
New insights into the role of intra-tumor genetic heterogeneity in carcinogenesis: identification of complex single gene variance within tumors


Abstract
Aim: Present cancer hypotheses are almost all based on the concept that accumulation of specific driver gene mutations cause carcinogenesis. The discovery of intra-tumor genetic heterogeneity (ITGH), has resulted in this hypothesis being modified by assuming that most of these ITGH mutations are in passenger genes. In addition, accumulating ITGH data on driver gene mutations have revealed considerable genotype/phenotype disconnects. This study proposes to investigate this disconnect by examining the nature and degree of ITGH in breast tumors.
Methods: ITGH was examined in tumors using next generation sequencing of up to 68,000 reads and analysis tools that allowed for identification of distinct minority variants within single genes, i.e., complex single gene variance (CSGV).
Results: CSGV was identified in the androgen receptor genes in all breast tumors examined.
Conclusion: Evidence of CSGV suggests that a selection - as opposed to a mutation - centric hypothesis could better explain carcinogenesis. Our hypothesis proposes that tumors develop by the selection of preexisting de novo mutations rather than just the accumulation of de novo mutations. Thus, the role of selection pressures, such as changes in tissue microenvironments will likely be critical to our understanding of tumor resistance as well as the development of more effective treatment protocols.
Keywords
Introduction
Current carcinogenesis hypotheses
The traditional understanding of carcinogenesis, that cancer cells accumulate somatic driver mutations that give them a growth advantage[1] is beginning to be questioned as data reveal the presence of driver gene mutations involved in carcinogenesis in normal tissues[2]. Further, a critical issue still to be elucidated is how these mutations create a gain-of-function in cells that results in them acquiring new oncogenic properties, rather than just the loss-of-function of factors that control cell growth and division. One indication as to why these properties might be more complicated than a simple case of excessive or distorted growth is that cancer genes are generally not over-expressed in the tissues from which the cancer develops[3]. For example, out of 130 highly specific-cancer genes only four are most highly expressed in the tissue from which the cancer originates[3]. Thus, other factors besides protein accumulation are likely to be involved. Compounding this conundrum is the observation that there are often different mutations in different cancer-associated genes in different cancer tissues[1]. Raising the question as to how these differences are related to the tissue specificity of certain cancer mutations.
Further, in a recent study looking for associations between specific cancer genes and specific cancer tissues some genes did not behave as expected[1]. The analyses suggested that both cell-intrinsic (i.e., genomic and epigenetic) and cell-extrinsic (i.e., environmental, both internal and external) factors could explain the differences in the cell type-specificity of cancer genes. For example, in breast cancer, specific external environmental factors have included estrogen receptor alpha (ER) activation by estradiol[4] and conversion of estrogen into genotoxic metabolites that can cause DNA double-strand breaks[5]. However, in most cases it has not been possible to associate any specific intrinsic or extrinsic factor with cancer tissue specificity. Underlying these fundamental questions is a growing awareness of substantial amounts of genetic heterogeneity not only within different types of cancer tissues[6], but within single tumor cancer tissues as well. These latter observations have been labelled as intra-tumor genetic heterogeneity (ITGH)[7].
Intra-tumor genetic heterogeneity
ITGH identified within breast tumors, has revealed numerous alterations in different genes, with the assumption that most mutations are in "passenger" genes[8], including studies using single cell sequencing techniques[9]. However, such studies have also not drawn many definitive conclusions as to precise roles of many of the "driver" genes in carcinogenesis. Genes being identified as drivers: (1) if they are either oncogenes or tumor suppressor genes; (2) if they function in some aspect of cell growth; (3) if their location are close to any of these types of genes[10]. Further, a recent paper noted that passenger genes can also have damaging effects on cancer progression[11].
We believe this confusion is partly because of a failure to investigate the nature and degree of genetic heterogeneity within single genes, a condition that we have labelled, complex single gene variance (CSGV), as opposed to just identifying mutations in different cancer-associated genes. Why this is important is that as natural selection is being increasingly identified as a critical process in cancer biology[12], there needs to be a better understanding of the nature of the genetic variation that is being subjected to selection.
Identification of single gene genetic heterogeneity
The question as to why genetic heterogeneity within individual genes has not been studied before is partially because the approach to identifying gene variants is based on using sequence analysis algorithms and tools that make it inherently difficult to identify CSGV. Essentially, they are designed to ignore or minimize the possibility that different mutations of an individual gene can exist in a single person's tissues. The assumption being that finding multiple variants of a single gene within an individual's tissues is highly unlikely and therefore if identified is likely the result of either PCR or sequencing errors. Indeed, almost all NGS analyses rely on the use of filters and other techniques such as sequence alignment tools to remove such variants[13]. These techniques further reduce the possibility of finding multiple mutations within an individual gene, as some are likely to be at very low frequencies, and will be present in only a small minority of cells within an individual tumor, as noted in a recent review of post-zygotic somatic mosaicism[14]. Therefore, one of the challenges of the study was to develop a sequencing analysis approach that allows for the identification of CSGV. Further, an important practical consideration for identifying CSGV is that it is increasingly becoming apparent that every driver gene mutation does not produce a cancer phenotype, with some driver mutations even being present in non-cancer tissues[15,16]. In the present study, we have used a sequencing approach that makes it easier to detect multiple mutations of the androgen receptor gene (AR) within individual breast tumors.
Androgen receptor and breast cancer
In the case of breast cancer (BC), the AR is more widely expressed than either estrogen receptor (ER) alpha or progesterone receptor (PR) genes, and so it is not surprising that the AR has become a significant marker in defining BC subtypes[17]. The AR has therefore started to be singled out as a possible therapeutic target, particularly in triple-negative [ER-/PR-/herceptin receptor (HER) 2-] BC (TNBC)[18,19]. Indeed, a large cohort study reported AR expression in 32% of TNBC cases[20]. In another study examining cases of ER-positive breast carcinoma, tumor cells changed after treatment from ER-dependent to AR-dependent, possibly explaining why such cells become resistant to aromatase inhibitor treatment[21]. At present, most studies have focused on AR expression during different BC stages, and, indeed, AR expression has been identified as a possible critical marker in predicting BC survival[22]. While androgen-based therapeutics have been used for over 50 years to treat BC[23]. The authors believe that to truly exploit potential AR related mechanisms to provide clinical therapeutic benefits, a more detailed understanding of AR variant distribution and frequency in BC tissues, i.e., AR CSGV, both before and throughout carcinogenesis, will be required.
Further, examining CSGV occurrence in other critical driver genes may help resolve the genotype-phenotype disconnects between the mutational status of putative cancer-associated genes and the occurrence and progression of cancer. For, if it is assumed that somatic clonal evolution is the mechanism driving carcinogenesis, then tissue microenvironments need to be able to select from different variants of individual genes. As the presence of a single variant would not allow cells and tissues sufficient flexibility to adapt to different selection pressures produced by different tissue microenvironments. Further, the ability to collect such data about all potential driver genes may well provide new insights into resistance to treatment as well as to treatment failures.
