
Androgen-AR axis in primary and metastatic prostate cancer: chasing steroidogenic enzymes for therapeutic intervention


Abstract
Androgens play an important role in prostate cancer (PCa) development and progression. Although androgen deprivation therapy remains the front-line treatment for advanced prostate cancer, patients eventually relapse with the lethal form of the disease. The prostate tumor microenvironment is characterised by elevated tissue androgens that are capable of activating the androgen receptor (AR). Inhibiting the steroidogenic enzymes that play vital roles in the biosynthesis of testosterone (T) and dihydrotestosterone (DHT) seems to be an attractive strategy for PCa therapies. Emerging data suggest a role for the enzymes mediating pre-receptor control of T and DHT biosynthesis by alternative pathways in controlling intratumoral androgen levels, and thereby influencing PCa progression. This supports the idea for the development of multi-targeting strategies, involving both dual and multiple inhibitors of androgen-metabolising enzymes that are able to affect androgen synthesis and signalling at different points in the biosynthesis. In this review, we will focus on CYP17A1, AKR1C3, HSD17B3 and SRD5A, as these enzymes play essential roles in all the three androgenic pathways. We will review also the AR as an additional target for the design of bifunctional drugs. Targeting intracrine androgens and AKR1C3 have potential to overcome enzalutamide and abiraterone resistance and improve survival of advanced prostate cancer patients.
Keywords
Introduction
Prostate cancer (PCa) is the most commonly diagnosed cancer in men and the second leading cause of death[1]. Androgens, which regulate normal prostate growth and function by interacting with the androgen receptor (AR), drive PCa growth and play a central role in PCa progression[2]. Individuals diagnosed with high-risk PCa are typically treated with surgery or a combination of radiation and androgen deprivation therapy (ADT) via chronic administration of gonadotropin-releasing hormone analogues, anti-androgens or a combination of these drugs [Table 1]. ADT is considered the standard choice of treatment for men with de novo or recurrent metastatic disease[3]. Initially, ADT provides palliation of symptoms, but the therapeutic effects of castration are usually short lived, with 70% of patients developing signs of disease progression within 2 years despite very low levels of circulating testosterone (T)[4,5]. Many patients will inevitably relapse and ultimately develop castration-resistant prostate cancer (CRPC), which is responsible for the vast majority of PCa mortalities. Although the mechanisms of resistance are multi-factorial, the androgen axis still plays a major role[6]. Evidence accumulated over the past decade clearly indicates that castration-resistant growth, to a large extent, is driven by continued AR signalling, despite castration resulting in only low levels of T in the serum. Emerging literature indicates a complex network of molecular players linked in part with amplification or mutations in androgen receptors allowing activation by progesterone, estrogens and androgen antagonists, generation of alternative splicing variants or with androgen neo-synthesis within the prostate tumour or adrenals[7-10]. Accordingly, both the management of PCa patients and complete abolition of androgens are difficult to achieve. Direct measurement of androgen levels in clinical samples from patients with CRPC reveal residual T (0.2-2.94 ng/g) and dihydrotestosterone (DHT, 0.36-2.19 ng/g) levels in tissue samples, respectively; nonetheless these levels are considered more than sufficient to activate the AR machinery and support tumour cell growth and survival[11]. Additionally, a number of studies have indicated several enzymes are able to facilitate the intratumoral neo-synthesis or conversion of circulating adrenal androgen precursors to the active AR ligands[12].
Therapies and approved drugs for PCa treatment according to its progression
| PCa progression | Therapy | Mechanisms | Drugs | Structure or number in the text | |
|---|---|---|---|---|---|
| Localised disease | Surgery | ||||
| Radiation | |||||
| ADT | GnRH agonists and antagonists | Buserelin | Synthetic peptide | ||
| Goserelin | Synthetic peptide | ||||
| Leuprolide | Synthetic peptide | ||||
| Triptorelin | Synthetic peptide | ||||
| Degarelix | Synthetic peptide | ||||
| Advanced PCa | ADT | AR antagonist | Steroidal | Cyproterone | 50 |
| Non steroidal | Flutamide | 51 | |||
| Nilutamide | 52 | ||||
| Bicalutamide | 53 | ||||
| CRPC | ADT | AR antagonist | Non steroidal | Enzalutamide | 54 |
| Androgen synthesis inhibitors (CYP17 inhibitors) | Abiraterone | 2 | |||
| Chemoterapy | Inductors of microtubule stabilization | Docetaxel | Taxane | ||
| Cabazitaxel | Taxane | ||||
| Metabolic radiation | Alpharadin | Radium-223 | |||
| Vaccine | Sipuleucel-T | - | |||
| Monoclonal antibody | Denosumab | - | |||
This review is focussed on outlining and discussing the key players in the steroidogenic pathway that is tightly linked with the AR activation.
The steroidogenic cascade involved in PCa
Under normal physiological conditions about 60% of androgens produced in the prostate come from circulating T synthesised from cholesterol in the testis. The remainder derives from dehydroepiandrosterone (DHEA) synthesised in the zona reticularis of the adrenal glands [Figure 1]. The prostate itself contributes to androgen anabolism by reducing testicular T to the more potent AR ligand DHT and converting DHEA to T and DHT [Figure 2]. The enzymes converting T to DHT are type 1 or 2 5α-reductase (SRD5A), the type 2 being the predominant isoform in prostate. This mechanism of production of DHT presumably allows the prostate to maintain constitutive levels of AR that are sufficient for activity in the luminal epithelium. The adrenal DHEA taken up by prostate cells as the sulphate derivative is reduced to androstenedione (AD) by a 3β-hydroxysteroid dehydrogenase type 1 (HSD3B1) expressed in prostate basal epithelialcells. This is followed by AD conversion to T by 17β hydroxysteroid dehydrogenase type 5 (HSD17B5). This enzyme is a member of the aldo-ketoreductase family, also known as AKR1C3 (aldo-keto reductase family 1, member 3), is somewhat different to the 17β reductases that are derived from the family of SDRs (short-chain dehydrogenase/reductase). By contrast, the synthesis of T in the testisis mediated by a SDR enzyme, named HSD17B3. In the normal prostate, AKR1C3 has been identified in stromal, endothelial and perineural cells, where its significance appears to be related to the ability to reduce prostaglandin D2 to F2 rather than to the synthesis of T, which can be assumed from the circulation.
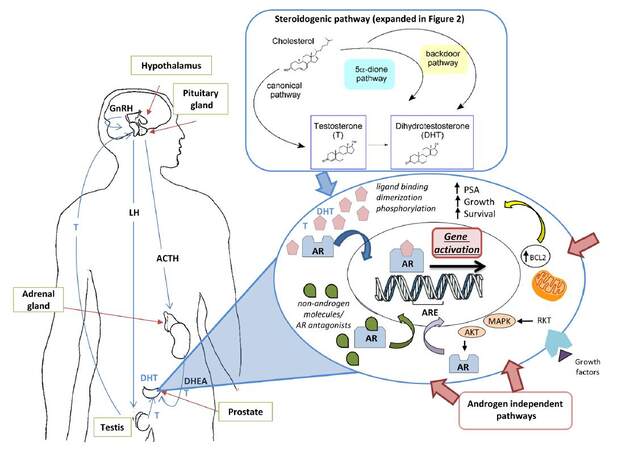
Figure 1. The production of androgens is regulated by the hypothalamic-pituitary-gonadal-adrenal axes. AR activation (dimerisation and phosphorylation) is regulated by both androgen-dependent (blue arrows) and androgen-independent pathways (red arrows). In the androgen-dependent pathway, T and DHT production is catalysed by the steroidogenic enzymes and occurs through the canonical, 5a-dione and backdoor pathways[24]. The androgen-independent pathway includes: (1) AR gain-function mutations; (2) activation by non-androgen steroids or androgen antagonists; (3) activation by non-steroid growth factors (receptor tyrosine kinases are activated and both AKT and MAPK pathways, producing a ligand-independent AR); and (4) increase of AR co-regulators. A parallel survival pathway, involving the anti-apoptotic protein BCL-2, also induces the cancer cell proliferation via bypassing the AR[183,184]. AR: androgen receptor; GnRH: gonadotropin-releasing hormone analogues; T: testosterone; DHT: dihydrotestosterone; ARE: androgen response element; DHEA: dehydroepiandrosterone; LH: luteinizing hormone; ACTH: adreno-cortico-tropic-hormone
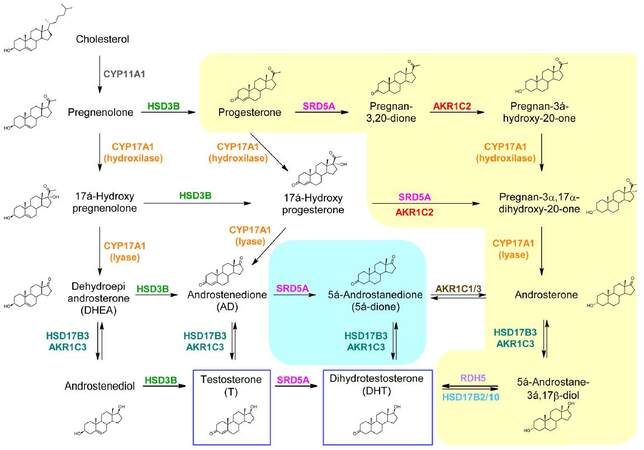
Figure 2. The principal and the two alternative androgen biosynthetic pathways: the canonical pathway is shown on white background, the backdoor pathway is shown on yellow background and the 5a-dione pathway on light blue background. In the squares, production of T and DHT in the development of PCa are shown. PCa: prostate cancer; T: testosterone; DHT: dihydrotestosterone; CYP17A1: cytochrome P450 17A1; HSD3B: 3b-hydroxysteroid dehydrogenase; HSD17B2/3/10: 17b-hydroxysteroid dehydrogenase type 2/3/10; SRD5A: steroid 5a-reductases; AKR1C1/2/3: aldo-keto reductase family 1, member 1/2/3; RDH5: retinol dehydrogenase type 5; AD: androstenedione
Intracellular levels of DHT are also regulated by phase I (reducing) and phase II (conjugating) enzymes that mediate DHT catabolism: AKR1C1 and AKR1C2 (reductive 3α-HSDs) convert DHT to 3α-androstanediol and 3β-androstanediol respectively, which are then glucuronidated by UDP glycosyltransferase UGT2B15 or UGT2B17[13]. 3α-androstanediol can be oxidised back to DHT by HSB17B6, which is expressed in prostatic stromal cells. In PCa patients that have received ADT, the presence of low levels of androgens, relative to high levels of T and DHT, can be maintained by intraprostatic synthesis, which essentially can occur through three putative synthetic pathways: the principal pathway is the classical or “canonical” de novo synthesis that initiates from cholesterol or other intermediates and results in T production. The two alternative pathways, “5α-dione” pathway and the “backdoor” pathway, allow direct synthesis of the AR ligand DHT without the requirement of T as intermediate.
The canonical pathway
This biosynthetic pathway is similar to that occurring in the testes. Androgens are known to be synthesised de novo starting from a number of precursor molecules absorbed from the circulation, including cholesterol, progesterone and adrenal DHEA. Starting from cholesterol, the first step is the conversion to C21 pregnenolone by the cholesterol aliphatic side-chain specific metabolic activity of mitochondrial CYP11A1. The next steps lead to the synthesis of AD by two different pathway branches where the intermediates progesterone or DHEA are formed by CYP17A1 or HSD3B respectively. The subsequent reduction of AD to T is catalysed by AKR1C3. T is further reduced to DHT by SRD5A enzymes. In patients with CRPC, DHEA, derived from the adrenal gland, is the predominant T precursor implicated in this pathway. The strongest evidence for the action of the canonical pathway in the prostate comes from a study published by Fankhauser et al.[14] which is focussed on the incubation of cultures of prostate samples from patients with benign prostatic hyperplasia (BPH), androgen-naive and/or hormone-refractory PCa with various precursor substrates including cholesterol, progesterone, AD, DHEA, and T. The results show the prominence of the conversion of AD to T, suggesting that the canonical pathway is the most pertinent T/DHT synthesis pathway in patients with PCa. These conclusions are supported by findings that expression of the HSD17B isoenzymes, and in particular AKR1C3, key enzymes responsible for the conversion of AD to T, are upregulated in tumour biopsy samples from patients with CRPC[15-17].
The 5α-dione pathway
The 5α-dione pathway allows PCa cells to generate the potent signalling androgen DHT without the need for T as a substrate. In this pathway, the order of reactions is reversed compared to the canonical biosynthesis: AD is initially 5α-reduced to 5α-androstanedione by SRD5A1 and then further reduced to DHT by HSD17B3[18,19]. In contrast, in the canonical biosynthesis AD is the substrate of HSD17B3 that is reduced to T which is then further reduced to DHT by SRD5A. The 5α-dione pathway was first described in 2011, and as such, fewer studies supporting this model are currently available compared with the other two models of androgen synthesis, although indirect evidence is available and supports the clinical relevance of this pathway too[15,17].
The backdoor androgen synthesis pathway
This biosynthetic pathway was originally identified in Tammar wallabies in 2003[20]. It was the first report to demonstrate that in the prostate the “backdoor” pathway was contributing to the synthesis of androgens without the need for androgenic precursors. Here, the progesterone produced by the same reactions as in the canonical pathway, is converted to androsterone by CYP17A1, SRD5A, and AKR1C2. These enzymes are responsible for converting AD to Tin a similar manner to HSD17B3 and AKR1C3 in the canonical pathway, resulting in the conversion of androsterone to 5α-androstane-3α,17β-diol. The final step of the pathway leading to DHT is catalysed by retinol dehydrogenase type 5 (RDH5)[21,22]. This enzyme, upregulated in mice with castration resistance[17,22], mediates a key step in DHT biosynthesis and is one of the few steroidogenic enzymes acting at a single point in the biosynthetic pathway. Studies in LNCaP xenografts indicate that the backdoor pathway might be dominant when tumours are treated with inhibitors of androgen synthesis, including ketoconazole and finasteride, which inhibit CYP17A1, and SRD5A2, respectively[23].
All these pathways (“canonical”, “backdoor” and “5α-dione” pathways, Figure 2), ultimately aim at generating the potent signalling androgen DHT. Involving mainly the same enzymes, they differ in terms of substrate preference and/or reaction sequence. Their occurrence and relative importance in the development and progression of PCa remains controversial, since the experimental evidence comes mainly from preclinical cell culture models, where different results are obtained depending on the cell lines studied or where more clinically-relevant biopsy samples have been used for analysis. The current understanding of androgen synthesis and the evidence for its role in castration resistance, either supporting or rebutting the relevance of each pathway to patients with PCa were recently extensively reviewed by Stuchbery et al.[24].
Inhibiting these steroidogenic enzymes that play vital roles in the biosynthesis of T and DHT seems to be an attractive strategy for the development of therapies for the treatment of PCa. The existence of alternative pathways in PCa leading to the synthesis of T and DHT supports the idea for the development of multi-targeting strategies, involving both dual and multiple inhibitors of androgen-metabolising enzymes that are able to affect androgen synthesis and signalling at different points in the biosynthesis. Therapeutic strategies aimed at more efficiently targeting the steroidogenic pathway could involve the concomitant use of inhibitors targeting two different enzymes or a unique dual-targeting inhibitor able to modulate more than one enzyme in the steroidogenesis pathway. A potential variation of this strategy involves the modulation of an androgen-metabolising enzyme and the AR by a bifunctional targeting molecule. In the following chapters, we will focus on CYP17A1, AKR1C3, HSD17B3 and SRD5A, as these enzymes play essential roles in all the three pathways mentioned above. In Table 2, their different expression level during the progression of PCa is described. Finally, we will review the AR as an additional target for the design of bifunctional drugs.
Different expression level of CYP17A1, AKR1C3, HSD17B3 and SRD5A enzymes during progression of PCa
| Enzyme | Presence | Ref. | |
|---|---|---|---|
| CYP17A1 | Expressed in all PCa and upregulated in CRPC | [15] | |
| AKR1C3 | Expressed 10-16 fold higher in several PCa cell lines with respect to healthy prostate cells and up to 3 fold in androgen responsive and androgen independent PCa cell xenografts upon androgen deprivation | [9] | |
| Upregulated in CRPC, both within the tumor microenvironment and in soft-tissue metastasis | [15-17,52,176,177] | ||
| HSD17B3 | Expressed almost exclusively in the testis, there are some reports of its over-expression in PCa tissues. HSD17B3 mRNA was increased over 30 fold in PCa biopsies and the enzyme has been shown to be upregulated 8-fold in LuCaP-23 and LuCAP-35 PCa cell lines, obtained from metastatic tissues of a patient resistant to castration therapy | [15,22] | |
| SRD5 | A1 | During PCa development its expression increases. A 2-4 fold increase of SRD5A1 expression, induced by activation of AR, has been observed in three androgen-responsive PCa cell lines | [88,89,178-181] |
| A2 | Predominant isoform expressed in the normal prostate. During PCa development, its expression decreases. AR represses SRD5A2 expression | ||
| A3 | Overexpressed in hormone-refractory PCa tissues | [182] | |
CYP17A1
Cytochrome P450 17A1 (CYP17A1, P450c17) plays a major role in the steroidogenic pathway that produces androgens and estrogens. It is expressed principally in the adrenal gland and gonads. In humans, the expression of CYP17A1 is driven by a complex interaction of different transcription factors (TFs) and, differently from rodents[25], it appears not directly influenced by epigenetic regulation[26,27]. Indeed, CpG islands, the sites of the epigenetic methylation, are absent in human CYP17A1 gene[25]. Indirect epigenetic control is however suggested by studies on the inductive effect of 5aza-dC on TFs required for CYP17A1 expression[28]. This membrane-bound protein has both 17α-hydroxylase and a 17,20-lyase activity. The 17α-hydroxylase activity is important for the production of the glucocorticoid cortisol, whereas the 17,20-lyase activity leads to androgen production[29]. The lyase activity is stimulated in a concerted fashion by cytochrome b5 and appears to be an allosteric function rather than via conventional electron transfer mechanism of this co-enzyme[30]. CYP17A1 is required in the three parallel pathways to catalyse the hydroxylation of the steroid ring carbon 17 of pregnenolone to form 17α-hydroxypregnenolone and progesterone to form 17α-hydroxyprogesterone (major product, Figure 2) and 16α-hydroxyprogesterone (minor product).The resulting metabolites undergo the 17,20-lyase reaction by the same enzyme involving the cleavage of the side-chain of the steroid nucleus in order to obtain DHEA and AD, respectively. The androsterone, precursor of DHT in the backdoor pathway is formed by CYP17A1 via metabolism of pregnan-3α-hydroxy-20-one. The need for this enzyme in all the metabolic pathways that allow and maintain the activation of the AR in the prostatic cells makes CYP17A1 one of the most important therapeutic targets in the biosynthesis pathway.
