


The combined analysis of solid and liquid biopsies provides additional clinical information to improve patient care

Abstract
Aim: To investigate if the genetic information provided by sequencing of both solid and liquid biopsies can shed light on tumor heterogeneity, and to understand the clinical usefulness of adding blood profiling to standard tissue analysis in cancer care.
Methods: Data from 351 patients with stage IV solid tumors for whom molecular profiling of their solid and liquid biopsies was available were studied, with a focus on the discrepant molecular information found between tissue and blood samples.
Results: In 86% of patients, solid and liquid biopsies provided different molecular information. Discrepant gene mutations with a functional impact on the corresponding protein were studied in detail. In 97% of cases, these additional mutations provided clinical value, mainly predicting sensitivity or resistance to targeted therapies. Specifically, 42% of the mutations found only in the liquid biopsy were directly predictive of approved therapies (80% targeted therapies), while 54% were inclusion criteria for molecularly-matched trials.
Conclusion: This study suggests that the addition of blood profiling should be considered in routine clinical oncology, especially for patients with metastatic disease where integrated analysis of solid and liquid biopsies provides a more complete characterization of tumor heterogeneity and provides valuable clinical information for patient treatment.
Keywords
Introduction
In recent years, the field of cancer therapy has evolved from a “one-size-fits-all” approach towards precision medicine, where therapeutic options are tailored specifically to each patient. This patient-tailored strategy is based on the molecular characterization of the tumor through biomarker analysis using tumor biopsy samples[1]. It is becoming clear that genetically different tumor subtypes need to be treated with distinct targeted approaches[2], for example the monoclonal antibody trastuzumab in HER2-positive breast cancers or vemurafenib in BRAF (V600E)-positive melanoma. Nonetheless, the use of targeted therapies is limited by either the presence of primary resistance or the development of acquired treatment resistance[2], and tumor heterogeneity has been clearly associated with such resistance[3]. The advent of deep sequencing studies has demonstrated that human cancers display both temporal (different genetic events taking place during the disease course) and spatial intratumor heterogeneity, harbouring subclones with both shared and unique genomic aberrations[4] that respond differently to targeted therapy. Spatial discrepancy can be explained by clonal heterogeneity within the primary tumor and by the presence of metastasis. Driven by the Darwinian model, during the metastatic process a selection of the “most efficient” clones occurs, due to external forces such as the treatment given to the patient or the tumor environment, for example the presence of hypoxia[5]. It has been reported that tumors with high levels of clonal heterogeneity may show poor prognosis[6].
As mentioned above, heterogeneity in cancer contributes to primary and acquired resistance[3], and that is why approaches providing a global vision of the genomic landscape of the tumor are important for selecting the most appropriate targeted therapy for each patient. Although solid biopsies are the standard way of tumor characterization and will continue to play a central role in cancer management[7], they show some limitations. One of them is that they may not capture tumor heterogeneity, as the aberrations found in a single solid biopsy can be different depending on the area where the sampling was performed, and this could lead to a biased characterization of the tumor that would influence therapy decision[4]. Fortunately, this limitation can be partially overcome by the use of liquid biopsies, such as the free circulating tumor DNA (ctDNA) in blood. ctDNA belongs to the pool of the total cell-free DNA (cfDNA) molecules; in individuals without cancer, the concentration of cfDNA is low, but tumor patients generally have significantly higher levels of cfDNA because of the high turnover of cancer cells. The ctDNA contains DNA mutations of both primary and metastatic lesions[7], since it is released from multiple tumor regions. Therefore, one potential advantage of ctDNA over tissue biopsies is the detection of molecular heterogeneity[8]; as such, ctDNA can harbour mutations that are undetected in the corresponding solid biopsy[9].
In this study we set out to determine the different genetic information revealed by solid and liquid biopsies, and examine the clinical utility of adding ctDNA profiling to the information obtained through tissue biopsies. To this end, we analysed data from 351 patients who had been previously characterized through sequencing of tissue and ctDNA samples.
