
Unmasking tumor heterogeneity and clonal evolution by single-cell analysis


Abstract
The intratumoral heterogeneity orchestrated by the tumor intrinsic and extrinsic mechanisms enable cancers to persist and spread notwithstanding the use of aggressive interventional therapies. The heterogeneity is revealed at multiple levels - at the level of individual tumor cells, in the cellular composition of tumor infiltrates and in the chemical microenvironment in which the cells reside. Deconvoluting the complex nature of the cell types present in the tumor, along with the homo and heterotypic interactions between different cell types can produce novel insights of biological and clinical relevance. However, most techniques analyze tumors at a gross level missing key inter-cell-type genotypic and phenotypic differences. The advent of single-cell sequencing has given an unprecedented opportunity to analyze the tumor at a resolution that not only captures the diversity of the cellular composition of a tumor but also provides information on the genetic, epigenetic and functional states of different cell types. In this review, we summarize the genesis of tumor heterogeneity, its impact on tumor growth and progression and their clinical consequences. We present an overview of the currently available platforms for isolation and sequencing of single tumor cells and provide evidence of its utility in precision medicine and personalized therapy.
Keywords
Introduction
A single cell is the ultimate denominator of a multicellular organism. In the progression of cancer, a single cell begins its journey to evolve into a malignant tumor cell and forms distinct subpopulations leading to intratumoral heterogeneity (ITH). Clonal diversity, the source of ITH, is the characteristics of all cancers and plays a critical role in cancer invasion, metastasis and development of resistance to targeted and non-targeted therapies[1-4]. Next-generation sequencing of bulk tumor tissues from many cancers has generated an unprecedented amount of multidimensional data bringing in novel insights into mechanisms of tumor initiation, progression and metastasis [Figure 1]. It has also unmasked the underlying deeper genotypic and phenotypic heterogeneity that exists between tumors belonging to the same cancer type. The ITH originating in the cancer genome can be revealed by deep exome and whole genome sequencing. However, transcriptome data from a complex mixture of cells derived from bulk tumor tissues fail to accurately elucidate the ITH, requiring technologies to study tumors at a single-cell resolution. Over the past ten years, there has been extraordinary progress in the development and application of single-cell analysis in cancer research as evidenced by the rise in publications describing different aspects of single-cell sequencing to characterize tumors at a deeper level [Figure 1]. In this review, we first introduce the concept of ITH and its clinical implications. Next, we outline new technologies enabling single-cell analysis with high sensitivity and finally provide examples of their applications in uncovering new perspectives in cancer diagnostics and treatment.
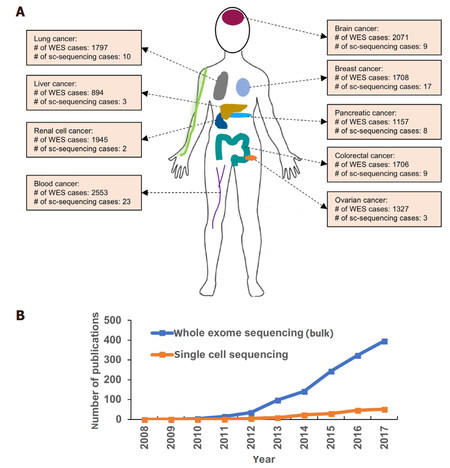
Figure 1. Application of whole exome sequencing (WES) and single-cell sequencing (sc-sequencing) to cancer research. A: Overview of patient cases to which WES and sc-sequencing were applied to characterize different types of human cancers to understand ITH and tumor microenvironment. The various types of cancers include liver cancer, lung cancer, renal cell cancer, blood cancer, brain cancer, breast cancer, pancreatic cancer, colorectal cancer and ovarian cancer compiled from public databases; B: the number of publications reporting applications of either whole exome or single-cell sequencing to cancer patients within the recent ten years. The key words “exome/single-cell sequencing” and “cancer patients” were used for searching articles from NCBI
Origin of ITH
ITH was first described by Fidler et al.[5] more than 30 years ago in murine models as a single tumor consisting of many cell subpopulations. However, this concept of heterogeneity in the composition of a tumor has now been expanded to include the genetic and molecular heterogeneity present within individual tumor cells and cells comprising the tumor microenvironment[6-9].
Genetic and epigenetic alterations
ITH arises as a result of both genetic and non-genetic changes in the tumor cells and the surrounding environment respectively [Figure 2][10]. Increased genetic instability as a result of mutations in DNA damage checkpoint control genes and DNA repair genes is one of the hallmarks of cancer and generates divergent clonal population of cells as the tumor grows over time[11,12]. With the significantly high rate of cancer cell divisions, events of random mutagenesis increase, leading to local and global genetic alterations, that influence the future course of tumor development and progression[13]. In addition, these genetic alterations create a hotbed for competition between clones driven by selection processes imposed by changes in the tumor microenvironment and by the use of therapies[14,15].
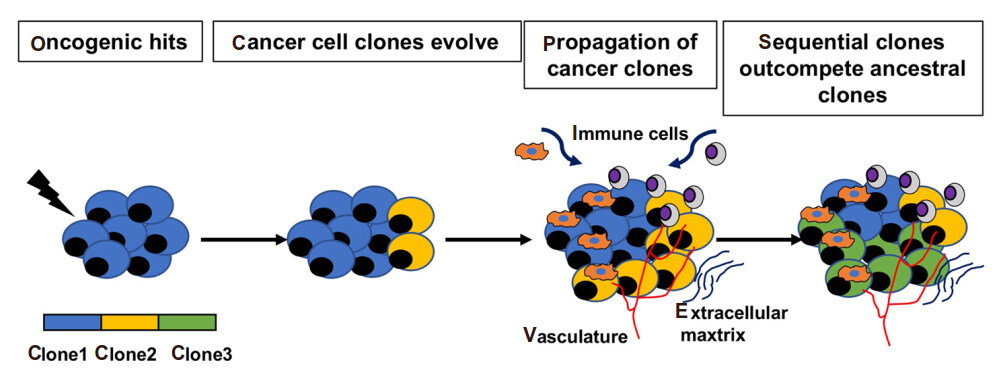
Figure 2. Origin of ITH. Upon certain oncogenic hits, some cells in the normal tissues undergo genetic alterations to generate cancer cells. ITH arises through clonal evolution in which cells are dictated by transcriptomic and epigenetic factors and the tumor microenvironment. Cancer clones (yellow) propagate and generate successive clones (green) which outcompete the ancestral ones
A vast majority of established driver mutations are clonal and arise early in the development of the tumor, however, subclonal de novo driver mutations may also arise in the later stages of tumorigenesis - to escape drug sensitivity and successful metastasis, for example[16]. In a recent UK-wide multi-center prospective longitudinal cohort study, “Tracking Renal Cell Cancer Evolution through therapy (TRACERx Renal)”, clonal phylogeny and evolutionary subtypes were elucidated by multi-region sampling on matched primary and metastasis biopsies from 100 renal cell carcinoma patients[17]. Subclonal driver mutations in the VHL and PBRM1 genes that were identified in the original tumor were absent in the widely disseminated metastatic tumor sites. Instead, these metastatic sites acquired loss of 9p and 14q mutations, suggesting that metastatic competence may not be driven by the founder driver mutations that established the primary tumor[17].
Tumor heterogeneity can also arise from epigenetic variations through DNA methylation that can profoundly modulate the open and closed conformation of chromatin in tumor cells, leading to gene expression alterations and phenotypic changes[18]. For example, the methylation status of the tumor suppressor gene CDKN2B can be used as a biomarker of response to treatment in multiple diseases[19]. However, heterogeneous methylation was observed in individual patients with acute myeloid leukemia, posing a challenge in using CDKN2B methylation as a biomarker[20]. Similarly, differential microRNA expression is known to affect the diversity of cellular phenotype within a single tumor by modulating the expression of target genes[21]. Subclonal expression of microRNAs (miRNA-21, miRNA-34a, miRNA-125, and miRNA-126) in prostate cancer is associated with diverse patient outcomes[22].
Cellular composition of tumors
Cell types present in the tumor stroma, such as immune cells, fibroblasts, vascular cells play a critical role in shaping the composition of tumors by secreting cytokines growth factors and extracellular matrix that changes the stiffness of the tumor tissue[23]. In a tumor microenvironment infiltrated by CD8 T cells at the tumor site is associated with increased overall survival, whereas myeloid-derived suppressor cells (MDSCs) possessing strong immune suppressive activity decreases overall survival[24]. The diversity of these functionally different immune cell types creates a heterogeneous tumor microenvironment and regulate tumor growth, metastasis and treatment response[25]. In addition, the distribution and density of the vasculature impact the supply of nutrients and oxygen selecting for tumor cells with specific metabolic phenotypes further contributing to tumor heterogeneity[26,27]. Tumor heterogeneity has a significant bearing on the management of disease as summarized in the next section.
Clinical impact of the ITH
Resistance to therapy
The resistance of tumors to therapies is often attributed to the presence of rare drug-resistant clones in the tumor before therapy or appears after treatment. An example of clonal resistance was observed in patients with anaplastic lymphoma kinase (ALK gene) rearranged non-small cell lung cancer (NSCLC) post treatment with ALK inhibitors[28]. Patients that developed drug resistance displayed a distinct spectrum of ALK resistance mutations in response to different generations of ALK inhibitors[28]. Particularly, ALKG1202R mutation is highly enriched in resistant tumors after treatment with second-generation ALK inhibitors, highlighting the significance of repeat biopsies and genotyping during the course of targeted therapy treatment[28]. In addition, studies investigating the mechanism of resistance of NSCLC tumors to EGFR tyrosine kinase inhibitors have revealed a variety of drug resistance mechanisms, including gatekeeper mutation T790M detected in > 50% of the EGFR TKI resistant tumors[29], amplification of MET receptor tyrosine kinase[30], activating mutation in PI3K pathway[31], and other uncharacterized mechanisms involving changes in the cellular phenotype. The appearance of a rare clonal population of tumor cells harboring drug resistance mutations or drug resistance phenotype can be captured by single-cell sequencing of the tumor and may not be discernible from whole tumor analysis, especially when present at a very low frequency. In an alternative model of drug resistance, resistant clones can be pre-existing in the tumor as a rare cell population and emerge post clearance of the drug-susceptible clones. In fact, in a study involving a cohort of 20 breast cancer patients, 8 out of 10 patients that did not show complete clearance of the tumor displayed unique somatic mutations in chemoresistant clones by single-cell sequencing. These mutations were pre-existing and were adaptively selected by the chemotherapy treatment[32]. It is possible to detect de novo or drug-induced resistant clones present at low frequency by ultra-deep exome sequencing, however, two critical pieces of information - number of cells harboring the mutation and the zygosity of the mutation - cannot be accurately assessed from the bulk sequencing.