To date, there are eight co-crystal structures of CYP17A1 complexed with an inhibitor or substrate and revealing the characteristic cytochrome P450 fold[31]. The crystal structure of CYP17A1 bound either to abiraterone [Figure 3A] or to galeterone (TOK-001), two clinically trailed CYP17A1 inhibitors (2 and 3, Figure 4), show that both inhibitors bind the haem iron at a 60° angle above the haeme plane while aligning their chemical structures against the central helix with the 3β-OH interacting with Asn 202 in the F helix[32].
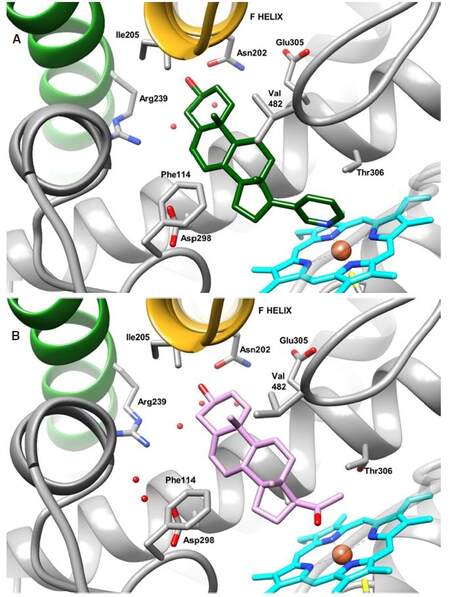
Figure 3. Structures of (A) CYP17A1 in complex with abiraterone (PDB ID 3RUK) and (B) CYP17A1 mutant A105L with substrate pregnenolone (PDB ID 4NKW). Abiraterone is depicted in green, pregnenolone in pink. Carbon atoms of the protein are depicted in grey, the haeme prosthetic group is coloured in blue. Nitrogen, oxygen and sulphur atoms are depicted in blue, red and yellow, respectively. Relevant water molecules are represented by red points
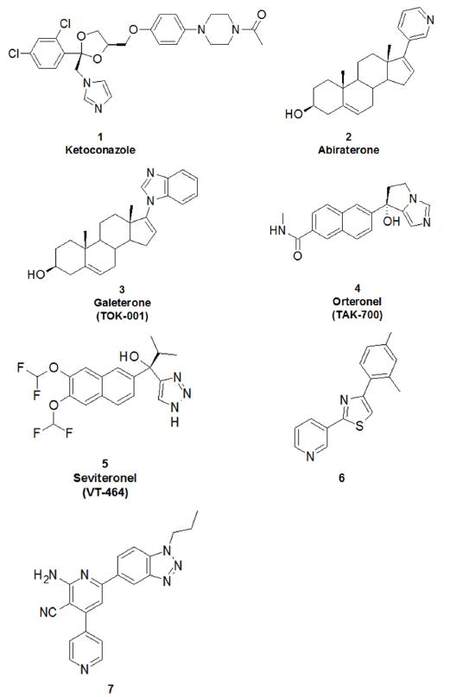
Figure 4. Chemical structures of selected CYP17A1 inhibitors
More recently the co-crystal structure of CYP17A1 mutant Ala105Leu in complex with hydroxylase substrates pregnenolone [Figure 3B], progesterone, 17,20-lyase substrates 17α-hydroxyprogesterone and 17α-hydroxypregnenolone, showed that the general orientation of all physiological substrates in the active site is quite similar to the one observed for abiraterone. Each substrate is aligned in a position that allows the formation of a hydrogen bond with the Asn202 side chain. The 17α-hydroxypregnenolone, a substrate of lyase activity, could also assume a second pose, that is closer to the catalytic iron and further away for Asn202, hence preventing the formation of a hydrogen bond as observed in the first position[33]. This observation could explain the substrate selectivity of the lyase reaction and the increased 17,20-lyase activity after the allosteric binding of cytochrome b5. NMR studies have already established that b5 binds differently to CYP17A1 depending on whether the substrate is pregnenolone or 17α-hydroxypregnenolone[34]. Cytochrome b5 could alter the positioning of 17α-hydroxypregnenolone to the second position, thus increasing the rate of the lyase reaction. These structural studies provide a rationale to increase our understanding of this enzyme’s dual hydroxylase and lyase activity and facilitate the design of inhibitors that may specifically interact with the androgen-generating lyase activity, ultimately leading to novel therapeutics with improved efficacy.
Several well-characterised CYP17A1 inhibitors have been discovered over the years for the treatment of advanced PCa [Figures 4 and 5] and several excellent reviews have been published on this topic[35]. Only abiraterone (2, Figure 4) has been approved for clinical use for the treatment of CRPC. Abiraterone, administered as an acetate prodrug, consists of a steroidal scaffold with a pyridin-3-yl moiety in position 17 that inhibits CYP17A1 through coordination to the haem iron[32]. This coordination obstructs the binding of endogenous substrates, leading to the competitive inhibition of CYP17A1. Recently, the steroidal CYP17A1 inhibitor galeterone (3, TOK-001)[36], has been shown to be three times more potent than abiraterone in CYP17 enzyme activity assays[37].
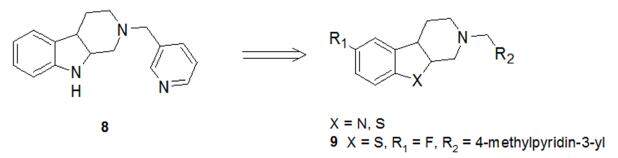
Figure 5. Design strategy of metal-binding inhibitors of CYP17A1
Together, the steroidal scaffold and the aromatic nitrogen-containing ring give to abiraterone a promiscuous profile with affinity toward steroid receptors and other CYP enzymes, which are likely to contribute to the undesirable side effects observed in patients receiving abiraterone treatment including liver dysfunction, characterised by elevated total bilirubin, aspartate aminotransferase and alanine aminotransferase[38].
Thus, these potential adverse effects of steroidal drugs triggered the efforts to develop nonsteroidal CYP17A1 inhibitors. Combinatorial synthesis programmes have been initiated by pharmaceutical companies to identify non-steroidal inhibitors to avoid the side effects associated with the steroidal scaffold and two such compounds, orteronel (4) and seviteronel (5), have been developed and are subject to clinical trials. Although, orteronel (TAK-700), an oralimidazole based inhibitor[39] had a 5-fold selectivity for 17,20-lyase activity in comparison with the 17α-hydroxylase activity of CYP17A1, it failed to increase overall survival in CRPC patients (NCT01193257).
Seviteronel (VT-464), anorally administered nonsteroidal CYP17A1 lyase inhibitor, is at the present under clinical development[40]. Similar to galeterone, seviteronel works downstream of abiraterone to inhibit CYP17A1 lyase and does not cause the same degree of mineralocorticoid production. This agent can therefore be administered without concomitant glucocorticoid administration, resulting in lack of associated toxicities (such as muscle wasting, skin friability, cushingoid features, and decreased bone mineral density).
Recently, Larsen and collaborators identified two novel non-steroidal and selective CYP17A1 inhibitors by virtual screening and reported the structural optimisation of one of these inhibitors, identifying compound 6 [Figure 4][41].
Compound 6, which like abiraterone also contains a pyridin-3-yl moiety, inhibited CYP17A1 with IC50 values of 230 and 500 nmol/L for the 17α-hydroxylase and 17,20-lyase reactions, respectively. The binding mode of compound 6 was determined by docking experiments, further refined by QM/MM optimisation. Compound 6 is a relatively non-polar compound with no hydrogen-bonding possibilities and, accordingly, no polar enzyme-inhibitor interactions were observed[41].
Subsequently, the combination of a structure-based virtual screening approach with density functional theory calculations was used to suggest newnon-steroidal compounds selective for CYP17A1[42]. This second study afforded the discovery of compound 7. In vitro assays in human H295R cells demonstrated that compounds 6 and 7 selectively inhibited CYP17A1 17α-hydroxylase (IC50 values of 830 and 52 nmol/L, respectively) and 17,20-lyase (IC50 values of 94 and 7.4 nmol/L, respectively) activities. Strong coordination of compound 7 to the haem iron is likely to be responsible for inhibition of both reactions. These compounds do not bind selected drug-metabolising cytochrome P450 enzymes or the steroidogenic CYP21A2, suggesting a reduced risk for undesirable side effects, especially on the corticosteroid production, consistent with data observed in vitro. Taken together, these data recommend compounds 6 and 7 as promising tools for the continued development of new drugs against PCa[42].
Structural analysis of the reported CYP17A1 inhibitors reveals that most of the inhibitors consist of two structural features. One is the metal-binding group that binds to the haem iron and the second is the scaffold that binds to the substrate pocket of CYP17A1. Based on this observation, recently Wang et al.[43] conducted a screen of compounds from an in-house metalloenzyme inhibitor library and identified compound 8 [Figure 5] to selectively inhibit rat CYP17A1 lyase with sub micromolar activity.
A preliminary modelling study indicated that compound 8 could fit nicely into the CYP17A1 binding pocket and maintain the key interactions with the residues of CYP17A1. The nitrogen of the pyridine and the tetrahydro-β-carboline core formed a coordination bond and hydrophobic interactions with haem group (iron atom) and hydrophobic pocket respectively. Since authors showed that there was unfilled space on the pyridine part in the active site cavity, they introduced substituent onto the pyridine ring to occupy this space and enhance the potency. These efforts led to the design and synthesis of a series of compounds bearing different substituted pyridine and pyrimidine moieties and evaluated their CYP17A1 activity. Of these analogues, the most potent compound was 9 [Figure 5], showing 1.5 fold greater potency against rat and human CYP17A1 protein than abiraterone. In NCI-H295R cells, the inhibitory effect of compound 9 on T production was also more potent than that of abiraterone at a concentration of 1 µmol/L. Further, it was shown that 9 reduced plasma T level in a dose-dependent manner in Sprague-Dawley rats and may be a lead compound for further preclinical studies.
AKR1C3
AKR1C3, also named HSD17B5, is a soluble enzyme member of the aldo-ketoreductase family, highly expressed in testes and extragonadal tissues such as basal cells of the prostate, adrenals and liver. Principally, it catalyses the NADPH dependent reduction of AD to T but is known to be involved with 3α-HSD, 20α-HSD, dihydrodiol dehydrogenase and prostaglandin synthase activities[44]. Compared to other HSD17B isoforms, AKR1C3 was the most abundant isoform expressed in several PCa cells and its expression is upregulated in CRPC [Table 2]. AKR1C3 plays a key role in producing DHT in each of the three pathways, since it can lead to the synthesis of DHT starting from AD and DHEA in the canonical pathway, from 5α-androstanedione in the 5α-dione pathway, and from androsterone in the backdoor pathway [Figure 2]. Elevated levels of expression of AKR1C3 in CPRC provide a mechanism to divert trace androgens that remain after ADT to the potent AR ligand DHT via these three pathways intratumourally and may indirectly also impact on CYP17A1 inhibitor or AR antagonist resistance mechanisms[9]. Furthermore, AKR1C3 has also been discovered to play a role in resistance to radiation therapy[45].
Because of its structural differences with HSD17B3, an enzyme belonging to SDR family and catalysing the same reaction of AKR1C3 in testis[46], AKR1C3 could be a good target for selective inhibition.
At present, there are more than 40 crystal structures of AKR1C3 in the 2017 International Union of Crystallography Protein Data Bank. The first crystal structure of AKR1C3 was reported by Lovering et al.[47] and revealed AKR1C3 as a typical aldo-keto reductase structure, with a catalytic pocket consisting mainly of loops A (116-143), B (217-238) and C (298-323). The ligand-binding pocket of AKR1C3 can be divided into five compartments as follows: an oxyanion site, a steroidchannel SC and subpockets SP1, SP2 and SP3 [Figure 6].
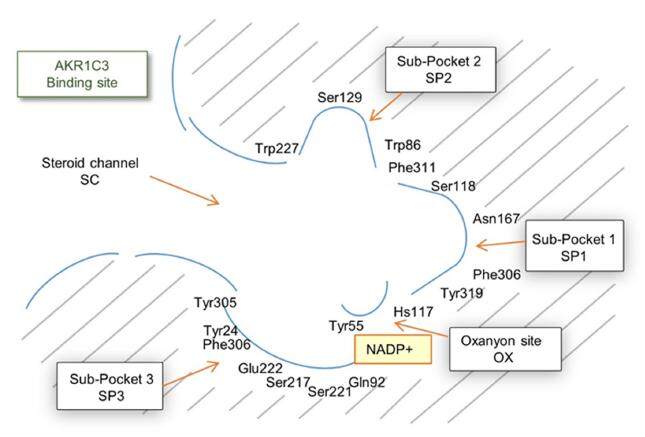
Figure 6. Close-up view of the AKR1C3 ligand-binding pocket. Illustrating the different compartments (the oxyanion site, the steroid channel and subpockets SP1, SP2 and SP3) that can be targeted with small molecules. NADP+ molecule is represented by a yellow square
The oxyanion site consists of the cofactor NADP± and the catalytic residues Tyr55 and His117, which are conserved among all AKR1C enzymes. The steroid channel is formed by Tyr24, Leu54, Ser129 and Trp227 and is open to solvent, guiding substrates into the oxyanion site. The SP1 pocket is located inside the ligand-binding pocket and is surrounded by Ser118, Asn167, Phe306, Phe311 and Tyr319. In contrast, the SP2 pocket is located in a shallow region surrounded by Trp86, Leu122, Ser129 and Phe311, while the SP3 pocket is located near the phosphate moiety of NADP+ and is surrounded by Tyr24, Glu192, Ser221 and Tyr305[48].
The structure of human AKR1C3 has been determined in complex with different substrates and inhibitors, which has enabled an excellent basis for the design of specific inhibitors. Selectivity is even more necessary with respect to AKR1C1 and AKR1C2, enzymes that have more than 86% of identity with AKR1C3, but inactivate DHT to 3β-androstanediol and to 3α-androstanediol respectively[49-51], thus decreasing the androgenic signalling. Between AKR1C3 inhibitors, several nonsteroidal anti-inflammatory drugs have been demonstrated to be very potent in inhibiting this enzyme. Some of them also exhibited good selectivity for the C3 isoform, e.g. indomethacin (10, Figure 7) and their binding mode within the ligand pocket has been investigated through X-ray crystallography[48].
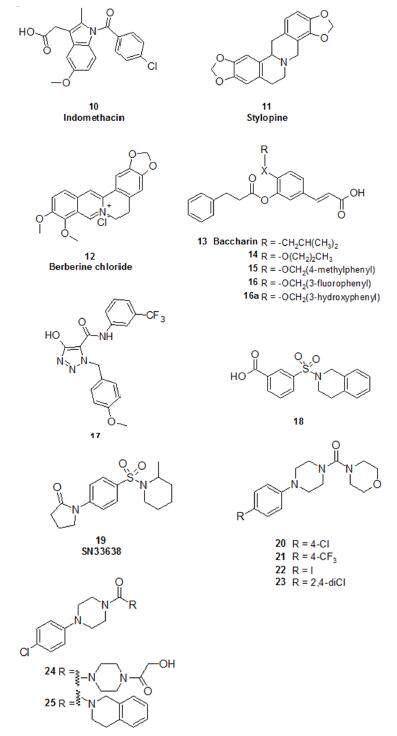
Figure 7. Examples of AKR1C3 inhibitors based on different chemical scaffolds
Discussion as to the use of AKR1C3 inhibitors to treat CRPC has been described in excellent reviews in 2011 and 2013[51,52]. Since that time, several groups have reported on the discovery of hit and lead compounds, and these will be briefly reviewed here.
Among natural inhibitors, Skarydova et al.[53] investigated the possible inhibitory effect of diverse types of isoquinoline alkaloids isolated from plant sources against the recombinant form of AKR1C3. Nineteen isoquinoline alkaloids were examined for their ability to inhibit AKR1C3 and as a result, stylopine (11, Figure 7) was demonstrated to be the most potent inhibitor among the tested compounds, demonstrating moderate selectivity towards AKR1C3.
In an attempt to identify potential AKR1C3 inhibitors based on known natural-based pharmacophores, Tian et al.[54] studied the blocking mechanism of berberine (2,3-methylenedioxy-9,10-dimenthoxyproto-berberine chloride; 12). This isoquinoline alkaloid screened from a traditional Chinese medicine monomer library, was shown to prevent AKR1C3-mediated intratumoral steroidogenesis incastrated nu/nu mice bearing subcutaneous LNCaP xenografts. The authors found that berberine inhibited AKR1C3-expressing 22Rv1 PCa cell proliferation and decreased cellular T formation in a dose-dependent manner, provided the experimental basis for the use of berberine as the lead compound for the further design, research, and development of AKR1C3 inhibitors.
Baccharin (3-prenyl-4-(dihydrocinnamoyloxy)cinnamic acid, 13) is a constituent in the ethanol extract of Brazilian propolis[55], which is a natural resinous substance collected by honeybees and has been used in alternative medicine to treat inflammation, liver disorders, and stomach ulcers. Recently Endo et al.[56] found that baccharin is a selective and potent inhibitor of AKR1C3, correlating with the antiproliferative effect of baccharin against human PC3 PCa cells. Baccharin was shown to exhibita 900-fold selectivity for AKR1C3 over the other three AKR1C isoforms. Due to its high inhibitory selectivity, baccharin represented a promising lead for the development of more potent and specific agents targeting AKR1C3. The structure activity relationship (SAR) of propolis-derived cinnamic acids suggested that the 3-prenyl moiety of baccharin is responsible for the selective binding to AKR1C3[56]. Endo et al.[46] also reported on the commercially available 3,4-dihydroxybenzaldehyde, derivatives configured with 3-aliphatic and aryl ethers instead of the 3-prenyl moiety. Within the series of aliphatic ethers, AKR1C3 inhibition was shown to decrease proportionally with increase in the aliphatic chain lengths. Compound 14, possessing an n-butyl ether, showed the highest inhibitory potency. Within the series of aromatic ethers, two benzyl ether derivatives, 15 and 16, showed an equivalent inhibitory potency to baccharin. The molecular docking of 15 in the crystal structure of AKR1C3 informed the design of a novel baccharin-based inhibitor (16a) with improved potency (Ki 6.4 nmol/L), which may be due to the introduction of a new interaction between the 3-hydroxyl group of the benzyl moiety of 16a and Tyr24 of the enzyme. The inhibitory selectivity of 16a for AKR1C3 over other human AKR1C isoforms was comparable or superior to that of baccharin. Additionally, 16a significantly decreased the cellular metabolism by AKR1C3 at much lower concentrations than baccharin.