Methods
Patient population
This work is a retrospective study evaluating 351 patients with stage IV solid tumors whose tissue and blood samples were tested from May 2016 to November 2017 using the OncoSTRAT&GO™ profiling solution (OncoDNA SA, Gosselies, Belgium), and who had failed at least one line of therapy before undergoing molecular profiling. In all cases the oncologist suggested this solution to the patient, who gave informed consent for the tumor analysis data to be published. For objectivity, all samples were included in our analysis without prior selection for age, cancer type, treatment, profiling results or follow-up data.
Samples
Matched tissue and blood samples from different tumor types were included in the analysis. The cancer types studied comprise breast (19.9%), colorectal (11.7%), lung cancer (11.4%), sarcoma (7.7%), ovarian (6.3%), prostate (6.3%), hepatobiliary (5.7%), brain (5.1%), pancreatic (5.1%), gastric (4.6%), carcinoma of unknown primary (3.4%), head and neck cancer (1.7%) and other cancers (11.1%). Solid biopsies were obtained as a formalin-fixed paraffin-embedded (FFPE) block from either the primary or the metastatic tumor and underwent review by a pathologist to determine if the criteria for sample acceptance were met: tumor tissue > 10% of the whole sample and tumor size > 5 mm2 in order to have enough tumor material, and lymphocyte invasion < 20% in the region where the tumor cells were located to avoid lymphocyte DNA contamination. In addition, tissue samples were macro-dissected to remove contaminating normal tissue. Blood was collected in two Cell-Free DNA BCT® CE tubes (Streck, La Vista, USA) and underwent visual inspection to determine that no hemolysis had occurred.
Sample preparation and next-generation sequencing
DNA was extracted from FFPE tissue and cfDNA was extracted from blood using the Qiagen DNA FFPE Tissue Kit or Qiagen DNA Blood Mini Kit (Qiagen, Valencia, USA) respectively. DNA quantity was measured using the Qubit 2.0 Fluorometer (Thermo Fisher Scientific, Waltham, USA). To identify somatic alterations in tumor samples, we used our proprietary solution OncoSTRAT&GO™ (OncoDNA, Gosselies, Belgium). This solution is built based on the Oncomine Comprehensive Panel v2 designed by ThermoFisher, which we updated in order to analyse genome regions or genes not included in that version but shown to be important in cancer. OncoSTRAT&GO™ is based on AmpliSeq technology, amplifying for next-generation sequencing (NGS) whole exons and hotspot mutations of 192 genes (gene panel for the solid biopsy part of OncoSTRAT&GO™) or hotspot mutations of 27 genes (gene panel for the liquid biopsy part of OncoSTRAT&GO™). Briefly, the targeted sequencing libraries were generated using the Ion AmpliSeq Library kit 2.0 according to the manufacturer’s instructions (Thermo Fisher Scientific, Waltham, USA). The starting material consisted of 10 ng DNA from FFPE or blood samples per pool of amplification (4 for the solid part and 2 for the liquid part, respectively). The primers used for amplification were partially digested by Pfu enzyme. The product of digestion was then ligated with corresponding barcoded adapters and purified using Ampure Beads (Beckman Coulter Inc., Indianapolis, USA). The product of purification was amplified for 5 more cycles and subsequently re-purified using Ampure Beads to generate the library sample. The quality of the libraries was assessed using the Qubit dsDNA HS Assay kit (Thermo Fisher Scientific, Waltham, USA). Ten pmol/L of each library was loaded into the IonChef system (Thermo Fisher Scientific, Waltham, USA) for the emulsion polymerase chain reaction. Libraries were then loaded into the sequencing chip that was placed in either the Personal Genome Machine, the Proton or the 5XL device (Thermo Fisher Scientific, Waltham, USA) depending on the required throughput. An average coverage of 1000× was generated in order to detect single-base substitutions down to 5% for the FFPE, and of 10000× to detect base substitutions down to 0.1% for blood samples.
