
Tumor heterogeneity and therapy resistance - implications for future treatments of prostate cancer

Abstract
Aim: To develop new therapies for prostate cancer, disease heterogeneity must be addressed. This includes patient variation, multi-focal disease, cellular heterogeneity, genomic changes and epigenetic modification. This requires more representative models to be used in more innovative ways.
Methods: This study used a panel of cell lines and primary prostate epithelial cell cultures derived from patient tissue. Several assays were used; alamar blue, colony forming assays, γH2AX and Ki67 immunofluorescence and comet assays. Ptychographic quantitative phase imaging (QPI), a label-free imaging technique, combined with Cell Analysis Toolbox software, was implemented to carry out real-time analysis of cells and to retrieve morphological, kinetic and population data.
Results: A combination of radiation and Vorinostat may be more effective than radiation alone. Primary prostate cancer stem-like cells are more resistant to etoposide than more differentiated cells. Analysis of QPI images showed that cell lines and primary cells differ in their size, motility and proliferation rate. A QPI signature was developed in order to identify two subpopulations of cells within a heterogeneous primary culture.
Conclusion: Use of primary prostate epithelial cultures allows assessment of therapies whilst taking into account cellular heterogeneity. Analysis of rare cell populations and embracing novel techniques may ultimately lead to identifying and overcoming treatment resistance.
Keywords
Introduction
Tumor heterogeneity and therapy resistance are two sides of the same coin; because there is tumor heterogeneity, therapy resistance is inevitable. There are many different kinds of heterogeneity [Figure 1], and so developing new treatments is an increasingly complex task. Not only do we have to consider the differences between patients, giving rise to the need and hope of patient stratification, but we also have to consider that the tumor that has been biopsied may be the larger more detectable one but not necessarily the most aggressive one. Alongside that, we have our dependence on hormone treatments for metastatic prostate cancer, whilst knowing that there are tumor cell subpopulations that are not responsive or develop acquired resistance to those treatments[1]. There is a poor choice of chemotherapy available for prostate cancer and it is typically a last resort, although some progress is being made in this area[2]. Even with all these known variations, only in recent years has the true complexity of prostate cancers emerged[3-6] with genomic and transcriptomic sequencing[7] as well as clonal tracking[8-10]. So, if we set out to test current treatments or develop novel therapies for prostate cancer, we must consider our current drug pipeline from bench to bedside; what models are used, do they take into account the different layers of heterogeneity and are they fit for purpose?
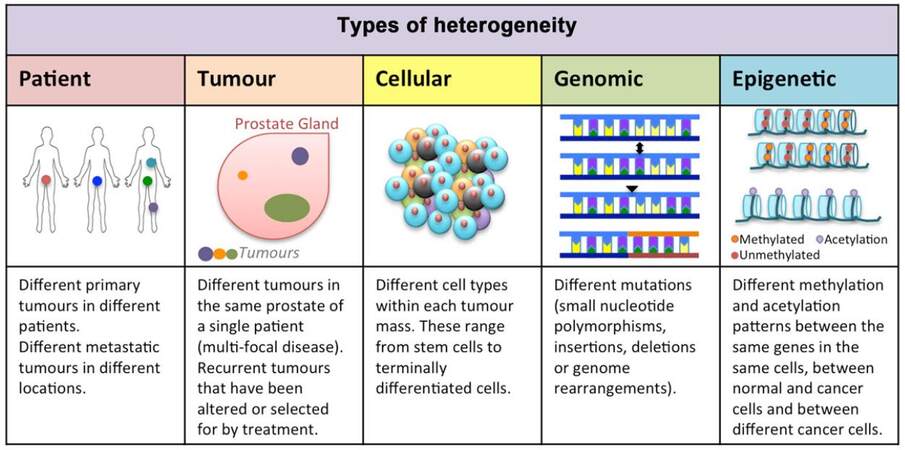
Figure 1. Heterogeneity in prostate cancer. When considering the task of improving current prostate cancer treatments or developing novel therapies, multiple types of heterogeneity have to be taken into account. These include patient tumor heterogeneity, multi-focal disease, intra-tumor cellular heterogeneity, genomic heterogeneity including mutations and gene fusions and finally epigenetic heterogeneity with inherent differences between cell populations but also the possibility of therapy-induced epigenetic changes
Here, we present a study that highlights the variation in results that can be acquired when using different cell line models, and also in comparison to primary prostate epithelial cells cultured from patient tissue. We consider how to tackle the cellular heterogeneity within tumors by assessing cell subpopulations rather than a heterogeneous mixture, as well as introducing a new technique that might be instrumental in assessing drug response whilst simultaneously taking into account cell heterogeneity.
Methods
Culturing of cell lines
PNT1a, PNT2-C2 and LNCaP cells were cultured in Roswell Park Memorial Institute medium (RPMI) with 10% fetal calf serum. BPH-1 cells were cultured in RPMI with 5% fetal calf serum[11]. PC3 cells were cultured in Hams-F12 media with 7% fetal calf serum. P4E6 cells were cultured in Keratinocyte Serum-Free medium (KSFM) with supplements (bovine pituitary extract 50 mg/mL and human recombinant epidermal growth factor 5 ng/mL) and with 2% fetal calf serum[12]. To all media, Glutamine (2 mmol/L) was added. No antibiotics were used in the media. Cells were grown in an incubator at 37 ℃ in a humidified atmosphere containing 5% CO2.
Culturing of primary prostate cells
Tumor tissue was obtained by targeted needle biopsy following radical prostatectomy. Following collection from the hospital, tissue was digested overnight in collagenase, followed by a trypsin digest. Primary prostate epithelial cells derived from patient tissue were cultured in stem cell media (SCM). SCM contains KSFM plus supplements (bovine pituitary extract 50 mg/mL and human recombinant epidermal growth factor 5 ng/mL) with the addition of 2 ng/mL stem cell factor, 100 ng/mL cholera toxin, 1 ng/mL granulocyte macrophage colony-stimulating factor and 2 ng/mL leukemia inhibitory factor. Cells were cultured on Biocoat collagen I 10 cm dishes with irradiated Sandoz inbred strain, thioguanine- and ouabain-resistance (STO) feeder cells. A detailed method of the whole procedure has been published[13]. Patient samples used in this study are listed in Table 1.