Since carboxylic acids are likely to be transported into cells by carrier-mediated processes rather than passive diffusion[57], there are potential advantages in finding non-carboxylate inhibitors[58,59]. Following this rationale, we have applied a scaffold hopping strategy replacing the benzoic acid moiety of flufenamic acid with an acidic hydroxyazolecarbonylic scaffold[60]. In particular, differently N-substituted hydroxylated triazoles were designed to simultaneously interact with both subpockets 1 and 2 in the active site of AKR1C3, larger for AKR1C3 than other AKR1Cs isoforms. Through computational design and iterative rounds of synthesis and biological evaluation, novel compounds were reported, sharing high selectivity (up to 230-fold) for AKR1C3 over 1C2 isoform and minimal COX1 and COX2 off-target inhibition. A docking study of compound 17, the most interesting compound of the series, suggested that its methoxybenzyl substitution has the ability to fit inside subpocket 2, being involved in π-π staking interaction with Trp227 (partial overlapping) and in a T-shape π-π staking with Trp86. This compound was also shown to diminish testosterone production in the AKR1C3-expressing 22RV1 prostate cancer cell line while synergistic effect was observed when 17 was administered in combination with abiraterone or enzalutamide.
Heinrich et al.[61] also reported on a non-carboxylate inhibitor class of phenylpyrrolidin-2-one derivatives, obtained modulating 18, an inhibitor deriving from a high-throughput screen[62]. This modulation afforded compound 19, named later as SN33638, that inhibited AKR1C3 without forming a direct interaction with the oxyanion hole in the active site. Furthermore, in a cell-based assay, 19 was shown to be more potent than the carboxylic acid analogue 18 (ratio IC50(enz)/IC50 (cell) was 0.48 for 18 vs. 8.5 for 19), suggesting a pharmacological disadvantage for the acids in PCa cells[61]. The authors explored the role of the sulphonamide substituent and probed its affinity within the enzyme hydrophobic pocket bound by residues Met120, Asn167, Tyr216, Phe306, Phe311, Tyr317, Pro318 and Tyr319 [Figure 8][61]. SAR studies of potent and selective non-carboxylate AKR1C3 inhibitor 19 showed that while the sulphonamide function was still as critical as in 18[62], there was much more tolerance for the sulphonamide substituent, with a range of monocyclic six-membered ring analogues retaining activity and AKR1C selectivity. Crystal structure studies show that the 2-pyrrolidinone was located in the SP3 pocket but did not bind to the oxyanion site, and variations in the position, co-planarity or electronic nature of the pyrrolidinone ring abolished or severely diminished activity. The effectiveness of compounds at inhibiting AKR1C3 activity in cells broadly correlated with their enzyme inhibitory activity[61].
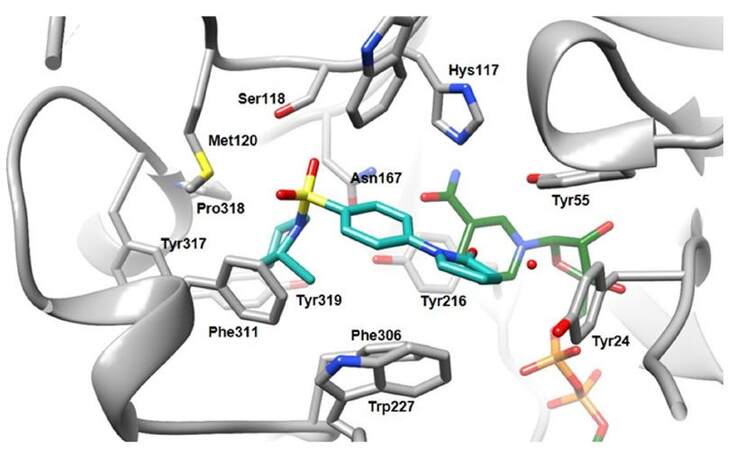
Figure 8. Structure of AKR1C3 in complex with SN33638 (19, PDB ID 4H7C). Carbon atoms of 19 are colored in blue, carbon atoms of cofactor NADP+ in green, carbon atoms of the protein are grey. Nitrogen, oxygen, sulphur and phosphorous atoms are depicted in blue, red, yellow and orange respectively. Relevant water molecules are represented by red points
Equipped by this powerful compound SN33638, Yin et al.[63] treated a panel of CRPC and ER-positive breast cancer cell lines, in the presence of hormone or prostaglandin precursors, prior to evaluation of cell proliferation and levels of 11β-prostaglandin F2α (11β-PGF2α), T production and PSA expression. Although SN33638 was shown to inhibit 11β-PGF2α formation, its ability to prevent T and 17β-estradiol production and their roles in CRPC and ER-positive breast cancer progression was limited due to AKR1C3-independent steroid hormone production. This is except in LAPC4 AKR1C3 cells, where the majority of T production was AKR1C3-dependent. These results suggested that inhibition of AKR1C3 is unlikely to produce therapeutic benefit in CRPC and ER-positive breast cancer patients, except possibly in the small subpopulation of CRPC patients with tumours that have upregulated AKR1C3 expression and are dependent on AKR1C3 to produce T required for growth. The study by Yin et al.[63] provided a valuable framework for future preclinical or clinical studies aimed at verifying this hypothesis that AKR1C3 inhibition suppresses tumour formation only in a selected population of CRPC patients expressing high levels of AKR1C3[64].
From the same high-throughput screen that enabled the discovery of SN33638 also came morpholino(phenylpiperazin-1-yl)methanone 20 [Figure 7], which was identified as a novel potent (IC50 = 100 nmol/L on isolated enzyme) AKR1C3-selective inhibitor without carboxylic function[65]. SAR studies of the new class of morpholino(phenylpiperazin-1-yl)methanone AKR1C3 inhibitors derived from 20 showed that these compounds bind selectively to AKR1C3 via the carbonyl oxygen of the central urea linker. This activity is favoured by lipophilic electron-withdrawing substituents on the phenyl ring (e.g. compounds 21-23) that probe specific regions of the SP1 pocket and H-bond acceptors on the other terminal ring and this was also supported by a QSAR study. Furthermore in the crystal structure of 20 bound to AKR1C3, the morpholine oxygen is within hydrogen bonding distance to a structured water molecule (HOH556), which is part of a network located in the SP3 pocket. The importance of it was examined via studying a series of derivatives in which the authors modulated the morpholine ring bound to urea. In this second series the complete inactivity of compounds bearing cyclohexane or phenylic ring suggested that there is a requirement for a secondary aliphatic nitrogen (i.e. a urea moiety) and an H-bond acceptor. It is possible also to replace the morpholine ring with larger substituents; e.g. compounds 24 and 25 derived from 20 were found active within this second series.
The discovery of AKR1C3 inhibitors with clinical potential has also been pursued by the pharmaceutical sector, including Astellas Pharmaceuticals and GTx-therapeutics. Both companies designed in vivo active compounds, namely ASP9521 (26, Figure 9) and GTx-560 (27), respectively[66,67]. However, only ASP9521 has so far been the subject of clinical evaluation. In a multi-centre phase I/II study, the compound was found to be orally bioavailable and well tolerated, but disappointingly without efficacy[68]. It is noteworthy, however, that 6/13 mCRPC patients discontinued treatment before the end-trial and patients were not preselected for AKR1C3 status. Also, none of the patients in the study had received prior treatment with abiraterone, so AKR1C3 expression may have been insufficient to observe significant inhibitory effects by ASP9521 in these patients.
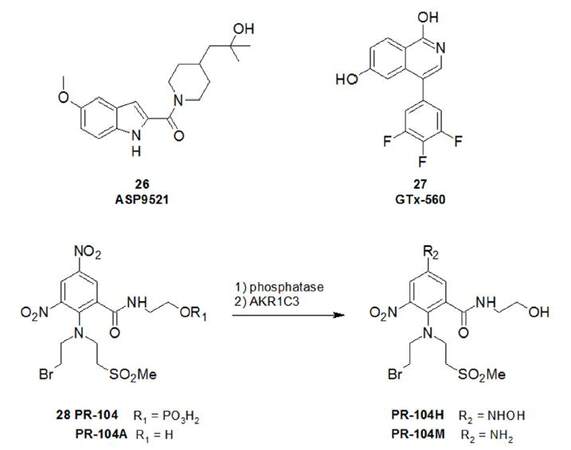
Figure 9. Examples of chemical structures of AKR1C3 inhibitors
ASP9521 remains the first and only rationalised AKR1C3-specific inhibitor to reach clinical evaluation. In addition, the non-selective AKR1C3 inhibitor indomethacin (10, Figure 7) has been used in combination with both enzalutamide[69] and abiraterone[70] in two different phase II clinical trials (NCT02935205 and NCT02849990, respectively).
The catalytic capacity of AKR1C3 has also been exploited in prodrug design and some work has focussed on the clinically evaluated bioreductive prodrug PR-104 (28, Figure 9)[71]. This prodrug that was originally designed to be bioreduced under hypoxic conditions to generate a DNA alkylating agent, has also been shown to be bioactivated by AKR1C3 in a hypoxia-independent manner to active species PR-104H and PR-104M. It is possible that a sub-population of patients with AKR1C3-expressing tumours could benefit by PR-104 treatment and hence expand the CRPC armamentarium of drugs[72].
HSD17B3
17β-hydroxysteroid dehydrogenase type 3 (17β-HSD3 or HSD17B3) is a microsomal enzyme member of the group of the NAD(P)(H) dependent oxidoreductases that catalyse the redox of hydroxyl/keto groups at position C17 of androgens and estrogens and in this manner regulate intracellular availability of steroid hormone ligands to their nuclear receptors. The 17β-hydroxysteroid dehydrogenases (HSD17Bs) belong to the short-chain dehydrogenase/reductase (SDR) superfamily, with the exception of HSD17B5 (AKR1C3), which is part of the aldo/ketoreductase family as already discussed.
Though HSD17B3 is expressed almost exclusively in the testis, there have been some reports of its over-expression in PCa tissues [Table 2]. In the testes,this enzyme catalyses the last step in the biosynthesis of T, by stereoselectively reducing the C17 ketone of AD using NADPH as cofactor. In PCa, HSD17B3 may operate like AKR1C3 and participate in all the three putative biosynthetic pathways [Figure 2]. In the canonical pathway, in addition to the reduction of AD to T, it could also reduce DHEA to androstenediol, while in the 5α-dione and the backdoor pathways it could also lead directly to DHT by reducing 5α-androstanedione or androsterone respectively [Figure 2][24]. A better understanding of what governs HSD17B3 and AKR1C3 in the PCa microenvironment could improve efforts to more effectively target these key enzymes in the steroidogenic biosynthetic pathway.
Due to the exclusive expression of HSD17B3 in the testes, selective inhibitors exerting effects equivalent to chemical castration may have potential as therapeutics for the treatment of PCa, and may be superior to the existing endocrine therapies based on a potential reduction in off-target effects. In addition, combination with an AKR1C3-selective inhibitor could possibly lead to more effective inhibition of the biosynthetic pathway and subsequent AR binding. Due to HSD17B3 being a membrane-bound protein, a crystal structure of HSD17B3 is not yet available. Nonetheless, as a member of the SDR family, HSD17B3 could share some highly conserved structural features, including the Rossmann fold, the cofactor binding site and the wide and easily accessible catalytic active site already demonstrated for the other six members of this family.
Several research groups have reported potent selective steroidal and nonsteroidal inhibitors as promising leads[73-76]. Recently, a review describing HSD17B3 as a target in hormone-dependent PCa therapy has been published[77], which described the main structure, function and reporting only a few examples of steroidal and non-steroidal inhibitors of HSD17B3. Here we provide a discussion of the most active nonsteroidal inhibitors developed to-date.
To aid structure-based drug design, some homology models of HSD17B3 have been developed[78-81]. A series of compounds based on the dibenzazepine scaffold was discovered in 2006 and compound 29 [Figure 10] was initially identified as promising hit compound and used as a lead to discover compound 30, which exerted picomolar activity in enzymatic as well as cellular (stably expressing 17β-HSD3 MDA-MB453 cells) assays[78]. This compound was very useful in helping the design of the subsequently discovered 17β-HSD3 inhibitors. In fact, Vicker et al.[79] built a homology model of 17β-HSD3 and used 30, as well as some structures described in Schering-Plough patents[82] (e.g. 31, Figure 10), to construct a pharmacophore and identified a potent compound STX2171 [32, IC50~200 nmol/L in the whole-cell 293-EBNA(HSD3) assay], that had only negligible activity against 17β-HSD2 (the enzyme that catalyses the reverse reaction) and was inactive against 17β-HSD1[83].
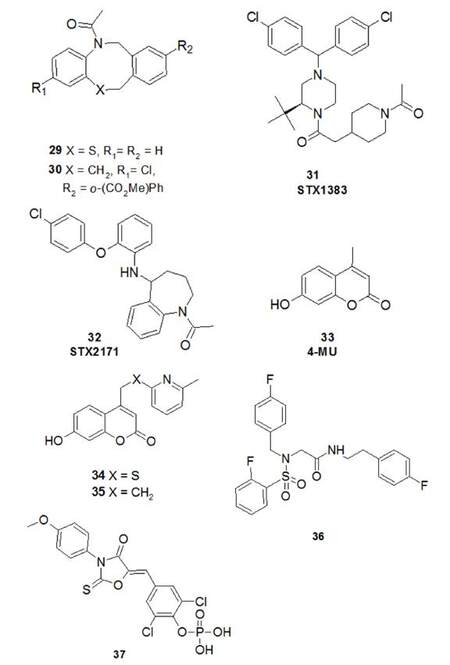
Figure 10. Structures of some nonsteroidal 17β-HSD3 inhibitors
STX2171 and 31 (later named STX1383) were also tested in a hormone-dependent PCa LNCaP(HSD3) xenografts, which were established in castrated male mice and using AD to stimulate tumour proliferation[83]. Both compounds were able to inhibit the proliferation of androgen-dependent prostate tumours (when stimulated by AD) and to reduce but not completely inhibit plasma T levels. An explanation of the incomplete abolition of plasma T levels can be found in the fact that also AKR1C3, prevalently expressed in the prostate, performs the conversion of AD to T.
In 2010, high-throughput screening led to the identification of 4-methylumbelliferone (4-MU, 33) as an inhibitor of HSD17B3[84]. The authors studied new 7-hydroxycoumarin derivatives of 4-MU and observed the most potent compounds carried substituents in the 4-position. Structures 34 and 35 exhibited low nanomolar inhibitory activity in HeLa cells expressing human 17b-HDS3 and selectivity versus other HSD17B isoenzymes and nuclear receptors.
Schuster et al.[81] rationalised the potential therapeutic opportunity of the concomitant inhibition of HSD17B3 and HSD17B5 because of their partly overlapping functions. They developed pharmacophore models for HSD17B types 3 and 5 and found interesting HSD17B 3/5 dual-targeting inhibitors with different selectivity profiles, although some of them were affected by weaker off-target activity against other HSD17B enzymes. For example, structure 36 [Figure 10] was able to reduce HSD17B3 and HSD17B5 activity by 56% and 58% at 2 µmol/L, respectively. Unfortunately, this compound was shown to also inhibit HSD17B1 by 20% at the same concentration. Although research for a dual inhibitor of HSD17B 3/5 enzymes needs deeper exploration, this approach could yield better compounds with clinical potential.
In 2012, Harada et al.[85] developed a phosphate ester prodrug 37 as an orally bioavailable HSD17B3 inhibitor. The potency of the active molecule (IC50 = 12 nmol/L in HeLa cells expressing human HSD17B3) rendered 37 capable of reducing plasma LHRH-induced T levels in a dose-dependent manner when administered orally to male Sprague-Dawley rats.
Interestingly, some environmental chemicals like benzophenone-1[80], tributyltin chloride and triphenyltin chloride[86] have been identified as HSD17B3 inhibitors, but their use is considered harmful to normal sexual development, since this enzyme plays an essential role in that process.
In the last decade, several steroidal and non-steroidal inhibitors of HSD17B3 have been designed and developed, but none of them has reached the clinic. One reason for this might be the difficulty in identifying an appropriate species to conduct the functional assays. Due to little sequence homology between human and other species isoforms, very potent inhibitors of the human enzyme show little activity toward HSD17Bs of other species, especially rodents[73,75,77]. Moreover, as AKR1C3, predominantly expressed in the prostate, performs the same biochemical conversions of HSD17B3, the in vivo HSD17B3 inhibition alone is not sufficient to completely abolish T levels in plasma. Additionally, the complexity and versatility of the steroidogenic pathways could bypass HSD17B3 inhibition in vivo, rendering these inhibitors not sufficiently efficacious in blocking tumour progression when tested alone.
SRD5A
Steroid 5α-reductases (SRD5A) are membrane-associated (microsomal) enzymes thatcatalyse the 5α-reduction of 3-oxo (3-keto), Δ4,5 C19/C21 steroids. The reaction involves a stereospecific, irreversible breakage of the double bond between carbons 4 and 5 with the aid of cofactor NADPH and the insertion of a hydride anion to the α face at carbon C-5 and a proton to the β face at position C-4. Examples of substrates are T, progesterone, AD, epitestosterone, cortisol, aldosterone and deoxycorticosterone.