Challenges in diagnostic and prognostic biomarker identification
Identifying clinically relevant diagnostic biomarkers are challenging given that the tumor is heterogeneous and diagnostic or prognostic biomarkers are not expressed uniformly in all cells and across longitudinal assessment periods [Figure 3]. For example, the divergent genetic landscape of metastatic cells can render biomarkers identified from primary tumors irrelevant [Figure 3][33].
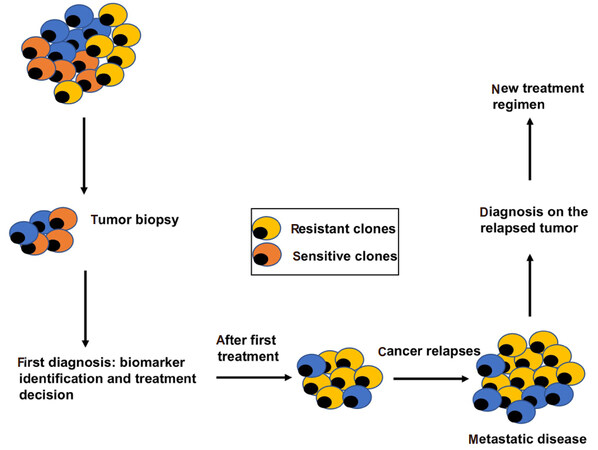
Figure 3. The clinical implications of tumor heterogeneity. Cancer diagnosis is commonly based on tumor biopsy, which is usually a small fraction of the total tumor mass and does not represent all subclones inside the tumor. Initial diagnosis is made based on the tumor biopsy. After the first-line treatment, dominant clones can be killed successfully whereas resistant clones persist and drive tumor progression. Metastasis may develop from the resistant clones that survive the initial treatment. New diagnosis needs to be made in order to apply the second-line treatment
In prostate cancer, ITH represents a major challenge for diagnostic and prognostic biomarker identification. Enhanced DNA ploidy and loss of PTEN, a tumor-suppressor gene, are critical prognostic markers of prostate cancer[34]. In a clinical study of 304 patients who underwent radical prostatectomy, a significant difference in DNA ploidy classification and loss of PTEN expression was observed by analyzing all tumor areas in comparison to a single biopsy sample, suggesting that the heterogeneous chromosomal alterations compromise the accuracy of histopathology analysis and confound disease prognosis[35]. Prognostic markers in ovarian cancer such as unique CpG methylation patterns have been suggested for progression-free survival as well as early disease recurrence following chemotherapy[36,37]. However, DNA methylation patterns are heterogeneous and occurs in both large and poorly defined genomic regions[20], posing a challenge in using CpG methylation as a biomarker. In a recent study by Rajaram et al.[38], a data-driven framework based on single-cell analysis has been reported that provides an estimate of the depth of sampling that may be minimally required to cover the full range of phenotypic heterogeneity for accurate biomarker discovery. Based on the analysis of 215 single-cell features, three replicates were sufficient to capture the heterogeneity for many features if they were defined by clear biomarkers without background noise[38]. For example, nuclear staining (the number of nuclei staining by DAPI: an easily detectable feature) requires 1-2 cores to capture the heterogeneity in > 90% of the patients, while 10 cores or more are needed to assess the heterogeneity of YAP transcription factor expression (a sparsely detectable feature)[38]. Therefore, both the complexity of the feature and the biomarkers that define the feature determine the number of samples required for studying heterogeneity[38].
Uncovering ITH by single-cell analysis
Single-cell analysis is a powerful tool to resolve ITH of solid tumors and to detect the genetic makeup of rare cancer cells such as circulating tumor cells (CTCs) to ultimately guide personalized treatment strategies. The sensitivity of detecting somatic variants or changes in gene expression at a single-cell level has improved dramatically over the years through the introduction of new technologies. Single-cell analysis workflow includes isolation of single cells, either from the tumor site or circulating tumor cells from the blood. Following tumor dissociation, single-cells can be obtained by serial dilution, flow cytometry or microfluidics technology and then sequenced at sufficient depth to capture the genetic changes.
Single-cell isolation methods from solid tumors
A major challenge in single-cell analysis is obtaining a viable cell sample from complex tumor tissues. Current methods include mechanical or enzymatic dissociation of tissues followed by isolation of single cells. Once the tissue is processed, multiple techniques to isolate single cells can be implemented [Figure 4]. A more labor-intensive technique of laser capture microdissection (LCM) is also a viable approach for single-cell isolation from sectioned tumor samples. One challenge for single-cell transcriptomics is the poor RNA quality extracted from archival tumor samples such as formalin-fixed paraffin-embedded (FFPE) samples[39]. However, with the Smart-3SEQ method , it is now feasible to perform single-cell RNA-seq on FFPE samples[39]. Additionally, recent advances using the SMART seq technology and cDNA synthesis methods using random priming (SMART-Seq Stranded Kit, Takara Inc.) have been beneficial in extracting reliable gene expression information from poor quality RNA from FFPE samples.
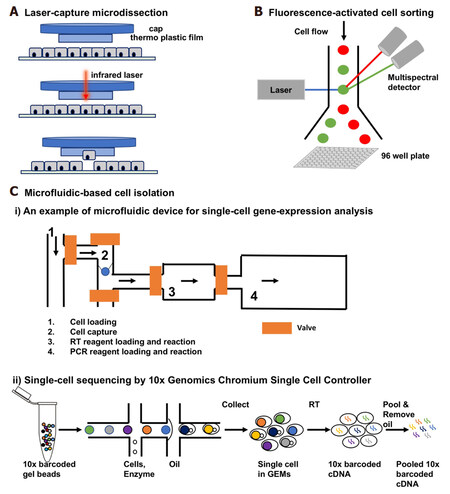
Figure 4. Different ways of single-cell isolation. A: Laser capture microdissection. A thermolabile polymer is placed on a tissue section on a glass slide. An infrared laser fires through the cap over the cells of interest to melt the film. The cell of interest adheres to the film, leaving the unwanted cells behind; B: fluorescence-activated cell sorting. A stream of single cells passes through an excitation laser beam and the fluorescent signal is analyzed by a multispectral detector. Single cells can be sorted into a 96 well plate; C: microfluidic-based single-cell isolation: i) An example showing a microfluidic device for single cell gene expression analysis (figure is adapted from White et al.[94,95], 2011): (1) loading of single cells; (2) capturing single cells; (3) reverse transcription; (4) PCR; ii) Gel Bead-in-EMulsions (GEMs) formation and barcoding of 10× Genomics single-cell sequencing platform (figure is adapted from 10× Genomics Inc). Single cell GEMs are generated by passing cells with enzyme mix, partitioning oil and 10× barcoded gel beads. After GEM formation, the gel bead is dissolved and the co-partitioned cell is lysed. Reverse transcription occurs inside GEMs and barcoded full-length cDNA is generated. After RT, the GEMs are broken and the cDNA is pooled prior to library preparation for sequencing
Single-cell isolation by mechanical or enzymatic dissociation
Conventionally, tumor tissues are dissociated into single cells by mechanical dissociation (e.g., meshing, trituration with a pipette/tip)[40-42] or by enzymatic dissociation[43-45] or a combination of both. Enzymes such as collagenase[41], DNase[46], trypsin[47] are commonly used for dissociating the cell-cell contacts and the extracellular matrix to generate single cell suspensions. The various dissociation methods may largely differ in their yield of viable cells[48,49], limiting their downstream applications. Therefore, tumor dissociation protocols optimized for different tumor types is a key gap that needs to be addressed for high-throughput single-cell analysis.
Single-cell isolation by LCM
To preserve the native properties of tumor cells shaped by the complex tumor microenvironment, LCM can be used to isolate tumor cells directly from sectioned tissues. It is a method to procure subpopulations of tissue cells under direct microscopic visualization by cutting away unwanted cells and obtain histologically pure cell population [Figure 4A][50]. A variety of downstream applications exist for microdissected cells such as DNA genotyping, RNA transcript profiling or cDNA library generation. Even though the majority of the studies take advantage of approximately 100-1000 dissected cells, LCM can also be used for single-cell isolation directly[51-53].
Isolation of rare CTCs
Currently, tumor biopsies are obtained to establish the diagnosis and determine whether the predictive biomarkers are consistent between the primary and the metastatic tumors. However, getting biopsies is invasive, expensive and not always feasible. Additionally, it is difficult to get biopsies of metastatic lesions or get repeat biopsies for difficult to access tumors. Analysis of disseminated tumor cells (DTCs) is a useful alternative to tumor biopsy in clinical setting for patient stratification, therapy selection and monitoring drug resistance during the course of treatment[54]. DTCs originate from the primary or metastatic tumors, extravasate into the bloodstream or lymphatics and carry genomic profiles of tumors from which they originate[55,56]. Disseminated cancer cells are usually detectable as CTCs in the circulation[54]. A small fraction of them that have reached to a secondary organ such as the bone marrow and lymph nodes is termed as DTCs[54]. Though for certain cancers, the presence of DTCs in distant organs is a strong predictive marker for cancer metastasis, the challenge with DTC isolation due to the invasive procedure is a deterrent in studying this population by single-cell sequencing. On the contrary, CTCs circulating in patient blood has proved to be a valuable resource for diagnostic and prognostic biomarker discovery[57], although distinguishing a DTC from a pool of CTCs is challenging.
CTCs contain signatures of tumor heterogeneity and carry the spectrum of somatic mutations present in both the primary and metastatic lesions in different cancers[55,56,58]. Because conventional molecular analysis of whole tumors provides genotype/phenotype information of the dominant clones or aggregated information of all clones, single-cell analysis of the CTCs is a potential solution to investigate heterogeneity. By isolating and sequencing single CTCs in the blood, it is possible to measure somatic mutations that are present at both the primary and metastatic tumor sites without performing an invasive core biopsy[59,60]. Two types of isolation methods - microfluidic-based and immunoaffinity-based are used for capturing CTCs.
Microfluidic-based cell isolation
The microfluidic platform can be used for single-step isolation of CTCs from unprocessed blood specimens[61,62]. As whole blood flows through the CTC-chip, individual CTCs are captured onto the microposts coated with anti-EpCAM antibody. This type of microfluidic processing enables high yield of pure CTCs[63]. Subsequent studies demonstrated the ability and reliability to isolate CTCs from patients with metastatic lung cancer using this CTC-chip to perform an EGFR mutational analysis[63]. An improved microfluidic CTC isolation platform, the herringbone (HB)-chip, is also developed by the same group[64]. The HB-chip uses calibrated microfluidic flow patterns to drive cells to come in contact with the antibody-coated walls of the device, thereby reducing cell collisions and improving target cell capture efficiency. A commercial microfluidic circuitry chip DEPArray System (Menarini Silicon Biosystems, Inc.) containing an array of individually controllable electrodes to create a dielectrophoretic (DEP) cage around each cell for single CTC isolation is also available[65]. Besides isolation of CTCs from blood, the microfluidic platform can also be used for single-cell isolation from other tissues[66,67]. For example, an innovative workflow using DEPArray system was established to examine tumor heterogeneity using FFPE samples, providing a solution for genetic analysis using minute archival clinical samples[68].