Methods
Laser capture microdissection and DNA extraction
Frozen tumors were obtained from a breast cancer tissue bank [Table 1] that had been set up with all the required experimental permissions and vetted by the Jewish General Hospital’s ethics board. Histological slides of 5-7 µm thick were prepared and stained using a standard hematoxylin/eosin protocol. To ensure the maximum purity of the cancer samples, following histo-pathological characterization by an expert pathologist, cells from cancer tumor areas were dissected by LCM using an AutoPix 100 (Molecular Devices, Sunnyvale, CA). An average of 2500 cells was dissected from each different section. Genomic DNA was extracted from the cells using a QIAamp DNA Micro kit (QIAGEN, Germantown, MD) following the manufacturer’s directions.
Clinical data
| Specimen No. | Age at diagnosis | Nuclear grade | Histology grade | Menopausal | T | N | M | TNM stage | ER | PR | HER2 |
|---|---|---|---|---|---|---|---|---|---|---|---|
| T-44 | 55 | III | III | + | pT2 | pN2a | pM1 | IV | + | + | + |
| T-102 | 78 | III | III | + | pT2 | pN3a | pM0 | IIIC | + | + | + |
| T-106 | 64 | II | II | + | pT1c | pN0 | pMx | I | + | + | - |
| T-112 | 60 | III | III | + | pT2 | pN0(i+) | pM0 | IIA | - | - | - |
| T-121 | 62 | I | I | + | pT1c | pN1a | pM0 | IIA | + | + | - |
| T-125 | 60 | II | II | + | pT1c | pN0(i-) | pM0 | I | + | + | - |
PCR amplification
Amplification of AR exons was carried out using the Fast Start High Fidelity PCR kit (Roche, Indianapolis, IN). PCR products were generated using 36 different pairs of fused primers designed to flank the AR sequences of exons 4-8, which has been shown to be the region of the AR that contains a high proportion of mutations, including those associated with cancer[24]. The primers also included the sequence of introns 3-8 [Table 2]. Each primer consisted of a 5’ overhang of 19 bp, a 3 bp patient-specific barcode, and a 20-27 bp AR-specific sequence. The 5’ overhang was used to facilitate emulsion PCR (em-PCR) and sequencing. The 3 bp barcode facilitated sample identification post sequencing, by allowing the pooling of different DNA samples for em-PCR. To ensure consistency three separate PCR preparations were prepared for each of the samples.
Primers used for sequencing
| Sample | Primers | Tag | Primer sequences |
|---|---|---|---|
| T-44 | 4A/882718B | AAC | GCCTCCCTCGCGCCATCAGAACATTCAAGTCTCTCTTCCTTC / GCCTTGCCAGCCCGCTCAGAACCAAAGGAGTCGGGCTGGTTGTT |
| T-102 | 4A/882718B | AAG | GCCTCCCTCGCGCCATCAGAAGATTCAAGTCTCTCTTCCTTC / GCCTTGCCAGCCCGCTCAGAAGCAAAGGAGTCGGGCTGGTTGTT |
| T-106 | 4A/882718B | ATG | GCCTCCCTCGCGCCATCAGATGATTCAAGTCTCTCTTCCTTC / GCCTTGCCAGCCCGCTCAGATGCAAAGGAGTCGGGCTGGTTGTT |
| T-112 | 4A/882718B | ATC | GCCTCCCTCGCGCCATCAGATCATTCAAGTCTCTCTTCCTTC / GCCTTGCCAGCCCGCTCAGATCCAAAGGAGTCGGGCTGGTTGTT |
| T-121 | 4A/882718B | ACA | GCCTCCCTCGCGCCATCAGACAATTCAAGTCTCTCTTCCTTC / GCCTTGCCAGCCCGCTCAGACACAAAGGAGTCGGGCTGGTTGTT |
| T-125 | 4A/882718B | ACT | GCCTCCCTCGCGCCATCAGACTATTCAAGTCTCTCTTCCTTC / GCCTTGCCAGCCCGCTCAGACTCAAAGGAGTCGGGCTGGTTGTT |
| T-44 | 605A/4B | AAC | GCCTCCCTCGCGCCATCAGAACGACAGTGTCACACATTGAAGGCTATG / GCCTTGCCAGCCCGCTCAGAACGGTCCATAGGAGCGTTCACT |
| T-102 | 605A/4B | AAG | GCCTCCCTCGCGCCATCAGAAGGACAGTGTCACACATTGAAGGCTATG / GCCTTGCCAGCCCGCTCAGAAGGGTCCATAGGAGCGTTCACT |
| T-106 | 605A/4B | ATG | GCCTCCCTCGCGCCATCAGATGGACAGTGTCACACATTGAAGGCTATG / GCCTTGCCAGCCCGCTCAGATGGGTCCATAGGAGCGTTCACT |
| T-112 | 605A/4B | ATC | GCCTCCCTCGCGCCATCAGATCGACAGTGTCACACATTGAAGGCTATG / GCCTTGCCAGCCCGCTCAGATCGGTCCATAGGAGCGTTCACT |
| T-121 | 605A/4B | ACA | GCCTCCCTCGCGCCATCAGACAGACAGTGTCACACATTGAAGGCTATG / GCCTTGCCAGCCCGCTCAGACAGGTCCATAGGAGCGTTCACT |
| T-125 | 605A/4B | ACT | GCCTCCCTCGCGCCATCAGACTGACAGTGTCACACATTGAAGGCTATG / GCCTTGCCAGCCCGCTCAGACTGGTCCATAGGAGCGTTCACT |
| T-44 | 5A/5B | ACC | GCCTCCCTCGCGCCATCAGACCTCTCTGCCCAACAGGGACTC / GCCTTGCCAGCCCGCTCAGACCATCACCACCAACCAGGTCTG |
| T-102 | 5A/5B | ACG | GCCTCCCTCGCGCCATCAGACGTCTCTGCCCAACAGGGACTC / GCCTTGCCAGCCCGCTCAGACGATCACCACCAACCAGGTCTG |
| T-106 | 5A/5B | AGA | GCCTCCCTCGCGCCATCAGAGATCTCTGCCCAACAGGGACTC / GCCTTGCCAGCCCGCTCAGAGAATCACCACCAACCAGGTCTG |
| T-112 | 5A/5B | AGC | GCCTCCCTCGCGCCATCAGAGCTCTCTGCCCAACAGGGACTC / GCCTTGCCAGCCCGCTCAGAGCATCACCACCAACCAGGTCTG |
| T-121 | 5A/5B | AGG | GCCTCCCTCGCGCCATCAGAGGTCTCTGCCCAACAGGGACTC / GCCTTGCCAGCCCGCTCAGAGGATCACCACCAACCAGGTCTG |
| T-125 | 5A/5B | TAA | GCCTCCCTCGCGCCATCAGTAATCTCTGCCCAACAGGGACTC / GCCTTGCCAGCCCGCTCAGTAAATCACCACCAACCAGGTCTG |
| T-44 | 6A/6B | TAT | GCCTCCCTCGCGCCATCAGTATCAATCAGAGACATTCCTCTGG / GCCTTGCCAGCCCGCTCAGTATAGTGGTCCTCTCTGAATCTC |
| T-102 | 6A/6B | TAG | GCCTCCCTCGCGCCATCAGTAGCAATCAGAGACATTCCTCTGG / GCCTTGCCAGCCCGCTCAGTAGAGTGGTCCTCTCTGAATCTC |
| T-106 | 6A/6B | TTA | GCCTCCCTCGCGCCATCAGTTACAATCAGAGACATTCCTCTGG / GCCTTGCCAGCCCGCTCAGTTAAGTGGTCCTCTCTGAATCTC |
| T-112 | 6A/6B | TTT | GCCTCCCTCGCGCCATCAGTTTCAATCAGAGACATTCCTCTGG / GCCTTGCCAGCCCGCTCAGTTTAGTGGTCCTCTCTGAATCTC |