Three isoenzymes of SRD5A, which are encoded by different genes (SRD5A1, SRD5A2, and SRD5A3), have been identified. All are involved in the conversion of T into DHT in the canonical pathway. In addition, SRD5A enzymes are also involved in the reduction of AD and progesterone or 17α-hydroxyprogesterone respectively to 5α-androstanedione and pregnan-3,20-dione or pregnan-3α,17α-dihydroxy-20-one. Therefore, these reductases appear to be key enzymes for the activation of both 5α-dione and backdoor pathways [Figure 2].
Even though these three isozymes are intrinsic membrane-bound proteins that catalyse the same reaction, they only share a limited degree of homology in protein sequence and possess distinctive biochemical properties. SRD5A1 is expressed in low levels in the prostate, and is relatively insensitive to finasteride, a 4-azasteroid enzyme inhibitor (38, Figure 11). SRD5A2 is expressed in high levels in the prostate and in many other androgen-sensitive tissues and is sensitive to finasteride. Instead, it remains controversial whether SRD5A3 enzyme activity is inhibited by finasteride or dutasteride, the latter a 4-azasteroid derivative effective at inhibiting both SRD5A1 and SRD5A2[87].
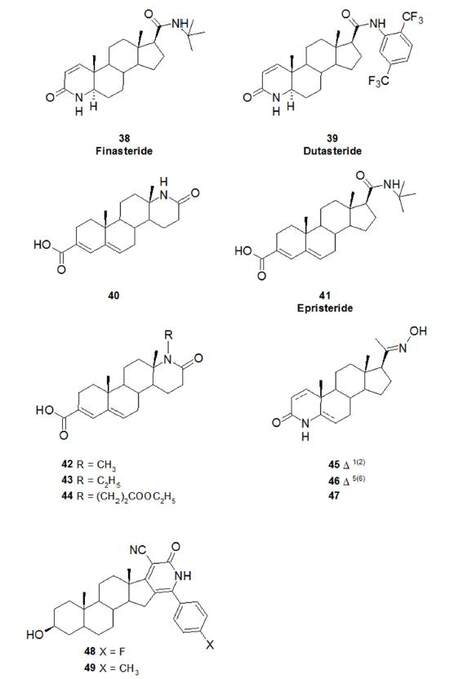
Figure 11. Structures of some 5a-reductase inhibitors
The inter- and intra-individual variability, the type and stages of progression of PCa and the methods used could explain the differences in the expression of the 3 isoenzymes observed in numerous reports [Table 2]. A summary of many studies that have discussed the distribution of SRD5A1-3 in different human tissues is tabulated in the review of Azzouni et al.[88], which extensively examines the basic biology of the SRD5A isoenzyme family. The different expression levels of SRD5A isoenzymes may confer response or resistance to 5a-reductase inhibitors and thus may have importance in PCa prevention. The mechanisms underlying androgen regulation of expression of the three different SRD5A isoenzymes in human prostate cells has been investigated by Li et al.[89]. The authors found that androgens regulate the mRNA levels of SRD5A isoenzymes in a cell type-specific manner, with regulation occuring at the transcriptional level and dependent on the AR. In addition, AR seems be recruited to a negative androgen response element (nARE) at the promoter of SRD5A3 in vivo and directly binds to the nARE in vitro.
Due to the unstable nature of these enzymes during purification, the crystal structures of both SRD5A1 and SRD5A2 are still unresolved. Despite this, a large number of molecules has been developed as SRD5A inhibitors over the past 40 years. Finasteride (38, Figure 11) and dutasteride (39) are the only two clinically used drugs, having been approved by the FDA for the treatment of BPH in 1992 and 2002, respectively[90,91]. Finasteride is a potent inhibitor of SRD5A2 with only weak in vitro activity versus SRD5A1 having IC50 value of 9.4 and 410 nmol/L on the isolated enzyme, respectively[92,93]. Dutasteride on the other hand, is a dual inhibitor of both SRD5A1 and SRD5A2 isozymes, with IC50 of 2.4 and 0.5 nmol/L respectively[94]. Both drugs are time-dependent competitive inhibitors and belong to the 4-azasteroids class of steroidal SRD5A inhibitors.
A detailed review of each category of inhibitors synthesised was published in 2010[93], and aids understanding of the structural features required for SRD5A inhibitory activity. The review discussed the need for more potent and less toxic inhibitors of SRD5A and suggested the use of molecules outside the steroidal template, as they can decrease the potential interaction with an additional enzyme or receptor of the steroidal endocrine system. Since 2010, only a few molecules have emerged as nonsteroidal SRD5A inhibitors[95]. In 2011, a hybrid molecule (40) was derived by merging structural features of finasteride and epristeride (41) as an inhibitor of SRD5A2. As epristeride behaves as an uncompetitive inhibitor[93,95,96], a hybrid compound from these two molecules could have propensity for exploiting both mechanisms. However, the relative potency of 40 was only 0.49 (compared with finasteride), and the authors concluded, from observing docking poses of 40 in the AKR1D1 active site, a surrogate of SRD5A2, that its inhibitory mechanism was the same as that of finasteride[97].
Aggarwal et al.[98] studied similar steroidal molecules (structures 42-44) able to inhibit the type II enzyme in the same range of concentration as finasteride. The carboxylic group at position-3 provided selective inhibition ofSRD5A2, as all the compounds of this series showed minimal inhibition against the type I enzyme.4-azasteroid-2-oximes (structures 45-47) were reported to be active against the SRD5A enzymes present on rat ventral prostate extract (both type 1 and the type 2 isozymes were present)[99].
Recently, Al-Mohizea et al.[100] prepared and performed pharmacological screening, including SRD5A inhibitory activities and antitumour properties (e.g. in LNCaP and PC-3 PC cell lines), of several steroids with a cyanopyridone heterocycle fused with its D-ring. The authors reported that these compounds had potent SRD5A inhibitory properties (in vivo assay with Sprague-Dawley rats). The best results were observed for the cyanopyridone structures with an oxygen bound to C3 (compounds 48-49, IC50 = 210 and 270 nmol/L, respectively when measured against rat SRD5A)[95,100].
Finasteride and dutasteride[90,91] have additionally been discussed in context of PCa prevention. Two clinical trials performed in the early 2000s in men at risk of developing PCa showed that the PCa incidence was significantly decreased in the treatment group, but the patients treated who were diagnosed with PCa had higher-grade tumors[101]. A retrospective study rejected the results of these clinical trials on PCa prevention and hence these drugs have not been FDA-approved yet for the prevention of PCa[102]. Subsequently, two clinical trials, (in phase II and IV, respectively) showed dutasteride to decrease the incidence of histopathological progression in patients with low-grade PCa and delay biochemical progression in patients who underwent radical prostatectomy or radiation therapy for localised PCa, respectively[103,104].
Androgen receptor
The AR is crucial for normal functioning of the prostate. As a member of the steroid hormone receptor family of ligand-activated nuclear transcription factors, it consists of four distinct functional domains, a poorly conserved N-terminal domain (NTD) with transcriptional activation function; a highly conserved deoxyribonucleic acid (DNA)-binding domain (DBD); and a moderately conserved ligand-binding domain (LBD). A short amino acid sequence called the “hinge region” separates the LBD from the DBD and also contains a part of a bipartite ligand-dependent nuclear localisation signal (NLS) for AR nuclear transport [Figure 12][105]. NTD contains a transactivation AF-1 region, with two transcription activation units (TAU1 and TAU5) and two motifs involved in protein-protein interactions and AR N/C interactions[6]. NTD contains a co-regulator binding surface, the disruption of which reduces the androgen-dependent proliferation and migration of PCa cells[106]. The LBD contains an activation AF-2 region, which is responsible for agonist-induced activity and androgen binding to induce conformational changes, which facilitates intra-and intermolecular interactions between the N-terminal and C-terminal domains and subsequently AR homo-dimerisation and nuclear translocation [Figure 12][107,108]. In the nucleus, ligand bound AR binds to specific recognition sequences known as “androgen response elements” (AREs) in the promoter and enhancer regions of target genes and recruits co-activators and co-repressors, which then modulate transcription of androgen-dependent proteins[109]. Under physiological conditions, both T and DHT can bind to and activate AR signalling [Figure 1][110,111]. Deregulated AR signaling is common during PCa development and CRPC progression. The ARs in tumour cells exposed to ADT undergo selective alterations that result in aberrant AR reactivation, which ultimately allows the AR pathway to remain active despite the shortage of androgenic ligands. AR amplification leads to AR overexpression, which is present in approximately 30% of CRPCs. Mutations in the AR gene occur in approximately 20% of CRPCs. Most significant AR mutations occur in the LBD, which increase the sensitivity and decrease the specificity of ligand binding[112]. Constitutively active splice variants (AR-Vs) are detected in PCa cell lines (e.g. LNCaP95, VCaP and 22Rv1) and in CRPC tissues. More than 20 AR-Vs have been reported, but only ARv567 and AR-V7 are considered to be clinically relevant because their levels of expression are correlated with CRPC and poor survival rates [Figure 12][6]. AR-V7, like other AR variants lacks an LBD, and via its nuclear localisation binds DNA independently, without androgen activation, regulating a unique set of target genes that facilitate mitosis in addition to the regular androgen-dependent genes that are activated by full-length ARs that promote disease progression[113].
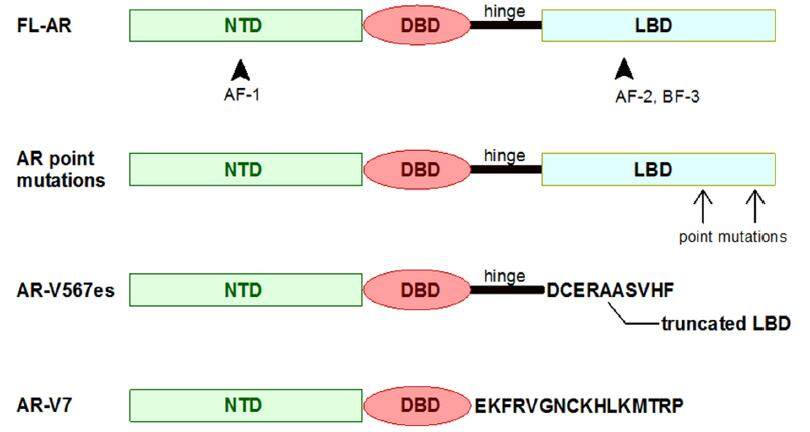
Figure 12. Domain organisation of full length AR (FL-AR) and some AR splice variants (AR-Vs). The four functional domains are indicated as follows: the N-terminal domain (NTD, green rectangles), the DNA-binding domain (DBD, red circles); ligand-binding domain (LBD, blue rectangles) and the "hinge region", separating the LBD from the DBD (black lines). For AR-Vs lacking the LBD and/or the hinge region, the amino acids outside the previously described domains are listed. AR: androgen receptor
As other steroid hormone receptors, also AR appear to be regulated by epigenetic mechanisms[27]. The first evidence of epigenetic regulation of AR came from comparing the hypermethylation of AR promoters in AR-deficient and AR-expressing cell lines (hyper- and hypomethylated, respectively)[114]. In human prostate cancer, a significant role of hypermethylation of AR genes has been suggested: AR hypermethylation was observed both in primary cell lines from PCa patients (20%) and in hormone-refractory prostate cancers (28%)[115]. Also other epigenetic mechanisms, such as histone acethylation/deacethylation, seem to participate in the regulation of AR-driven genes[116].
The literature is abundant with research articles and reviews concerning the development of AR-based therapy for PCa. The AR is a validated therapeutic target for PCa and five molecules have already been approved by the FDA (cyproterone acetate 50, flutamide 51, nilutamide 52, bicalutamide 53, enzalutamide 54, Figure 13)[117] while several others are currently under preclinical/clinical development. Among papers of considerable interest on AR, we recommend the following for further reading: the report of Lu et al.[118] that describes the mechanism of function of AR and its targetable domains, the review by Imamura and Sadar[6], which focuses on AR-related mechanisms of resistance and AR antagonist therapeutic agents undergoing clinical trials, and finally the review by Martinez-Ariza and Hulme[117], that encompasses non-ligand-binding protein modulators of the AR.
Figure 13. Androgen receptor ligand-binding protein antagonists approved by FDA or in clinical trials
Unfortunately, most CRPC patients treated with AR-antagonist therapy will eventually develop resistance and succumb to the disease. Mechanisms of resistance to these drugs include modification of the AR, AR gain-of-function point mutations, truncated AR isoforms and constitutively-active AR splice variants[6]. Accordingly, new agents to target these alternative ARs through novel mechanisms of action should lead to intensified research in the PCa community and lead to new drugs with clinical potential.
AR-antagonists can be classified based on their ability to interact with different domains of the AR. Approved drugs and similar structures under development show affinity for the LBD. These molecules, also named traditional AR antagonists, compete with androgens in binding the AR and prevent formation of the AF-2 (activation function-2) hydrophobic groove inside the LBD and its interaction with co-regulators. However, in some cases, the AR can still dimerise and become nuclear, as observed with enzalutamide using confocal micrographs[119]. Apalutamide (55, Figure 13) and darolutamide (56, OMD-201) are two molecules under evaluation in phase 3 clinical trials in patients with non-metastatic CRPC (NCT01946204 and NCT02200614, respectively). Apalutamide shows high structural similarity to enzalutamide, but achieves the same therapeutic response as enzalutamide at a lower dose in a LNCaP xenograft mouse model and does not induce AR nuclear translocation or DNA binding[120]. Darolutamide is characterised by a different chemical scaffold from its cognate antagonists, and is able to antagonise AR mutants F876L, W741L and T877A[121].
Seviteronel (5, Figure 4), a non-steroidal CYP17A1 inhibitor with 17,20-lyase selectivity (see above), has been found to show AR-antagonist activity independent of CYP17A1 enzyme inhibition, with evidence of direct binding to the AR LBD[40]. Similarly, also galeterone (3, Figure 4) is a competitive AR antagonist mediated by binding of the drug to the steroid-binding pocket of AR and concomitantly inhibiting T biosynthesis through inhibition of CYP17A1 lyase activity[36]. In addition, galeterone targets the LBP of mutated T878A AR. The authors also reported on PSA reduction in LNCaP and VCaP cell lines, an effect that was partially reversed upon addition of DHT in a dose-dependent manner[122,123].
The SAR for AF-2 targeting have been extensively studied[124-126], and X-ray structures of AR-LBD in complex with T (PDB: 2AM9), R-bicalutamide (PDB codes: 1Z95 and 4OJB, Figure 14 or hydroxyflutamide (PDB: 2AX6) and other ligands have been resolved[127]. Essentially, the compounds consist of three structural parts: the first part is usually an aromatic ring substituted with a nitrile and an electron withdrawn group interacting with Arg752, Phe764, and Gln711 [Figure 14], the second part is a nitrogen-containing moiety such as an amide function or a heterocyclic ring, and the third part, often connected to the second through a short linker, is different in nature in various antagonists but nonetheless important in suppressing the agonistic effect[118].
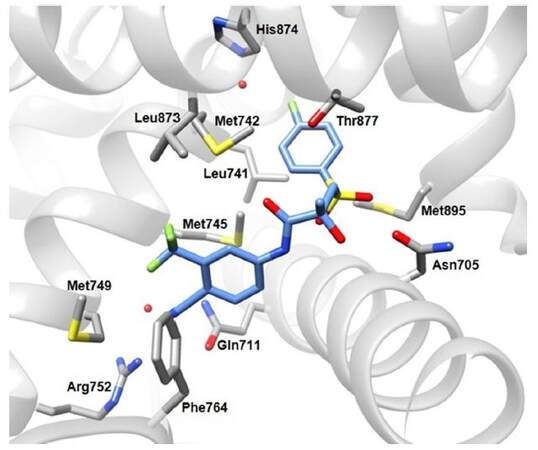
Figure 14. Crystal structure of Trp741Leu AR-LBD in complex with R-bicalutamide (PDB ID 4OJB). Carbon atoms of R-bicalutamide are coloured in blue, the AR is grey. Nitrogen, oxygen, sulphur, atoms are depicted in blue, red and yellow respectively. Relevant water molecules are represented by red points. AR: androgen receptor; LBD: ligand-binding domain
Most of these agents are derivatives of already approved non-steroidal AR antagonists, but there is a need to discover broader chemotypes, distinct from known scaffolds to avoid cross-resistance with these compounds. In this regard, Kandil et al.[128] synthesised umbelliferone derivatives merging two independent in silico pharmacophores based on virtual screening studies. Putative binding modes of compound 57 [Figure 15], the most active of the series (IC50 = 0.93 in 22Rv1 cells), within the antagonistic AR-LBD showed hydrogen bond interactions with key amino acids Arg752, Gln711 (with the lactone carbonyl group), Thr877 (with the terminal carbonyl group) and Asn705 (with the methylene group). Another interesting example of an innovative structureis represented by the molecule synthesised by Johnson et al.[129] (compound 58), though only the PSA luciferase assay was conducted to evaluate biological activity. BMS-641988 (59) is a non-steroidal compound disclosed in 2015 with high binding affinity for the AR (Ki = 1.7 nmol/L), efficacious in a CWR22-BMSLD1 PCa xenograft model with superior efficacy to bicalutamide. Due to its acceptable preclinical safety profile both in vitro and in vivo, it was selected for clinical development and the outcomes of two Phase I studies in patients with CRPC (NCT00644488 and NCT00326586) have been published[130].
Figure 15. Selected competitive hormone antagonists under development
Recently, new derivatives[131] of DIMN (60), a potent and well characterised AR antagonist interacting with the LBD were designed and synthesised[132]. Some of these derivatives exhibited higher AR antagonistic activity than DIMN itself and bicalutamide, even with DHT co-treatment, and higher inhibitory effects on LNCaP cells proliferation. Compounds 61 and 62 bear long, linear and hydrophobic side chains on the tetrahydroisoquinoline moiety, while 63 carries an additional bulky group such as a phenyl ring [Figure 15]. Their potency in inhibiting LNCaP cells (IC50 range: 0.35-1.01 µmol/L) was shown to be superior to DIMN (IC50 = 4.46 µmol/L); this indicates that the occupation of a cone-shaped cavity, located near Thr877 (interacting with ethereal oxygen from docking studies proposed by the authors) increases bioactivity of the series.