Primary processing of next-generation sequencing data and identification of putative somatic mutations
The data generated from the FFPE and blood samples were first aligned to the human reference sequence and annotated using the Consensus Coding DNA Sequences, RefSeq, and Ensembl databases. NGS data were then analysed using the Torrent Suite Software (Thermo Fisher Scientific, Waltham, USA). Next, somatic mutations were identified with the Variant Caller 4.0 software (Thermo Fisher Scientific, Waltham, USA) using the somatic high stringency parameters to ensure sufficient coverage of the analysed bases and to exclude mapping and sequencing errors [Supplementary Tables 1 and 2]. Genetic aberration analysis was focussed on single-base substitutions, small insertions and deletions. Candidate somatic alterations were further filtered based on: coverage of > 100 in solid biopsy analysis; a forward-reverse ratio of 10%, 90%; the exclusion of intronic and silent changes; and the retention of mutations resulting in missense mutations, nonsense mutations, frameshifts, or splice site alterations in the protein coding region. A manual visual inspection step was used to further remove artefactual changes.
Somatic high-stringency parameters used during the analysis of next-generation sequencing data for the solid biopsy part of OncoSTRAT&GOTM
 |
Somatic high-stringency parameters used during the analysis of next-generation sequencing data for the liquid biopsy part of OncoSTRAT&GOTM
 |
Clinical value of the detected aberrations
To evaluate the impact of the variants on the function of the proteins and on clinical benefit, a literature search was performed to identify in vitro studies, Food and Drug Administration (FDA) labels, guidelines and published retrospective and prospective clinical studies pertaining to genomic alterations in each gene and their association with functional impact on the protein and outcomes in cancer patients. Based on this research, variants were classified into four categories.
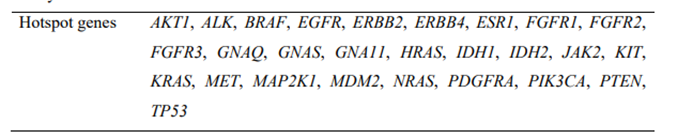
For the purposes of this study, the data shown refer only to the 27 genes of the panel comprising the liquid biopsy part of OncoSTRAT&GO™ [Supplementary Table 3], which are also included in the solid biopsy panel.
The 27 genes analysed by next-generation sequencing for this study
 |
Results
We analysed data from 351 patients who were molecularly characterized from May 2016 to November 2017 using the OncoSTRAT&GO™ profiling solution, which combines the analysis by NGS of genetic variants in the solid and liquid (blood) biopsies.
Different genetic information revealed in the solid vs. the liquid biopsy
Patients with no variant detected in the tissue or the blood biopsy represented 11% of the total population and were excluded from further analysis. We classified the remaining patients (n = 313) into four categories according to the mutation discrepancies found between their solid and liquid biopsies [Figure 1A]. This analysis showed that 41% of the patients carried mutations that were detected only in the solid biopsy, while for 4% of the cases, variants were found only in the blood. On the other hand, 41% of samples had partially shared mutations, and only 14% of the samples fully shared the gene variants in both biopsies [Figure 2]. These results suggest that in 86% of the patients, solid and liquid biopsies provide different information regarding genetic alterations.

Figure 1. Patient and variant categories. (A) Patient categories. Fully shared: patient having all the variants (1&2) detected in both the solid and liquid biopsies; partially shared: patient having different variants detected in the solid and/or liquid biopsies (4&5), and some variants that are shared (3); only in solid: patient having variants (6&7) only detected in the solid biopsy; only in liquid: patient having variants (8&9) detected only in the liquid biopsy; (B) Variant categories. Shared: variants that are detected in both the solid and liquid biopsy. These variants can be from "fully shared" patients or common variants from "partially shared" patients; solid: variants that are detected only in the solid biopsy. These can be from "only solid" patients or variants present only in tissue biopsy in "partially shared" patients; liquid: variants that are detected only in the liquid biopsy. These can be from "only liquid" patients or variants present only in blood in "partially shared" patients

Figure 2. Patient distribution according to discrepancy between solid and liquid biopsies. Patients were classified according to . The percentage of "fully shared", "partially shared", "only in solid" and "only in liquid" patients was then calculated
Since the category “partially shared” includes patients who share variants in the two biopsies, we further performed a classification at the variant level by grouping the mutations in three categories: shared, solid and liquid [Figure 1B]. This analysis indicated that in the majority of the cases (60%), tissue and blood biopsies analysis showed discrepant patterns: 51% of the variants were detected only in the solid biopsy, while mutations detected only in the blood, which can provide information about spatial tumor heterogeneity, represented 9% of the total [Figure 3].