| T-121 | 6A/6B | TTC | GCCTCCCTCGCGCCATCAGTTCCAATCAGAGACATTCCTCTGG / GCCTTGCCAGCCCGCTCAGTTCAGTGGTCCTCTCTGAATCTC |
| T-125 | 6A/6B | TTG | GCCTCCCTCGCGCCATCAGTTGCAATCAGAGACATTCCTCTGG / GCCTTGCCAGCCCGCTCAGTTGAGTGGTCCTCTCTGAATCTC |
| T-44 | 7A/7B | TCA | GCCTCCCTCGCGCCATCAGTCATGTGGTCAGAAAACTTGGTG / GCCTTGCCAGCCCGCTCAGTCATGGCTCTATCAGGCTGTTCTC |
| T-102 | 7A/7B | TCT | GCCTCCCTCGCGCCATCAGTCTTGTGGTCAGAAAACTTGGTG / GCCTTGCCAGCCCGCTCAGTCTTGGCTCTATCAGGCTGTTCTC |
| T-106 | 7A/7B | TCC | GCCTCCCTCGCGCCATCAGTCCTGTGGTCAGAAAACTTGGTG / GCCTTGCCAGCCCGCTCAGTCCTGGCTCTATCAGGCTGTTCTC |
| T-112 | 7A/7B | TCG | GCCTCCCTCGCGCCATCAGTCGTGTGGTCAGAAAACTTGGTG / GCCTTGCCAGCCCGCTCAGTCGTGGCTCTATCAGGCTGTTCTC |
| T-121 | 7A/7B | TGA | GCCTCCCTCGCGCCATCAGTGATGTGGTCAGAAAACTTGGTG / GCCTTGCCAGCCCGCTCAGTGATGGCTCTATCAGGCTGTTCTC |
| T-125 | 7A/7B | TGG | GCCTCCCTCGCGCCATCAGTGGTGTGGTCAGAAAACTTGGTG / GCCTTGCCAGCCCGCTCAGTGGTGGCTCTATCAGGCTGTTCTC |
| T-44 | 8A/8B | ATT | GCCTCCCTCGCGCCATCAGATTACCTCCTTGTCACCCTGTTT / GCCTTGCCAGCCCGCTCAGATTAAGGCACTGCAGAGGAGTAG |
| T-102 | 8A/8B | AGT | GCCTCCCTCGCGCCATCAGAGTACCTCCTTGTCACCCTGTTT / GCCTTGCCAGCCCGCTCAGAGTAAGGCACTGCAGAGGAGTAG |
| T-106 | 8A/8B | TGT | GCCTCCCTCGCGCCATCAGTGTACCTCCTTGTCACCCTGTTT / GCCTTGCCAGCCCGCTCAGTGTAAGGCACTGCAGAGGAGTAG |
| T-112 | 8A/8B | TGC | GCCTCCCTCGCGCCATCAGTGCACCTCCTTGTCACCCTGTTT / GCCTTGCCAGCCCGCTCAGTGCAAGGCACTGCAGAGGAGTAG |
| T-121 | 8A/8B | TGG | GCCTCCCTCGCGCCATCAGTGGACCTCCTTGTCACCCTGTTT / GCCTTGCCAGCCCGCTCAGTGGAAGGCACTGCAGAGGAGTAG |
| T-125 | 8A/8B | CCG | GCCTCCCTCGCGCCATCAGCCGACCTCCTTGTCACCCTGTTT / GCCTTGCCAGCCCGCTCAGCCGAAGGCACTGCAGAGGAGTAG |
Ultra-deep pyrosequencing (next generation sequencing)
After conventional PCR amplification, the DNA from each sample was quantified by PicoGreen® dsDNA Assay (Invitrogen, Carlsbad, CA) and pooled equimolarly (em). For optimal em-PCR, the theoretical distribution ratio of beads and ssDNA is 1:1 for the clonal amplification. Based on this ratio, the initial eight em-PCR reactions were performed to determine the optimal ratio for em-PCR, based on bead recovery percentage (which was between 10%-15%). After the em-PCR reaction, the micro-reactors were broken and the beads captured by filtration. The biotin-labeled amplicon-positive beads were enriched using Streptavidin magnetic beads and then single stranded. The DNA beads were pre-incubated with DNA polymerase, sequencing primer and single strand binding protein (SSB), and then distributed into the wells on a PicoTiterPlate™ optical faceplate (454, Branford, CT), that contained 1.6 million wells. After adding the DNA beads and enzymatic beads (ATP sulfurylase and luciferase), the packing beads were layered onto the wells and the plate centrifuged for bead deposition. The signal processing and base-callings were performed using the software package from 454 (Branford, CT)[25].
The sequence reads that passed quality control were aligned to the AR reference sequence (NM_000044.2) mRNA sequence of Homo sapiens androgen receptor, transcript variant 1 using a BLAST-based approach to determine the direction of each read; exons 4-8 were examined. To determine the likelihood of identifying PCR and sequencing errors, which is known that the 454 sequencing technology can generate[26], special care was taken in sequencing homopolymeric regions, which can generate spontaneous insertions/deletions. However, as the study only sequenced exons 4-8 of the AR, that do not contain any homopolymeric regions, such errors were unlikely be a problem.
Sequence analysis
The sequencing data was aligned using MAFFT version 7.050, a multiple sequence alignment software. The data was then filtered by the length of each read, only reads that were the expected length were retained. The mode of the length of the total reads was used to imply expected length. Since sequencing errors are known to depend on position within the read, with more errors occurring near the end of each read, we further filtered the data by retaining only the sequence between the fifth and one hundred and fiftieth bp. All variants in the data sets were then identified.
Results
The samples were analyzed by ultra-deep sequencing at a depth of up to 68,000 reads for each sample [Table 3]. The analyses revealed 53 exonic mutations [Table 4]. These included 20 mutations in exon 4, 11 mutations in exon 5, 10 mutations in exon 6, 4 mutations in exon 7, and 8 mutations in exon 8. It was noted that a significant number of the mutations (18 out of 53) had previously been identified as either associated with androgen insensitivity syndrome (AIS) (11 mutations) or prostate cancer (7 mutations). Twenty-one mutations occurred in several of the tumor samples, with 4 of the mutations occurring in at least 4 of the tumor samples. The distribution of the mutations in each tumor was unique, resulting in a different set of AR variants being present in each of the tumors [Figure 1].