Immunoaffinity-based cell isolation
The CellSearch Circulating Tumor Cell Kit (Menarini Silicon Biosystems, Inc.) is based on ferrofluid- and fluorochrome-couple antibodies with high binding affinities for the EpCAM antigen of CTCs. After immunomagnetic capture and enrichment, CTCs in peripheral blood are detected and enumerated as measured by fluorescence intensity. ITH has been reported for PIK3CA and TP53 mutations in metastatic breast cancer using a combination of CellSearch and DEPArray technologies[69,70]. CTCs can also be purified and enriched using an immunomagnetic enrichment device termed MagSweeper[71]. Using this technique, high level of heterogeneity among individual CTCs was detected in the blood of metastatic breast cancer patients[72].
Isolation of single cells using Fluorescence-activated cell sorting
Flow cytometry using fluorescence-activated cell sorting is a powerful method of isolating single cells that share the same marker from liquid suspensions. Cells passing through the lasers emit optical signals enabling their separation and capture from other cells that lack the signal[73,74]. Single cells can be sorted individually onto a 96 well plate format [Figure 4B]. Alternatively, a serial dilution can be performed using the sorted cell suspensions into a 96 well plate such that each well contains a single cell. Downstream sequencing can be performed using a 96 well plate format.
Isolated single cells can be interrogated by a variety of genomic technologies for deeper genotype-phenotype characterization. Significant technological advancement summarized in the next section is producing novel insights into the biology of the disease and applications in the clinic.
Downstream analysis of single cells
Single-cell genomics
The work-flow of single-cell sequencing involves amplification of genomic DNA or RNA transcripts to produce enough material for library construction. The earliest method of sequencing DNA from single-cells combined flow-sorting cells by DNA ploidy followed by single-nucleus sequencing by degenerative-oligonucleotide-PCR technique[74,75]. However, this method failed to generate genome-wide single nucleotide variants due to low coverage of ~6%[74,75]. A non-PCR-based multiple-displacement DNA amplification method using Phi29 enzyme and random hexamers [Table 1] produced good genome coverage with high sequence fidelity in multiple single-cell studies[58,76-79]. Another amplification method - multiple annealing and looping-based amplification cycles (MALBAC) reduced whole-genome amplification bias and improved genome coverage [Table 1]. In the MALBAC method, limited isothermal amplification using degenerate primers, followed by PCR amplification produced 93% genome coverage for a single cell and both copy-number variations and single nucleotide variations were detected[80]. Amplification bias is a serious limitation in single-cell sequencing, which can reduce the accuracy of genomic information from single-cell genomes[81]. Statistical models have been developed to calibrate allelic bias in single-cell whole-genome amplification to reduce the sequencing artifacts[81].
Techniques for single-cell analysis
| Methods | Example | Advantage | Disadvantage | Ref. | |
|---|---|---|---|---|---|
| Genome | DOP-PCR | High-throughput, high coverage | Amplification bias, allelic dropout | [74,75] | |
| MDA | High-throughput, even coverage | Amplification bias, allelic dropout | [58,76-79] | ||
| MALBAC | High-throughput, even coverage | Amplification bias, allelic dropout | [80] | ||
| Transcriptome | MMLV | Smart-seq | Full-length transcript, amplify quickly | Weak 3’ bias | [83,84] |
| IVT | CEL-Seq | Full-length transcript, specificity, ratio fidelity | 3’ bias, low efficiency | [87,88] | |
| Phi29 DNA polymerase | Full-length transcript, high efficiency, low bias | No strand specificity | [89,90] |
Single-cell transcriptomics
The first study of single-cell RNA transcriptome of mouse blastomere detected novel splice junctions and expression of more genes than previous microarray studies[82]. However, this method was found to have a strong 3’ bias due to the inefficiency of first-strand cDNA synthesis by reverse transcriptase. To overcome this problem, Smart-seq technique was developed using MMLV reverse transcriptase with template switching activity [Table 1][83,84]. This Smart-seq method utilizes an intrinsic property of MMLV to add three to four cytosines specifically to the 3’ end of the first cDNA strand, which is subsequently used to anchor a universal PCR primer for amplification[85]. In a single-cell RNA-seq of CTCs from melanoma patients, Smart-seq has improved read coverage across transcripts despite increased noise in gene expression estimates[83]. Moreover, distinct gene expression patterns including candidate biomarkers for melanoma CTCs were reported in this study[83].
In vitro transcription (IVT) -based linear RNA amplification uses T7 RNA polymerase to produce transcripts with high specificity and low error rate [Table 1], it has the drawback of lower efficiency and is biased towards the 3’ end of input transcripts[86]. CEL-Seq method of pooling cells and libraries reduced some of the limitations of IVT and was used to capture differential gene expression in two-cell stage embryo of C. elegans[87,88].
The third strategy used Phi29 DNA polymerase for cDNA library generation from single cells [Table 1][89,90]. RNA is reverse transcribed, circularized and then amplified using Phi29 polymerase which preserves full-length transcript coverage. Additionally, random primers can be incorporated to generate cDNA, making this method suitable for prokaryotes[89].
A combined method of single-cell isolation and single-cell sequencing
Microfluidic devices for single-cell isolation coupled with single-cell RT-qPCR or whole transcriptome has been developed by multiple groups[91-93]. A good example is a microfluidic device developed by White et al.[94,95] capable of performing high precision RT-qPCR measurements of gene expression from hundreds of single cells per run. This device combines cell loading, cell lysis, reverse transcription and quantitative PCR in one cell processing unit [Figure 4Ci][94,95]. Once cells are loaded, a single cell is trapped in a cell capture chamber[Figure 4Ci][94,95]. After cell lysis, the transcript target is reverse transcribed before being injected into the PCR chamber[94]. Master mixes for RT and qPCR are loaded onto the common feed channel sequentially to enable each reaction step. A similar device, featuring additional cell processing chambers and sample elution capabilities has been released as a commercial product (Fluidigm C1) in 2012. Since then, an increasing number of studies investigated ITH using Fluidigm’s microfluidic device[96-98].
Efforts to reduce amplification bias by incorporating unique molecular identifiers before transcriptome amplification are ongoing[99]. A novel technique termed Drop-seq uses the microfluidic chamber to isolate single cells followed by labeling RNA of individual cells with a different barcode, allowing pooling of cDNA during sequencing thereby greatly improving the multiplexing efficiency[100]. Applying Drop-seq to mouse retinal bipolar cells resulted in the identification of different types of neurons by matching molecular expression to cell morphology[101]. A similar technique was commercialized by 10× Genomics Inc [Figure 4Cii] in 2016. The 10x platform applies unique barcodes to separately index each cell by partitioning thousands of cells into Gel Bead-in-Emulsions. Libraries are generated and sequenced and the 10x barcodes are used to associate individual reads back to the individual cells. The platform can profile up to 10,000 cells from a complex mixture of different cell types.
Applications of single-cell sequencing
Recent technical advances have enabled generation of unprecedented amount of information on genomics and transcriptomics at the single-cell level [Table 2]. Compared to bulk transcriptomics data obtained from tumor tissues, single-cell RNA-seq allows capturing of the gene expression profile from individual cells of heterogenous origin, which is a significant advantage over bulk sequencing that captures the average gene expression of a sample. Secondly, for the samples with limited amount of material, single-cell analysis is a good alternative to characterize the genotype. Taking CTCs for an example, mutations identified in CTCs are also present in the primary tumor and may be found in the metastatic lesions[55], suggesting that single-cell analysis on CTCs is an effective option to non-invasively monitor cancer progression and predict metastatic risk. Last but not the least, single-cell analysis facilitates researchers to dissect tumor heterogeneity at a much higher resolution than before. For example, the degree of karyotypic anomalies in human cancer is associated with tumor progression and therapeutic response to cancer treatment[102]. However, current karyotypic analysis methods rely on a small fraction of dividing mitotic subpopulations in the sample and do not provide in-depth information on copy number variations (CNV)[102,103]. Single-cell whole genome sequencing offers a significant advantage over traditional methods in analyzing karyotypic anomalies and CNVs at a much higher resolution.