Figure 3. Variant distribution according to discrepancy between solid and liquid biopsy. Variants detected in the samples across different cancer types were classified according to . The percentage of "shared", "solid" and "liquid" was then calculated
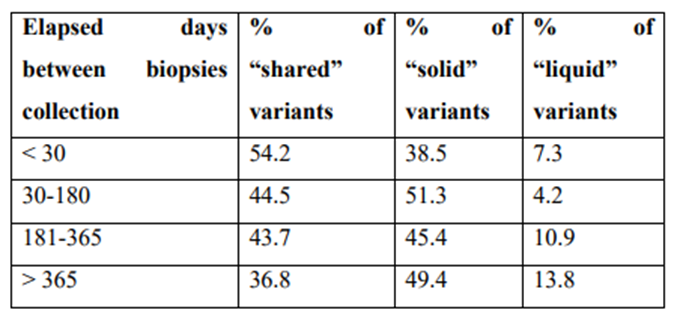
Next, we studied how the distribution of these three variant categories could be influenced by the time elapsed between the collection of the solid biopsy and the blood biopsy. As expected, the percentage of “shared” variants decreased progressively as the time elapsed between sampling increased. The opposite trend was observed for the percentage of “solid” and “liquid” [Supplementary Table 4]. In fact, when the solid biopsy was collected more than one year before the blood sample, the number of shared variants was only one third that of recent biopsies (less than 30 days), while the number of “solid” or “liquid” variants increased by around one-third and double, respectively. These data confirm that the mutations in a tumor change over time, and that this temporal heterogeneity can be demonstrated by comparing the solid and liquid biopsies at different collection times.
Percentage of shared, solid and liquid variants according to time elapsed between collection of solid and liquid biopsies
 |
Therapeutic implication of addressing tumor heterogeneity
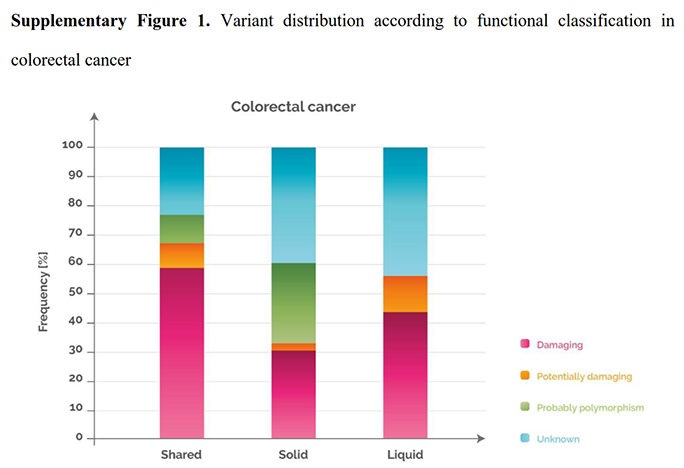
In order to understand the biological and clinical implications of the variants identified in the tissue and blood analyses, we grouped the mutations into four different categories based on their impact on the function of the protein, and then sorted them according to the discrepancy between solid and liquid biopsy. 48%, 23% and 28% of the variants detected in “shared”, “solid” and “liquid”, were damaging [Figure 4; see legend for explanation of functional classification]. These percentages were cancer dependent. For example, when analysing the colorectal cancer samples separately, the percentage of damaging variants present in “shared”, “solid” and “liquid” rose to 59%, 31% and 44%, respectively [Supplementary Figure 1].