Number of sequencing reads
| Exon | Patient | |||||
|---|---|---|---|---|---|---|
| T-44 | T-102 | T-106 | T-112 | T-121 | T-125 | |
| 4A | 37,704 | 68,001 | 33,819 | 15,660 | 20,289 | 59,399 |
| 4B | 2884 | 1317 | 3882 | n/a | 2862 | 6640 |
| 5 | 4206 | 3765 | 4488 | 3705 | 4460 | 3763 |
| 6 | 9612 | 2683 | 4198 | 3108 | 1434 | 2853 |
| 7 | 19,248 | 7104 | 3729 | 1260 | 6188 | 2993 |
| 8 | 3443 | 3836 | 4430 | 1569 | 1662 | 1795 |
Summary of androgen receptor exonic mutations
| Codon | WT NT | Mutant | Context | WT AA | AA change | Patients tumor | Disease phenotype | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| T-44 | T-102 | T-106 | T-112 | T-121 | T-125 | |||||||
| Exon 4A | Number of reads | |||||||||||
| 37,704 | 68,001 | 33,819 | 15,660 | 20,289 | 59,399 | |||||||
| Number of mutants | ||||||||||||
| 630 | 1888C | T | CC | Arg | Trp | 68 | 72 | |||||
| 647 | 1941C | T | TC | Ser | Ser | 40 | 118 | 21 | 23 | 107 | ||
| 649 | 1947C | T | AC | Thr | Thr | 66 | ||||||
| 650 | 1950C | T | AC | Thr | Thr | 31 | 73 | PCa | ||||
| 652 | 1955C | T | C CC | Pro | Pro | 67 | ||||||
| 658 | 1972C | T | ACC | Gln | Stop | 20 | 63 | CAIS | ||||
| 672 | 2015C | T | CC | Pro | Pro | 21 | 60 | |||||
| Exon 4B | Number of reads | |||||||||||
| 2884 | 1317 | 3882 | n/a | 2862 | 6640 | |||||||
| Number of mutants | ||||||||||||
| 672 | 2015C | T | CC | Pro | Pro | 4 | ||||||
| 678 | 2021C | T | C | Leu | Leu | 3 | 9 | |||||
| 683 | 2047C | T | Pro | Ser | 2 | |||||||
| 689 | 2065G | A | Gly | Arg | 4 | |||||||
| 694 | 2080C | T | C | Gln | Stop | 4 | 4 | 4 | 7 | CAIS | ||
| 695 | 2084C | T | C | Pro | Leu | |||||||
| 695 | 2085C | T | CC | Pro | Pro | 7 | 3 | |||||
| 696 | 2086G | A | Asp | Asn | 4 | CAIS | ||||||
| 697 | 2091C | T | TC | Ser | Ser | 4 | 5 | 6 | ||||
| 705 | 2113C | T | C | Leu | Ser | 2 | ||||||
| 708 | 2124G | A | CT | Leu | Leu | 4 | ||||||
| 709 | 2125G | A | CTG | Gly | Arg | 2 | CAIS | |||||
| 715 | 2141A | AA (ins A) | GT | His | fs | 6 | ||||||
| Exon 5 | Number of reads | |||||||||||
| 4206 | 3765 | 4488 | 3705 | 4460 | 3763 | |||||||
| Number of mutants | ||||||||||||
| 727 | 2180G | A | C C | Arg | His | 5 | ||||||
| 731 | 2191G | A | GT | Val | Ala | 6 | 4 | 7 | ||||
| 734 | 2200C | T | GAC | Gln | Stop | 5 | 4 | CAIS | ||||
| 739 | 2218T | TT (ins T) | AT | Gln | fs | 10 | 6 | 4 | ||||
| 742 | 2225G | A | T | Trp | Stop | 7 | 9 | 7 | 6 | PCa | ||
| 743 | 2229G | A | AT | Met | Ile | 5 | 6 | PAIS | ||||
| 744 | 2231G | A | ATG | Gly | Arg | 5 | CAIS | |||||
| 746 | 2238G | A | AT | Met | Ile | 4 | 4 | |||||
| 749 | 2246C | T | G | Ala | Val | 8 | PCa | |||||
| 750 | 2250G | A | AT | Met | Ile | 4 | 4 | PCa | ||||
| 752 | 2255G | A | T | Trp | Stop | 5 | PCa | |||||
| Exon 6 | Number of reads | |||||||||||
| 9612 | 2683 | 4198 | 3108 | 1434 | 2853 | |||||||
| Number of mutants | ||||||||||||
| 775 | 2323C | T | TAC | Arg | Cys | 11 | 4 | CAIS | ||||
| 779 | 2337C | T | TC | Ser | Ser | 4 | ||||||
| 780 | 2338C | CC (ins C) | TCC | Arg | Pro fs | 3 | ||||||
| 786 | 2354T | C | G | Val | Ala | 3 | ||||||
| 787 | 2359C | T | GTC | Arg | Stop | 21 | 3 | PCa, CAIS | ||||
| 796 | 2390G | A | TTT | Gly | Arg | 3 | 5 | 3 | ||||
| 797 | 2391G | A | T | Trp | Stop | 3 | CAIS | |||||
| 799 | 2395C | T | CTC | Gln | Stop | 20 | 6 | 6 | 5 | 5 | CAIS | |
| 801 | 2403C | T | AC | Thr | Thr | 12 | ||||||
| 802 | 2405C | T | ACC C | Pro | Leu | 3 | ||||||
| Exon 7 | Number of reads | |||||||||||
| 19,248 | 7104 | 3729 | 1260 | 6188 | 2993 | |||||||
| Number of mutants | ||||||||||||
| 824 | 2471A | AA (ins A) | AAA A | Asn Gln | Lys fs | 5 | ||||||
| 825 | 2472T | TA (ins A) | AAA AA | Gln | fs | 21 | ||||||
| 825 | 2473C | CA (ins A) | AAA AAT | Gln Lys | Gln Lys fs | 4 | ||||||
| Exon 8 | Number of reads | |||||||||||
| 3443 | 3836 | 4430 | 1569 | 1662 | 1795 | |||||||
| Number of mutants | ||||||||||||
| 880 | 2638T | TT (ins T) | ACT TT | Asp | Stop | 4 | ||||||
| 887 | 2661G | A | AT | Met | Ile | 2 | PCa | |||||
| 890 | 2670G | A | GT | Val | Val | 2 | 2 | |||||
| 893 | 2678C | T | TTT C | Pro | Leu | 2 | CAIS | |||||
| 893 | 2678C | CC (ins C) | TTT C | Pro | Pro fs | 2 | ||||||
| 893 | 2679G | A | TTT CC | Pro | Pro | 2 | ||||||
| 905 | 2715C | CC (ins C) | GTG CC | Pro | Pro fs | 3 | ||||||
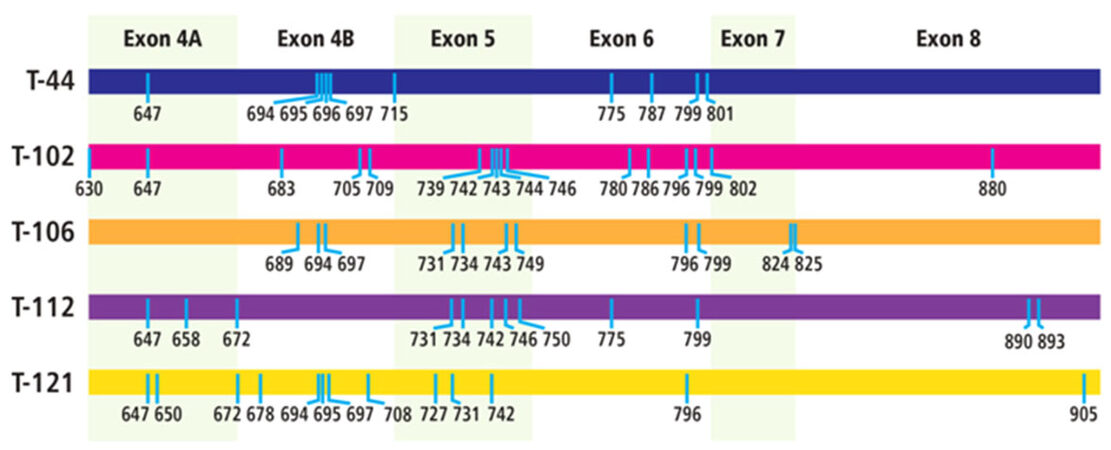
Figure 1. AR exonic mutations present in each of the tumor samples. T- refers to individual tumor samples. AR refers to codon within which mutations were found
Discussion
Do CSGVs really exist?