Overview of single-cell studies on analyzing ITH
| Tumor type | Sample type | Method | Description | Ref. |
|---|---|---|---|---|
| Colorectal cancer | CTC | DNA-seq | mutation profiling, clonal evolution | [55] |
| Prostate cancer | CTC | DNA-seq | genetic lineage | [58] |
| Breast cancer | CTC | RNA-seq | transcriptome profiling | [72] |
| Breast cancer | primary tumor | DNA-seq | clonal diversity | [75] |
| Melanoma | CTC | RNA-seq | transcriptome profiling | [83] |
| Leukemia | primary tumor | DNA-seq | mutation profiling, clonal evolution | [97] |
| Glioblastoma multiforme | primary tumor | RNA-seq | clonal evolution | [106] |
| Acute myeloid leukemia | primary tumor | DNA-seq | mutation profiling, clonal evolution | [105] |
| Breast cancer | primary tumor | DNA-seq | copy number evolution, clonal evolution | [74] |
| Breast cancer | primary tumor | DNA-seq | copy number evolution, clonal evolution | [77] |
| Acute myeloid leukemia | primary tumor | DNA-seq | clonal evolution | [109] |
| Kidney cancer | primary tumor | DNA-seq | mutation profiling | [76] |
| Bladder cancer | primary tumor | DNA-seq | mutation profiling, clonal evolution | [110] |
| Colon cancer | primary tumor | DNA-seq | clonal evolution | [111] |
| Acute myeloid leukemia | primary tumor | DNA-seq | clonal evolution | [112] |
| Chronic lymphocytic leukemia | primary tumor | DNA-seq, RNA-seq | genotype-phenotype relationship clonal evolution, mutation profiling | [113] |
| Lung cancer | CTC | DNA-seq | copy number evolution | [56] |
| Pancreatic ductal adenocarcinoma | CTC | RNA-seq | phenotype characterization | [115] |
| Glioblastoma | primary tumor | RNA-seq | transcriptional profiling, phenotype characterization | [43] |
| Glioblastoma | primary tumor | DNA-seq | EGFR evolution | [116] |
| B cell leukemia | primary tumor | DNA-seq | karyotype heterogeneity | [117] |
| Myeloproliferative neoplasm | primary tumor | DNA-seq | mutation profiling, clonal evolution | [78] |
| Melanoma | CTC | DNA-seq | mutation profiling, copy number evolution | [118] |
| Breast cancer | CTC | RNA-seq | transcriptome profiling | [120] |
| Various cancers | primary tumor | RNA-seq | TCR repertoire analysis | [124,126] |
| Liver cancer | primary tumor | RNA-seq | characterization of T cell functional states | [130] |
| Breast cancer | primary tumor | RNA-seq | tumor microenvironment characterization | [132] |
| Prostate cancer | CTC | RNA-seq | heterogeneity in signaling pathways | [136] |
| Prostate cancer | CTC | DNA-seq | copy number evolution | [137] |
| Breast cancer | primary tumor | DNA-seq, RNA-seq | clonal evolution, transcriptome profiling | [32] |
Understanding tumor evolution
Tumor evolution is a dynamic process and describes the emergence of cancer cell subpopulations under environmental pressure. As the tumor grows, each generation of cells acquire novel somatic mutations that provide cells with survival advantages thereby determining the overall fitness of the clonal population[104]. Waves of clonal expansion and contraction driven by changes in the tumor microenvironment govern the life cycle of a tumor. Single-cell sequencing can potentially identify low abundance clones carrying driver mutations, which can be further leveraged to refine therapeutic strategies. Although low abundance driver mutations are possible to detect by deep exome sequencing, the fraction of cells carrying the mutation, or the zygosity of the change (relevant for loss of function mutations in tumor suppressor genes) are hard to estimate without single cell sequencing. A computational approach to map single-cell mutational profile from exome sequencing was successfully used to chart the chronological acquisition of mutations and create a phylogenic map of tumor evolution in both glioblastoma multiforme and secondary acute myeloid leukemia (AML)[105,106]. A similar analysis in breast cancer identified three clonal populations in the primary tumor of which only one clone was present in the metastatic lesion[74]. This observation supports the hypothesis that rare clones present in the primary tumor harbor genetic signatures of metastasis even before they have spread and colonized distant sites[74,107,108]. In a follow-up breast cancer study, aneuploidy rearrangements were shown to occur early in tumor evolution, which remained highly stable as the tumor grew, whereas, point mutations generated clonal diversity[77]. A similar pattern is observed in lymphoblastic leukemia patients where recurrent translocations appear earlier than structural nucleotide variants[109]. This suggests that large structural alterations offer selective advantage early during tumor growth followed by accumulation of mutations producing clonal diversity. This is supported by the finding that subclonal populations arise more frequently in tumors with high mutational burdens such as bladder and colon cancer, but not in tumors with low mutational burden such as renal cell carcinoma[76,110,111]. A clonal progression of multiple mutations was mapped in hematopoietic stem cells of AML patients, suggesting the clonal evolution of AML genomes from founder mutations[112]. An interesting finding from single-cell analysis is that phenotypic diversity fails to recapitulate genotypic diversity detected in subclones strongly implicating that a large proportion of genotypic variation may lack functional consequences, appearing and disappearing without contributing to tumor evolution[113].
Disease diagnosis and therapeutic stratification of patients
Modern cancer treatment relies heavily on accurate molecular and immuno/histopathological tissue analysis of needle biopsies or surgically resected tissues for diagnosis. Tumor heterogeneity often confounds accuracy of disease diagnostics by subsampling a subset of tumor cells that may not represent the whole tumor. This calls for obtaining multiregional and longitudinal samples to guide therapeutic intervention, which is often not routine. High-resolution single-cell analysis of tumor samples or CTCs can aid in refining diagnostic parameters and patient stratification.
In a single-cell sequencing study of CTCs from metastatic lung cancer, patients who share the same subtype of lung cancer displayed similar patterns of copy number variations in their CTCs, providing a potential biomarker of CTC-based cancer diagnostics[56]. In pancreatic cancer, pancreas epithelial cells can be present in the blood at pre-cancerous stages in pancreatic ductal adenocarcinoma patients[114]. In another study, single-cell sequencing analysis on CTCs obtained from pancreatic ductal adenocarcinoma patients identified a macrophage-pancreatic tumor cell fusion product with high proliferative and metastatic potential[115]. These studies suggest that early detection of these pancreatic epithelial cells in the blood stream can serve as an important diagnostic tool for pancreatic cancer detection[114,115].
The treatment of glioblastomas, an aggressive type of brain tumor has benefited from single-cell sequencing because of a high degree of tumor heterogeneity harboring a diverse population of cells with a large spectrum of stemness, differentiation states, and variable proliferative capacity[43]. By applying single-cell sequencing to EGFR-amplified glioblastomas, novel EGFR truncation variants were identified[116]. In vitro and in vivo functional studies revealed that a specific EGFR variant (EGFRvII, deletion of exons 14 and 15) was sensitive to EGFR inhibitors, which are currently in clinical trials[116]. In chromosomally unstable B cell leukemia patients, different degrees of karyotypic abnormalities were detected by single-cell whole genome sequencing, which bulk sequencing failed to detect. Because karyotypic abnormalities associate with poor clinical outcome in multiple cancers[102], the degree of karyotypic anomalies assessed by single-cell sequencing can be utilized as an important readout for stratifying patient risk[117]. Single-cell analysis has identified novel mutations in JAK2-negative myeloproliferative neoplasm such as SESN2 and NTRK1, chronic lymphocytic leukemia such as LCP1 and WNK1 and chromosomal abnormalities in melanoma such as chromosomal 12 amplification[78,113,118], opening up opportunities to target these neoplasms. For example, NTRK1 encodes a tyrosine kinase receptor and inhibitors are available to target its NTRK1 gene fusions that results in constitutive activation of the kinase[119]. For patients who are JAK2 mutation negative but harbor NTRK1 mutation, it is tempting to speculate that NTRK1 can be a target for the treatment of myeloproliferative neoplasm.
Disease monitoring and prognostic biomarkers
Cancer heterogeneity in part is driven by selection pressure that arises during drug treatment. Capturing this dynamic heterogeneity at the genetic and cellular composition level prior to, during, or post-treatment is crucial in assessing drug efficacy and predicting patient survival. Single-cell analysis is an extremely powerful tool to capture the dynamic events at a molecular level for disease monitoring and in predicting prognostic biomarkers. Below are few examples of the application of single-cell sequencing in developing prognostic and predictive biomarkers.
CTC analysis
Single-cell analysis of CTCs can provide prognostic markers in several cancers. Microfluidics-based RNA sequencing has aided identification of CTC clusters held together by the cell junction component plakoglobin that mediate intercellular adhesion. Presence of high levels of CTC clusters over single CTCs correlated with poor prognosis indicating their role in the metastatic spread of cancer[120]. Indeed, heterogonous expression of plakoglobin in the primary tumor supports the evidence that tightly adhered groups of cells from the primary tumors serve as the precursors to CTC clusters in circulation. Thus, single-cell identification of plakoglobin-positive clonal cell populations of tumor cells in conjunction with the presence of CTC clusters in the patient blood is a potent prognostic marker of breast cancer metastasis[120].
TCR repertoire analysis
Anti-tumor immunity is largely driven by antigen-specific CD8 T cells, which recognize tumor-derived neoantigenic peptides complexed with human leukocyte antigen also referred to as major histocompatibility complex (MHC) in mouse, to mount an anti-tumor immune response[121]. Adoptive cell therapy using autologous tumor infiltrating lymphocytes (TILs) has been shown to be effective for the treatment of multiple cancers[122,123]. The anti-tumor effects observed post T cell therapy are associated with the activation of neoantigen reactive T cells[122]. To improve the efficacy of the T cell therapy, engineering TILs to express the neoantigen-specific TCR can be a promising next-generation immunotherapy drug[124]. However, to develop these engineered T cells, identifying paired sequences of both TCR a and b chains from the vast repertoire of TCRs is a challenge. One way to overcome this challenge is to perform, single-cell TCR profiling to obtain paired TCR α/β sequence information[125]. Using patient samples, neoantigen specific CD8 T cells were clonally expanded in vitro and multiple paired TCR sequences were identified by single-cell analysis[124]. Importantly, the transduced T cells expressing TCRs recognized the neoantigen presented by autologous antigen-presenting cells[124]. Another study using single-cell TCR repertoire analysis revealed that clonally expanded CD8 T cells were antigen-specific and showed cytotoxic activity against tumors in mouse models[126]. Intriguingly, the combination of 10x Genomics’ single cell TCR sequencing platform coupled to gene expression holds enormous potential for assessing and monitoring patient response to cancer vaccines and immunotherapy drugs.
Monitoring the functional state of CD8 T cells
In the tumor microenvironment, the ability of CD8 T cells to secrete pro-inflammatory cytokines and exert cytotoxic function can be compromised during persistent immune activation[127]. Such exhausted CD8 T cells differ profoundly from memory CD8 T cells and co-express multiple co-inhibitory immune checkpoint regulators such as PD-1, LAG-3, and TIM-3 and lack successful anti-tumor immune response[127,128]. Even though various checkpoint inhibitors show clinical efficacy by unleashing cytotoxic T cells activity, a large fraction of patients fails to respond to these immunotherapies[129]. Therefore, a detailed understanding of the mechanisms of CD8 T cell exhaustion is required. Further, since the transcriptional signatures of T cell exhaustion are closely intertwined with their activated T cell state, single-cell analysis is an optimal approach to identify biomarkers specific to T cell dysfunction. In a single-cell RNA-seq analysis of T cells from hepatocellular carcinoma patients, 11 unique T cell subsets were identified based on their molecular and functional properties[130]. Exhaustion signature gene LAYN was identified and associated with inhibition of IFN-g production[130]. A single-cell RNA-seq of CD8 tumor-infiltrating lymphocytes from murine tumor models has also aided identification of novel molecular pathways of T cell exhaustion that is uncoupled from T cell activation[131].
Profiling of immune suppressive cell types present in the tumor microenvironment
Single-cell transcriptome profiling enables characterization of the complex tumor microenvironment with its heterogeneous mixture of tumor cells along with stromal and immune cells[132]. Targeting of immunosuppressive cell types in the tumor microenvironment can sometimes be key to the efficacy of checkpoint inhibitors such as anti-CTLA-4 therapy. A variety of cell types including T regulatory cells (Tregs), tumor-associated macrophages, type 2 NKT cells, M2 macrophages and MDSCs enforce immune suppression in the tumor helping tumor cells to survive anti-tumor immune attack[133]. Identifying MDSCs has been challenging from bulk sequencing data due to the absence of unique MDSC markers. In addition, the presence of over 10 different myeloid subsets further complicates bioinformatics analysis[134]. Tregs are potent immune modulators and assessing their frequency, phenotype, and function at tissue sites has been profoundly challenging due to the fact that majority of the defining markers like CD25, FOXP3 and CTLA4 are also present in effector T cells[135]. Single-cell analysis of tumor infiltrated immune cells can help circumvent some of these hurdles in tumor characterization. In a recent single-cell analysis study tumor cells from 11 breast cancer patients, cancer cells were separated from immune cells based on their copy number variations[132]. Analysis of the immune cell fraction revealed the presence of immunosuppressive macrophages of M2 phenotype and activated T effector cells. Interestingly, the T cells also expressed markers of T cell exhaustion such LAG3 and TIGIT suggesting that they could be targeted by immune checkpoint inhibitors[132].