Figure 4. Variant distribution according to functional classification and discrepancy between solid and liquid biopsy. Variants detected only in solid, only in liquid or in both were analysed for their functional impact on the corresponding protein, and were classified into 4 categories as follows: (1) damaging: a variant for which several published studies demonstrated a functional impact on the protein (activating or inhibiting) and where clinical information is also available confirming the impact; (2) potentially damaging: a variant for which only one publication has shown a functional impact based on an in vitro model and for which no clinical information is available; (3) unknown: a variant for which there are no publications associated with a functional impact and that is not known as a single nucleotide polymorphism in the NCBI dbSNP database; (4) polymorphism: a variant identified in the NCBI dbSNP database as a polymorphic variant with a minor allele frequency of at least 1%. NCBI: National Center for Biotechnology Information; dbSNP: Single Nucleotide Polymorphism Database
Variant distribution according to functional classification in colorectal cancer
 |
The genes showing the highest frequencies of damaging variants in the three categories were TP53, KRAS and PIK3CA, although the order varied: in “shared”, KRAS was the gene with the most damaging mutations (28%), followed by PIK3CA (24%) and TP53 (23%), while in “solid” and “liquid” the gene with a higher frequency of damaging variants was TP53 (31% and 34%), followed by KRAS (26% and 25%) and PIK3CA (16% and 13%, respectively) [Figure 5].

Figure 5. Damaging variant distribution per gene and type of biopsy. The distribution of the damaging variants identified in was analysed according to whether they were detected in (A) shared, (B) solid or (C) liquid
To address whether these mutations could have a clinical impact on the patients, we analysed the potential clinical benefit associated with each damaging variant detected in the samples. In 97% of the cases, the damaging mutations found solely in the solid or in the liquid biopsy were predictive of either sensitivity (82%) or resistance (15%) to specific cancer treatments - mainly targeted therapies (98%). When we studied in more detail the damaging variants detected in “liquid”, we observed that 96% were clinically actionable: 42% were directly predictive of approved therapies in the indicated cancer type, either targeted therapies (80%) or hormone therapies (20%, aromatase inhibitors), whereas 54% were inclusion criteria for trials using targeted therapies in molecularly selected patients (which were actively recruiting at the time of submission) [Table 1]. These data highlight that the integrated analysis of both biopsy samples provides valuable clinical information that could guide the use of cancer therapy.
Damaging variants detected in the "liquid" category and their clinical implication according to cancer type
| Cancer type analysed | Gene | No. of variants | Therapeutic value |
|---|---|---|---|
| Breast cancer HR+ | ESR1 | 2 | Resistance to aromatase inhibitors[10] |
| KRAS | 1 | Phase 2 trial (NCT02576444) | |
| TP53 | 3 | Phase 2 trial (NCT02576444) | |
| Cholangiocarcinoma | PIK3CA | 1 | Phase 2 trial (NCT02465060) |
| Colorectal cancer | KRAS | 4 | Resistance to anti-EGFR antibodies[11] |
| PIK3CA | 1 | Resistance to anti-EGFR antibodies[12] | |
| TP53 | 2 | Phase 2 trial (NCT02576444) | |
| Endometrial carcinoma | TP53 | 1 | Phase 2 trial (NCT02576444) |
| Gastric cancer | PIK3CA | 1 | Phase 2 trial (NCT02465060) |
| GBM | BRAF | 1 | Phase 2 trial (NCT02465060) |
| GIST | KIT | 1 | Resistance to KIT/PDGFRA-tyrosine kinase inhibitors[13] |
| NSCLC | EGFR | 1 | Resistance to EGFR-tyrosine kinase inhibitors[14] |
| GNAS | 1 | Unknown | |
| MAP2K1 | 1 | Sensitivity to MEK inhibitors[15] | |
| Pancreatic cancer | KRAS | 1 | Phase 2 trial (NCT02576444) |
| Prostate cancer | TP53 | 2 | Phase 2 trial (NCT02576444) |
Discussion
This study compared the discrepant distribution of mutations found in the molecular profiling of solid vs. liquid biopsies of metastatic cancer patients in order to better understand tumor heterogeneity. This analysis highlighted that the addition of ctDNA testing to tissue profiling might increase therapeutic value and could better guide oncologists in precision medicine.