Before discussing the results, it seems reasonable to address the controversy with regards to whether intra-tissue genetic heterogeneity really exists, particularly as it has been identified not just within tumors, but within normal tissues as well[27,28]. Indeed, questions have been raised as to the possible role of methodological errors in generating genetic heterogeneity in both tumors[29] and tissues in general[30]. To address these questions, it is important to discuss the sequence analysis tools used in our NGS protocols. In traditional sequencing approaches, coverage is based on genome mapping approaches, which use a theoretical redundancy in coverage based on the expression LN/G, where L is the read length, N is the number of reads and G is the haploid genome length[31]. Unfortunately, many factors can result in unequal coverage that produces gaps or much lower coverage than expected[32]. Further, problems such as the choice of alignment algorithms means that even the best mapping algorithms cannot align all reads to a reference genome[33]. As the cost of sequencing has come down, so has the depth of sequencing increased, and this has had a profound effect on the sensitivity of sequencing and the ability to detect rare mutations accurately[34]. Experimental data has confirmed that the major factors that influence detection sensitivity are read depth and experimental precision[34]. Indeed, it would appear possible to accurately detect mutations at a frequency of as low as 0.1%, provided there is sufficient read depth[34]. Somewhat surprisingly, the use of filters used to eliminate false reads etc. does not necessarily prevent low frequency mutations from being detected[35]. Indeed, if used correctly they can in fact enhance the ability to detect low frequency mutations, and in cases of tumor genetic heterogeneity, such an ability is likely to be extremely important[35]. In the case of the present study we believe we have adopted a sufficiently precise sequencing technique that we can use a 0.1% cutoff value to identify the mutations present in our breast tumor samples.
Importance of identifying changing frequencies of driver gene variants during carcinogenesis
At present, identification of ITGH has solely been based on whether specific driver gene variants have been present within cancer tissues, but their frequencies have generally not been assessed. This is because it has been assumed that such variants are present in most tumor cells and are therefore responsible for the cancer phenotype, so that ITGH just reflects the complex genetic makeup of individual tumors, but that the basic mutation-centric paradigm still applies. However, evidence that driver gene mutations can also be present in normal tissues has considerably confused the role of these driver genes in carcinogenesis. We believe that identifying cases of CSGV is likely to be helpful in resolving the phenotype/genotype disconnect, because the data will reveal the actual frequency of the variants and put them in context within a tumor. In a previous study examining an AR CAG repeat length polymorphism in breast tumors, changes in the frequency of these polymorphisms in normal and cancer tissues from individual tumors, as well as in matching blood samples were investigated. This revealed the distribution frequencies of different length AR CAG repeat variants associated with carcinogenesis[6]. A similar approach applied to analyzing driver gene CSGV is likely to give further information to help elucidate the significant genetic events of carcinogenesis.
How can identifying CSGV in tumors contribute to our understanding of cancer genetics?
Clearly, the presence of CSGV within cancer tissues clashes with our present understanding that carcinogenesis is the result of “purifying” selection pressure on single gene variants in a tumor that eventually will lead to removal of all the non-selected variants of that gene[36]. This argument in turn justifies being satisfied with the identification of a single variant per gene, and therefore to ignore any other low frequency variants within the same gene, on the assumption that they must be artifacts, possibly due to PCR or sequencing errors. The recognition that a selection of different single gene variants can remain in individual tumors, is clearly not in line with our present understanding of the occurrence and distribution of cancer mutations. However, our present results would question the validity of this understanding as CSGV were identified in the AR within all 6 breast tumors examined and suggests that the role of mutations in carcinogenesis is more complex than previously thought.
How can identifying CSGV help in understanding treatment resistance?
First, it suggests a mechanism to explain how some tumors can become rapidly resistant to treatment by proposing the existence of genetic variants that can be selected for in genes that have been targeted by chemotherapy. Indeed, the selection of such variants could be a response to ensure the survival of cells that contained the targeted gene as postulated by the atavistic model[37], which considers resistance of cancer cells to treatment as one of their major characteristics. Second, it places much more emphasis on understanding the role of selection pressures generated by different tissue microenvironments on carcinogenesis[38,39]. It also suggests that analyzing the makeup of tissue microenvironments may facilitate the recognition of specific factors involved in the selection of cancer-associated variants.
A different paradigm to explain carcinogenesis
The principle of “parsimony” has underwritten our understanding of science since the middle of the 19th century by telling us to choose the simplest scientific explanation that fits (all) the observed evidence. In studying the genetics of cancer this has been reflected in our belief that identifying common gene mutations present in tumor tissues is one of the keys to understanding the ontology of solid tumors. However, the validity of this concept is being challenged by accumulating evidence of genetic diversity within individual tumors, which this study has further expanded by revealing evidence of AR CSGV in breast tumors. As noted previously, current cancer hypotheses are almost all based on the concept that accumulation of specific de novo individual driver mutations within specific tissues can result in carcinogenesis. However, the lack of a consistent relationship between driver mutations and cancer types and the discovery of the presence of many different driver mutant genes within the same types of cancer tissues has resulted in complex genetic profiles. These have effectively meant that many of these driver gene mutations have been reduced to risk factors, albeit with significant clinical implications, rather than gene mutations that are directly responsible for carcinogenesis.
Interestingly, such phenotype/genotype lack of precision has been found not just in multifactorial diseases such as cancer, but in locus specific genetic disorders as well. For example, in certain locus specific diseases a significant number of individuals that exhibit the disease phenotype do not have a mutation in the putative disease-causing gene, such as in the case of androgen insensitivity syndrome[24] and PKU[40]. Further, a review of genotype-phenotype relationships in a wide range of genetic diseases has revealed many cases of reduced or even zero penetrance[41]. While whole genome sequencing studies have found individuals that can have well known disease-causing gene mutations but do not exhibit the disease phenotypes[42] including cancer-associated genes in healthy individuals[43].
Other recent evidence has further complicated the genetics of cancer, by revealing the effect on cancer phenotypes of processes such as epigenetic regulation, DNA and RNA editing, cellular differentiation hierarchies, gene expression stochasticity and protein-protein interactions[44]. However, their roles are not well defined at present, as in many cases these factors are analyzed as separate events, rather than studying their integrated effect on the selection pressures of the complete tissue microenvironment[45].
One possible hypothesis we have previously proposed is that while intra-tissue genetic heterogeneity may provide the genetic underpinnings for carcinogenesis. It is tumor microenvironment selection pressure on preexisting de novo mutations that is the carcinogenic trigger, rather than just the accumulation of de novo mutations[46]. We have further postulated that these mutations occur early in human embryogenesis[45], as has now been suggested in another recent study[47].