Understanding mechanisms of disease resistance
Resistance to chemotherapy and molecularly targeted therapies is a major barrier to achieving long-term benefit to treatment. ITH arising from diverse cell subpopulations with distinct molecular features produce varying levels of drug sensitivity and resistance[16]. Retrospective analysis of CTCs from patients who had developed resistance to inhibitors of the androgen receptor (AR) showed higher activation of non-canonical Wnt pathway beside altered expression and mutations in AR compared to untreated patients[136,137]. In castrate-resistant prostate cancers high content single-cell longitudinal profiling of CTCs from a patient undergoing chemotherapy and targeted therapy revealed a selective clonal expansion of cells with AR amplification supporting the adaptive model of therapy resistance evolution[137]. Similar observation of selective clonal persistence was seen in breast cancer patients treated with chemotherapy. In this study, single-cell sequencing post-chemotherapy revealed transcriptional reprogramming of resistant signatures, elucidating the mechanism of therapy resistance[32].
Based on aforementioned studies, an accurate assessment of ITH by single-cell sequencing using multiregional, longitudinal sampling is essential to understand the mechanism of drug resistance and facilitate the development of more effective therapies.
Future directions
With the development of precision microfluidic devices and sequencing technologies, single-cell analysis has transformed our understanding of ITH and clonal evolution. Single-cell genomics promises to deconvolute complex biological processes in cancer, reveal epigenetic alterations and monitor the evolution of metastatic and treatment resistance clones. By applying single-cell sequencing to different experimental systems, such as cells in culture, patient-derived xenografts, murine models and analysis of human tumors, novel diagnostics and therapies can be developed. A major hurdle in single-cell sequencing is the high cost of the technology. Moreover, the volume and complexity of single-cell sequencing datasets exceed that of the traditional bulk sequencing, calling for better statistical algorithms to deconvolute the data. Additional caution should be given on the transcriptome coverage and number of cells taken for single-cell analysis to ensure the accuracy of gene expression distribution estimates. Future breakthroughs in developing cost-effective sequencing methods and powerful data analysis pipeline for single-cell sequencing are likely to expand the scope of this technology beyond cancer to other diseases.
Declarations
Authors’ contributionsManuscript drafting: Shi X, Chakraborty P, Chaudhuri A
Availability of data and materialsNot applicable.
Financial support and sponsorshipThis work was supported by MedGenome Inc.
Conflicts of interestAll authors are full-time employees of MedGenome Inc.
Ethical approval and consent to participateNot applicable.
Consent for publicationNot applicable.
Copyright© The Author(s) 2018.
REFERENCES
1. Shah SP, Roth A, Goya R, Oloumi A, Ha G, Zhao Y, Turashvili G, Ding J, Tse K, Haffari G, Bashashati A, Prentice LM, Khattra J, Burleigh A, Yap D, Bernard V, McPherson A, Shumansky K, Crisan A, Giuliany R, Heravi-Moussavi A, Rosner J, Lai D, Birol I, Varhol R, Tam A, Dhalla N, Zeng T, Ma K, Chan SK, Griffith M, Moradian A, Cheng SWG, Morin GB, Watson P, Gelmon K, Chia S, Chin SF, Curtis C, Rueda OM, Pharoah PD, Damaraju S, Mackey J, Hoon K, Harkins T, Tadigotla V, Sigaroudinia M, Gascard P, Tlsty T, Costello JF, Meyer IM, Eaves CJ, Wasserman WW, Jones S, Huntsman D, Hirst M, Caldas C, Marra MA, Aparicio S. The clonal and mutational evolution spectrum of primary triple-negative breast cancers. Nature 2012;486:395.
2. Gerlinger M, Rowan AJ, Horswell S, Larkin J, Endesfelder D, Gronroos E, Martinez P, Matthews N, Stewart A, Tarpey P, Varela I, Phillimore B, Begum S, McDonald NQ, Butler A, Jones D, Raine K, Latimer C, Santos CR, Nohadani M, Eklund AC, Spencer-Dene B, Clark G, Pickering L, Stamp G, Gore M, Szallasi Z, Downward J, Futreal PA, Swanton C. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. New Engl J Med 2012;366:883-92.
3. Davis A, Gao R, Navin N. Tumor evolution: Linear, branching, neutral or punctuated? Biochim Biophys Acta, Rev Cancer 2017;1867:151-61.
4. McGranahan N, Swanton C. Biological and therapeutic impact of intratumor heterogeneity in cancer evolution. Cancer Cell 2015;27:15-26.
5. Fidler IJ, Hart IR. Biological diversity in metastatic neoplasms: origins and implications. Science 1982;217:998-1003.
6. Friedl P, Alexander S. Cancer invasion and the microenvironment: plasticity and reciprocity. Cell 2011;147:992-1009.
7. McGranahan N, Swanton C. Clonal heterogeneity and tumor evolution: past, present, and the future. Cell 2017;168:613-28.
8. Swanton C. Intratumor heterogeneity: evolution through space and time. Cancer Res 2012;72:4875-82.
9. Jamal-Hanjani M, Thanopoulou E, Peggs KS, Quezada SA, Swanton C. Tumour heterogeneity and immune-modulation. Curr Opin Pharm 2013;13:497-503.
10. Nassar A, Radhakrishnan A, Cabrero IA, Cotsonis GA, Cohen C. Intratumoral heterogeneity of immunohistochemical marker expression in breast carcinoma: a tissue microarray-based study. Appl Immunohistochem Mol Morphol 2010;18:433-41.
11. Negrini S, Gorgoulis VG, Halazonetis TD. Genomic instability—an evolving hallmark of cancer. Nat Rev Mol Cell Biol 2010;11:220.
13. Caswell DR, Swanton C. The role of tumour heterogeneity and clonal cooperativity in metastasis, immune evasion and clinical outcome. BMC Med 2017;15:133.
15. Venkatesan S, Swanton C, Taylor BS, Costello JF. Treatment-induced mutagenesis and selective pressures sculpt cancer evolution. Cold Spring Harb Perspect Med 2017;7:a026617.
16. Dagogo-Jack I, Shaw AT. Tumour heterogeneity and resistance to cancer therapies. Nat Rev Clin Oncol 2017;15:81.
17. Turajlic S, Xu H, Litchfield K, Rowan A, Chambers T, Lopez JI, Nicol D, O'Brien T, Larkin J, Horswell S, Stares M, Au L, Jamal-Hanjani M, Challacombe B, Chandra A, Hazell S, Eichler-Jonsson C, Soultati A, Chowdhury S, Rudman S, Lynch J, Fernando A, Stamp G, Nye E, Jabbar F, Spain L, Lall S, Guarch R, Falzon M, Proctor I, Pickering L, Gore M, Watkins TBK, Ward S, Stewart A, DiNatale R, Becerra MF, Reznik E, Hsieh JJ, Richmond TA, Mayhew GF, Hill SM, McNally CD, Jones C, Rosenbaum H, Stanislaw S, Burgess DL, Alexander NR, Swanton C. Tracking cancer evolution reveals constrained routes to metastases: TRACERx Renal. Cell 2018;173:581-94.e12.
18. Wilting RH, Dannenberg JH. Epigenetic mechanisms in tumorigenesis, tumor cell heterogeneity and drug resistance. Drug Resist Updat 2012;15:21-38.
19. Brakensiek K, Wingen LU, Länger F, Kreipe H, Lehmann U. Quantitative high-resolution CpG island mapping with pyrosequencing TM reveals disease-specific methylation patterns of the CDKN2B gene in myelodysplastic syndrome and myeloid leukemia. Clin Chem 2006; doi: 10.1373/clinchem.2007.072629.
20. Candiloro ILM, Mikeska T, Hokland P, Dobrovic A. Rapid analysis of heterogeneously methylated DNA using digital methylation-sensitive high resolution melting: application to the CDKN2B (p15) gene. Epigenetics & Chromatin 2008;1:7.
21. Petrovic N, Ergün S, Isenovic ER. Levels of microRNA heterogeneity in cancer biology. Mol Diagn Ther 2017;21:511-23.
22. Zedan AH, Blavnsfeldt SG, Hansen TF, Nielsen BS, Marcussen N, Pleckaitis M, Osther PJS, Sørensen FB. Heterogeneity of miRNA expression in localized prostate cancer with clinicopathological correlations. PLoS One 2017;12:e0179113.
23. Li H, Fan X, Houghton J. Tumor microenvironment: the role of the tumor stroma in cancer. J Cell Biochem 2007;101:805-15.
24. Kumar V, Patel S, Tcyganov E, Gabrilovich DI. The nature of myeloid-derived suppressor cells in the tumor microenvironment. Trends Immunol 2016;37:208-20.
25. Joyce JA, Fearon DT. T cell exclusion, immune privilege, and the tumor microenvironment. Science 2015;348:74-80.
26. Tlsty TD, Coussens LM. Tumor stroma and regulation of cancer development. Annu Rev Pathol Mech Dis 2006;1:119-50.
27. Hanahan D, Coussens LM. Accessories to the crime: functions of cells recruited to the tumor microenvironment. Cancer Cell 2012;21:309-22.
28. Gainor JF, Dardaei L, Yoda S, Friboulet L, Leshchiner I, Katayama R, Dagogo-Jack I, Gadgeel S, Schultz K, Singh M, Chin E, Parks M, Lee D, DiCecca RH, Lockerman E, Huynh T, Logan J, Ritterhouse LL, Le LP, Muniappan A, Digumarthy S, Channick C, Keyes C, Getz G, Dias-Santagata D, Heist RS, Lennerz J, Sequist LV, Benes CH, Iafrate AJ, Mino-Kenudson M, Engelman JA, Shaw AT. Molecular mechanisms of resistance to first- and second-generation ALK inhibitors in ALK-rearranged lung cancer. Cancer Discovery 2016;6:1118-33.
29. Pao W, Miller VA, Politi KA, Riely GJ, Somwar R, Zakowski MF, Kris MG, Varmus H. Acquired resistance of lung adenocarcinomas to gefitinib or erlotinib is associated with a second mutation in the EGFR kinase domain. PLoS Med 2005;2:e73.