The results show that in the majority of the cases, the information obtained by sequencing tumor tissue DNA and ctDNA is complimentary. We observed a higher percentage of mutations detected only in the solid biopsy (51%) compared to only in the liquid or to shared variants. One factor that can explain this is the tumor location, as it has been previously demonstrated that different cancer types shed different amounts of DNA into the blood[16]. Bettegowda et al.[16] showed that ctDNA was detectable in 100% of patients with stage IV colorectal cancer, while less than 10% of patients with advanced gliomas harboured detectable ctDNA (as the blood-brain barrier could prevent the entry of ctDNA into the circulation). In accordance with this, in our analysis we found that 56% of the variants were shared between solid and liquid biopsies in colorectal cancer patients, while in glioblastoma multiforme patients, 89% of the mutations were found in “solid”, 11% only in liquid biopsies and none in “shared” (data not shown). This confirms that the location of the tumor has an impact on the utility of ctDNA. A second factor that can explain why a high percentage of variants were only present in tissue DNA is the temporal heterogeneity, which is influenced by patient-specific selective pressures[17] such as the prescribed treatments and fluctuations in tumor microenvironment. In fact, the percentage of “shared” mutations markedly decreased when the time space of the collection dates increased. This temporal heterogeneity provides interesting information about how the subclonal heterogeneity of a tumor evolves over the course of treatment, with some clones persisting or disappearing while new clones appear[18]. Note that this temporal analysis might not always provide useful information about the current treatments that would be of clinical benefit to the patient, as the current molecular status of the tumor is the critical factor in therapy personalization.
On the other hand, 9% of the variants were detected only in the blood, which are a reflection of spatial heterogeneity. This intratumor heterogeneity occurs either within different regions of the same tumor mass, or between the primary tumor and its metastases[4]. Moreover, different metastatic sites may also harbour different molecular features[19]. Previous studies have reported the existence of mutations that were only found in ctDNA but not in the corresponding tissue[7]. Since ctDNA can be shed into the blood from the primary tumor and/or the metastases, it can potentially provide tumor information from all cancer sites[4]. On the other side, spatial heterogeneity cannot be fully determined with a single-site solid biopsy.
We observed that on average across cancer types, “solid” and “liquid” variants with a demonstrated functional impact on the protein (damaging) were around 25% of all the mutations found, although the frequency was different depending on the cancer type. The genes that harboured the highest number of damaging variants were TP53, KRAS and PIK3CA. TP53 is a key tumor suppressor that responds to several cellular stress signals by promoting different responses, such as cell cycle arrest, senescence or apoptosis[20]. TP53 mutations have been found in almost every cancer type, and its inactivation is a common event in the tumorigenesis process[21]. KRAS is also one of the most frequently mutated genes in many cancers. It plays an important role in the regulation of cell division and activating mutations can lead to cell transformation because they impair the ability of the KRAS protein to switch between the “on” and the “off” state. Finally, PIK3CA, another commonly mutated oncogene in cancer[22], is involved in many cellular processes, such as cell growth and proliferation and, when mutated, the increased kinase activity of PIK3CA protein contributes to cellular transformation[23].
What are the clinical implications of analysing the discrepancy between the damaging variants found solely in the solid and solely in the liquid biopsy? In almost all the cases, these mutations were clinically actionable, meaning that they provided information about tumor sensitivity or resistance to approved or investigational targeted therapies. When we took into consideration only the spatial heterogeneity (“liquid” category), we again found a very high frequency of clinically actionable variants (96%), most of them either related to approved targeted therapies - for example KRAS mutations in colorectal cancer are associated with resistance to anti-EGFR antibodies[11], and MAP2K1 mutations in non-small cell lung cancer are related to sensitivity to MEK inhibitors[15] - or to actively recruiting trials of targeted therapies, such as NCI-MATCH (NCT02465060). This emphasizes that the addition of ctDNA profiling to the analysis of solid biopsies, the current gold standard for tumor molecular characterization, can provide valuable extra information for oncologists, either for the prescription of an approved treatment or for enrolling them in relevant clinical trials. In fact, the utility of ctDNA analysis for tumor characterization and for guiding treatment choice is being increasingly recognized, as reflected by the first FDA approval of a blood-based companion diagnostic to guide targeted therapy in non-small cell lung cancer (cobas EGFR Mutation Test v2).