Patient samples
| Sample | Operation | Patient age (years) | Diagnosis |
|---|---|---|---|
| 209/12 LA | RP | 64 | Normal |
| 329/13 R | RP | 53 | Normal |
| 434/14 LM | RP | 68 | Normal |
| 048/11 | RP | - | Gl6 (3+3) |
| 018/11 | RP | - | Gl7 |
| 035/11 | RP | - | Gl7 (3+4) |
| 054/11 | RP | 58 | Gl7 (3+4) |
| 665 | RP | 53 | Gl7 (3+4) |
| 049/11 | RP | - | Gl7 (3+4) |
| 087/11 | RP | 68 | Gl7 (3+4) |
| 031/10 | RP | - | Gl7 (3+4) |
| 034/11 | RP | - | Gl7 (3+4) |
| 517/15 RM | RP | 65 | Gl7 (3+4) |
| 329/13 L | RP | 53 | Gl7 (3+4) |
| 209/12 RA | RP | 64 | Gl7 (4+3) |
| 545/15 LB | RP | 69 | Gl7 (4+3) |
| 307/13 LB | RP | 65 | Gl7 (4+3) |
| 545/15 RM | RP | 69 | Gl7 (4+3) |
Selection of stem cells, transit amplifying cells and committed basal cells
Following trypsinisation of primary cultures, cells were first selected using collagen adherence. stem cells (SC) and transit amplifying (TA) cells are α2β1integrinhi and committed basal (CB) cells are α2β1integrinlo. A stringent selection of TA cells can be achieved with 5 min adherence to collagen I. Any non-adherent cells can be passed on to another plate, then any cells not adhered after 20 min are the committed basal cells. A slightly less stringent selection of TA cells can be achieved with a 20 min adherence where any non-adherent cells represent the committed basal population. This latter selection can be used when trying to achieve maximum stem cell (α2β1integrinhi/CD133+) yield. To select stem cells, positive selection using a CD133 microbead kit (Miltenyi Biotec) was used[14].
Ethics approval and patient consent
Patient samples were collected with ethical permission from Castle Hill Hospital (Cottingham, Hull) (ethics number: 07/H1304/121). Use of patient tissue was approved by the Local Research Ethics Committees. Patients gave informed consent and all patient samples were anonymized.
Alamar blue assay
The alamarBlue® reagent (ThermoFisher scientific) was used as an assessment of cell viability. Briefly, cells were plated at 5,000 cells per well in a 96-well plate and treated with drug. Radiation of cells was carried out prior to plating. The alamar blue assay was carried out 24-72 h post-treatment. Cells were in 200 μL and a 1:10 dilution of alamar blue reagent was added. Fluorescence was measured on a plate reader 2 h after addition of reagent.
Colony forming assay
Selected cells (SC and TA) were plated at 100-500 cells per well on a collagen I-coated 6-well plate and treated with 30 µmol/L etoposide or an appropriate dilution of DMSO for 45 min at 37 ℃, washed twice with phosphate buffered saline (PBS) and fresh SCM was added to each well. Cells were kept at 37 ℃ and SCM was changed every second day. An appropriate amount of irradiated feeder cells were added to keep the wells confluent. After 6-14 days SCM was removed and cells were washed once with PBS then stained with crystal violet (1% crystal violet, 10% ethanol in PBS) for 20 min, and after a final PBS wash, the number of colonies was determined. Colonies with < 32 cells and ≥ 32 cells (5 population doublings) were counted. Colony forming assays were also carried out with radiation and Vorinostat treatment. For combination treatments, cells were treated with 0.625, 2.5 or 10 µmol/L of Vorinostat for 30 min then treated with a range of radiation doses.
Treatments with radiation and drugs
An RS2000 X-Ray Biological Irradiator was used, which contains a Comet MXR-165 X-Ray Source (Rad-Source Technologies Inc. GA, USA). A range of radiation doses were administered with a dose rate of 0.02 or 0.08 Gy/s. Addition of Vorinostat (Cambridge Bioscience) was carried out at three concentrations: low, 0.625 μmol/L; medium, 2.5 μmol/L; high, 10 μmol/L.
Comet assays
The comet assay was carried out as previously described[15,16]. Briefly, drug-treated cells were resuspended in 25 μL of PBS and mixed with 225 μL of low melting point agarose. Following mixing, the cells and agarose were spread onto a glass slide that had been pre-coated with 1% agarose in PBS. A clean coverslip was placed on top until the cell-agarose mixture had set. Slides were placed in lysis buffer overnight and then incubated in alkaline solution for 40 min at 4 ℃ then electrophoresed at 23V/300 mA in the alkaline solution for 40 min on ice. This was followed by two washes in neutralising buffer. SYBR Gold was applied at a concentration of 1:10,000 in TE buffer to stain the DNA. Following collection of images on a fluorescent microscope (Nikon Eclipse TE300), comets were quantified using CometScore freeware (TriTek Corp, VA, USA).
Immunofluorescence
Immunocytochemistry was carried out to stain selected populations for DNA damage [γH2AX - anti-phospho-Histone H2A.X (Ser139) clone JBW301, Millipore, UK], proliferation (Ki67 - ab15580, abcam) and a cell marker (CD49b - anti-human CD49b:RPE, Serotec MCA743PET). Primary cells were plated at 10,000 cells per well in collagen I coated 8-well chamber slides or in the case of rare stem cells, all stem cells collected were plated on the slide. Staining of γH2AX was carried out as described previously[15]. Fixation for CD49b and Ki67 staining was with 4% paraformaldehyde, with no permeabilisation step when staining CD49b and with permeabilisation using 0.3% Triton X-100 for Ki67 staining. Alexa Fluor secondary antibodies (goat anti-mouse and goat anti-rabbit) with fluorescent tags were used at a concentration of 1:1000.
Flow cytometry
Flow cytometry was used to measure expression levels of CD49b on primary cells. All cell populations (WP, TA and CB) were harvested and resuspended in 300 μL MACs buffer. Control (REA control (I)-APC) and target (CD49b-APC human clone REA188) antibodies were used (Miltenyi Biotec). Ten μL of antibody was added and incubated with rotation for 10 min at 4 ℃. Cells were washed, resuspended in MACs buffer and analyzed by flow cytometry including a cell only control to set gates.
Image capture using ptychography and image analysis using cell analysis toolbox
Quantitative phase imaging (QPI) was carried out using a VL21 Live Cell Imaging System (Phase Focus Limited, Sheffield, UK), which utilises a method known as ptychography in image formation. The high contrast images generated by the system are label-free and exempt from focal drift, allowing extended time-lapse imaging[17-19]. The high-contrast nature of the images facilitates automated individual cell segmentation and tracking with the Cell Analysis Toolbox® software, which outputs extensive and specific feature measurements for each cell. As a result, data analysis can include information on cell populations in addition to individual cell information such as cell morphology and cell kinetics.