30. Engelman JA, Zejnullahu K, Mitsudomi T, Song Y, Hyland C, Park JO, Lindeman N, Gale CM, Zhao X, Christensen J, Kosaka T, Holmes AJ, Rogers AM, Cappuzzo F, Mok T, Lee C, Johnson BE, Cantley LC, Jänne PA. MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science 2007;316:1039-43.
31. Eng J, Woo KM, Sima CS, Plodkowski A, Hellmann MD, Chaft JE, Kris MG, Arcila ME, Ladanyi M, Drilon A. Impact of concurrent PIK3CA mutations on response to EGFR tyrosine kinase inhibition in EGFR-mutant lung cancers and on prognosis in oncogene-driven lung adenocarcinomas. J Thorac Oncol 2015;10:1713-9.
32. Kim C, Gao R, Sei E, Brandt R, Hartman J, Hatschek T, Crosetto N, Foukakis T, Navin NE. Chemoresistance evolution in triple-negative breast cancer delineated by single-cell sequencing. Cell 2018;173:879-93.e13.
33. Turner NC, Reis-Filho JS. Genetic heterogeneity and cancer drug resistance. Lancet Oncol 2012;13:e178-85.
34. Danielsen HE, Pradhan M, Novelli M. Revisiting tumour aneuploidy—the place of ploidy assessment in the molecular era. Nat Rev Clin Oncol 2016;13:291.
35. Cyll K, Ersvær E, Vlatkovic L, Pradhan M, Kildal W, Avranden Kjær M, Kleppe A, Hveem TS, Carlsen B, Gill S, Löffeler S, Haug ES, Wæhre H, Sooriakumaran P, Danielsen HE. Tumour heterogeneity poses a significant challenge to cancer biomarker research. Br J Cancer 2017;117:367.
36. Wei SH, Chen CM, Strathdee G, Harnsomburana J, Shyu CR, Rahmatpanah F, Shi H, Ng SW, Yan PS, Nephew KP, Brown R, Huang THM. Methylation microarray analysis of late-stage ovarian carcinomas distinguishes progression-free survival in patients and identifies candidate epigenetic markers. Clin Cancer Res 2002;8:2246-52.
37. Wei SH, Balch C, Paik HH, Kim YS, Baldwin RL, Liyanarachchi S, Li L, Wang Z, Wan JC, Davuluri RV, Karlan BY, Gifford G, Brown R, Kim S, Huang THM, Nephew KP. Prognostic DNA methylation biomarkers in ovarian cancer. Clin Cancer Res 2006;12:2788-94.
38. Rajaram S, Heinrich LE, Gordan JD, Avva J, Bonness KM, Witkiewicz AK, Malter JS, Atreya CE, Warren RS, Wu LF, Altschuler SJ. Sampling strategies to capture single-cell heterogeneity. Nat Methods 2017;14:967.
39. Foley JW, Zhu C, Jolivet P, Zhu SX, Lu P, Meaney MJ, West RB. Gene-expression profiling of single cells from archival tissue with laser-capture microdissection and Smart-3SEQ. bioRxiv 2018; doi: 10.1101/207340.
40. Hayashida Y, Partida GJ, Ishida AT. Dissociation of retinal ganglion cells without enzymes. J Neurosci Methods 2004;137:25-35.
41. Gomez GG, Kruse CA. Isolation and culture of human brain tumor cells. Cancer Cell Culture. Methods Mol Med: Springer; 2004. pp. 101-9.
42. Louis SA, Mak CK, Reynolds BA. Methods to culture, differentiate, and characterize neural stem cells from the adult and embryonic mouse central nervous system. Methods Mol Med: Springer; 2013. pp. 479-506.
43. Patel AP, Tirosh I, Trombetta JJ, Shalek AK, Gillespie SM, Wakimoto H, Cahill DP, Nahed BV, Curry WT, Martuza RL, Louis DN, Rozenblatt-Rosen O, Suvà ML, Regev A, Bernstein BE. Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science 2014;344:1396-401.
44. Yan Y, Xu Y, Gao YY, Zong ZH, Zhang Q, Li C, Wang HQ. Implication of 14-3-3ε and 14-3-3θ/τ in proteasome inhibition-induced apoptosis of glioma cells. Cancer Sci 2013;104:55-61.
45. Panchision DM, Chen HL, Pistollato F, Papini D, Ni HT, Hawley TS. Optimized flow cytometric analysis of central nervous system tissue reveals novel functional relationships among cells expressing CD133, CD15, and CD24. Stem Cells 2007;25:1560-70.
46. Galli R, Binda E, Orfanelli U, Cipelletti B, Gritti A, De Vitis S, Fiocco R, Foroni C, Dimeco F, Vescovi A. Isolation and characterization of tumorigenic, stem-like neural precursors from human glioblastoma. Cancer Res 2004;64:7011-21.
47. Singh SK, Hawkins C, Clarke ID, Squire JA, Bayani J, Hide T, Henkelman RM, Cusimano MD, Dirks PB. Identification of human brain tumour initiating cells. Nature 2004;432:396.
48. Wolters GH, Vos-Scheperkeuter GH, Lin HC, van Schilfgaarde R. Different roles of class I and class II Clostridium histolyticum collagenase in rat pancreatic islet isolation. Diabetes 1995;44:227-33.
50. Espina V, Wulfkuhle JD, Calvert VS, VanMeter A, Zhou W, Coukos G, Geho DH, Petricoin III EF, Liotta LA. Laser-capture microdissection. Nat Protoc 2006;1:586.
51. Obiakor H, Sehgal D, Dasso JF, Bonner RF, Malekafzali A, Mage RG. A comparison of hydraulic and laser capture microdissection methods for collection of single B cells, PCR, and sequencing of antibody VDJ. Anal Biochem 2002;306:55-62.
52. Suarez-Quian C, Goldstein S, Pohida T, Smith P, Peterson J, Wellner E, Ghany M, Bonner R. Laser capture microdissection of single cells from complex tissues. BioTechniques 1999;26:328-35.
53. Keays KM, Owens GP, Ritchie AM, Gilden DH, Burgoon MP. Laser capture microdissection and single-cell RT-PCR without RNA purification. J Immunol Methods 2005;302:90-8.
54. Dasgupta A, Lim AR, Ghajar CM. Circulating and disseminated tumor cells: harbingers or initiators of metastasis? Mol Oncol 2017;11:40-61.
55. Heitzer E, Auer M, Gasch C, Pichler M, Ulz P, Hoffmann EM, Lax S, Waldispuehl-Geigl J, Mauermann O, Lackner C, Höfler G, Eisner F, Sill H, Samonigg H, Pantel K, Riethdorf S, Bauernhofer T, Geigl JB, Speicher MR. Complex tumor genomes inferred from single circulating tumor cells by array-CGH and next-generation sequencing. Cancer Res 2013;73:2965-75.
56. Ni X, Zhuo M, Su Z, Duan J, Gao Y, Wang Z, Zong C, Bai H, Chapman AR, Zhao J, Xu L, An T, Ma Q, Wang Y, Wu M, Sun Y, Wang S, Li Z, Yang X, Yong J, Su XD, Lu Y, Bai F, Xie XS, Wang J. Reproducible copy number variation patterns among single circulating tumor cells of lung cancer patients. Proc Natl Acad Sci U S A 2013;110:21083-8.
57. Rantalainen M. Application of single-cell sequencing in human cancer. Brief Funct Genomics 2018;17:273-82.
58. Lohr JG, Adalsteinsson VA, Cibulskis K, Choudhury AD, Rosenberg M, Cruz-Gordillo P, Francis JM, Zhang CZ, Shalek AK, Satija R, Trombetta JJ, Lu D, Tallapragada N, Tahirova N, Kim S, Blumenstiel B, Sougnez C, Lowe A, Wong B, Auclair D, Van Allen EM, Nakabayashi M, Lis RT, Lee GSM, Li T, Chabot MS, Ly A, Taplin ME, Clancy TE, Loda M, Regev A, Meyerson M, Hahn WC, Kantoff PW, Golub TR, Getz G, Boehm JS, Love JC. Whole-exome sequencing of circulating tumor cells provides a window into metastatic prostate cancer. Nat Biotechnol 2014;32:479.
60. Gabriel MT, Calleja LR, Chalopin A, Ory B, Heymann D. Circulating tumor cells: a review of non-EpCAM-based approaches for cell enrichment and isolation. Clin Chem 2016;62:571-81.
61. Nagrath S, Sequist LV, Maheswaran S, Bell DW, Irimia D, Ulkus L, Smith MR, Kwak EL, Digumarthy S, Muzikansky A, Ryan P, Balis UJ, Tompkins RG, Haber DA, Toner M. Isolation of rare circulating tumour cells in cancer patients by microchip technology. Nature 2007;450:1235.
62. Stott SL, Lee RJ, Nagrath S, Yu M, Miyamoto DT, Ulkus L, Inserra EJ, Ulman M, Springer S, Nakamura Z, Moore AL, Tsukrov DI, Kempner ME, Dahl DM, Wu CL, Iafrate AJ, Smith MR, Tompkins RG, Sequist LV, Toner M, Haber DA, Maheswaran S. Isolation and characterization of circulating tumor cells from patients with localized and metastatic prostate cancer. Sci Trans Med 2010;2:25ra3-ra3.
63. Maheswaran S, Sequist LV, Nagrath S, Ulkus L, Brannigan B, Collura CV, Inserra E, Diederichs S, Iafrate AJ, Bell DW, Digumarthy S, Muzikansky A, Irimia D, Settleman J, Tompkins RG, Lynch TJ, Toner M, Haber DA. Detection of mutations in EGFR in circulating lung-cancer cells. New Engl J Med 2008;359:366-77.
64. Stott SL, Hsu CH, Tsukrov DI, Yu M, Miyamoto DT, Waltman BA, Rothenberg SM, Shah AM, Smas ME, Korir GK, Floyd FP, Gilman AJ, Lord JB, Winokur D, Springer S, Irimia D, Nagrath S, Sequist LV, Lee RJ, Isselbacher KJ, Maheswaran S, Haber DA, Toner M. Isolation of circulating tumor cells using a microvortex-generating herringbone-chip. Proc Natl Acad Sci U S A 2010;107:18392-7.
65. Morimoto A, Mogami T, Watanabe M, Iijima K, Akiyama Y, Katayama K, Futami T, Yamamoto N, Sawada T, Koizumi F, Koh Y. High-density dielectrophoretic microwell array for detection, capture, and single-cell analysis of rare tumor cells in peripheral blood. PLoS One 2015;10:e0130418.
66. Lin CH, Chang HC, Hsu CH. A microfluidic platform for high-throughput single-cell Isolation and culture. J Vis Exp 2016.