Limitations of this study include the lack of information about patient follow-up regarding oncologists’ treatment decisions and patient outcomes. Strengths include the number of patients and the cancer types analysed, as well as having addressed the importance of understanding tumor heterogeneity in the clinical setting.
In conclusion, this study suggests that the combination of recent solid and liquid biopsies provides the most comprehensive and therapeutically valuable characterization of the heterogeneity of the patient’s tumor, which cannot be achieved by performing only one type of biopsy, and that the inclusion of ctDNA profiling should be considered in routine oncology care especially for cancers where targeted therapies are approved or in development.
Declarations
AcknowledgmentsWe thank all the clinicians and patients who provided the biopsy samples used in the study.
Authors’ contributionsConceived and designed the study: Laes JF
Worked on the acquisition of data, the writing of the article and the analysis and interpretation of data: all authors
Reviewed and agreed on the manuscript before submission: all authors
Data source and availabilityDue to privacy reasons, the data of this study cannot be open for readers. Nonetheless, if a researcher asks for data, they could be shared upon the signature of a confidentiality agreement.
Financial support and sponsorshipThis study was sponsored by OncoDNA SA. The costs associated with the development and publishing of the manuscript were provided by OncoDNA SA.
Conflicts of interestAll authors are employees of OncoDNA. Ghitti G and Laes JF report ownership of OncoDNA shares.
Patient consentPatients gave informed consent for their tumor analysis data to be published.
Ethics approvalThis study is not approved by IRB; however, all patients signed an informed consent before their samples underwent molecular profiling according to Belgian law.
Copyright© The Author(s) 2018.
REFERENCES
1. La Thangue NB, Kerr DJ. Predictive biomarkers: a paradigm shift towards personalized cancer medicine. Nat Rev Clin Oncol 2011;8:587-96.
3. Zahreddine H, Borden KL. Mechanisms and insights into drug resistance in cancer. Front Pharmacol 2013;4:1-8.
4. Seoane J, De Mattos-Arruda L. The challenge of intratumour heterogeneity in precision medicine. J Intern Med 2014;276:41-51.
5. Junttila MR, de Sauvage FJ. Influence of tumour micro-environment heterogeneity on therapeutic response. Nature 2013;501:346-54.
6. Turner NC, Reis-Filho JS. Genetic heterogeneity and cancer drug resistance. Lancet Oncol 2012;13:e178-85.
7. Yi X, Ma J, Guan Y, Chen R, Yang L, Xia X. The feasibility of using mutation detection in ctDNA to assess tumor dynamics. Int J Cancer 2017;140:2642-7.
8. Diaz LJ, Bardelli A. Liquid biopsies: genotyping circulating tumor DNA. J Clin Oncol 2014;32:579-86.
9. Wan JCM, Massie C, Garcia-Corbacho J, Mouliere F, Brenton JD, Caldas C, Pacey S, Baird R, Rosenfeld N. Liquid biopsies come of age: towards implementation of circulating tumour DNA. Nat Rev Cancer 2017;17:223-38.
11. Bronte G, Silvestris N, Castiglia M, Galvano A, Passiglia F, Sortino G, Cicero G, Rolfo C, Peeters M, Bazan V, Fanale D, Giordano A, Russo A. New findings on primary and acquired resistance to anti-EGFR therapy in metastatic colorectal cancer: do all roads lead to RAS? Oncotarget 2015;6:24780-96.
12. Therkildsen C, Bergmann TK, Henrichsen-Schnack T, Ladelund S, Nilbert M. The predictive value of KRAS, NRAS, BRAF, PIK3CA and PTEN for anti-EGFR treatment in metastatic colorectal cancer: a systematic review and meta-analysis. Acta Oncol 2014;53:852-64.
13. Liegl B, Kepten I, Le C, Zhu M, Demetri GD, Heinrich MC, Fletcher CD, Corless CL, Fletcher JA. Heterogeneity of kinase inhibitor resistance mechanisms in GIST. J Pathol 2008;216:64-74.