Statistical analysis
Alamar blue assays were performed in triplicate and data presented as % cell viability with percentage standard error. Significance calculations were carried out using the unpaired, nonparametric Mann-Whitney U-test. The P values indicating statistical significance are displayed (*P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.001).
Results
A combination of Radiation and Vorinostat treatment on a panel of cell lines and on primary prostate cells shows a varied response and reduces colony forming ability of primary prostate cells more effectively than either treatment alone.
A previous investigation had shown that prostate cancer stem-like cells were more radio-resistant than progenitor (TA) cells and more differentiated (CB) cells from primary prostate epithelial cells cultured from patient tissue[15]. Pre-treatment with a low dose of a histone modifier, Trichostatin A, resulted in radio sensitisation of the stem-like cells, which was observed as an increase in DNA damage and a decrease in colony forming ability. To follow on from this, a clinically approved histone modifier, Vorinostat, was tested in combination with radiation treatment. First, a panel of cell lines including normal prostate (PNT1a), benign (BPH-1), localized cancer (P4E6) and metastatic cancer (PC3), were tested using alamar blue assays to measure viability following treatment with a combination of seven drug concentrations (0.156/0.3125/0.625/1.25/2.5/5/10 μmol/L) and six radiation doses (2, 5, 10, 25, 50, 75 Gy) with measurements taken at 24, 48 and 72 h. The percentage viability of each of the highest doses alone and in combination in the cell line panel is shown in Figure 2A-C. There is a significant decrease in viability in all cell lines with the combination treatment compared to drug only, however the effect on PC3 cells is minimal, whilst the effect on the cell line derived from the localized cancer, P4E6, is most significant. Viability of primary prostate epithelial cell cultures (n = 6) was then measured following single and combination treatments [Figure 2D and E]. There was a significant reduction in viability with the combination treatment in normal and cancer cells, however the cancer cells showed less of a reduction in viability. We have previously shown that radiation can cause senescence of primary prostate epithelial cells rather than cell death, and so the small reduction in viability as measured by alamar blue could be because the cells are senescing rather than dying[20]. Therefore, we tested the effect of three drug doses with and without 2 Gy of radiation on colony formation [Figure 2F]. As previously seen, 2 Gy of radiation results in a 50% surviving fraction. We used 0.625, 2.5 and 10 μmol/L of Vorinostat, with and without 2 Gy radiation. Drug alone only reduced the surviving fraction by 10%-50% with patient variability observed. The combination treatments reduced surviving fraction by 65%-95%.
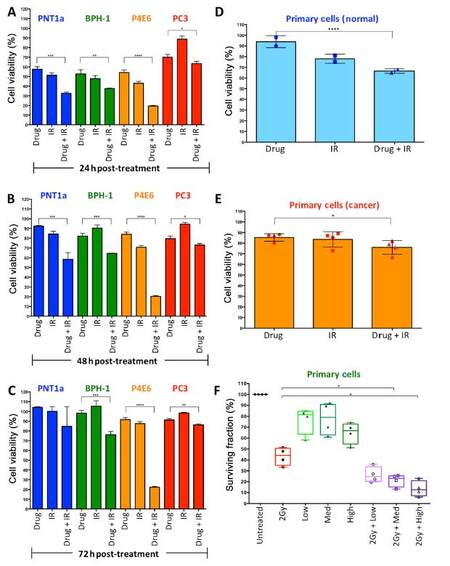
Figure 2. A combination of radiation treatment and Vorinostat has varied effects on viability and colony forming ability in a panel of cell lines and primary prostate epithelial cells. PNT1a, BPH-1, P4E6 and PC3 cells were treated with Vorinostat (10 µmol/L) or radiation (75 Gy) or both and measured using alamar blue assay at 24 h (A), 48 h (B) and 72 h (C) post-treatment. Primary epithelial cultures from six patients, normal (2 samples) (D) and prostate cancer (4 samples) (E) were treated with Vorinostat (10 µmol/L) or radiation (75 Gy) or both and measured using alamar blue assay at 24 h post-treatment. Each symbol represents a different patient sample; (F) primary epithelial cultures from four patients were treated with 2 Gy radiation or three concentrations of Vorinostat (low: 0.625 µmol/L, medium: 2.5 µmol/L, high: 10 µmol/L) or both and assessed for colony forming ability 10-14 days after growth. Colony forming ability is presented as % surviving fraction
Cancer stem-like cells from patient tumor tissue are more resistant to etoposide than the progenitor cells due to a quiescent phenotype.
Previous studies had identified the cancer stem-like cells of primary prostate epithelial cell cultures as being more radiation-resistant[15]. One report showed that stem-like cells from the Du145 cell line were more resistant to etoposide[21]. However, there is currently no experimental evidence determining the effect of chemotherapeutic agents specifically on primary prostate cancer stem-like cells. Therefore, the colony forming ability of selected subpopulations of primary prostate epithelial cells, including stem-like cells and TA cells, were analyzed following treatment with etoposide [Figure 3A]. Cancer stem-like cells showed increased ability to form colonies compared to TA cells post-treatment. Since etoposide is known to cause DNA damage, this was measured in two ways, comet assays [Figure 3B] and γH2AX foci [Figure 3C]. Both methods of measurement showed that stem-like cells sustained less DNA damage following etoposide treatment. Finally, Ki67 staining was carried out, and this indicated that TA cells were more proliferative (50%-90% Ki67-positive cells) than stem-like cells (10%-60% Ki67-positive cells), with patient variability being observed [Figure 3D].
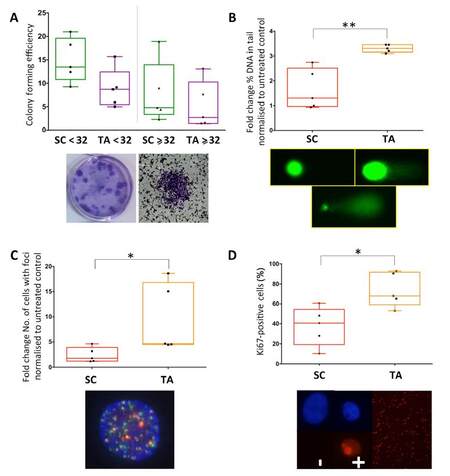
Figure 3. Prostate cancer stem-like cells (SC) sustain less DNA damage and form more colonies than progenitor cells following etoposide treatment, which correlates with less proliferation. Cancer SC and transit amplifying (TA) cells were selected from primary prostate epithelial cell cultures, treated with 30 μmol/L of etoposide and assessed for (A) colony forming ability, (B) comet assay, (C) γH2AX foci formation and (D) Ki67 expression. Each symbol represents a patient sample
Use of QPI to compare growth and proliferation of cell lines to primary prostate epithelial cells cultured from patient tissue.