We believe that this hypothesis is supported by the presence of genetic heterogeneity in both cancer and normal tissues, as well as by the evidence of non-genomic, often environmental factors as risk factors for cancer. Indeed, the complexity of post-zygotic variation[14] has only added to the importance of variant selection due to environmental factors within tissue microenvironments in determining cancer phenotypes[48]. A detailed examination of the arguments favoring a selection-centric paradigm has been given in a recent paper[49], which the identification of AR CSGV in breast tumors has further strengthened.
How the identification of CSGV could affect approaches to cancer treatment
Based on many cases of individual-gene genetic heterogeneity that have recently been identified in normal as well as cancer tissue, it seems reasonable to believe that CSGV is likely to also occur in normal tissue. The presence of multiple variants within single genes at low frequencies in normal tissue and cells prior to tissue becoming cancerous would further strengthen the selection-centric paradigm of carcinogenesis. This paradigm could also better explain many observations in which, environmental factors that are clearly non-mutagenic, i.e., diet, exercise, etc., can somehow direct mutations in specific “driver” genes[50]. Thus, “healthy” lifestyle factors can result in the selection of environments that are “cancer resistant”, while other environments identified as “cancer causing”, that are often man-made, can lead to cancer[51]. CSGV could then simply explain a “cancer resistant” environment as one that selects for pre-existing wild-type gene variants and a “cancer causing” environment as one that selects for pre-existing oncogenic gene variants.
Based partially on the principle of parsimony discussed previously, success of species, tissues or cells, has always been considered to eventually result in a specific species, tissues or cells eliminating the competition. However, in the case of CSGV this clearly does not seem to be the case, as while gene variants may not be selected, they are not eliminated entirely either. Thus, in the case of cancer, just destroying the cancer cells and not changing the conditions that allow for them to be preferentially selected, is possibly going to allow other cancer cells with different gene variants to eventually be selected, as the environmental conditions that selected cells with oncogenic properties have not been altered. Our present approach to cancer treatment of removing cancer cells, does of course not preclude the possibility of cancer recurring. However, the presence of CSGV would suggest an approach to cancer treatment that in addition to removing the cancer would also seek to select the normal tissue and cells that are always present within cancer tissues, although normally only as a very small minority of cells. This new treatment approach would therefore require that cancer tissue microenvironments be returned to conditions that would once again select for normal cells, although this is clearly not a simple task.
Recently, more attention has started to be given to the carcinogenic role of the tumor microenvironment including in both tumorigenesis[52] and differential tissue responses to therapy[53]. These studies have begun to analyze and reveal some of the tumor micro-environmental factors that may play a critical role in carcinogenesis. Naturally, these data are also likely to help reveal the tissue micro-environmental properties within normal, non-cancer tissues. However, our understanding of what constitutes tissue-specific micro-environment conditions is still very incomplete. Also, it is highly likely that individuals will have their own set of micro-environmental, chemical and biological conditions, so it will be necessary to analyze their tissue microenvironments in considerable detail. Clearly, cells and tissues exist in complex three-dimensional environments, which include both extra- and intracellular environments. To analyze these microenvironments new technologies are being developed, including atomic force microscopy[54], quantitative extracellular matrix proteomics[55], and single cell multiomics[56] that are being used to create complex databases of tissue micro-environmental factors that will hopefully facilitate the identification of those significant factors that allow for the selection of normal as opposed to cancer cells.
However, at first glance there appears to be the same underlying problem with this approach as the one that has characterized attempts to analyze the genomic and post-genomic events that cause cells to become oncogenic. Namely, the inability to identify the critical oncogenic events involved because we can only measure conditions before and after a cell becomes cancerous. However, the tissue micro-environmental conditions that result in normal cells being selected do not suffer from this drawback, as normal cells remain dominant in tissue over relatively long periods of time, presumably because they are subject to relatively consistent tissue micro-environmental conditions. Nevertheless, it is important to note that tissue microenvironments are likely to be highly individualized, so that even within an individual different tissue microenvironments might exist around different tissues.
Conclusion
Before the discovery of ITGH and now CSGV, the novel approach to cancer treatment that we are suggesting would have never been considered. However, if it is proven that cancer-associated genes within tumors as well as normal tissue consistently exhibit CSGV. Then a treatment approach that includes the goal of reselecting normal tissues by adjusting the tissue microenvironment, would seem to be the logical way to ensure that cancer treatments finally result in the permanent elimination of cancer.
Decarations
Authors’ contributions
Design: Gottlieb B
Literature research: Gottlieb B
Sequencing: Babrzadeh F, Wang C, Gharizadeh B
Analysis of data: Oros KK, Greenwood CMT
Tissue and DNA preparation: Alvarado C
Tumor samples: Basik M
Manuscript writing: Gottlieb B
Manuscript editing: Beitel LK, Trifiro M
Availability of data and materials
Data is available from Dr. Bruce Gottlieb. Materials are unavailable.
Financial support and sponsorship
This study was supported by a grant to BG from the Weekend to End Breast Cancer Fund of the Jewish General Hospital, Montreal, Quebec, Canada.
Conflicts of interest
All authors declare that there are no conflicts of interest.
Ethical approval and consent to participate
This study was approved by the ethical review board of Jewish General Hospital.
Consent for publication
Not applicable.
Copyright
© The Author(s) 2018.
REFERENCES
1. Lawrence MS, Stojanov P, Mermel CH, Robinson JT, Garraway LA, Golub TR, Myerson M, Gabriel SB, Lander ES, Getz G. Discovery and saturation analysis of cancer genes across 21 tumor types. Nature 2014;505:495-501.
2. Martincorena I, Roshan A, Gerstung M, Van Loo P, McLaren S, Wedge DC, Fullam A, Alexandrov LB, Tubio JM, Stebbings L, Menzies A, Widaa S, Stratton MR, Jones PH, Campbell PJ. Tumor evolution. High burden and pervasive selection of somatic mutations in normal human skin. Science 2015;348:880-6.
3. Schaefer MH, Serrano L. Cell type-specific properties and environment shape tissue specificity of cancer genes. Sci Rep 2016;6:2707.
4. Williamson LM, Lees-Miller SP. Estrogen receptor a-mediated transcription induces cell cycle-dependent DNA double-strand breaks. Carcinogenesis 2011;32:279-85.
5. Savage KI, Matchett KB, Barros EM, Cooper KM, Irwin GW, Gorski JJ, Orr KS, Vohhodina J, Kavanagh JN, Madden AF, Powell A, Manti L, McDade SS, Park BH, Prise KM, McIntosh SA, Salto-Tellez M, Richard DJ, Elliott CT, Harkinet DP. BRCA1 deficiency exacerbates estrogen-induced DNA damage and genomic instability. Cancer Res 2014;74:2773-84.
6. Kleppe M, Levine RL. Tumor heterogeneity confounds and illuminates: assessing the implications. Nat Med 2014;20:342-4.
7. Gottlieb B, Alvarado C, Wang C, Gharizadeh B, Babrzadeh F, Richards B, Basik M, Beitel LK, Trifiro M. Making sense of intra-tumor genetic heterogeneity: altered frequency of androgen receptor CAG repeat length variants in breast cancer tissues. Hum Mutat 2013;34:610-8.
8. Ng CK, Scultheis AM, Bidard FC, Wegelt B, Reis-Filho JS. Breast cancer genomics from microarray to massively parallel sequencing: paradigms and new insights. J Natl Cancer Inst 2015;107:djv015.