14. Arcila ME, Nafa K, Chaft JE, Rekhtman N, Lau C, Reva BA, Zakowski MF, Kris MG, Ladanyi M. EGFR exon 20 insertion mutations in lung adenocarcinomas: prevalence, molecular heterogeneity, and clinicopathologic characteristics. Mol Cancer Ther 2013;12:220-9.
15. Diamond EL, Durham BH, Haroche J, Yao Z, Ma J, Parikh SA, Wang Z, Choi J, Kim E, Cohen-Aubart F, Lee SC, Gao Y, Micol JB, Campbell P, Walsh MP, Sylvester B, Dolgalev I, Aminova O, Heguy A, Zappile P, Nakitandwe J, Ganzel C, Dalton JD, Ellison DW, Estrada-Veras J, Lacouture M, Gahl WA, Stephens PJ, Miller VA, Ross JS, Ali SM, Briggs SR, Fasan O, Block J, Héritier S, Donadieu J, Solit DB, Hyman DM, Baselga J, Janku F, Taylor BS, Park CY, Amoura Z, Dogan A, Emile JF, Rosen N, Gruber TA, Abdel-Wahab O. Diverse and targetable kinase alterations drive histiocytic neoplasms. Cancer Discov 2016;6:154-65.
16. Bettegowda C, Sausen M, Leary RJ, Kinde I, Wang Y, Agrawal N, Bartlett BR, Wang H, Luber B, Alani RM, Antonarakis ES, Azad NS, Bardelli A, Brem H, Cameron JL, Lee CC, Fecher LA, Gallia GL, Gibbs P, Le D, Giuntoli RL, Goggins M, Hogarty MD, Holdhoff M, Hong SM, Jiao Y, Juhl HH, Kim JJ, Siravegna G, Laheru DA, Lauricella C, Lim M, Lipson EJ, Marie SK, Netto GJ, Oliner KS, Olivi A, Olsson L, Riggins GJ, Sartore-Bianchi A, Schmidt K, Shih LM, Oba-Shinjo SM, Siena S, Theodorescu D, Tie J, Harkins TT, Veronese S, Wang TL, Weingart JD, Wolfgang CL, Wood LD, Xing D, Hruban RH, Wu J, Allen PJ, Schmidt CM, Choti MA, Velculescu VE, Kinzler KW, Vogelstein B, Papadopoulos N, Diaz LA Jr. Detection of circulating tumor DNA in early- and late-stage human malignancies. Sci Transl Med 2014;6:1-11.
17. Welch DR. Tumor heterogeneity - a 'contemporary concept' founded on historical insights and predictions. Cancer Res 2016;76:4-6.
18. González S, Volkova N, Beer P, Gerstung M. Immuno-oncology from the perspective of somatic evolution. Semin Cancer Biol 2017; doi: 10.1016/j.semcancer.2017.12.001.
19. Kidess E, Jeffrey S. Circulating tumor cells versus tumor-derived cell- free DNA: rivals or partners in cancer care in the era of single-cell analysis? Genome Med 2013;5:1-4.
20. Bieging K, Mello S, Attardi L. Unravelling mechanisms of p53-mediated tumour suppression. Nat Rev Cancer 2014;14:359-70.
21. Rivlin N, Brosh R, Oren M, Rotter V. Mutations in the p53 tumor suppressor gene: important milestones at the various steps of tumorigenesis. Genes Cancer 2011;2:466-74.
22. Samuels Y, Waldman T. Oncogenic mutations of PIK3CA in human cancers. Curr Top Microbiol Immunol 2010;347:21-41.
Cite This Article
How to Cite
Download Citation
Export Citation File:
Type of Import
Tips on Downloading Citation
Citation Manager File Format
Type of Import
Direct Import: When the Direct Import option is selected (the default state), a dialogue box will give you the option to Save or Open the downloaded citation data. Choosing Open will either launch your citation manager or give you a choice of applications with which to use the metadata. The Save option saves the file locally for later use.
Indirect Import: When the Indirect Import option is selected, the metadata is displayed and may be copied and pasted as needed.
About This Article
Copyright













Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.