Recently, a new class of AR modulators bearing the triazole core has been proposed, which are able to exert antiproliferative effects on LNCaP-AR cells and on CW22Rv1 cells, which constitutively expresses high levels of AR-V7. Compound 64 was the most effective compound of this series and was also evaluated in vivo using CW22Rv1 xenografts, demonstrating superior activity to enzalutamide in this model[133].
BF-3 is another targetable binding domain located at the surface of the AR, where it controls the allosteric modulation of AF-2[134]. Notably, the mutation which occurred in the AF-2of LBD will not alter or weaken the binding of antagonists in the BF-3 site. Interestingly, flufenamic acid (65, Figure 16), that has also the ability to inhibit AKR1C3, can bind BF-3 with moderate affinity (range of activity: 10-50 µmol/L)[135]. Among small molecules inhibiting this domain and described in recent reviews[117,118,125,126], compound 66 displays excellent anti-androgen potency, antiproliferative activity against androgen-sensitive (LNCaP) and enzalutamide-resistant (MR49F) PCa cell lines, and effective inhibition of tumour growth in vivo, in both LNCaP and MR49F xenograft models[136]. The data are very promising in highlighting the therapeutic relevance of the BF-3 groove in AR function. Recently, Zhang et al.[137] designed a conjugate of thiosalicylamide and the BF-3 binding small molecule tolfenamic acid. This molecule, named YZ03 (67), enhanced acetylation of endogenous AR at Lys720 residue, critical for protein-protein interaction with the FXXLF coactivator peptide binding.
Figure 16. Selected examples of BF-3 site modulators
The AR NTD contains amino acids 1-558 and is an intrinsically disordered region. Activation function-1 (AF-1) is a protein binding domain known to bind different co-regulators and its low sequence identity with other nuclear receptors renders it an appealing target for selective small molecule inhibitors[117]. The most studied AR NTD modulators are EPI compounds that have been shown to be bound covalently to AF-1 and to inhibit AR nuclear translocation. Importantly, also constitutively-active AR splice variants lacking LBD are inhibited by EPI analogues where traditional AR antagonists are ineffective[138]. EPI-001 (68), discovered by Andersen et al.[139] by functional assay screening of marine sponge extracts, is shown in Figure 17; its stereoisomers EPI-002-EPI-005 have also been isolated and evaluated later. The potential therapeutic benefits of EPI have been demonstrated using different human PCa cell lines and xenograft models in castrated male mice[140]; a small molecule belonging to this class (EPI-506, a prodrug analogue of EPI-002[141]) is now under clinical evaluation in a phase I/II study in men with mCRPC that have disease progression after enzalutamide and/or abiraterone treatment (NCT02606123).
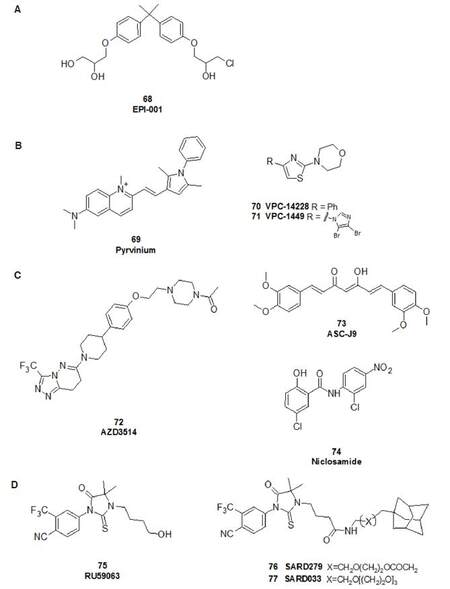
Figure 17. Selected compounds that act as: (A) NTD modulators; (B) DBD inhibitors; (C) indirect inhibitors of AR and AR variants; and (D) selective androgen receptor downregulator (SARD) compounds. AR: androgen receptor; DBD: DNA-binding domain; NTD: N-terminal domain
The DBD is responsible for mediating interactions with AREs. Encouragingly, the 3D crystal structure of the rat AR DNA-binding domain has been obtained (PDB: 1R4I)[142]. Some structures able to interact with this domain and prevent its interaction with DNA and are shown in Figure 17. Pyrvinium (69), an antihelmintic (and its hydrogenated analogue 1,2,3,4-tetrahydropyrvinium[143]), was able to bind at the interface of the DBD dimer (inhibiting also AR splice variants lacking the LBD) and inhibit cell lines derived from both bone and prostate[144]. Although the results are promising, some doubts have been expressed about the binding site on the DBD involved in the AR inhibition by 69[117,145].
Insights into AR DBD inhibition were provided by Li et al.[145] through the study of two molecules (70 and 71) identified through a virtual screening campaign and subsequent medicinal chemistry investigations. Both compounds exhibited nanomolar potency against the AR and effectively inhibited the growth of enzalutamide-resistant cells. Their binding modes were corroborated by mutagenesis experiments, confirming interaction of these inhibitors with residues Gln592 and Tyr594 of AR DBD. Compound 71 was also evaluated in LNCaP xenografts in mice, causing comparable reductions in tumour volume to enzalutamide[146].
In summary, much effort has gone into AR-targeted drug design and in particular the LBD has been pursued as a target. However, mutated forms of AR, especially constitutively active AR variants, need to be considered as an aspect of AR-related resistance mechanisms and, for this reason, targeted with novel inhibitors. Accordingly, we look with particular interest to molecules targeting co-regulator binding regions and include AF-2 and BF-3 pockets (still within LBD, but their inhibition is able to overcome gain-of-function point mutations induced by AF-2 inhibitors), the NTD and the DND, or molecules able to inhibit AR through an indirect mechanism.
Degradation of all forms of ARs are emerging as an advantageous therapeutic paradigm for the more effective treatment of PCa in the context of AR mutations that confer resistance to second-generation AR antagonists. The compounds able to degrade the AR are classified as Selective Androgen Receptor Degraders (SARD). A first-in-class non-steroidal SARD, AZD3514 (72, AstraZeneca, Macclesfield, UK, Figure 17) was developed and was shown to downregulate the AR[147]. AZD3514, which binds the AR LBD and prevents its ligand-driven nuclear translocation, promotes down-regulation of AR levels. It has been clinically evaluated, but disappointingly only moderate anti-tumour activity in patients with advanced CRPC was observed; it was also shown to be poorly tolerated, with nausea and vomiting being the main toxicities[148]. In the 2012, Yamashita et al.[149] identified that ASC-J9 (73), also named as dimethylcurcumin, functioned as an AR degradation enhancer for full length AR and AR splicing variants. ASC-J9 is the first anti-AR compound discovered that selectively degrades AR in selective cells[150].
Niclosamide (74), an anthelmintic drug, has been foundable to inhibit AR-V7 transcription activity and downregulate its protein expression. Both in vitro and in vivo PCa tumour growth were reduced upon treatment with 74 and it has been suggested that it acts by promoting AR-V7 degradation via a proteasome-dependent pathway[151]. Furthermore, the combination of niclosamide and enzalutamide resulted in significant inhibition of enzalutamide-resistant tumour growth, suggesting that niclosamide enhances enzalutamide therapy and overcomes enzalutamide resistance in CRPC cells[151]. A phase I study of niclosamide in combination with enzalutamide in men with CRPC is currently under investigation (NCT02532114).
Also galaterone (3, Figure 4), a known CYP17 inhibitor and AR antagonist, promotes the proteasomal degradation of both AR and its ligand-independent variants AR-V7 and Arv567es[152,153].
Hydrophobic tagging technology has also been employed to degrade the AR. Bradbury et al.[154] showed that molecules containing hydrophobic regions linked to small-molecule AR ligands induce AR degradation, reduce expression of AR target genes and inhibit proliferation in androgen-dependent PCa cell lines. By appending the alkylfluoryl chain of fulvestrant onto DHT, a selective SARD compound was discovered. As a parallel strategy to the rational design of a SARD, Gustafson et al.[155] appended an adamantyl moiety to the AR agonist RU59063 (75) via a short PEG linker to create SARD279 and SARD033 (76 and 77, Figure 17). They found that this addition switched the agonist into a pure antagonist capable of degrading AR protein (half-maximal degradation at 1 μmol/L; maximal degradation of 95%). Moreover, this SARD was also able to inhibit proliferation of a model castration-resistant PCa cell line resistant to enzalutamide. These results suggest that selective AR degradation may be an effective therapeutic prostate tumour strategy in the context of AR mutations that confer resistance to second-generation AR antagonists[156].
Co-administration of inhibitors and antagonists targeting the AR axis
Androgen biosynthesis prevention and AR signalling inhibition, should in principle produce blockade of the AR axis. As these pathways are implicated in the progression of CRPC, these concurrent therapeutic actions should both reduce the incidence of resistance and increase therapeutic efficacy. Such a potential powerful combination strategy could replace the current PCa treatment paradigm of sequentially adding agents at the time of disease progression. Many drug combinations targeting the AR axis are described in literature. Unfortunately, the use of dutasteride in combination with bicalutamide for advanced PCa has not been as successful. Dutasteride plus bicalutamide in patients with progressive non-metastatic PCa did not delay further progression compared to only bicalutamide[157] while dutasteride in combination with abiraterone as well as enzalutamide is currently in a phase II clinical trial where the outcome is pending at the time of writing[158]. Recently Liu et al.[159] showed overexpression of AKR1C3 to confer resistance to enzalutamide. Furthermore, the combination of indomethacin, an AKR1C3 inhibitor, and enzalutamide resulted in significant inhibition of enzalutamide-resistant tumour growth. These results suggested that AKR1C3 activation is a critical resistance mechanism associated with enzalutamide resistance. Accordingly, the dual targeting of intracrine androgens and AKR1C3 promises to overcome enzalutamide resistance and improve survival of advanced PCa patients. Subsequently, the same research group reported that treatment of abiraterone-resistant cells with indomethacin overcomes resistance and enhances abiraterone therapy both in vitro and in vivo by reducing the levels of intracrine androgens and diminishing AR transcriptional activity[160]. Furthermore, these studies provide preclinical proof-of-principle for starting clinical trials focussed on investigating the combination of using indomethacin with enzalutamide, or with abiraterone for advanced PCa[69,161].
Bifunctional inhibitors and antagonists targeting the AR axis
Despite the highly significant therapeutic relevance of combination therapies, potential advantages of a targeted therapy based on a single drug that modulates the activity of multiple targets over combination therapy are: (1) a more predictable pharmacokinetic profile; (2) a lower probability of developing target-based resistance[162]; (3) a superior safety profile; and (4) a minimised risk of adverse effects[162,163].
One of the first bifunctional non-steroidal small molecules studied by Chen et al.[164] as therapeutic leads for CRPC was an N-(aryl)amino-benzoate inhibitor (77, Figure 18). The authors exploited the observation that some flufenamic acid analogues with AKR1C3 inhibitory activity also acted as AR antagonists[165] and subsequently synthesised a second generation of AKR1C3 inhibitors in which the key features were the inclusion of an additional ring on the phenylamino ring. The derivative 3-[(4-nitronaphthalen-1-yl)amino] benzoic acid (77, Figure 18A) retained nanomolar potency and selective inhibition of AKR1C3 but also acted as an AR antagonist. It inhibited 5a-dihydrotestosterone-stimulated AR reporter gene activity with an IC50 = 4.7 µmol/L and produced a concentration-dependent reduction in AR levels in PCa cells. The in vitro and cell-based effects of compound 77 makes it a promising lead for the development of dual acting agent for CRPC.
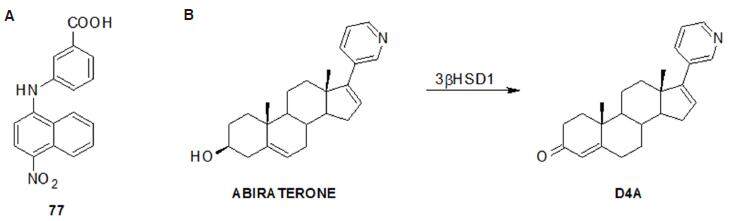
Figure 18. (A) Compound 77, analogue of flufenamic acid; (B) metabolism of abiraterone to D4A[170]
As mentioned above, the CYP17A1 inhibitor galeterone not only inhibits the enzyme but is also a competitive AR antagonist and causes degradation of the AR and its variants AR-V7 and Arv567es[153,166-168]. Furthermore, galeterone also impaired AR binding to DNA and selectively up-regulated degradation of the mutated T878A AR protein[122,123]. For its multi-funtional activity, three different clinical studies have been initiated with galeterone. A phase I clinical trial has been completed, while a phase II clinical trial (ARMOR2) is still ongoing in CRPCa patients. A phase III clinical trial (ARMOR3-V7) has started recruiting CRPCa patients that specifically express AR-V7. Unfortunately ARMOR3-SV was terminated as it failed to meet its primary endpoint of demonstrating an improvement in radiographic progression-free survival (rPFS) for galeterone versus enzalutamide in AR-V7 positive mCRPC[169].
Another interesting polyfunctional drug is D4A, a metabolite of abiraterone. Li et al.[170] recently showed that abiraterone is converted to D4A in mice and patients with PCa [Figure 18B]. D4A inhibits CYP17A1, HSD3B and SRD5A, which are required for DHT synthesis. In particular, D4A is approximately 10-fold more potent than abiraterone at blocking the conversion of [3H]DHEA to AD by 3β-HSD in LNCaP and VCaP cells.
D4A and abiraterone similarly block conversion of [3H]pregnenolone to DHEA by CYP17A1, as shown by a study in cells expressing CYP17A1[170]. To determine the effect of D4A on endogenously expressed SRD5A, LAPC4 cells, which exhibit robust SRD5A enzymatic activity[18], were treated with D4A, abiraterone or enzalutamide respectively and cultured in the presence of [3H]AD (the preferred natural substrate of SRD5A1)[18]. D4A (10 mmol/L) almost completely blocked conversion of AD to 5α-androstanedione and other 5a-reduced androgens, whereas abiraterone and enzalutamide had no detectable effect, even at a concentration of 100 mmol/L. Abiraterone has been reported to have modest affinity for AR, particularly in the presence of mutations in the ligand-binding domain[171]. To determine how conversion from abiraterone to the 3-keto structure of D4A affects drug affinity for AR, Li et al.[170] performed a competition assay. The affinity of D4A for mutant (expressed in LNCaP) and wild- type (expressed in LAPC4) AR is greater than that of abiraterone and comparable to that of enzalutamide, and greater than bicalutamide. D4A also has more potent anti-tumour activity against xenograft tumours than abiraterone. These findings suggest that direct treatment with D4A potentially could be more clinically effective than abiraterone treatment.
Conclusion
Inter-patient heterogeneity and distinct patterns of abnormal expression and regulation of steroidogenic enzymes contribute to PCa patient relapse. As discussed in this review, the many enzymes involved in the steroidogenic pathway provide obstacles and opportunities for researchers engaged in developing better drugs. Currently, hormone therapy remains first choice for patients with advanced PCa, either as alone or in combination with chemotherapy. Androgen-dependent and independent production is central to fuelling PCa growth, and the biosynthetic steroidogenic pathway plays a vital role in the former. Great progress in PCa biology and drug design have enabled effective therapies to be used clinically, while several promising preclinical strategies are underway. Newer drugs such as abiraterone have performed well in several clinical studies, but although able to extend overall survival rates, it rarely has curative power. The lack of long-lasting therapeutic effects of abiraterone and ADT may in part be linked with androgen-independent pathways and crosstalk to signal transduction pathways and in part to mutations to the AR[172]. An ever-increasing number of studies over the past decade have provided insight into prostate cancer biology and it is becoming apparent that new chemotypes and new drug combination strategies are required to target the heterogeneous prostate microenvironment more effectively. Accordingly, future drug discovery should be focussed on multi-targeting agents that inhibit several steps in the biosynthetic steroidogenic pathway, or degrader drugs that eradicate the AR (normal or mutated) to prevent it fuelling PCa growth. Whereas the former requires multifactorial drug design and appropriate multifunctional in vitro models, the latter may be a strategy to be implemented in the clinic within a shorter timeframe. In parallel efforts, an attractive route to better therapeutic outcomes is to conduct clinical trials that explore the possibility of using certain types of drugs at a much earlier disease state. Indeed, this is the thinking behind the STAMPEDE trial, which tests a number of additional therapies, given alongside first-line ADT and is discussed by Malcolm Mason in another review “Getting better at treating prostate cancer: what clinicians should want from scientists” in this themed prostate cancer issue. PCa patients who have suffered relapse with bone metastasis currently have poor overall survival rates, with only bisphosphonates available for palliative treatment. Obstacles in obtaining bone biopsies have halted our understanding of how we can effectively treat PCa patients suffering from bone metastases. Generally, the PCa microenvironment is known to be under oxidative stress and indeed this might have a significant impact on how steroidogenic enzymes respond within the bone microenvironment. Evidence points to the intra-tumoural synthesis of T and DHT is minimal, yet high expression levels of certain steroidogenic enzymes (SRD5A1, AKR1C2, AKR1C3, and HSD17B10) have been detected in a smaller number of bone metastases when compared to non-malignant prostate and primary prostate tumor tissue[173]. Furthermore, in a recent study AR amplification was not observed in bone metastases from previously untreated PC patients, but was detected in about half of metastatic samples from patients with CRPC. This AR amplification was associated with increased AR and AR-V7 expression and a particularly poor prognosis[174].
Steroidogenic enzymes are dependent on NAD/NADH co-factor activity and hence better knowledge of the Warburg effect in the bone microenvironment might also be important. Additionally, enhanced understanding of how other cytochrome P450 or aldehyde dehydrogenase isoforms exist and cooperate in retinoic acid production, stem cell maintenance or in inflammatory response may also help to understand significant differences between localised and metastasised PCa. As osteoblasts regulate the intratumoral steroidogenesis of CRPC in bone, targeting osteoblasts may therefore be important in the development of new therapeutic approaches[175]. To improve on our strategies for therapeutic intervention, we need better models for evaluating new compounds, including co-culture systems, multicellular spheroids, patient-derived xenografts and organoids. Discovery of highly selective chemical probes to investigate steroidogenic/metabolic pathways should yield new drugs that more effectively target the AR axis, which can be employed in combination with other drugs employed in PCa management.
Declarations
AcknowledgmentsWe thank Dr. Davide Bonanni for production of the crystallographic images, Prof. Franca Viola for helpful discussions and revision of the manuscript and Dr. Robert A. Falconer for proofreading the final manuscript.