9. Wang Y, Waters J, Leung ML, Unruh Roh W A, Shi X, Chen K, Scheet Vattathil S P, Liang H, Multani A, Zhang H, Zhao R, Michor F, Meric-Bernstam F, Navin NE. Clonal evolution in breast cancer revealed by single nucleus genome sequencing. Nature 2014;512:155-60.
10. Raphael BJ, Dobson JR, Oesper L, Vandin F. Identifying driver mutations in sequenced cancer genomes: computational approaches to enable precision medicine. Genome Med 2014;6:5.
11. McFarland CA, Yaglom YA, Wojtkowiak JW, Scott JG, Morse DL, Sherman MY, Mirny LA. The damaging effect of passenger mutations on cancer progression. Cancer Res 2017;77:4763-72.
12. Fortunato A, Boddy A, Mallo D, Aktipis A, Maley CC, Pepper JW. Natural selection in cancer biology: from molecular snowflakes to trait hallmarks. Cold Spring Harb Perspect Med 2017;7:a029652.
13. Schirmer M, Ijaz UZ, D'Amore R, Hall N, Sloan WT, Quince C. Insight into biases and sequencing errors for amplicon sequencing with the Illumina MiSeq platform. Nucleic Acids Res 2015;43:e37.
14. Forsberg LA, Gisselsson D, Damanski JP. Mosaicism in health and disease - clones picking up speed. Nat Rev Genet 2017;18:128-42.
15. Hoang ML, Kinde I, Tomasetti C, Wyatt McMahon K, Rosenquist TA, Grollman AP, Kinzler KW, Vogelstein B, Papadopoulos N. Genome-wide quantification of rare somatic mutations in normal human tissues using massively parallel sequencing. PNAS 2016;113:9846-51.
16. Anglesio MS, Papadopoulos N, Ayhan A, Nazeran TM, Noë M, Horlings HM, Lum A, Jones S, Senz J, Seckin T, Ho J, Wu RC, Lac V, Ogawa H, Tessier-Cloutier B, Alhassan R, Wang A, Wang Y, Cohen JD, Wong F, Hasanovic A, Orr N, Zhang M, Popoli M, McMahon W, Wood LD, Mattox A, Allaire C, Segars J, Williams C, Tomasetti C, Boyd N, Kinzler KW, Gilks CB, Diaz L, Wang TL, Vogelstein B, Yong PJ, Huntsman DG, Shih IM. Cancer-associated mutations in endometriosis without cancer. N Engl J Med 2017;376:1835-48.
17. Guedj M, Marisa I, de Renies A, Orsetti B, Schiappa R, Bibeau F, MacGrogan G, Lerebours F, Finetti P, Longy M, Bertheau P, Bertrand F, Bonnet F, Martin AL, Feugeas JP, Bièche I, Lehmann-Che J, Lidereau R, Birnbaum D, Bertucci F, de Thé H, Theillet C. A refined molecular taxonomy of breast cancer. Oncogene 2012;31:1196-206.
18. Lehnann BD, Bauer JA, Chen X, Sanders ME, Chakravarthy AB, Shyr Y, Pietenpol JA. Identification of human triple-negative breast cancer subtypes and preclinical models for selection of targeted therapies. J Clin Invest 2011;121:2750-67.
19. Hirschfield KM, Ganesan S. Triple-negative breast cancer: molecular subtypes and targested therapy. Curr Opin Obstet Gyncol 2014;26:34-40.
20. Collins LC, Cole KS, Marotti JD, Hu R, Schnitt SJ, Tamimi RM. Androgen expression in breast cancer in relation to molecular phenotype: results from the Nurses Health Study. Mod Pathol 2011;24:924-31.
21. Fujii R, Hanamura T, Sizuki T, Gohno T, Shibahara Y, Niwa T, Yamaguchi Y, Ohnuki K, Kakugawa Y, Hirakawa H, Ishida T, Sasano H, Ohuchi N, Hayashi S. Increased androgen receptor activity and cell proliferation in aromatase inhibitor-resistant breast carcinoma. J Steroid Biochem Mol Biol 2014;144:513-22.
22. Peters KM, Edwards SL, Nair SS, French JD, Bailry PJ, Salkield K, Stein S, Wagner S, Francis GD, Clark SJ, Brown MA. Androgen receptor expression predicts cancer survival: the role of genetic and epigenetic events. BMC Cancer 2012;12:132.
23. MacNamara KM, Moore NL, Hickey TE, Sasano H, Tiley WD. Complexities of androgen receptor signaling in breast cancer. Endocr Relat Cancer 2014;21:T161-81.
24. Gottlieb B, Beitel LK, Nadarajah A, Paliouras M, Trifiro M. The androgen receptor gene mutations database (ARDB): 2012 update. Hum Mutat 2012;33:887-94.
25. Brockman W, Alvarez P, Young S, Garber M, Gainnoukos G, Lee WI, Russ C, Lander ES, Nusbaum C, Jaffe DB. Quality scores and SNP detection in sequencing-by-sequencing systems. Genome Res 2008;19:767-70.
26. Shao W, Boltz VF, Spindler JE, Kearney MF, Maldarelli F, Mellors JW, Stewart C, Volfovsky N, Levitsky A, Stephens RM, Coffin JM. Analysis of 454 sequencing error rate, error sources, and artifact recombination for detection of low-frequency drug resistance mutations in HIV-1 DNA. Retrovirology 2013;10:18.
27. Behjati S, Huch M, van Boxtel R, Karthaus W, Wedge DC, Tamuri AU, Martincorena I, Petljak M, Alexandrov LB, Gundem G, Patrick S, Tarpey PS, Roerink S, Blokker J, Maddison M, Mudie L, Robinson B, Nik-Zainal S, Campbell P, Goldman N, van de Wetering M, Cuppen E, Clevers H, Stratton MR. Genome sequencing of normal cells reveals developmental lineages and mutational processes. Nature 2014;513:422-5.
28. Gulukota K, Helseth DL Jr, Khadekar JD. Direct observation of genomic heterogeneity through local haplotyping analysis. BMC Genomics 2014;15:418.
29. Makohon-Moore AP, Zhang M, Reiter JG, Bozic I, Allen B, Kundu D, Chatterjee K, Wong F, Jiao Y, Kohutek ZA, Hong J, Attiyeh M, Javier B, Wood LD, Hruban RH, Nowak MA, Papadopoulos N, Kinzler KW, Vogelstein B, Iacobuzio-Donahue CA. Limited heterogeneity of known driver gene mutations among the metastases of individual patients with pancreatic cancer. Nat Genet 2017;49:358-66.
30. Chen L, Liu P, Evans TC Jr, Ettwiller L. DNA damage is a major cause of sequencing errors, directly confounding variant identification. Science 2017;355:752-6.
31. Lander ES, Waterman MS. Genomic mapping by fingerprinting random clones: a mathematical analysis. Genomics 1988;2:231-9.
32. Sims D, Sudbery I, Ilott NE, Heger A, Ponting CP. Sequencing depth and coverage: key considerations in genomic analysis. Nat Rev Genet 2014;15:121-32.
33. Hatem A, Bozdag D, Toland AE, Catalyurek UV. Benchmarking short sequence mapping tools. BMC Bioinformatics 2013;14:184.