What these results and previous studies have shown us is that the model that is used can strongly impact the conclusions. This is why we advocate the use of a panel of cell lines, with an understanding of the origin of those cell lines, such that the relevance of the result can be best understood. Cell lines are excellent tools to establish methods and make an initial determination of mechanism of action and effectiveness of a compound. However, our hypothesis is that use of patient-derived primary prostate epithelial cell cultures is more clinically relevant and is more representative of intra-and inter-tumor heterogeneity[13,15,22,23]. Cell lines are usually characterized by expression of certain markers, for example whether they are androgen receptor positive or negative[24,25]. However, an alternative strategy to compare the different cell types might be to look at cell behavior. In order to do this we used a ptychographic QPI label-free imaging technique. We used a panel of cell lines from a variety of sources, PNT2-C2 (normal), BPH-1 (benign), P4E6 (localized cancer), PC3 (bone metastasis), LNCaP (lymph node metastasis) and compared them to a primary culture [Figure 4A]. A 72-h time-lapse experiment was performed (images collected every 6 min). Segmentation and tracking of every cell was carried out, during which each cell track is assigned an identification number [Figure 4B]. The movement of every cell was tracked and measured over time, and tracks were observed as 2D [Figure 4C] and 3D representations [Figure 4D]. The representations demonstrate that the automated tracking procedure used by the Cell Analysis Toolbox (CAT) software is capable of following individual cells. For example with BPH-1 cells a doublet of cells circled round and round each other, which is observed as a spiral over time [Figure 4E]. Morphological measurements (e.g. area, thickness, volume, radius, sphericity) and kinetic measurements (speed, displacement, meandering index) can be extracted from CAT. Of note, cells in the primary cultures are significantly larger on average than all the cell lines [Figure 4F]. In addition, most of the cell lines were much less motile than the primary culture apart from P4E6, which is the closest cell line to a primary culture [Supplementary Video 1]. Even though the primary cells are significantly more motile, which can be measured as mean speed of a track [Figure 4G], they actually show slower growth and proliferation than the cell lines. Indeed, there is a range of growth and proliferation rates in all cell lines measured. The growth and proliferation rate is one example of a unique QPI measurement that takes into account the whole population rather than individual cells and in this case is represented as total dry mass over time[17][Figure 4H].
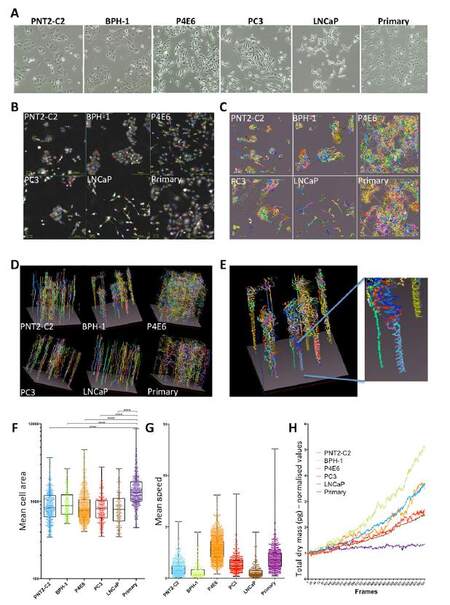
Figure 4. Label-free quantitative phase imaging (QPI) shows that primary prostate cultures divide less frequently than cell lines but undertake significantly more movement in 2D culture. A panel of prostate cell lines was grown alongside a primary prostate epithelial culture in a 6-well dish and time-lapse imaging was carried out. (A) Brightfield images of each cell type; (B) QPI images of each cell type with cell segmentation outlines (colored lines) and cell tracking ID (colored numbers) shown; (C) 2D representation of tracking of each cell type (X-axis, x position; Y-axis, y position); (D) 3D representation of tracking of each cell type (as for 2D but including a Z-axis, time); (E) close up view of the 3D rendition showing the trace of two cells spinning round each other; (F) mean cell area is plotted for each cell type. Each dot represents a single cell track; (G) mean speed is plotted for each cell type. Each dot represents a single cell track; (H) the total dry mass of each frame of the time-lapse video is plotted, which is indicative of cell growth and proliferation
Supplementary Video 1
Ptychographic label-free imaging can distinguish between cell populations in heterogeneous primary epithelial cell cultures.
Although ptychographic QPI can measure detailed morphological and kinetic measurements to distinguish between different cell populations, the power of the technique is to harness these individual cell measurements to take into account cell heterogeneity. We sought to determine whether QPI can distinguish between cell populations within a primary prostate epithelial cell culture. We already know that within these cultures, which have a predominantly basal epithelial cell phenotype, there are three subtypes; rare stem-like cells - CD133+/α2β1 integrinhi, TA cells - CD133-/α2β1 integrinhi and CB cells - α2β1 integrinlo. First, we enriched for TA and CB cells using rapid collagen adherence to select the TA cells (which also contains the rare stem cell population). Immunofluorescence staining highlights the high expression of α2β1 integrin in TA cells and the low expression in CB cells [Figure 5A]. Staining of the whole population shows a mixture of cells with different fluorescent intensities. Staining the cells with CD49b and analyzing by flow cytometry also shows the separation of the two populations [Figure 5B]. After selection, QPI was carried out [Figure 5C] and an analysis using the CAT was completed. A QPI signature was established for each cell type [Figure 5D and E], indicating that CB cells had a larger mass and size [Figure 5D and E] than the TA cells. The TA cells had a higher value relating to cell sphericity compared to the CB cells [Figure 5F] Significantly, once these parameters were established, a heterogeneous (unselected) culture of primary prostate epithelial cells was analyzed. The area measurement from the ptychographic signatures of each cell type was applied to the images of the mixed culture and the software was able to identify TA and CB cells within the culture [Figure 5G].