Authors’ contributionsLiterature search: A.C. Pippione, D. Boschi
Drafting and writing the manuscript: A.C. Pippione, D. Boschi, S. Oliaro-Bosso
Manuscript’s revision and supervised: K. Pors, S. Oliaro-Bosso, M.L. Lolli
Financial support and sponsorshipAuthors acknowledge support in part from University of Turin (Ricerca Locale grant 2014 and 2015).
Conflicts of interestThere are no conflicts of interest.
Patient consentNot applicable.
Ethics approvalNot applicable.
Copyright© The Author(s) 2017.
REFERENCES
2. Nelson PS, Clegg N, Arnold H, Ferguson C, Bonham M, White J, Hood L, Lin B. The program of androgen-responsive genes in neoplastic prostate epithelium. Proc Natl Acad Sci U S A 2002;99:11890-5.
3. Saad F, Fizazi K. Androgen deprivation therapy and secondary hormone therapy in the management of hormone-sensitive and castration-resistant prostate cancer. Urology 2015;86:852-61.
4. Huggins C, Stevens RE, Hodges CV. Studies on prostatic cancer. II. The effects of castration on advanced carcinoma of the prostate gland. Arch Surg 1941;43:209.
5. James ND, Spears MR, Clarke NW, Dearnaley DP, De Bono JS, Gale J, Hetherington J, Hoskin PJ, Jones RJ, Laing R, Lester JF, McLaren D, Parker CC, Parmar MK, Ritchie AW, Russell JM, Strebel RT, Thalmann GN, Mason MD, Sydes MR. Survival with newly diagnosed metastatic prostate cancer in the "docetaxel era": data from 917 patients in the control arm of the STAMPEDE trial (MRC PR08, CRUK/06/019). Eur Urol 2015;67:1028-38.
6. Imamura Y, Sadar MD. Androgen receptor targeted therapies in castration-resistant prostate cancer: bench to clinic. Int J Urol 2016;23:654-65.
7. Seisen T, Roupret M, Gomez F, Malouf GG, Shariat SF, Peyronnet B, Spano JP, Cancel-Tassin G, Cussenot O. A comprehensive review of genomic landscape, biomarkers and treatment sequencing in castration-resistant prostate cancer. Cancer Treat Rev 2016;48:25-33.
8. Kumagai J, Hofland J, Erkens-Schulze S, Dits NF, Steenbergen J, Jenster G, Homma Y, de Jong FH, van Weerden WM. Intratumoral conversion of adrenal androgen precursors drives androgen receptor-activated cell growth in prostate cancer more potently than de novo steroidogenesis. Prostate 2013;73:1636-50.
9. Penning TM. Mechanisms of drug resistance that target the androgen axis in castration resistant prostate cancer (CRPC). J Steroid Biochem Mol Biol 2015;153:105-13.
10. Sakai M, Martinez-Arguelles DB, Aprikian AG, Magliocco AM, Papadopoulos V. De novo steroid biosynthesis in human prostate cell lines and biopsies. Prostate 2016;76:575-87.
11. Titus MA, Schell MJ, Lih FB, Tomer KB, Mohler JL. Testosterone and dihydrotestosterone tissue levels in recurrent prostate cancer. Clin Cancer Res 2005;11:4653-7.
12. Zhang W, Meng Y, Liu N, Wen XF, Yang T. Insights into chemoresistance of prostate cancer. Int J Biol Sci 2015;11:1160-70.
13. Cai C, Balk SP. Intratumoral androgen biosynthesis in prostate cancer pathogenesis and response to therapy. Endocr Relat Cancer 2011;18:R175-82.
14. Fankhauser M, Tan Y, Hong MKH, Nguyen A, Macintyre G, Haviv I, Pedersen JS, Costello AJ, Hovens CM, Corcoran NM. Canonical androstenedione reduction is the predominant source of signaling androgens in hormone-refractory prostate cancer. Clin Cancer Res 2014;20:5547-57.
15. Montgomery RB, Mostaghel EA, Vessella R, Hess DL, Kalhorn TF, Higano CS, True LD, Nelson PS. Maintenance of intratumoral androgens in metastatic prostate cancer: a mechanism for castration-resistant tumor growth. Cancer Res 2008;68:4447-54.
16. Miller WL, Auchus RJ. The molecular biology, biochemistry, and physiology of human steroidogenesis and its disorders. Endocr Rev 2011;32:81-151.
17. Stanbrough M, Bubley GJ, Ross K, Golub TR, Rubin MA, Penning TM, Febbo PG, Balk SP. Increased expression of genes converting adrenal androgens to testosterone in androgen-independent prostate cancer. Cancer Res 2006;66:2815-25.
18. Chang KH, Li R, Papari-Zareei M, Watumull L, Zhao YD, Auchus RJ, Sharifi N. Dihydrotestosterone synthesis bypasses testosterone to drive castration-resistant prostate cancer. Proc Natl Acad Sci U S A 2011;108:13728-33.
19. Campbell TJ, Tindall DJ, Figg WD. Dihydrotestosterone synthesis from adrenal precursors does not involve testosterone in castration-resistant prostate cancer. Cancer Biol Ther 2012;13:237-8.
20. Wilson JD, Auchus RJ, Leihy MW, Guryev OL, Estabrook RW, Osborn SM, Shaw G, Renfree MB. 5alpha-androstane-3alpha,17beta-diol is formed in tammar wallaby pouch young testes by a pathway involving 5alpha-pregnane-3alpha,17alpha-diol-20-one as a key intermediate. Endocrinology 2003;144:575-80.
22. Locke JA, Guns ES, Lubik AA, Adomat HH, Hendy SC, Wood CA, Ettinger SL, Gleave ME, Nelson CC. Androgen levels increase by intratumoral de novo steroidogenesis during progression of castration-resistant prostate cancer. Cancer Res 2008;68:6407-15.
23. Locke JA, Nelson CC, Adomat HH, Hendy SC, Gleave ME, Guns ES. Steroidogenesis inhibitors alter but do not eliminate androgen synthesis mechanisms during progression to castration-resistance in LNCaP prostate xenografts. J Steroid Biochem Mol Biol 2009;115:126-36.
24. Stuchbery R, McCoy PJ, Hovens CM, Corcoran NM. Androgen synthesis in prostate cancer: do all roads lead to Rome? Nat Rev Urol 2017;14:49-58.
25. Missaghian E, Kempna P, Dick B, Hirsch A, Alikhani-Koupaei R, Jegou B, Mullis PE, Frey BM, Fluck CE. Role of DNA methylation in the tissue-specific expression of the CYP17A1 gene for steroidogenesis in rodents. J Endocrinol 2009;202:99-109.
26. Chung BC, Picado-Leonard J, Haniu M, Bienkowski M, Hall PF, Shively JE, Miller WL. Cytochrome P450c17 (steroid 17 alpha-hydroxylase/17,20 lyase): cloning of human adrenal and testis cDNAs indicates the same gene is expressed in both tissues. Proc Natl Acad Sci U S A 1987;84:407-11.
27. Martinez-Arguelles DB, Papadopoulos V. Epigenetic regulation of the expression of genes involved in steroid hormone biosynthesis and action. Steroids 2010;75:467-76.
28. Fluck CE, Miller WL. GATA-4 and GATA-6 modulate tissue-specific transcription of the human gene for P450c17 by direct interaction with Sp1. Mol Endocrinol 2004;18:1144-57.
29. Yoshimoto FK, Auchus RJ. The diverse chemistry of cytochrome P450 17A1 (P450c17, CYP17A1). J Steroid Biochem Mol Biol 2015;151:52-65.
30. Estrada DF, Laurence JS, Scott EE. Substrate-modulated cytochrome P450 17A1 and cytochrome b5 interactions revealed by NMR. J Biol Chem 2013;288:17008-18.
31. Manenda MS, Hamel CJ, Masselot-Joubert L, Picard ME, Shi R. Androgen-metabolizing enzymes: a structural perspective. J Steroid Biochem Mol Biol 2016;161:54-72.
32. DeVore NM, Scott EE. Structures of cytochrome P450 17A1 with prostate cancer drugs abiraterone and TOK-001. Nature 2012;482:116-9.
33. Petrunak EM, DeVore NM, Porubsky PR, Scott EE. Structures of human steroidogenic cytochrome P450 17A1 with substrates. J Biol Chem 2014;289:32952-64.
34. Estrada DF, Skinner AL, Laurence JS, Scott EE. Human cytochrome P450 17A1 conformational selection: modulation by ligand and cytochrome b5. J Biol Chem 2014;289:14310-20.
35. Gomez L, Kovac JR, Lamb DJ. CYP17A1 inhibitors in castration-resistant prostate cancer. Steroids 2015;95:80-7.
36. Njar VCO, Brodie AMH. Discovery and development of galeterone (TOK-001 or VN/124-1) for the treatment of all stages of prostate cancer. J Med Chem 2015;58:2077-87.
37. Handratta VD, Vasaitis TS, Njar VC, Gediya LK, Kataria R, Chopra P, Newman D Jr, Farquhar R, Guo Z, Qiu Y, Brodie AM. Novel C-17-heteroaryl steroidal CYP17 inhibitors/antiandrogens: synthesis, in vitro biological activity, pharmacokinetics, and antitumor activity in the LAPC4 human prostate cancer xenograft model. J Med Chem 2005;48:2972-84.
38. Ryan CJ, Smith MR, de Bono JS, Molina A, Logothetis CJ, de Souza P, Fizazi K, Mainwaring P, Piulats JM, Ng S, Carles J, Mulders PF, Basch E, Small EJ, Saad F, Schrijvers D, Van Poppel H, Mukherjee SD, Suttmann H, Gerritsen WR, Flaig TW, George DJ, Yu EY, Efstathiou E, Pantuck A, Winquist E, Higano CS, Taplin ME, Park Y, Kheoh T, Griffin T, Scher HI, Rathkopf DE; COU-AA-302 Investigators. Abiraterone in metastatic prostate cancer without previous chemotherapy. N Engl J Med 2013;368:138-48.
39. Kaku T, Hitaka T, Ojida A, Matsunaga N, Adachi M, Tanaka T, Hara T, Yamaoka M, Kusaka M, Okuda T, Asahi S, Furuya S, Tasaka A. Discovery of orteronel (TAK-700), a naphthylmethylimidazole derivative, as a highly selective 17,20-lyase inhibitor with potential utility in the treatment of prostate cancer. Bioorg Med Chem 2011;19:6383-99.
40. Toren PJ, Kim S, Pham S, Mangalji A, Adomat H, Guns ES, Zoubeidi A, Moore W, Gleave ME. Anticancer activity of a novel selective CYP17A1 inhibitor in preclinical models of castrate-resistant prostate cancer. Mol Cancer Ther 2015;14:59-69.
41. Larsen M, Hansen CH, Rasmussen TB, Islin J, Styrishave B, Olsen L, Jorgensen FS. Structure-based optimisation of non-steroidal cytochrome P450 17A1 inhibitors. Chem Commun (Camb) 2017;53:3118-21.
42. Bonomo S, Hansen CH, Petrunak EM, Scott EE, Styrishave B, Jorgensen FS, Olsen L. Promising tools in prostate cancer research: selective non-steroidal cytochrome P450 17A1 inhibitors. Sci Rep 2016;6:29468.
43. Wang M, Fang Y, Gu S, Chen F, Zhu Z, Sun X, Zhu J. Discovery of novel 1,2,3,4-tetrahydrobenzo[4, 5]thieno[2, 3-c]pyridine derivatives as potent and selective CYP17 inhibitors. Eur J Med Chem 2017;132:157-72.
44. Penning TM, Burczynski ME, Jez JM, Lin HK, Ma H, Moore M, Ratnam K, Palackal N. Structure-function aspects and inhibitor design of type 5 17beta-hydroxysteroid dehydrogenase (AKR1C3). Mol Cell Endocrinol 2001;171:137-49.
45. Sun SQ, Gu X, Gao XS, Li Y, Yu H, Xiong W, Yu H, Wang W, Li Y, Teng Y, Zhou D. Overexpression of AKR1C3 significantly enhances human prostate cancer cells resistance to radiation. Oncotarget 2016;7:48050-8.
46. Endo S, Hu D, Matsunaga T, Otsuji Y, El-Kabbani O, Kandeel M, Ikari A, Hara A, Kitade Y, Toyooka N. Synthesis of non-prenyl analogues of baccharin as selective and potent inhibitors for aldo-keto reductase 1C3. Bioorg Med Chem 2014;22:5220-33.
47. Lovering AL, Ride JP, Bunce CM, Desmond JC, Cummings SM, White SA. Crystal structures of prostaglandin D2 11-ketoreductase (AKR1C3) in complex with the nonsteroidal anti-inflammatory drugs flufenamic acid and indomethacin. Cancer Res 2004;64:1802-10.
48. Flanagan JU, Yosaatmadja Y, Teague RM, Chai MZL, Turnbull AP, Squire CJ. Crystal structures of three classes of non-steroidal anti-inflammatory drugs in complex with aldo-keto reductase 1C3. PLoS One 2012;7:e43965.
49. Penning TM, Burczynski ME, Jez JM, Hung CF, Lin HK, Ma H, Moore M, Palackal N, Ratnam K. Human 3alpha-hydroxysteroid dehydrogenase isoforms (AKR1C1-AKR1C4) of the aldo-keto reductase superfamily: functional plasticity and tissue distribution reveals roles in the inactivation and formation of male and female sex hormones. Biochem J 2000;351:67-77.
50. Burczynski ME, Harvey RG, Penning TM. Expression and characterization of four recombinant human dihydrodiol dehydrogenase isoforms: oxidation of trans-7, 8-dihydroxy-7,8-dihydrobenzo[a]pyrene to the activated o-quinone metabolite benzo[a]pyrene-7,8-dione. Biochemistry 1998;37:6781-90.
51. Byrns MC, Jin Y, Penning TM. Inhibitors of type 5 17beta-hydroxysteroid dehydrogenase (AKR1C3): overview and structural insights. J Steroid Biochem Mol Biol 2011;125:95-104.
52. Adeniji AO, Chen M, Penning TM. AKR1C3 as a target in castrate resistant prostate cancer. J Steroid Biochem Mol Biol 2013;137:136-49.
53. Skarydova L, Hofman J, Chlebek J, Havrankova J, Kosanova K, Skarka A, Hostalkova A, Plucha T, Cahlikova L, Wsol V. Isoquinoline alkaloids as a novel type of AKR1C3 inhibitors. J Steroid Biochem Mol Biol 2014;143:250-8.
54. Tian Y, Zhao L, Wang Y, Zhang H, Xu D, Zhao X, Li Y, Li J. Berberine inhibits androgen synthesis by interaction with aldo-keto reductase 1C3 in 22Rv1 prostate cancer cells. Asian J Androl 2016;18:607-12.
55. Akao Y, Maruyama H, Matsumoto K, Ohguchi K, Nishizawa K, Sakamoto T, Araki Y, Mishima S, Nozawa Y. Cell growth inhibitory effect of cinnamic acid derivatives from propolis on human tumor cell lines. Biol Pharm Bull 2003;26:1057-9.
56. Endo S, Matsunaga T, Kanamori A, Otsuji Y, Nagai H, Sundaram K, El-Kabbani O, Toyooka N, Ohta S, Hara A. Selective inhibition of human type-5 17beta-hydroxysteroid dehydrogenase (AKR1C3) by baccharin, a component of Brazilian propolis. J Nat Prod 2012;75:716-21.
57. Ogihara T, Tamai I, Tsuji A. Structural characterization of substrates for the anion exchange transporter in Caco-2 cells. J Pharm Sci 1999;88:1217-21.
58. Pippione AC, Dosio F, Ducime A, Federico A, Martina K, Sainas S, Frolund B, Gooyit M, Janda KD, Boschi D, Lolli ML. Substituted 4-hydroxy-1,2,3-triazoles: synthesis, characterization and first drug design applications through bioisosteric modulation and scaffold hopping approaches. Medchemcomm 2015;6:1285-92.
59. Sainas S, Pippione AC, Giorgis M, Lupino E, Goyal P, Ramondetti C, Buccinna B, Piccinini M, Braga RC, Andrade CH, Andersson M, Moritzer AC, Friemann R, Mensa S, Al-Kadaraghi S, Boschi D, Lolli ML. Design, synthesis, biological evaluation and X-ray structural studies of potent human dihydroorotate dehydrogenase inhibitors based on hydroxylated azole scaffolds. Eur J Med Chem 2017;129:287-302.
60. Pippione AC, Giraudo A, Bonanni D, Carnovale IM, Marini E, Cena C, Costale A, Zonari D, Pors K, Sadiq M, Boschi D, Oliaro-Bosso S, Lolli ML. Hydroxytriazole derivatives as potent and selective aldo-keto reductase 1C3 (AKR1C3) inhibitors discovered by bioisosteric scaffold hopping approach. Eur J Med Chem 2017;139:936-46.
61. Heinrich DM, Flanagan JU, Jamieson SM, Silva S, Rigoreau LJ, Trivier E, Raynham T, Turnbull AP, Denny WA. Synthesis and structure-activity relationships for 1-(4-(piperidin-1-ylsulfonyl)phenyl)pyrrolidin-2-ones as novel non-carboxylate inhibitors of the aldo-keto reductase enzyme AKR1C3. Eur J Med Chem 2013;62:738-44.
62. Jamieson SM, Brooke DG, Heinrich D, Atwell GJ, Silva S, Hamilton EJ, Turnbull AP, Rigoreau LJ, Trivier E, Soudy C, Samlal SS, Owen PJ, Schroeder E, Raynham T, Flanagan JU, Denny WA. 3-(3,4-Dihydroisoquinolin-2(1H)-ylsulfonyl)benzoic acids: highly potent and selective inhibitors of the type 5 17-beta-hydroxysteroid dehydrogenase AKR1C3. J Med Chem 2012;55:7746-58.
63. Yin YD, Fu M, Brooke DG, Heinrich DM, Denny WA, Jamieson SM. The activity of SN33638, an inhibitor of AKR1C3, on testosterone and 17beta-estradiol production and function in castration-resistant prostate cancer and ER-positive breast cancer. Front Oncol 2014;4:159.
64. Zhou W, Limonta P. AKR1C3 inhibition therapy in castration-resistant prostate cancer and breast cancer: lessons from responses to SN33638. Front Oncol 2014;4:162.
