK, Escargueil AE, Skladanowski A. Resistance mechanisms associated with altered intracellular distribution of anticancer agents. Pharmacol Ther 2000;85:217-29.
120. Lopes-Rodrigues V, Seca H, Sousa D, Sousa E, Lima RT, Vasconcelos MH. The network of P-glycoprotein and microRNAs interactions. Int J Cancer 2014;135:253-63.
121. Corcoran C, Rani S, O'Brien K, O'Neill A, Prencipe M, Sheikh R, Webb G, McDermott R, Watson W, Crown J, O'Driscoll L. Docetaxel-resistance in prostate cancer: evaluating associated phenotypic changes and potential for resistance transfer via exosomes. PloS One 2012;7:e50999.
122. Zhang FF, Zhu YF, Zhao QN, Yang DT, Dong YP, Jiang L, Xing WX, Li XY, Xing H, Shi M, Chen Y, Bruce IC, Jin J, Ma X. Microvesicles mediate transfer of P-glycoprotein to paclitaxel-sensitive A2780 human ovarian cancer cells, conferring paclitaxel-resistance. Eur J Pharmacol 2014;738:83-90.
123. Lopes-Rodrigues V, Di Luca A, Mleczko J, Meleady P, Henry M, Pesic M, Cabrera D, van Liempd S, Lima RT, O'Connor R, Falcon-Perez JM, Vasconcelos MH. Identification of the metabolic alterations associated with the multidrug resistant phenotype in cancer and their intercellular transfer mediated by extracellular vesicles. Sci Rep 2017;7:44541.
124. Altadill T, Campoy I, Lanau L, Gill K, Rigau M, Gil-Moreno A, Reventos J, Byers S, Colas E, Cheema AK. Enabling metabolomics based biomarker discovery studies using molecular phenotyping of exosome-like vesicles. PloS One 2016;11:e0151339.
125. Brauer HA, Makowski L, Hoadley KA, Casbas-Hernandez P, Lang LJ, Romàn-Pèrez E, D'Arcy M, Freemerman AJ, Perou CM, Troester MA. Impact of tumor microenvironment and epithelial phenotypes on metabolism in breast cancer. Clin Cancer Res 2013;19:571-85.
126. Zhao H, Yang L, Baddour J, Achreja A, Bernard V, Moss T, Marini JC, Tudawe T, Seviour EG, San Lucas FA, Alvarez H, Gupta S, Maiti SN, Cooper L, Peehl D, Ram PT, Maitra A, Nagrath D. Tumor microenvironment derived exosomes pleiotropically modulate cancer cell metabolism. Elife 2016;5:e10250.
128. Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science 2009;324:1029-33.
129. Welton JL, Webber JP, Botos LA, Jones M, Clayton A. Ready-made chromatography columns for extracellular vesicle isolation from plasma. J Extracell Vesicles 2015;4:27269.
130. Duijvesz D, Versluis CY, van der Fels CA, Vredenbregt-van den Berg MS, Leivo J, Peltola MT, Bangma CH, Pettersson KS, Jenster G. Immuno-based detection of extracellular vesicles in urine as diagnostic marker for prostate cancer. Int J Cancer 2015;137:2869-78.
131. Wang J, De Veirman K, Faict S, Frassanito MA, Ribatti D, Vacca A, Menu E. Multiple myeloma exosomes establish a favourable bone marrow microenvironment with enhanced angiogenesis and immunosuppression. J Pathol 2016;239:162-73.
132. Al-Nedawi K, Meehan B, Micallef J, Lhotak V, May L, Guha A, Rak J. Intercellular transfer of the oncogenic receptor EGFRvIII by microvesicles derived from tumour cells. Nat Cell Biol 2008;10:619-24.
133. Al-Nedawi K, Meehan B, Kerbel RS, Allison AC, Rak J. Endothelial expression of autocrine VEGF upon the uptake of tumor-derived microvesicles containing oncogenic EGFR. Proc Natl Acad Sci U S A 2009;106:3794-9.
134. Taraboletti G, D'Ascenzo S, Giusti I, Marchetti D, Borsotti P, Millimaggi D, Giavazzi R, Pavan A, Dolo V. Bioavailability of VEGF in tumor-shed vesicles depends on vesicle burst induced by acidic pH. Neoplasia 2006;8:96-103.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Shephard AP, Yeung V, Clayton A, Webber JP. Prostate cancer exosomes as modulators of the tumor microenvironment. J Cancer Metastasis Treat 2017;3:288-301. http://dx.doi.org/10.20517/2394-4722.2017.32
AMA Style
Shephard AP, Yeung V, Clayton A, Webber JP. Prostate cancer exosomes as modulators of the tumor microenvironment. Journal of Cancer Metastasis and Treatment. 2017; 3: 288-301. http://dx.doi.org/10.20517/2394-4722.2017.32
Chicago/Turabian Style
Shephard, Alex P., Vincent Yeung, Aled Clayton, Jason P. Webber. 2017. "Prostate cancer exosomes as modulators of the tumor microenvironment" Journal of Cancer Metastasis and Treatment. 3: 288-301. http://dx.doi.org/10.20517/2394-4722.2017.32
ACS Style
Shephard, AP.; Yeung V.; Clayton A.; Webber JP. Prostate cancer exosomes as modulators of the tumor microenvironment. J. Cancer. Metastasis. Treat. 2017, 3, 288-301. http://dx.doi.org/10.20517/2394-4722.2017.32
About This Article
Special Issue
Copyright

Data & Comments
Data


 Cite This Article 20 clicks
Cite This Article 20 clicks

 Like This Article 7
likes
Like This Article 7
likes






Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.