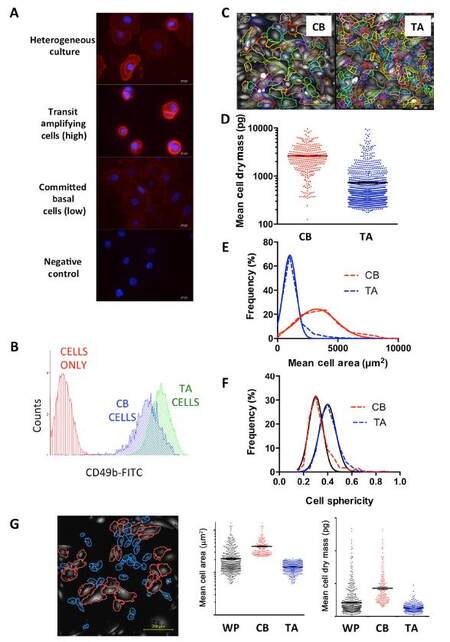
Figure 5. Signatures of two populations of cells within primary prostate cultures can be characterized from quantitative phase imaging (QPI) data and used to identify different cell populations within heterogeneous cultures. (A) Immunofluorescence of CD49b in a mixed culture of cells, transit amplifying (TA) cells and committed basal (CB) cells; (B) flow cytometry of TA and CB cells using the CD49b surface marker; (C) QPI images of TA and CB cells showing cell segmentation outlines (colored lines). Data from QPI analysis of each cell type was measured including (D) mean cell dry mass, (E) mean cell area and (F) cell sphericity; (G) analysis of a mixed culture of cells with gates applied to separate out the two cell populations on the basis of cell area. Data from the whole population (WP) and each cell type was measured and plotted as mean cell area and mean cell dry mass
Discussion
These studies highlight that the use of a single cell line is insufficient to make a conclusion about efficacy and mechanism of action of a treatment. In addition, using a panel of cell lines may also not be a great improvement because results from experiments in cell lines have been seen here and in other studies to be quite different from primary cells[26-28]. Cells in primary cultures have compensatory signaling pathways that have been lost in cell lines, and so an inhibitor that works well in cell lines may be less effective or ineffective in primary cultures[28]. This is one explanation for such high attrition rate in the drug pipeline; weak, incomplete, unrepresentative or inappropriate models. Also, it has previously been shown that the DNA methylation profile is quite different in cell lines compared to primary cells and indeed between different primary cell subpopulations[29,30], thus impacting how cells respond to various treatments. Indeed, epigenetic changes can also be induced in response to treatments such as radiation, which relates to radioresistance and radiosensitization[31]. The use of primary cells from both normal and cancerous patient tissue as part of the drug pipeline may be at least part of the solution. Indeed, use of this model, in vitro primary cell culture, was critical in the development of an oncolytic adenovirus for prostate cancer[32-34], which is currently in clinical trials.
Results presented here and previous studies looking at cancer stem-like cells[15] suggest that a combination treatment of Vorinostat and radiation may be more effective in treating prostate cancer than radiation alone. Since Vorinostat is already clinically-approved[35], a move to clinical trials for the combination treatment could be swift. However, before this could happen, a prognostic indicator and/or a measurement output, other than overall survival, to show any differential response of the combination treatment would be required. This could be something similar to the PORTOS score; predictor of response to postoperative radiotherapy in prostate cancer[36]. Alternatively, a pre-treatment prognostic gene signature could be of use to decide which patients would benefit from a new combination treatment[37]. We carried out a preliminary analysis to measure gene expression, using “DNA damage signaling pathway” polymerase chain reaction arrays, in primary cultures. Cell subpopulations (SC/TA/CB) selected and enriched from primary cultures derived from different disease states (benign prostatic hyperplasia, Gleason 7 prostate cancers and high Gleason prostate cancers) were used, both untreated and treated with radiation (2 Gy). The results illustrated variation between patients, between disease state, and between each cell type (SC, TA, CB). Exploring the heterogeneity of gene expression between disease states and between cell types with and without treatment may ultimately lead to novel drug targets being exploited[38,39].
This is the first report of chemotherapy resistance of cancer stem-like cells from primary prostate epithelial cultures. This study only shows the resistance to etoposide, however we anticipate that this would also be true for other chemotherapeutic drugs that act as cell cycle inhibitors since it appears that the reduced proliferation rate of the stem cells is acting as a resistance mechanism. This result also highlights the need to enrich and/or sort for subpopulations of cells within the patient cell cultures[22] to observe the response of rare populations of cells, since they can be masked when looking at the whole population.
The use of QPI illustrates behavioral differences between cell lines and primary cells. By making measurements encompassing morphological, kinetic and population data a cell signature for each cell type can be established. One significant observation is the larger size of primary cells. Also, the different growth and proliferation rates of the cell lines and primary cells will impact the length of time for drugs to take effect. In addition, it will be of interest to explore the meaning behind the increased cell motility of the primary cultures. Since, ptychography is able to identify heterogeneous populations within a culture, the hope for this technique is to use analysis post-treatment to observe cell behavior in real-time and to detect any inherently resistant cells within the heterogeneous primary cultures.
Going forward, using primary prostate epithelial cultures, as part of the lab to clinic pipeline, has many advantages including patient variation, current and follow-up pathology, correlation with patient outcome, representation of modern disease, close-to-patient, clinically relevant and less adapted to tissue culture conditions than cell lines. The model is also flexible since the cultures with typically a basal phenotype can be pushed to differentiate and express luminal markers[40,41] and 3D spheroid culture is also possible[42]. Even in the 2D culture, microenvironment studies can be carried out using the STO feeder cells as a stromal mimic. This technique has been used to elegant effect, where STO feeder cells were engineered to express human IL-4, and this resulted in an increase in clonogenic potential of primary prostate epithelial cells through the STAT6 signaling pathway[43].
Several recent studies using primary prostate epithelial cultures have shown the heterogeneity of response to current and novel treatments including autophagy[20,26], necrosis[27], cell differentiation[44,45], apoptosis, DNA damage[15,27], cell cycle arrest and senescence[20][Figure 6]. Several of these can act as a crossroads for a cell resulting in cell survival or cell death and if we are able to predict which response may occur then we may be able to manipulate it towards cell death. It is not appropriate to totally rely on endpoint assays; we cannot be satisfied with a 90% reduction in cell viability without questioning what is happening in the other 10% of cells, and indeed characterizing these cells relative to the whole population. If we can identify mechanisms of resistance in bulk populations as well as rare cell populations we will be more able to design biologically relevant combination treatments. In addition, there shouldn’t be too much reliance on a single model. All models have their advantages and limitations; the important thing is to acknowledge them rather than to ignore them. In terms of primary prostate cultures, heterogeneity provides an advantage to testing therapies rather than a confounding factor. If we are able to use techniques such as QPI to measure each individual cell as a data point we should be able to tease apart the variation in cell responses to different treatments as well as identifying and characterizing resistant cells. Ultimately, the hope is that this could lead to more targeted use of current drugs as well as better testing of novel treatments prior to clinical trials.