67. Zhang Q, Wang T, Zhou Q, Zhang P, Gong Y, Gou H, Xu J, Ma B. Development of a facile droplet-based single-cell isolation platform for cultivation and genomic analysis in microorganisms. Sci Rep 2017;7:41192.
68. Bolognesi C, Forcato C, Buson G, Fontana F, Mangano C, Doffini A, Sero V, Lanzellotto R, Signorini G, Calanca A, Sergio M, Romano R, Gianni S, Medoro G, Giorgini G, Morreau H, Barberis M, Corver WE, Manaresi N. Digital sorting of pure cell populations enables unambiguous genetic analysis of heterogeneous formalin-fixed paraffin-embedded tumors by next generation sequencing. Sci Rep 2016;6:20944.
69. Fernandez SV, Bingham C, Fittipaldi P, Austin L, Palazzo J, Palmer G, Alpaugh K, Cristofanilli M. TP53 mutations detected in circulating tumor cells present in the blood of metastatic triple negative breast cancer patients. Breast Cancer Res 2014;16:445.
70. Pestrin M, Salvianti F, Galardi F, De Luca F, Turner N, Malorni L, Pazzagli M, Di Leo A, Pinzani P. Heterogeneity of PIK3CA mutational status at the single cell level in circulating tumor cells from metastatic breast cancer patients. Mol Oncol 2015;9:749-57.
71. Talasaz AH, Powell AA, Huber DE, Berbee JG, Roh KH, Yu W, Xiao W, Davis MM, Pease RF, Mindrinos MN, Jeffrey SS, Davis RW. Isolating highly enriched populations of circulating epithelial cells and other rare cells from blood using a magnetic sweeper device. Proc Natl Acad Sci U S A 2009;106:3970-5.
72. Powell AA, Talasaz AH, Zhang H, Coram MA, Reddy A, Deng G, Telli ML, Advani RH, Carlson RW, Mollick JA, Sheth S, Kurian AW, Ford JM, Stockdale FE, Quake SR, Pease RF, Mindrinos MN, Bhanot G, Dairkee SH, Davis RW, Jeffrey SS. Single cell profiling of circulating tumor cells: transcriptional heterogeneity and diversity from breast cancer cell lines. PLoS One 2012;7:e33788.
73. Hulett HR, Bonner WA, Barrett J, Herzenberg LA. Cell sorting: automated separation of mammalian cells as a function of intracellular fluorescence. Science 1969;166:747-9.
74. Navin N, Kendall J, Troge J, Andrews P, Rodgers L, McIndoo J, Cook K, Stepansky A, Levy D, Esposito D. Tumour evolution inferred by single-cell sequencing. Nature 2011;472:90.
75. Baslan T, Kendall J, Ward B, Cox H, Leotta A, Rodgers L, Riggs M, D'Italia S, Sun G, Yong M, Miskimen K, Gilmore H, Saborowski M, Dimitrova N, Krasnitz A, Harris L, Wigler M, Hicks J. Optimizing sparse sequencing of single cells for highly multiplex copy number profiling. Genome Res 2015;25:714-24.
76. Xu X, Hou Y, Yin X, Bao L, Tang A, Song L, Li F, Tsang S, Wu K, Wu H, He W, Zeng L, Xing M, Wu R, Jiang H, Liu X, Cao D, Guo G, Hu X, Gui Y, Li Z, Xie W, Sun X, Shi M, Cai Z, Wang B, Zhong M, Li J, Lu Z, Gu N, Zhang X, Goodman L, Bolund L, Wang J, Yang H, Kristiansen K, Dean M, Li Y, Wang J. Single-cell exome sequencing reveals single-nucleotide mutation characteristics of a kidney tumor. Cell 2012;148:886-95.
77. Wang Y, Waters J, Leung ML, Unruh A, Roh W, Shi X, Chen K, Scheet P, Vattathil S, Liang H, Multani A, Zhang H, Zhao R, Michor F, Meric-Bernstam F, Navin NE. Clonal evolution in breast cancer revealed by single nucleus genome sequencing. Nature 2014;512:155.
78. Hou Y, Song L, Zhu P, Zhang B, Tao Y, Xu X, Li F, Wu K, Liang J, Shao D, Wu H, Ye X, Ye C, Wu R, Jian M, Chen Y, Xie W, Zhang R, Chen L, Liu X, Yao X, Zheng H, Yu C, Li Q, Gong Z, Mao M, Yang X, Yang L, Li J, Wang W, Lu Z, Gu N, Laurie G, Bolund L, Kristiansen K, Wang J, Yang H, Li Y, Zhang X, Wang J. Single-cell exome sequencing and monoclonal evolution of a JAK2-negative myeloproliferative neoplasm. Cell 2012;148:873-85.
79. Leung ML, Wang Y, Waters J, Navin NE. SNES: single nucleus exome sequencing. Genome Biol 2015;16:55.
80. Zong C, Lu S, Chapman AR, Xie XS. Genome-wide detection of single-nucleotide and copy-number variations of a single human cell. Science 2012;338:1622-6.
81. Zhang CZ, Adalsteinsson VA, Francis J, Cornils H, Jung J, Maire C, Ligon KL, Meyerson M, Love JC. Calibrating genomic and allelic coverage bias in single-cell sequencing. Nat Commun 2015;6:6822.
82. Tang F, Barbacioru C, Wang Y, Nordman E, Lee C, Xu N, Wang X, Bodeau J, Tuch BB, Siddiqui A, Lao K, Surani MA. mRNA-Seq whole-transcriptome analysis of a single cell. Nat Methods 2009;6:377.
83. Ramsköld D, Luo S, Wang YC, Li R, Deng Q, Faridani OR, Daniels GA, Khrebtukova I, Loring JF, Laurent LC, Schroth GP, Sandberg R. Full-Length mRNA-Seq from single cell levels of RNA and individual circulating tumor cells. Nat Biotechnol 2012;30:777-82.
84. Picelli S, Faridani OR, Björklund ÅK, Winberg G, Sagasser S, Sandberg R. Full-length RNA-seq from single cells using Smart-seq2. Nat Protoc 2014;9:171.
85. Zhu Y, Machleder E, Chenchik A, Li R, Siebert P. Reverse transcriptase template switching: A SMART? approach for full-length cDNA library construction. BioTechniques 2001;30:892-7.
86. Morris J, Singh JM, Eberwine JH. Transcriptome analysis of single cells. J Vis Exp 2011;(50):2634.
87. Jaitin DA, Kenigsberg E, Keren-Shaul H, Elefant N, Paul F, Zaretsky I, Mildner A, Cohen N, Jung S, Tanay A, Amit I. Massively parallel single cell RNA-Seq for marker-free decomposition of tissues into cell types. Science (New York, NY) 2014;343:776-9.
88. Hashimshony T, Wagner F, Sher N, Yanai I. CEL-Seq: single-cell RNA-Seq by multiplexed linear amplification. Cell Rep 2012;2:666-73.
89. Kang Y, Norris MH, Zarzycki-Siek J, Nierman WC, Donachie SP, Hoang TT. Transcript amplification from single bacterium for transcriptome analysis. Genome Res 2011;21:925-35.
90. Pan X, Durrett RE, Zhu H, Tanaka Y, Li Y, Zi X, Marjani SL, Euskirchen G, Ma C, LaMotte RH, Park IH, Snyder MP, Mason CE, Weissman SM. Two methods for full-length RNA sequencing for low quantities of cells and single cells. Proc Natl Acad Sci U S A 2013;110:594-9.
91. Sanchez-Freire V, Ebert AD, Kalisky T, Quake SR, Wu JC. Microfluidic single-cell real-time PCR for comparative analysis of gene expression patterns. Nat Protoc 2012;7:829.
92. Streets AM, Zhang X, Cao C, Pang Y, Wu X, Xiong L, Yang L, Fu Y, Zhao L, Tang F. Microfluidic single-cell whole-transcriptome sequencing. Proc Natl Acad Sci U S A 2014;111:7048-53.
93. Toriello NM, Douglas ES, Thaitrong N, Hsiao SC, Francis MB, Bertozzi CR, Mathies RA. Integrated microfluidic bioprocessor for single-cell gene expression analysis. Proc Natl Acad Sci U S A 2008;105:20173-8.
94. White AK, VanInsberghe M, Petriv I, Hamidi M, Sikorski D, Marra MA, Piret J, Aparicio S, Hansen CL. High-throughput microfluidic single-cell RT-qPCR. Proc Natl Acad Sci U S A 2011;108:13999-4004.
95. White A, Heyries K, Doolin C, Vaninsberghe M, Hansen C. High-throughput microfluidic single-cell digital polymerase chain reaction. Anal Chem 2013;85:7182-90.
96. Dalerba P, Kalisky T, Sahoo D, Rajendran PS, Rothenberg ME, Leyrat AA, Sim S, Okamoto J, Johnston DM, Qian D, Zabala M, Bueno J, Neff NF, Wang J, Shelton AA, Visser B, Hisamori S, Shimono Y, van de Wetering M, Clevers H, Clarke MF, Quake SR. Single-cell dissection of transcriptional heterogeneity in human colon tumors. Nat Biotechnol 2011;29:1120.
97. Potter NE, Ermini L, Papaemmanuil E, Cazzaniga G, Vijayaraghavan G, Titley I, Ford A, Campbell P, Kearney L, Greaves M. Single-cell mutational profiling and clonal phylogeny in cancer. Genome Res 2013;23:2115-25.
98. Lawson DA, Bhakta NR, Kessenbrock K, Prummel KD, Yu Y, Takai K, Zhou A, Eyob H, Balakrishnan S, Wang CY, Yaswen P, Goga A, Werb Z. Single-cell analysis reveals a stem-cell program in human metastatic breast cancer cells. Nature 2015;526:131.
99. Islam S, Zeisel A, Joost S, La Manno G, Zajac P, Kasper M, Lönnerberg P, Linnarsson S. Quantitative single-cell RNA-seq with unique molecular identifiers. Nat Methods 2014;11:163.
100. Macosko Evan Z, Basu A, Satija R, Nemesh J, Shekhar K, Goldman M, Tirosh I, Bialas Allison R, Kamitaki N, Martersteck Emily M, Trombetta John J, Weitz David A, Sanes Joshua R, Shalek Alex K, Regev A, McCarroll Steven A. Highly parallel genome-wide expression profiling of individual cells using nanoliter droplets. Cell 2015;161:1202-14.
101. Shekhar K, Lapan SW, Whitney IE, Tran NM, Macosko EZ, Kowalczyk M, Adiconis X, Levin JZ, Nemesh J, Goldman M, McCarroll SA, Cepko CL, Regev A, Sanes JR. Comprehensive classification of retinal bipolar neurons by single-cell transcriptomics. Cell 2016;166:1308-23.e30.