65. Flanagan JU, Atwell GJ, Heinrich DM, Brooke DG, Silva S, Rigoreau LJ, Trivier E, Turnbull AP, Raynham T, Jamieson SM, Denny WA. Morpholylureas are a new class of potent and selective inhibitors of the type 5 17-beta-hydroxysteroid dehydrogenase (AKR1C3). Bioorg Med Chem 2014;22:967-77.
66. Kikuchi A, Furutani T, Azami H, Watanabe K, Niimi T, Kamiyama Y, Kuromitsu S, Baskin-Bey E, Heeringa M, Ouatas T, Enjo K. In vitro and in vivo characterisation of ASP9521: a novel, selective, orally bioavailable inhibitor of 17beta-hydroxysteroid dehydrogenase type 5 (17betaHSD5; AKR1C3). Invest New Drugs 2014;32:860-70.
67. Yepuru M, Wu Z, Kulkarni A, Yin F, Barrett CM, Kim J, Steiner MS, Miller DD, Dalton JT, Narayanan R. Steroidogenic enzyme AKR1C3 is a novel androgen receptor-selective coactivator that promotes prostate cancer growth. Clin Cancer Res 2013;19:5613-25.
68. Loriot Y, Fizazi K, Jones RJ, Van den Brande J, Molife RL, Omlin A, James ND, Baskin-Bey E, Heeringa M, Baron B, Holtkamp GM, Ouatas T, De Bono JS. Safety, tolerability and anti-tumour activity of the androgen biosynthesis inhibitor ASP9521 in patients with metastatic castration-resistant prostate cancer: multi-centre phase I/II study. Invest New Drugs 2014;32:995-1004.
69. Enzalutamide and indomethacin in treating patients with recurrent or metastatic hormone-resistant prostate cancer. In: ClinicalTrials.gov; Identifier: NCT02935205. Available from: https://clinicaltrials.gov/ct2/show/NCT02935205. [Last accessed on 13 Nov 2017].
70. A phase II neoadjuvant study of ARN-509, abiraterone acetate, prednisone, degarelix and indomethacin in men with localized prostate cancer pre-prostatectomy. Available from: http://www.centerwatch.com/clinical-trials/listings/98162/stage-iii-prostate-adenocarcinoma-phase-ii-neoadjuvant-study/?&radius=50. [Last accessed on 13 Nov 2017].
71. Guise CP, Abbattista MR, Singleton RS, Holford SD, Connolly J, Dachs GU, Fox SB, Pollock R, Harvey J, Guilford P, Donate F, Wilson WR, Patterson AV. The bioreductive prodrug PR-104A is activated under aerobic conditions by human aldo-keto reductase 1C3. Cancer Res 2010;70:1573-84.
72. Manesh DM, El-Hoss J, Evans K, Richmond J, Toscan CE, Bracken LS, Hedrick A, Sutton R, Marshall GM, Wilson WR, Kurmasheva RT, Billups C, Houghton PJ, Smith MA, Carol H, Lock RB. AKR1C3 is a biomarker of sensitivity to PR-104 in preclinical models of T-cell acute lymphoblastic leukemia. Blood 2015;126:1193-202.
73. Marchais-Oberwinkler S, Henn C, Moller G, Klein T, Negri M, Oster A, Spadaro A, Werth R, Wetzel M, Xu K, Frotscher M, Hartmann RW, Adamski J. 17beta-Hydroxysteroid dehydrogenases (17beta-HSDs) as therapeutic targets: protein structures, functions, and recent progress in inhibitor development. J Steroid Biochem Mol Biol 2011;125:66-82.
74. Poirier D. 17beta-Hydroxysteroid dehydrogenase inhibitors: a patent review. Expert Opin Ther Pat 2010;20:1123-45.
75. Day JM, Tutill HJ, Purohit A. 17ss-hydroxysteroid dehydrogenase inhibitors. Minerva Endocrinol 2010;35:87-108.
76. Olusanjo MS, Ahmed S. Inhibitors of 17-hydroxysteroid dehydrogenase type 3 (17-beta-HSD 3). Drugs Future 2009;34:555.
77. Ning X, Yang Y, Deng H, Zhang Q, Huang Y, Su Z, Fu Y, Xiang Q, Zhang S. Development of 17β-hydroxysteroid dehydrogenase type 3 as a target in hormone-dependent prostate cancer therapy. Steroids 2017;121:10-6.
78. Fink BE, Gavai AV, Tokarski JS, Goyal B, Misra R, Xiao HY, Kimball SD, Han WC, Norris D, Spires TE, You D, Gottardis MM, Lorenzi MV, Vite GD. Identification of a novel series of tetrahydrodibenzazocines as inhibitors of 17beta-hydroxysteroid dehydrogenase type 3. Bioorg Med Chem Lett 2006;16:1532-6.
79. Vicker N, Sharland CM, Heaton WB, Gonzalez AM, Bailey HV, Smith A, Springall JS, Day JM, Tutill HJ, Reed MJ, Purohit A, Potter BV. The design of novel 17beta-hydroxysteroid dehydrogenase type 3 inhibitors. Mol Cell Endocrinol 2009;301:259-65.
80. Nashev LG, Schuster D, Laggner C, Sodha S, Langer T, Wolber G, Odermatt A. The UV-filter benzophenone-1 inhibits 17beta-hydroxysteroid dehydrogenase type 3: virtual screening as a strategy to identify potential endocrine disrupting chemicals. Biochem Pharmacol 2010;79:1189-99.
81. Schuster D, Kowalik D, Kirchmair J, Laggner C, Markt P, Aebischer-Gumy C, Strohle F, Moller G, Wolber G, Wilckens T, Langer T, Odermatt A, Adamski J. Identification of chemically diverse, novel inhibitors of 17beta-hydroxysteroid dehydrogenase type 3 and 5 by pharmacophore-based virtual screening. J Steroid Biochem Mol Biol 2011;125:148-61.
82. Guzi TJ, Liu Y-T, Doll RJ, Saksena A, Girijavallabhan VM, Pachter JA. Preparation of 6,11-dihydro-5H-benzo[5,6]cyclohepta[1,2-b]pyridine derivatives as 17 beta-hydroxysteroid dehydrogenase type 3 inhibitors for the treatment of androgen dependent diseases. Kenilworth, USA: Schering Corporation; 2004. p. 72.
83. Day JM, Foster PA, Tutill HJ, Schmidlin F, Sharland CM, Hargrave JD, Vicker N, Potter BV, Reed MJ, Purohit A. STX2171, a 17beta-hydroxysteroid dehydrogenase type 3 inhibitor, is efficacious in vivo in a novel hormone-dependent prostate cancer model. Endocr Relat Cancer 2013;20:53-64.
84. Harada K, Kubo H, Tomigahara Y, Nishioka K, Takahashi J, Momose M, Inoue S, Kojima A. Coumarins as novel 17beta-hydroxysteroid dehydrogenase type 3 inhibitors for potential treatment of prostate cancer. Bioorg Med Chem Lett 2010;20:272-5.
85. Harada K, Kubo H, Abe J, Haneta M, Conception A, Inoue S, Okada S, Nishioka K. Discovery of potent and orally bioavailable 17β-hydroxysteroid dehydrogenase type 3 inhibitors. Bioorg Med Chem 2012;20:3242-54.
86. Ohno S, Nakajima Y, Nakajin S. Triphenyltin and tributyltin inhibit pig testicular 17beta-hydroxysteroid dehydrogenase activity and suppress testicular testosterone biosynthesis. Steroids 2005;70:645-51.
87. Titus MA, Li Y, Kozyreva OG, Maher V, Godoy A, Smith GJ, Mohler JL. 5α-reductase type 3 enzyme in benign and malignant prostate. Prostate (Hoboken, NJ, U S) 2014;74:235-49.
88. Azzouni F, Godoy A, Li Y, Mohler J. The 5 alpha-reductase isozyme family: a review of basic biology and their role in human diseases. Adv Urol 2012;2012:530121.
89. Li J, Ding Z, Wang Z, Lu JF, Maity SN, Navone NM, Logothetis CJ, Mills GB, Kim J. Androgen regulation of 5alpha-reductase isoenzymes in prostate cancer: implications for prostate cancer prevention. PLoS One 2011;6:e28840.
90. Gormley GJ, Stoner E, Bruskewitz RC, Imperato-McGinley J, Walsh PC, McConnell JD, Andriole GL, Geller J, Bracken BR, Tenover JS, Vaughan ED, Pappas F, Taylor A, Binkowitz B, Ng J. The effect of finasteride in men with benign prostatic hyperplasia. 1992. J Urol 2002;167:1102-8.
91. Roehrborn CG, Boyle P, Nickel JC, Hoefner K, Andriole G. Efficacy and safety of a dual inhibitor of 5-alpha-reductase types 1 and 2 (dutasteride) in men with benign prostatic hyperplasia. Urology 2002;60:434-41.
92. Faller B, Farley D, Nick H. Finasteride: a slow-binding 5 alpha-reductase inhibitor. Biochemistry 1993;32:5705-10.
93. Aggarwal S, Thareja S, Verma A, Bhardwaj TR, Kumar M. An overview on 5alpha-reductase inhibitors. Steroids 2010;75:109-53.
94. Clark RV, Hermann DJ, Cunningham GR, Wilson TH, Morrill BB, Hobbs S. Marked suppression of dihydrotestosterone in men with benign prostatic hyperplasia by dutasteride, a dual 5alpha-reductase inhibitor. J Clin Endocrinol Metab 2004;89:2179-84.
95. Salvador JA, Pinto RM, Silvestre SM. Steroidal 5alpha-reductase and 17alpha-hydroxylase/17,20-lyase (CYP17) inhibitors useful in the treatment of prostatic diseases. J Steroid Biochem Mol Biol 2013;137:199-222.
96. Holt DA, Levy MA, Oh HJ, Erb JM, Heaslip JI, Brandt M, Lan-Hargest HY, Metcalf BW. Inhibition of steroid 5 alpha-reductase by unsaturated 3-carboxysteroids. J Med Chem 1990;33:943-50.
97. Yao Z, Xu Y, Zhang M, Jiang S, Nicklaus MC, Liao C. Discovery of a novel hybrid from finasteride and epristeride as 5α-reductase inhibitor. Bioorg Med Chem Lett 2011;21:475-8.
98. Aggarwal S, Thareja S, Bhardwaj TR, Haupenthal J, Hartmann RW, Kumar M. Synthesis and biological evaluation of novel unsaturated carboxysteroids as human 5alpha-reductase inhibitors: a legitimate approach. Eur J Med Chem 2012;54:728-39.
99. Kim S, Kim Y-U, Ma E. Synthesis and 5α-reductase inhibitory activity of C21 steroids having 1,4-diene or 4,6-diene 20-ones and 4-azasteroid 20-oximes. Molecules 2012;17:355-68.
100. Al-Mohizea AM, Al-Omar MA, Abdalla MM, Amr AG. 5alpha-reductase inhibitors, antiviral and anti-tumor activities of some steroidal cyanopyridinone derivatives. Int J Biol Macromol 2012;50:171-9.
101. Lacy JM, Kyprianou N. A tale of two trials: the impact of 5α-reductase inhibition on prostate cancer (Review). Oncol Lett 2014;8:1391-6.
102. FDA Drug Safety Communication: 5-alpha reductase inhibitors (5-ARIs) may increase the risk of a more serious form of prostate cancer. Available from: https://www.fda.gov/Drugs/DrugSafety/ucm258314.htm. [Last accessed on 13 Nov 2017].
103. Fleshner NE, Lucia MS, Egerdie B, Aaron L, Eure G, Nandy I, Black L, Rittmaster RS. Dutasteride in localised prostate cancer management: the REDEEM randomised, double-blind, placebo-controlled trial. Lancet 2012;379:1103-11.
104. Schroder F, Bangma C, Angulo JC, Alcaraz A, Colombel M, McNicholas T, Tammela TL, Nandy I, Castro R. Dutasteride treatment over 2 years delays prostate-specific antigen progression in patients with biochemical failure after radical therapy for prostate cancer: results from the randomised, placebo-controlled Avodart After Radical Therapy for Prostate Cancer Study (ARTS). Eur Urol 2013;63:779-87.
105. Ramalingam S, Ramamurthy VP, Njar VC. Dissecting major signaling pathways in prostate cancer development and progression: Mechanisms and novel therapeutic targets. J Steroid Biochem Mol Biol 2017;166:16-27.
106. Blessing AM, Ganesan S, Rajapakshe K, Ying Sung Y, Reddy Bollu L, Shi Y, Cheung E, Coarfa C, Chang JT, McDonnell DP, Frigo DE. Identification of a novel coregulator, SH3YL1, that interacts with the androgen receptor N-terminus. Mol Endocrinol 2015;29:1426-39.
107. Dubbink HJ, Hersmus R, Verma CS, van der Korput HA, Berrevoets CA, van Tol J, Ziel-van der Made AC, Brinkmann AO, Pike AC, Trapman J. Distinct recognition modes of FXXLF and LXXLL motifs by the androgen receptor. Mol Endocrinol 2004;18:2132-50.
108. Saporita AJ, Zhang Q, Navai N, Dincer Z, Hahn J, Cai X, Wang Z. Identification and characterization of a ligand-regulated nuclear export signal in androgen receptor. J Biol Chem 2003;278:41998-2005.
109. Shang Y, Myers M, Brown M. Formation of the androgen receptor transcription complex. Mol Cell 2002;9:601-10.
110. Lonergan PE, Tindall DJ. Androgen receptor signaling in prostate cancer development and progression. J Carcinog 2011;10:20.
111. Green SM, Mostaghel EA, Nelson PS. Androgen action and metabolism in prostate cancer. Mol Cell Endocrinol 2012;360:3-13.
112. Kahn B, Collazo J, Kyprianou N. Androgen receptor as a driver of therapeutic resistance in advanced prostate cancer. Int J Biol Sci 2014;10:588-95.
113. Shafi AA, Yen AE, Weigel NL. Androgen receptors in hormone-dependent and castration-resistant prostate cancer. Pharmacol Ther 2013;140:223-38.
114. Jarrard DF, Kinoshita H, Shi Y, Sandefur C, Hoff D, Meisner LF, Chang C, Herman JG, Isaacs WB, Nassif N. Methylation of the androgen receptor promoter CpG island is associated with loss of androgen receptor expression in prostate cancer cells. Cancer Res 1998;58:5310-4.
115. Nakayama T, Watanabe M, Suzuki H, Toyota M, Sekita N, Hirokawa Y, Mizokami A, Ito H, Yatani R, Shiraishi T. Epigenetic regulation of androgen receptor gene expression in human prostate cancers. Lab Invest 2000;80:1789-96.
116. Schulz WA, Hoffmann MJ. Epigenetic mechanisms in the biology of prostate cancer. Semin Cancer Biol 2009;19:172-80.
117. Martinez-Ariza G, Hulme C. Recent advances in allosteric androgen receptor inhibitors for the potential treatment of castration-resistant prostate cancer. Pharm Pat Anal 2015;4:387-402.
118. Lu X, Dun K, Wang Y, Yang Y, You Q, Li Z. Recent androgen receptor antagonists in prostate cancer. Mini Rev Med Chem 2014;14:655-63.
119. Tran C, Ouk S, Clegg NJ, Chen Y, Watson PA, Arora V, Wongvipat J, Smith-Jones PM, Yoo D, Kwon A, Wasielewska T, Welsbie D, Chen CD, Higano CS, Beer TM, Hung DT, Scher HI, Jung ME, Sawyers CL. Development of a second-generation antiandrogen for treatment of advanced prostate cancer. Science 2009;324:787-90.
120. Clegg NJ, Wongvipat J, Joseph JD, Tran C, Ouk S, Dilhas A, Chen Y, Grillot K, Bischoff ED, Cai L, Aparicio A, Dorow S, Arora V, Shao G, Qian J, Zhao H, Yang G, Cao C, Sensintaffar J, Wasielewska T, Herbert MR, Bonnefous C, Darimont B, Scher HI, Smith-Jones P, Klang M, Smith ND, De Stanchina E, Wu N, Ouerfelli O, Rix PJ, Heyman RA, Jung ME, Sawyers CL, Hager JH. ARN-509: a novel antiandrogen for prostate cancer treatment. Cancer Res 2012;72:1494-503.
121. Fizazi K, Albiges L, Loriot Y, Massard C. ODM-201: a new-generation androgen receptor inhibitor in castration-resistant prostate cancer. Expert Rev Anticancer Ther 2015;15:1007-17.
122. Yu Z, Cai C, Gao S, Simon NI, Shen HC, Balk SP. Galeterone prevents androgen receptor binding to chromatin and enhances degradation of mutant androgen receptor. Clin Cancer Res 2014;20:4075-85.
123. Bastos DA, Antonarakis ES. Galeterone for the treatment of advanced prostate cancer: the evidence to date. Drug Des Devel Ther 2016;10:2289-97.
124. Biron E, Bedard F. Recent progress in the development of protein-protein interaction inhibitors targeting androgen receptor-coactivator binding in prostate cancer. J Steroid Biochem Mol Biol 2016;161:36-44.
125. Ran F, Xing H, Liu Y, Zhang D, Li P, Zhao G. Recent developments in androgen receptor antagonists. Arch Pharm (Weinheim) 2015;348:757-75.
126. Tice CM, Zheng YJ. Non-canonical modulators of nuclear receptors. Bioorg Med Chem Lett 2016;26:4157-64.
127. Lallous N, Dalal K, Cherkasov A, Rennie PS. Targeting alternative sites on the androgen receptor to treat castration-resistant prostate cancer. Int J Mol Sci 2013;14:12496-519.
128. Kandil S, Westwell AD, McGuigan C. 7-Substituted umbelliferone derivatives as androgen receptor antagonists for the potential treatment of prostate and breast cancer. Bioorg Med Chem Lett 2016;26:2000-4.
129. Johnson JK, Skoda EM, Zhou J, Parrinello E, Wang D, O'Malley K, Eyer BR, Kazancioglu M, Eisermann K, Johnston PA, Nelson JB, Wang Z, Wipf P. Small molecule antagonists of the nuclear androgen receptor for the treatment of castration-resistant prostate cancer. ACS Med Chem Lett 2016;7:785-90.