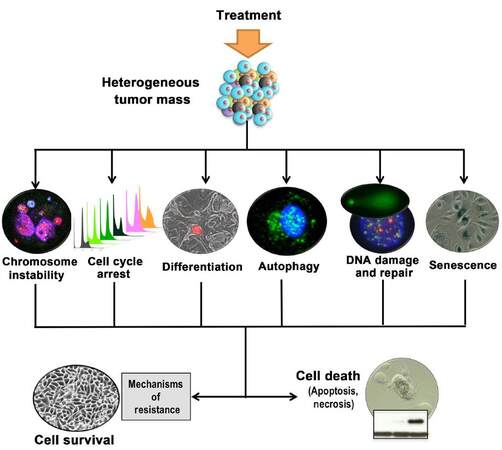
Figure 6. Consequences of treatment; paths to resistance or death. Inducing cell death in cancer cells is not a single pathway and initial treatments may push cells to many different outcomes. To overcome cell thresholds and safeguards and push cells towards cell death, other stimuli may be required. This could involve sensitisers to make the initial treatment more effective or it could include inhibitors to prevent activation of cell survival mechanisms. Heterogeneity of response dictates that combination treatments are likely to be more effective
Declarations
AcknowledgmentsOur sincere appreciation goes to the patients from Castle Hill Hospital, Cottingham, for providing prostate tissue, and also goes to Prof. Simon Hayward for BPH-1 cells.
Authors’ contributionsConceived the overall study and wrote the manuscript: F.M. Frame
Designed the graphics in Figures 1 and 6 and executed the work in Figure 2: F.M. Frame
Conceived and executed the experiments in Figures 4 and 5: F.M. Frame, A.R. Noble
Provided technical expertise integral to completion of the experimental works: H.F. Walker
Conceived and carried out the data in Figure 3: S. Klein
Provided technical expertise for the Quantitative Phase Imaging work: R. Suman, R. Kasprowicz
Arranged permission, collection and delivery of patient samples: V.M. Mann
Provided patient material and be the liaison between the laboratory and the clinic: M.S. Simms
Oversaw all work and was awarded the funding for this study: N.J. Maitland
Financial support and sponsorshipFMF, ARN, HFW and VMM are funded by a PCUK Innovation Award - RIA15-ST2-022. SK was supported by a White Rose Fund studentship.
Conflicts of interestRK and RS are employees of PhaseFocus Ltd. This company has provided the VL21 microscope for use by FMF and AN. RK and RS have provided technical knowledge and support.
Patient consentPatients gave informed consent and all patient samples were anonymized.
Ethics approvalPatient samples were collected with ethical permission from Castle Hill Hospital (Cottingham, Hull) (Ethics Number: 07/H1304/121). Use of patient tissue was approved by the Local Research Ethics Committees.
Copyright© The Author(s) 2017.
REFERENCES
1. El-Amm J, Aragon-Ching JB. The changing landscape in the treatment of metastatic castration-resistant prostate cancer. Ther Adv Med Oncol 2013;5:25-40.
2. Hwang C. Overcoming docetaxel resistance in prostate cancer: a perspective review. Ther Adv Med Oncol 2012;4:329-40.
3. Barbieri CE, Demichelis F, Rubin MA. Molecular genetics of prostate cancer: emerging appreciation of genetic complexity. Histopathology 2012;60:187-98.
4. Boutros PC, Fraser M, Harding NJ, de Borja R, Trudel D, Lalonde E, Meng A, Hennings-Yeomans PH, McPherson A, Sabelnykova VY, Zia A, Fox NS, Livingstone J, Shiah YJ, Wang J, Beck TA, Have CL, Chong T, Sam M, Johns J, Timms L, Buchner N, Wong A, Watson JD, Simmons TT, P'ng C, Zafarana G, Nguyen F, Luo X, Chu KC, Prokopec SD, Sykes J, Dal Pra A, Berlin A, Brown A, Chan-Seng-Yue MA, Yousif F, Denroche RE, Chong LC, Chen GM, Jung E, Fung C, Starmans MH, Chen H, Govind SK, Hawley J, D'Costa A, Pintilie M, Waggott D, Hach F, Lambin P, Muthuswamy LB, Cooper C, Eeles R, Neal D, Tetu B, Sahinalp C, Stein LD, Fleshner N, Shah SP, Collins CC, Hudson TJ, McPherson JD, van der Kwast T, Bristow RG. Spatial genomic heterogeneity within localized, multifocal prostate cancer. Nat Genet 2015;47:736-45.
5. Fraser M, Berlin A, Bristow RG, van der Kwast T. Genomic, pathological, and clinical heterogeneity as drivers of personalized medicine in prostate cancer. Urol Oncol 2015;33:85-94.
6. Wei L, Wang J, Lampert E, Schlanger S, DePriest AD, Hu Q, Gomez EC, Murakam M, Glenn ST, Conroy J, Morrison C, Azabdaftari G, Mohler JL, Liu S, Heemers HV. Intratumoral and intertumoral genomic heterogeneity of multifocal localized prostate cancer impacts molecular classifications and genomic prognosticators. Eur Urol 2017;71:183-92.
7. Beltran H, Yelensky R, Frampton GM, Park K, Downing SR, MacDonald TY, Jarosz M, Lipson D, Tagawa ST, Nanus DM, Stephens PJ, Mosquera JM, Cronin MT, Rubin MA. Targeted next-generation sequencing of advanced prostate cancer identifies potential therapeutic targets and disease heterogeneity. Eur Urol 2013;63:920-6.
8. Baca SC, Prandi D, Lawrence MS, Mosquera JM, Romanel A, Drier Y, Park K, Kitabayashi N, MacDonald TY, Ghandi M, Van Allen E, Kryukov GV, Sboner A, Theurillat JP, Soong TD, Nickerson E, Auclair D, Tewari A, Beltran H, Onofrio RC, Boysen G, Guiducci C, Barbieri CE, Cibulskis K, Sivachenko A, Carter SL, Saksena G, Voet D, Ramos AH, Winckler W, Cipicchio M, Ardlie K, Kantoff PW, Berger MF, Gabriel SB, Golub TR, Meyerson M, Lander ES, Elemento O, Getz G, Demichelis F, Rubin MA, Garraway LA. Punctuated evolution of prostate cancer genomes. Cell 2013;153:666-77.