34. Flaherty P, Natsoulis G, Muralidharan O, Winters M, Buenrostro J, Bell J, Brown S, Holodniy M, Zhang N, Ji HP. Ultrasensitive detection of rare mutations using next-generation targeted sequencing. Nucleic Acids Res 2012;40:e2.
35. Cibulskis K, Lawrence MS, Carter SL, Sivachenko A, Jaffe D, Sougnez C, Gabriel S, Meyerson M, Lander ES, Getz G. Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat Biotech 2013;31:213-9.
36. Ostrow SL, Barshir R, DeGregori J, Yeger-Lotem E, Hershberg R. Cancer evolution is associated with pervasive positive selection on globally expressed genes. PLoS Genet 2014;10:e1004239.
37. Thomas F, Ujvan B, Renaud F, Vincent M. Cancer adaptations: atavism, de novo selection, or something in between? Bioessays 2017;39:1700039.
38. Polyak K. Tumor heterogeneity confounds and illuminates: a case of Darwinian tumor evolution. Nature Med 2014;20:344-6.
40. Zhu T, j Ye, Han I, Qiu QW, Zhang H, Liang L, Gu X. Variations in genotype-phenotype correlations in phenylalanine hydroxylate deficiency in Chinese Han population. Gene 2013;529:80-7. in Chinese
41. Cooper DN, Krawczak M, Polychronakos C, Tyler-Smith C, Kehrer-Sawatzki H. Where genotype is not predictive of phenotype: towards an understanding of the molecular basis of reduced penetrance in human inherited disease. Hum Genet 2013;132:1077-130.
42. Chen R, Shi L, Hakenberg J, Naughton B, Sklar P, Zhang J, Zhou H, Tian L, Prakash O, Lemire M, Sleiman P, Cheng WH, Chen W, Shah H, Shen Y, Fromer M, Omberg L, Deardorff MA, Zackai E, Bobe JR, Levin E, Hudson TJ, Groop L, Wang J, Hakonarson H, Wojcicki A, Diaz GA, Edelmann L, Schadt EE, Friend SH. Analysis of 589,306 genomes identifies individuals resilient to severe Mendelian childhood diseases. Nat Biotechnol 2016;34:531-8.
43. Youseef O, Knuuttila A, Piirla P, Bohling T, Sargadi V, Knuutila S. Presence of cancer-associated mutations in exhaled breath condensates of healthy individuals by next generation sequencing. Oncotarget 2017;8:18166-76.
44. Calado F, Siva-Santos B, Norell H. Intra-tumor heterogeneity going beyond genetics. FEBS J 2016;283:2245-58.
45. Gottlieb B, Beitel LK, Trifiro M. Changing genetic paradigms: creating next-generation genetic databases as tools to understand the emerging complexities of genotype/phenotype relationships. Hum Genomics 2014;8:9.
46. Gottlieb B, Beitel LK, Alvarado C, Trifiro MA. Selection and mutation in the "new" genetics: an emerging hypothesis. Hum Genet 2010;127:491-501.
47. Ju YS, Martincorena I, Gerstung M, Petljak M, Alexandrov LB, Rahbari R, Wedge DC, Davies HR, Ramakrishna M, Fullam A, Martin S, Alder C, Patel N, Gamble S, O'Meara S, Giri DD, Sauer T, Pinder SE, Purdie CA, Å Borg, Stunnenberg H, van de Vijver M, Tan BK, Caldas C, Tutt A, Ueno NT, van 't Veer LJ, Martens JW, Sotiriou C, Knappskog S, Span PN, Lakhani SR, Eyfjörd JE, Børresen-Dale AL, Richardson A, Thompson AM, Viari A, Hurles ME, Nik-Zainal S, Campbell PJ, Stratton MR. Somatic mutations reveal asymmetric cellular dynamics in the early human embryo. Nature 2017;543:714-8.
48. Liggett LA, DeGregori J. Changing mutational and adaptive landscapes and the genesis of cancer. Biochem Biophys Acta 2017;1867:84-94.
49. Scott J, Marusyk A. Somatic clonal evolution: a selection-centric perspective. Biochem Biophys Acta 2017;1867:139-50.
50. Kerr J, Anderson C, Lippman SM. Physical activity, sedentary behavior, diet and cancer: an update and emerging new evidence. Lancet Oncol 2017;18:e457-71.
51. Hochberg ME, Noble RJ. A framework for how environment contributes to cancer risk. Ecology Letters 2017;20:117-34.
52. Wang M, Zhao J, Zhang L, Wei F, Lian Y, Wu Y, Gong Z, Zhang S, Zhou J, Cao K, Li X, Xiong W, Li G, Zeng Z. Role of tumor microenvironment in tumorigenesis. J Cancer 2017;8:761-73.
53. Hirata E, Sahai E. Tumor microenvironment and differential responses to therapy. Cold Spring Harb Perspect Med 2017;7:a026781.
54. Jorba I, Uriarte JJ, Campillo N, Farre R, Navajas D. Probing micromechanical properties of the extracellular matrix of soft tissues by atomic force microscopy. J Cell Physiol 2017;232:19-26.
55. Goddard ET, Ryan C, Hill RC, Barrett A, Betts C, Guo Q, Maller O, Borges VF, Hansen KC, Schedin P. Quantitative extracellular matrix proteomics to study mammary and liver tissue microenvironments. Intl J Biochem Cell Biol 2016;81:223-32.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Gottlieb B, Babrzadeh F, Oros KK, Alvarado C, Wang C, Gharizadeh B, Basik M, Greenwood CMT, Beitel LK, Trifiro M. New insights into the role of intra-tumor genetic heterogeneity in carcinogenesis: identification of complex single gene variance within tumors. J Cancer Metastasis Treat 2018;4:37. http://dx.doi.org/10.20517/2394-4722.2018.26
AMA Style
Gottlieb B, Babrzadeh F, Oros KK, Alvarado C, Wang C, Gharizadeh B, Basik M, Greenwood CMT, Beitel LK, Trifiro M. New insights into the role of intra-tumor genetic heterogeneity in carcinogenesis: identification of complex single gene variance within tumors. Journal of Cancer Metastasis and Treatment. 2018; 4: 37. http://dx.doi.org/10.20517/2394-4722.2018.26
Chicago/Turabian Style
Gottlieb, Bruce, Farbod Babrzadeh, Kathleen Klein Oros, Carlos Alvarado, Chunlin Wang, Baback Gharizadeh, Mark Basik, Celia M.T. Greenwood, Lenore K. Beitel, Mark Trifiro. 2018. "New insights into the role of intra-tumor genetic heterogeneity in carcinogenesis: identification of complex single gene variance within tumors" Journal of Cancer Metastasis and Treatment. 4: 37. http://dx.doi.org/10.20517/2394-4722.2018.26
ACS Style
Gottlieb, B.; Babrzadeh F.; Oros KK.; Alvarado C.; Wang C.; Gharizadeh B.; Basik M.; Greenwood CMT.; Beitel LK.; Trifiro M. New insights into the role of intra-tumor genetic heterogeneity in carcinogenesis: identification of complex single gene variance within tumors. J. Cancer. Metastasis. Treat. 2018, 4, 37. http://dx.doi.org/10.20517/2394-4722.2018.26
About This Article
Special Issue
Copyright

Data & Comments
Data


 Cite This Article 1 clicks
Cite This Article 1 clicks

 Like This Article 0
likes
Like This Article 0
likes






Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.