130. Balog A, Rampulla R, Martin GS, Krystek SR, Attar R, Dell-John J, DiMarco JD, Fairfax D, Gougoutas J, Holst CL, Nation A, Rizzo C, Rossiter LM, Schweizer L, Shan W, Spergel S, Spires T, Cornelius G, Gottardis M, Trainor G, Vite GD, Salvati ME. Discovery of BMS-641988, a novel androgen receptor antagonist for the treatment of prostate cancer. ACS Med Chem Lett 2015;6:908-12.
131. Zhao C, Choi YH, Khadka DB, Jin Y, Lee KY, Cho WJ. Design and synthesis of novel androgen receptor antagonists via molecular modeling. Bioorg Med Chem 2016;24:789-801.
132. Yang SH, Song CH, Van HT, Park E, Khadka DB, Gong EY, Lee K, Cho WJ. SAR based design of nicotinamides as a novel class of androgen receptor antagonists for prostate cancer. J Med Chem 2013;56:3414-8.
133. Ferroni C, Pepe A, Kim YS, Lee S, Guerrini A, Parenti MD, Tesei A, Zamagni A, Cortesi M, Zaffaroni N, De Cesare M, Beretta GL, Trepel JB, Malhotra SV, Varchi G. 1,4-substituted triazoles as nonsteroidal anti-androgens for prostate cancer treatment. J Med Chem 2017;60:3082-93.
134. Buzon V, Carbo LR, Estruch SB, Fletterick RJ, Estebanez-Perpina E. A conserved surface on the ligand binding domain of nuclear receptors for allosteric control. Mol Cell Endocrinol 2012;348:394-402.
135. Estebanez-Perpina E, Arnold LA, Nguyen P, Rodrigues ED, Mar E, Bateman R, Pallai P, Shokat KM, Baxter JD, Guy RK, Webb P, Fletterick RJ. A surface on the androgen receptor that allosterically regulates coactivator binding. Proc Natl Acad Sci U S A 2007;104:16074-9.
136. Munuganti RS, Hassona MD, Leblanc E, Frewin K, Singh K, Ma D, Ban F, Hsing M, Adomat H, Lallous N, Andre C, Jonadass JP, Zoubeidi A, Young RN, Guns ET, Rennie PS, Cherkasov A. Identification of a potent antiandrogen that targets the BF3 site of the androgen receptor and inhibits enzalutamide-resistant prostate cancer. Chem Biol 2014;21:1476-85.
137. Zhang Y, Mantravadi PK, Jobbagy S, Bao W, Koh JT. Antagonizing the androgen receptor with a biomimetic acyltransferase. ACS Chem Biol 2016;11:2797-802.
138. Myung JK, Banuelos CA, Fernandez JG, Mawji NR, Wang J, Tien AH, Yang YC, Tavakoli I, Haile S, Watt K, McEwan IJ, Plymate S, Andersen RJ, Sadar MD. An androgen receptor N-terminal domain antagonist for treating prostate cancer. J Clin Invest 2013;123:2948-60.
139. Andersen RJ, Mawji NR, Wang J, Wang G, Haile S, Myung JK, Watt K, Tam T, Yang YC, Banuelos CA, Williams DE, McEwan IJ, Wang Y, Sadar MD. Regression of castrate-recurrent prostate cancer by a small-molecule inhibitor of the amino-terminus domain of the androgen receptor. Cancer Cell 2010;17:535-46.
140. Antonarakis ES, Antonarakis ES, Luo J, Chandhasin C, Osbourne E, Perabo F, Sadar MD. Targeting the N-terminal domain of the androgen receptor: a new approach for the treatment of advanced prostate cancer. Oncologist 2016;21:1427-35.
142. Shaffer PL, Jivan A, Dollins DE, Claessens F, Gewirth DT. Structural basis of androgen receptor binding to selective androgen response elements. Proc Natl Acad Sci U S A 2004;101:4758-63.
143. Diamond M, Jones J, Renslo A. Androgen receptor inhibitors, including tetrahydropyrvinium derivatives and benzoxazole compounds, and their therapeutic use. Oakland, USA: University of California; 2009:67.
144. Lim M, Otto-Duessel M, He M, Su L, Nguyen D, Chin E, Alliston T, Jones JO. Ligand-independent and tissue-selective androgen receptor inhibition by pyrvinium. ACS Chem Biol 2014;9:692-702.
145. Li H, Ban F, Dalal K, Leblanc E, Frewin K, Ma D, Adomat H, Rennie PS, Cherkasov A. Discovery of small-molecule inhibitors selectively targeting the DNA-binding domain of the human androgen receptor. J Med Chem 2014;57:6458-67.
146. Dalal K, Roshan-Moniri M, Sharma A, Li H, Ban F, Hassona MD, Hsing M, Singh K, LeBlanc E, Dehm S, Tomlinson Guns ES, Cherkasov A, Rennie PS. Selectively targeting the DNA-binding domain of the androgen receptor as a prospective therapy for prostate cancer. J Biol Chem 2014;289:26417-29.
147. Loddick SA, Ross SJ, Thomason AG, Robinson DM, Walker GE, Dunkley TP, Brave SR, Broadbent N, Stratton NC, Trueman D, Mouchet E, Shaheen FS, Jacobs VN, Cumberbatch M, Wilson J, Jones RD, Bradbury RH, Rabow A, Gaughan L, Womack C, Barry ST, Robson CN, Critchlow SE, Wedge SR, Brooks AN. AZD3514: a small molecule that modulates androgen receptor signaling and function in vitro and in vivo. Mol Cancer Ther 2013;12:1715-27.
148. Omlin A, Jones RJ, van der Noll R, Satoh T, Niwakawa M, Smith SA, Graham J, Ong M, Finkelman RD, Schellens JH, Zivi A, Crespo M, Riisnaes R, Nava-Rodrigues D, Malone MD, Dive C, Sloane R, Moore D, Alumkal JJ, Dymond A, Dickinson PA, Ranson M, Clack G, de Bono J, Elliott T. AZD3514, an oral selective androgen receptor down-regulator in patients with castration-resistant prostate cancer - results of two parallel first-in-human phase I studies. Invest New Drugs 2015;33:679-90.
149. Yamashita S, Lai KP, Chuang KL, Xu D, Miyamoto H, Tochigi T, Pang ST, Li L, Arai Y, Kung HJ, Yeh S, Chang C. ASC-J9 suppresses castration-resistant prostate cancer growth through degradation of full-length and splice variant androgen receptors. Neoplasia 2012;14:74-83.
150. Lai KP, Huang CK, Chang YJ, Chung CY, Yamashita S, Li L, Lee SO, Yeh S, Chang C. New therapeutic approach to suppress castration-resistant prostate cancer using ASC-J9 via targeting androgen receptor in selective prostate cells. Am J Pathol 2013;182:460-73.
151. Liu C, Lou W, Zhu Y, Nadiminty N, Schwartz CT, Evans CP, Gao AC. Niclosamide inhibits androgen receptor variants expression and overcomes enzalutamide resistance in castration-resistant prostate cancer. Clin Cancer Res 2014;20:3198-210.
152. Purushottamachar P, Kwegyir-Afful AK, Martin MS, Ramamurthy VP, Ramalingam S, Njar VC. Identification of novel steroidal androgen receptor degrading agents inspired by galeterone 3β-imidazole carbamate. ACS Med Chem Lett 2016;7:708-13.
153. Kwegyir-Afful AK, Ramalingam S, Purushottamachar P, Ramamurthy VP, Njar VC. Galeterone and VNPT55 induce proteasomal degradation of AR/AR-V7, induce significant apoptosis via cytochrome c release and suppress growth of castration resistant prostate cancer xenografts in vivo. Oncotarget 2015;6:27440-60.
154. Bradbury RH, Hales NJ, Rabow AA, Walker GE, Acton DG, Andrews DM, Ballard P, Brooks NA, Colclough N, Girdwood A, Hancox UJ, Jones O, Jude D, Loddick SA, Mortlock AA. Small-molecule androgen receptor downregulators as an approach to treatment of advanced prostate cancer. Bioorg Med Chem Lett 2011;21:5442-5.
155. Gustafson JL, Neklesa TK, Cox CS, Roth AG, Buckley DL, Tae HS, Sundberg TB, Stagg DB, Hines J, McDonnell DP, Norris JD, Crews CM. Small-molecule-mediated degradation of the androgen receptor through hydrophobic tagging. Angew Chem Int Ed Engl 2015;54:9659-62.
156. Lai AC, Crews CM. Induced protein degradation: an emerging drug discovery paradigm. Nat Rev Drug Discov 2017;16:101-14.
157. Chu FM, Sartor O, Gomella L, Rudo T, Somerville MC, Hereghty B, Manyak MJ. A randomised, double-blind study comparing the addition of bicalutamide with or without dutasteride to GnRH analogue therapy in men with non-metastatic castrate-resistant prostate cancer. Eur J Cancer 2015;51:1555-69.
158. Hamid AR, Verhaegh GW, Smit FP, van Rijt-van de Westerlo C, Armandari I, Brandt A, Sweep FC, Sedelaar JP, Schalken JA. Dutasteride and enzalutamide synergistically suppress prostate tumor cell proliferation. J Urol 2015;193:1023-9.
159. Liu C, Lou W, Zhu Y, Yang JC, Nadiminty N, Gaikwad NW, Evans CP, Gao AC. Intracrine androgens and AKR1C3 activation confer resistance to enzalutamide in prostate cancer. Cancer Res 2015;75:1413-22.
160. Liu C, Armstrong CM, Lou W, Lombard A, Evans CP, Gao AC. Inhibition of AKR1C3 activation overcomes resistance to abiraterone in advanced prostate cancer. Mol Cancer Ther 2017;16:35-44.
161. Androgen receptor antagonist ARN-509, abiraterone acetate, prednisone, degarelix, and indomethacin in treating patients with localized prostate cancer before surgery. Available from: https://www.bioportfolio.com/resources/trial/162934/Androgen-Receptor-Antagonist-ARN-509-Abiraterone-Acetate-Prednisone-Degarelix-and-Indomethacin-in.html. [Last accessed on 13 Nov 2017].
162. Anighoro A, Bajorath J, Rastelli G. Polypharmacology: challenges and opportunities in drug discovery. J Med Chem 2014;57:7874-87.
163. Boschi D, Giorgis M, Cena C, Talniya NC, Di Stilo A, Morini G, Coruzzi G, Guaita E, Fruttero R, Gasco A. Multitarget drugs: synthesis and preliminary pharmacological characterization of zileuton analogues endowed with Dual 5-LO inhibitor and NO-dependent activities. Chem Med Chem 2010;5:1444-9.
164. Chen M, Adeniji AO, Twenter BM, Winkler JD, Christianson DW, Penning TM. Crystal structures of AKR1C3 containing an N-(aryl)amino-benzoate inhibitor and a bifunctional AKR1C3 inhibitor and androgen receptor antagonist. Therapeutic leads for castrate resistant prostate cancer. Bioorg Med Chem Lett 2012;22:3492-7.
165. Feau C, Arnold LA, Kosinski A, Zhu F, Connelly M, Guy RK. Novel flufenamic acid analogues as inhibitors of androgen receptor mediated transcription. ACS Chem Biol 2009;4:834-43.
166. Purushottamachar P, Godbole AM, Gediya LK, Martin MS, Vasaitis TS, Kwegyir-Afful AK, Ramalingam S, Ates-Alagoz Z, Njar VC. Systematic structure modifications of multitarget prostate cancer drug candidate galeterone to produce novel androgen receptor down-regulating agents as an approach to treatment of advanced prostate cancer. J Med Chem 2013;56:4880-98.
167. Bruno RD, Vasaitis TS, Gediya LK, Purushottamachar P, Godbole AM, Ates-Alagoz Z, Brodie AMH, Njar VC. Synthesis and biological evaluations of putative metabolically stable analogs of VN/124-1 (TOK-001): head to head anti-tumor efficacy evaluation of VN/124-1 (TOK-001) and abiraterone in LAPC-4 human prostate cancer xenograft model. Steroids 2011;76:1268-79.
168. Vasaitis T, Belosay A, Schayowitz A, Khandelwal A, Chopra P, Gediya LK, Guo Z, Fang HB, Njar VC, Brodie AM. Androgen receptor inactivation contributes to antitumor efficacy of 17{alpha}-hydroxylase/17,20-lyase inhibitor 3beta-hydroxy-17-(1H-benzimidazole-1-yl)androsta-5,16-diene in prostate cancer. Mol Cancer Ther 2008;7:2348-57.
169. Tokai Pharmaceuticals Announces Clinical Update. Available from: http://www.businesswire.com/news/home/20160726005553/en/Tokai-Pharmaceuticals-Announces-Clinical-Update. [Last accessed on 13 Nov 2017].
170. Li Z, Bishop AC, Alyamani M, Garcia JA, Dreicer R, Bunch D, Liu J, Upadhyay SK, Auchus RJ, Sharifi N. Conversion of abiraterone to D4A drives anti-tumour activity in prostate cancer. Nature 2015;523:347-51.
171. Richards J, Lim AC, Hay CW, Taylor AE, Wingate A, Nowakowska K, Pezaro C, Carreira S, Goodall J, Arlt W, McEwan IJ, de Bono JS, Attard G. Interactions of abiraterone, eplerenone, and prednisolone with wild-type and mutant androgen receptor: a rationale for increasing abiraterone exposure or combining with MDV3100. Cancer Res 2012;72:2176-82.
172. Buttigliero C, Tucci M, Bertaglia V, Vignani F, Bironzo P, Di Maio M, Scagliotti GV. Understanding and overcoming the mechanisms of primary and acquired resistance to abiraterone and enzalutamide in castration resistant prostate cancer. Cancer Treat Rev 2015;41:884-92.
173. Jernberg E, Thysell E, Bovinder YE, Rudolfsson S, Crnalic S, Widmark A, Bergh A, Wikstrom P. Characterization of prostate cancer bone metastases according to expression levels of steroidogenic enzymes and androgen receptor splice variants. PLoS One 2013;8:e77407.
174. Djusberg E, Jernberg E, Thysell E, Golovleva I, Lundberg P, Crnalic S, Widmark A, Bergh A, Brattsand M, Wikström P. High levels of the AR-V7 splice variant and co-amplification of the golgi protein coding YIPF6 in AR amplified prostate cancer bone metastases. Prostate 2017;77:625-38.
175. Hagberg Thulin M, Nilsson ME, Thulin P, Céraline J, Ohlsson C, Damber J-E, Welén K. Osteoblasts promote castration-resistant prostate cancer by altering intratumoral steroidogenesis. Mol Cell Endocrinol 2016;422:182-91.
176. Hofland J, van Weerden WM, Dits NF, Steenbergen J, van Leenders GJ, Jenster G, Schroder FH, de Jong FH. Evidence of limited contributions for intratumoral steroidogenesis in prostate cancer. Cancer Res 2010;70:1256-64.
177. Pfeiffer MJ, Smit FP, Sedelaar JP, Schalken JA. Steroidogenic enzymes and stem cell markers are upregulated during androgen deprivation in prostate cancer. Mol Med 2011;17:657-64.
178. Audet-Walsh E, Yee T, Tam IS, Giguere V. Inverse regulation of DHT synthesis enzymes 5alpha-reductase types 1 and 2 by the androgen receptor in prostate cancer. Endocrinology 2017;158:1015-21.
179. Luo J, Dunn TA, Ewing CM, Walsh PC, Isaacs WB. Decreased gene expression of steroid 5 alpha-reductase 2 in human prostate cancer: implications for finasteride therapy of prostate carcinoma. Prostate 2003;57:134-9.
180. Thomas LN, Lazier CB, Gupta R, Norman RW, Troyer DA, O'Brien SP, Rittmaster RS. Differential alterations in 5alpha-reductase type 1 and type 2 levels during development and progression of prostate cancer. Prostate 2005;63:231-9.
181. Bjelfman C, Soderstrom TG, Brekkan E, Norlen BJ, Egevad L, Unge T, Andersson S, Rane A. Differential gene expression of steroid 5 alpha-reductase 2 in core needle biopsies from malignant and benign prostatic tissue. J Clin Endocrinol Metab 1997;82:2210-4.
182. Uemura M, Tamura K, Chung S, Honma S, Okuyama A, Nakamura Y, Nakagawa H. Novel 5 alpha-steroid reductase (SRD5A3, type-3) is overexpressed in hormone-refractory prostate cancer. Cancer Sci 2008;99:81-6.
183. Feldman BJ, Feldman D. The development of androgen-independent prostate cancer. Nat Rev Cancer 2001;1:34-45.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Pippione AC, Boschi D, Pors K, Oliaro-Bosso S, Lolli ML. Androgen-AR axis in primary and metastatic prostate cancer: chasing steroidogenic enzymes for therapeutic intervention. J Cancer Metastasis Treat 2017;3:328-61. http://dx.doi.org/10.20517/2394-4722.2017.44
AMA Style
Pippione AC, Boschi D, Pors K, Oliaro-Bosso S, Lolli ML. Androgen-AR axis in primary and metastatic prostate cancer: chasing steroidogenic enzymes for therapeutic intervention. Journal of Cancer Metastasis and Treatment. 2017; 3: 328-61. http://dx.doi.org/10.20517/2394-4722.2017.44
Chicago/Turabian Style
Pippione, Agnese C., Donatella Boschi, Klaus Pors, Simonetta Oliaro-Bosso, Marco L. Lolli. 2017. "Androgen-AR axis in primary and metastatic prostate cancer: chasing steroidogenic enzymes for therapeutic intervention" Journal of Cancer Metastasis and Treatment. 3: 328-61. http://dx.doi.org/10.20517/2394-4722.2017.44
ACS Style
Pippione, AC.; Boschi D.; Pors K.; Oliaro-Bosso S.; Lolli ML. Androgen-AR axis in primary and metastatic prostate cancer: chasing steroidogenic enzymes for therapeutic intervention. J. Cancer. Metastasis. Treat. 2017, 3, 328-61. http://dx.doi.org/10.20517/2394-4722.2017.44
About This Article
Special Issue
Copyright

Data & Comments
Data


 Cite This Article 23 clicks
Cite This Article 23 clicks

 Like This Article 0
likes
Like This Article 0
likes






Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.