9. Cooper CS, Eeles R, Wedge DC, Van Loo P, Gundem G, Alexandrov LB, Kremeyer B, Butler A, Lynch AG, Camacho N, Massie CE, Kay J, Luxton HJ, Edwards S, Kote-Jarai Z, Dennis N, Merson S, Leongamornlert D, Zamora J, Corbishley C, Thomas S, Nik-Zainal S, Ramakrishna M, O'Meara S, Matthews L, Clark J, Hurst R, Mithen R, Bristow RG, Boutros PC, Fraser M, Cooke S, Raine K, Jones D, Menzies A, Stebbings L, Hinton J, Teague J, McLaren S, Mudie L, Hardy C, Anderson E, Joseph O, Goody V, Robinson B, Maddison M, Gamble S, Greenman C, Berney D, Hazell S, Livni N, Group IP, Fisher C, Ogden C, Kumar P, Thompson A, Woodhouse C, Nicol D, Mayer E, Dudderidge T, Shah NC, Gnanapragasam V, Voet T, Campbell P, Futreal A, Easton D, Warren AY, Foster CS, Stratton MR, Whitaker HC, McDermott U, Brewer DS, Neal DE. Analysis of the genetic phylogeny of multifocal prostate cancer identifies multiple independent clonal expansions in neoplastic and morphologically normal prostate tissue. Nat Genet 2015;47:367-72.
10. Gundem G, Van Loo P, Kremeyer B, Alexandrov LB, Tubio JM, Papaemmanuil E, Brewer DS, Kallio HM, Hognas G, Annala M, Kivinummi K, Goody V, Latimer C, O'Meara S, Dawson KJ, Isaacs W, Emmert-Buck MR, Nykter M, Foster C, Kote-Jarai Z, Easton D, Whitaker HC, Group IPU, Neal DE, Cooper CS, Eeles RA, Visakorpi T, Campbell PJ, McDermott U, Wedge DC, Bova GS. The evolutionary history of lethal metastatic prostate cancer. Nature 2015;520:353-7.
11. Hayward SW, Dahiya R, Cunha GR, Bartek J, Deshpande N, Narayan P. Establishment and characterization of an immortalized but non-transformed human prostate epithelial cell line: BPH-1. In Vitro Cell Dev Biol Anim 1995;31:14-24.
12. Maitland NJ, Macintosh CA, Hall J, Sharrard M, Quinn G, Lang S. In vitro models to study cellular differentiation and function in human prostate cancers. Radiat Res 2001;155:133-42.
13. Frame FM, Pellacani D, Collins AT, Maitland NJ. Harvesting human prostate tissue material and culturing primary prostate epithelial cells. Methods Mol Biol 2016;1443:181-201.
14. Collins AT, Habib FK, Maitland NJ, Neal DE. Identification and isolation of human prostate epithelial stem cells based on alpha(2)beta(1)-integrin expression. J Cell Sci 2001;114:3865-72.
15. Frame FM, Pellacani D, Collins AT, Simms MS, Mann VM, Jones GD, Meuth M, Bristow RG, Maitland NJ. HDAC inhibitor confers radiosensitivity to prostate stem-like cells. Br J Cancer 2013;109:3023-33.
16. Sturmey RG, Hawkhead JA, Barker EA, Leese HJ. DNA damage and metabolic activity in the preimplantation embryo. Hum Reprod 2009;24:81-91.
17. Kasprowicz R, Suman R, O'Toole P. Characterising live cell behaviour: traditional label-free and quantitative phase imaging approaches. Int J Biochem Cell Biol 2017;84:89-95.
18. Marrison J, Raty L, Marriott P, O'Toole P. Ptychography -- a label free, high-contrast imaging technique for live cells using quantitative phase information. Sci Rep 2013;3:2369.
19. Suman R, Smith G, Hazel KE, Kasprowicz R, Coles M, O'Toole P, Chawla S. Label-free imaging to study phenotypic behavioural traits of cells in complex co-cultures. Sci Rep 2016;6:22032.
20. Frame FM, Savoie H, Bryden F, Giuntini F, Mann VM, Simms MS, Boyle RW, Maitland NJ. Mechanisms of growth inhibition of primary prostate epithelial cells following gamma irradiation or photodynamic therapy include senescence, necrosis, and autophagy, but not apoptosis. Cancer Med 2016;5:61-73.
21. Yan J, Tang D. Prostate cancer stem-like cells proliferate slowly and resist etoposide-induced cytotoxicity via enhancing DNA damage response. Exp Cell Res 2014;328:132-42.
22. Risbridger GP, Taylor RA. Patient-derived prostate cancer: from basic science to the clinic. Horm Cancer 2016;7:236-40.
23. Collins AT, Berry PA, Hyde C, Stower MJ, Maitland NJ. Prospective identification of tumorigenic prostate cancer stem cells. Cancer Res 2005;65:10946-51.
24. Sobel RE, Sadar MD. Cell lines used in prostate cancer research: a compendium of old and new lines -- part 2. J Urol 2005;173:360-72.
25. Sobel RE, Sadar MD. Cell lines used in prostate cancer research: a compendium of old and new lines -- part 1. J Urol 2005;173:342-59.
26. Ulukaya E, Frame FM, Cevatemre B, Pellacani D, Walker H, Mann VM, Simms MS, Stower MJ, Yilmaz VT, Maitland NJ. Differential cytotoxic activity of a novel palladium-based compound on prostate cell lines, primary prostate epithelial cells and prostate stem cells. PLoS One 2013;8:e64278.
27. Hirst AM, Simms MS, Mann VM, Maitland NJ, O'Connell D, Frame FM. Low-temperature plasma treatment induces DNA damage leading to necrotic cell death in primary prostate epithelial cells. Br J Cancer 2015;112:1536-45.
28. Butler DE, Marlein C, Walker HF, Frame FM, Mann VM, Simms MS, Davies BR, Collins AT, Maitland NJ. Inhibition of the PI3K/AKT/mTOR pathway activates autophagy and compensatory Ras/Raf/MEK/ERK signalling in prostate cancer. Oncotarget 2017;8:56698-713.
29. Pellacani D, Kestoras D, Droop AP, Frame FM, Berry PA, Lawrence MG, Stower MJ, Simms MS, Mann VM, Collins AT, Risbridger GP, Maitland NJ. DNA hypermethylation in prostate cancer is a consequence of aberrant epithelial differentiation and hyperproliferation. Cell Death Differ 2014;21:761-73.
30. Pellacani D, Packer RJ, Frame FM, Oldridge EE, Berry PA, Labarthe MC, Stower MJ, Simms MS, Collins AT, Maitland NJ. Regulation of the stem cell marker CD133 is independent of promoter hypermethylation in human epithelial differentiation and cancer. Mol Cancer 2011;10:94.
31. Peitzsch C, Cojoc M, Hein L, Kurth I, Mabert K, Trautmann F, Klink B, Schrock E, Wirth MP, Krause M, Stakhovsky EA, Telegeev GD, Novotny V, Toma M, Muders M, Baretton GB, Frame FM, Maitland NJ, Baumann M, Dubrovska A. An epigenetic reprogramming strategy to resensitize radioresistant prostate cancer cells. Cancer Res 2016;76:2637-51.
32. Adamson RE, Frazier AA, Evans H, Chambers KF, Schenk E, Essand M, Birnie R, Mitry RR, Dhawan A, Maitland NJ. In vitro primary cell culture as a physiologically relevant method for preclinical testing of human oncolytic adenovirus. Hum Gene Ther 2012;23:218-30.
33. Schenk E, Essand M, Kraaij R, Adamson R, Maitland NJ, Bangma CH. Preclinical safety assessment of Ad[I/PPT-E1A], a novel oncolytic adenovirus for prostate cancer. Hum Gene Ther Clin Dev 2014;25:7-15.
34. Schenk E, Essand M, Bangma CH, Consortium GF, Barber C, Behr JP, Briggs S, Carlisle R, Cheng WS, Danielsson A, Dautzenberg IJ, Dzojic H, Erbacher P, Fisher K, Frazier A, Georgopoulos LJ, Hoeben R, Kochanek S, Koppers-Lalic D, Kraaij R, Kreppel F, Lindholm L, Magnusson M, Maitland N, Neuberg P, Nilsson B, Ogris M, Remy JS, Scaife M, Schooten E, Seymour L, Totterman T, Uil TG, Ulbrich K, Veldhoven-Zweistra JL, de Vrij J, van Weerden W, Wagner E, Willemsen R. Clinical adenoviral gene therapy for prostate cancer. Hum Gene Ther 2010;21:807-13.
35. Frew AJ, Johnstone RW, Bolden JE. Enhancing the apoptotic and therapeutic effects of HDAC inhibitors. Cancer Lett 2009;280:125-33.
36. Zhao SG, Chang SL, Spratt DE, Erho N, Yu M, Ashab HA, Alshalalfa M, Speers C, Tomlins SA, Davicioni E, Dicker AP, Carroll PR, Cooperberg MR, Freedland SJ, Karnes RJ, Ross AE, Schaeffer EM, Den RB, Nguyen PL, Feng FY. Development and validation of a 24-gene predictor of response to postoperative radiotherapy in prostate cancer: a matched, retrospective analysis. Lancet Oncol 2016;17:1612-20.
37. Yang L, Taylor J, Eustace A, Irlam J, Denley H, Hoskin PJ, Alsner J, Buffa FM, Harris AL, Choudhury A, West CML. A gene signature for selecting benefit from hypoxia modification of radiotherapy for high risk bladder cancer patients. Clin Cancer Res 2017;23:4761-8.
38. Birnie R, Bryce SD, Roome C, Dussupt V, Droop A, Lang SH, Berry PA, Hyde CF, Lewis JL, Stower MJ, Maitland NJ, Collins AT. Gene expression profiling of human prostate cancer stem cells reveals a pro-inflammatory phenotype and the importance of extracellular matrix interactions. Genome Biol 2008;9:R83.
39. Kroon P, Berry PA, Stower MJ, Rodrigues G, Mann VM, Simms M, Bhasin D, Chettiar S, Li C, Li PK, Maitland NJ, Collins AT. JAK-STAT blockade inhibits tumor initiation and clonogenic recovery of prostate cancer stem-like cells. Cancer Res 2013;73:5288-98.
40. Frame FM, Hager S, Pellacani D, Stower MJ, Walker HF, Burns JE, Collins AT, Maitland NJ. Development and limitations of lentivirus vectors as tools for tracking differentiation in prostate epithelial cells. Exp Cell Res 2010;316:3161-71.
41. Swift SL, Burns JE, Maitland NJ. Altered expression of neurotensin receptors is associated with the differentiation state of prostate cancer. Cancer Res 2010;70:347-56.
42. Lang SH, Anderson E, Fordham R, Collins AT. Modeling the prostate stem cell niche: an evaluation of stem cell survival and expansion in vitro. Stem Cells Dev 2010;19:537-46.
43. Nappo G, Handle F, Santer FR, McNeill RV, Seed RI, Collins AT, Morrone G, Culig Z, Maitland NJ, Erb HHH. The immunosuppressive cytokine interleukin-4 increases the clonogenic potential of prostate stem-like cells by activation of STAT6 signalling. Oncogenesis 2017;6:e342.
44. Oldridge EE, Walker HF, Stower MJ, Simms MS, Mann VM, Collins AT, Pellacani D, Maitland NJ. Retinoic acid represses invasion and stem cell phenotype by induction of the metastasis suppressors RARRES1 and LXN. Oncogenesis 2013;2:e45.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Frame FM, Noble AR, Klein S, Walker HF, Suman R, Kasprowicz R, Mann VM, Simms MS, Maitland NJ. Tumor heterogeneity and therapy resistance - implications for future treatments of prostate cancer. J Cancer Metastasis Treat 2017;3:302-14. http://dx.doi.org/10.20517/2394-4722.2017.34
AMA Style
Frame FM, Noble AR, Klein S, Walker HF, Suman R, Kasprowicz R, Mann VM, Simms MS, Maitland NJ. Tumor heterogeneity and therapy resistance - implications for future treatments of prostate cancer. Journal of Cancer Metastasis and Treatment. 2017; 3: 302-14. http://dx.doi.org/10.20517/2394-4722.2017.34
Chicago/Turabian Style
Frame, Fiona M., Amanda R. Noble, Sandra Klein, Hannah F. Walker, Rakesh Suman, Richard Kasprowicz, Vin M. Mann, Matt S. Simms, Norman J. Maitland. 2017. "Tumor heterogeneity and therapy resistance - implications for future treatments of prostate cancer" Journal of Cancer Metastasis and Treatment. 3: 302-14. http://dx.doi.org/10.20517/2394-4722.2017.34
ACS Style
Frame, FM.; Noble AR.; Klein S.; Walker HF.; Suman R.; Kasprowicz R.; Mann VM.; Simms MS.; Maitland NJ. Tumor heterogeneity and therapy resistance - implications for future treatments of prostate cancer. J. Cancer. Metastasis. Treat. 2017, 3, 302-14. http://dx.doi.org/10.20517/2394-4722.2017.34
About This Article
Special Issue
Copyright

Data & Comments
Data


 Cite This Article 32 clicks
Cite This Article 32 clicks

 Like This Article 13
likes
Like This Article 13
likes






Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.