102. McGranahan N, Burrell RA, Endesfelder D, Novelli MR, Swanton C. Cancer chromosomal instability: therapeutic and diagnostic challenges:'Exploring aneuploidy: the significance of chromosomal imbalance'review series. EMBO reports 2012;13:528-38.
103. Bakker B, van den Bos H, Lansdorp PM, Foijer F. How to count chromosomes in a cell: an overview of current and novel technologies. Bioessays 2015;37:570-7.
104. Gerlinger M, Horswell S, Larkin J, Rowan AJ, Salm MP, Varela I, Fisher R, McGranahan N, Matthews N, Santos CR, Martinez P, Phillimore B, Begum S, Rabinowitz A, Spencer-Dene B, Gulati S, Bates PA, Stamp G, Pickering L, Gore M, Nicol DL, Hazell S, Futreal PA, Stewart A, Swanton C. Genomic architecture and evolution of clear cell renal cell carcinomas defined by multiregion sequencing. Nat Genet 2014;46:225.
105. Hughes AEO, Magrini V, Demeter R, Miller CA, Fulton R, Fulton LL, Eades WC, Elliott K, Heath S, Westervelt P, Ding L, Conrad DF, White BS, Shao J, Link DC, DiPersio JF, Mardis ER, Wilson RK, Ley TJ, Walter MJ, Graubert TA. Clonal architecture of secondary acute myeloid leukemia defined by single-cell sequencing. PLoS Genet 2014;10:e1004462.
106. Müller S, Liu SJ, Di Lullo E, Malatesta M, Pollen AA, Nowakowski TJ, Kohanbash G, Aghi M, Kriegstein AR, Lim DA. Single-cell sequencing maps gene expression to mutational phylogenies in PDGF- and EGF-driven gliomas. Mol Syst Biol 2016;12:889.
107. Ramaswamy S, Ross KN, Lander ES, Golub TR. A molecular signature of metastasis in primary solid tumors. Nat Genet 2003;33:49.
108. Scheel C, Onder T, Karnoub A, Weinberg RA. Adaptation versus selection: the origins of metastatic behavior. Cancer Res 2007;67:11476-80.
109. Gawad C, Koh W, Quake SR. Dissecting the clonal origins of childhood acute lymphoblastic leukemia by single-cell genomics. Proc Natl Acad Sci U S A 2014;111:17947-52.
110. Li Y, Xu X, Song L, Hou Y, Li Z, Tsang S, Li F, Im KM, Wu K, Wu H, Ye X, Li G, Wang L, Zhang B, Liang J, Xie W, Wu R, Jiang H, Liu X, Yu C, Zheng H, Jian M, Nie L, Wan L, Shi M, Sun X, Tang A, Guo G, Gui Y, Cai Z, Li J, Wang W, Lu Z, Zhang X, Bolund L, Kristiansen K, Wang J, Yang H, Dean M, Wang J. Single-cell sequencing analysis characterizes common and cell-lineage-specific mutations in a muscle-invasive bladder cancer. GigaScience 2012;1:12.
111. Yu C, Yu J, Yao X, Wu WKK, Lu Y, Tang S, Li X, Bao L, Li X, Hou Y, Wu R, Jian M, Chen R, Zhang F, Xu L, Fan F, He J, Liang Q, Wang H, Hu X, He M, Zhang X, Zheng H, Li Q, Wu H, Chen Y, Yang X, Zhu S, Xu X, Yang H, Wang J, Zhang X, Sung JJY, Li Y, Wang J. Discovery of biclonal origin and a novel oncogene SLC12A5 in colon cancer by single-cell sequencing. Cell Res 2014;24:701.
112. Jan M, Snyder TM, Corces-Zimmerman MR, Vyas P, Weissman IL, Quake SR, Majeti R. Clonal evolution of preleukemic hematopoietic stem cells precedes human acute myeloid leukemia. Sci Trans Med 2012;4:149ra18.
113. Wang L, Fan J, Francis JM, Georghiou G, Hergert S, Li S, Gambe R, Zhou CW, Yang C, Xiao S, Cin PD, Bowden M, Kotliar D, Shukla SA, Brown JR, Neuberg D, Alessi DR, Zhang CZ, Kharchenko PV, Livak KJ, Wu CJ. Integrated single-cell genetic and transcriptional analysis suggests novel drivers of chronic lymphocytic leukemia. Genome Res 2017;27:1300-11.
114. Rhim AD, Thege FI, Santana SM, Lannin TB, Saha TN, Tsai S, Maggs LR, Kochman ML, Ginsberg GG, Lieb JG, Chandrasekhara V, Drebin JA, Ahmad N, Yang YX, Kirby BJ, Stanger BZ. Detection of circulating pancreas epithelial cells in patients with pancreatic cystic lesions. Gastroenterology 2014;146:647-51.
115. Clawson GA, Matters GL, Xin P, McGovern C, Wafula E, dePamphilis C, Meckley M, Wong J, Stewart L, D'Jamoos C, Altman N, Imamura Kawasawa Y, Du Z, Honaas L, Abraham T. "Stealth dissemination" of macrophage-tumor cell fusions cultured from blood of patients with pancreatic ductal adenocarcinoma. PLoS One 2017;12:e0184451.
116. Francis JM, Zhang CZ, Maire CL, Jung J, Manzo VE, Adalsteinsson VA, Homer H, Haidar S, Blumenstiel B, Pedamallu CS, Ligon AH, Love JC, Meyerson M, Ligon KL. EGFR variant heterogeneity in glioblastoma resolved through single-nucleus sequencing. Cancer discovery 2014;4:956-71.
117. Bakker B, Taudt A, Belderbos ME, Porubsky D, Spierings DCJ, de Jong TV, Halsema N, Kazemier HG, Hoekstra-Wakker K, Bradley A, de Bont ESJM, van den Berg A, Guryev V, Lansdorp PM, Colomé-Tatché M, Foijer F. Single-cell sequencing reveals karyotype heterogeneity in murine and human malignancies. Genome Biol 2016;17:115.
118. Ruiz C, Li J, Luttgen MS, Kolatkar A, Kendall JT, Flores E, Topp Z, Samlowski WE, McClay E, Bethel K, Ferrone S, Hicks J, Kuhn P. Limited genomic heterogeneity of circulating melanoma cells in advanced stage patients. Phys Biol 2015;12:016008.
119. Amatu A, Sartore-Bianchi A, Siena S. NTRK gene fusions as novel targets of cancer therapy across multiple tumour types. ESMO open 2016;1:e000023.
120. Aceto N, Bardia A, Miyamoto David T, Donaldson Maria C, Wittner Ben S, Spencer Joel A, Yu M, Pely A, Engstrom A, Zhu H, Brannigan Brian W, Kapur R, Stott Shannon L, Shioda T, Ramaswamy S, Ting David T, Lin Charles P, Toner M, Haber Daniel A, Maheswaran S. Circulating tumor cell clusters are oligoclonal precursors of breast cancer metastasis. Cell 2014;158:1110-22.
121. Medler TR, Cotechini T, Coussens LM. Immune response to cancer therapy: mounting an effective antitumor response and mechanisms of resistance. Trends Cancer 2015;1:66-75.
122. Tran E, Ahmadzadeh M, Lu YC, Gros A, Turcotte S, Robbins PF, Gartner JJ, Zheng Z, Li YF, Ray S, Wunderlich JR, Somerville RP, Rosenberg SA. Immunogenicity of somatic mutations in human gastrointestinal cancers. Science 2015;350:1387-90.
123. Rosenberg SA, Yang JC, Sherry RM, Kammula US, Hughes MS, Phan GQ, Citrin DE, Restifo NP, Robbins PF, Wunderlich JR, Morton KE, Laurencot CM, Steinberg SM, White DE, Dudley ME. Durable complete responses in heavily pretreated patients with metastatic melanoma using T-cell transfer immunotherapy. Clin Cancer Res 2011;17:4550-7.
124. Lu YC, Zheng Z, Robbins PF, Tran E, Prickett TD, Gartner JJ, Li YF, Ray S, Franco Z, Bliskovsky V, Fitzgerald PC, Rosenberg SA. An efficient single-cell RNA-seq approach to identify neoantigen-specific T cell receptors. Mol Ther 2018;26:379-89.
125. Rosati E, Dowds CM, Liaskou E, Henriksen EKK, Karlsen TH, Franke A. Overview of methodologies for T-cell receptor repertoire analysis. BMC Biotechnol 2017;17:61.
126. Shitaoka K, Hamana H, Kishi H, Hayakawa Y, Kobayashi E, Sukegawa K, Piao X, Lyu F, Nagata T, Sugiyama D, Nishikawa H, Tanemura A, Katayama I, Murahashi M, Takamatsu Y, Tani K, Ozawa T, Muraguchi A. Identification of tumoricidal TCRs from tumor-infiltrating lymphocytes by single-cell analysis. Cancer Immunology Research 2018;6:378-88.
127. Baitsch L, Fuertes-Marraco SA, Legat A, Meyer C, Speiser DE. The three main stumbling blocks for anticancer T cells. Trends Immunol 2012;33:364-72.
128. Anderson AC, Joller N, Kuchroo VK. Lag-3, Tim-3, and TIGIT: co-inhibitory receptors with specialized functions in immune regulation. Immunity 2016;44:989-1004.
129. Restifo NP, Smyth MJ, Snyder A. Acquired resistance to immunotherapy and future challenges. Nat Rev Cancer 2016;16:121.
130. Zheng C, Zheng L, Yoo JK, Guo H, Zhang Y, Guo X, Kang B, Hu R, Huang JY, Zhang Q, Liu Z, Dong M, Hu X, Ouyang W, Peng J, Zhang Z. Landscape of infiltrating T xells in liver cancer revealed by single-cell sequencing. Cell 2017;169:1342-56.e16.
131. Singer M, Wang C, Cong L, Marjanovic ND, Kowalczyk MS, Zhang H, Nyman J, Sakuishi K, Kurtulus S, Gennert D, Xia J, Kwon JYH, Nevin J, Herbst RH, Yanai I, Rozenblatt-Rosen O, Kuchroo VK, Regev A, Anderson AC. A distinct gene module for dysfunction uncoupled from activation in tumor-infiltrating T cells. Cell 2016;166:1500-11.e9.
132. Chung W, Eum HH, Lee HO, Lee KM, Lee HB, Kim KT, Ryu HS, Kim S, Lee JE, Park YH. Single-cell RNA-seq enables comprehensive tumour and immune cell profiling in primary breast cancer. Nat Commun 2017;8:15081.
133. Najjar YG, Finke JH. Clinical perspectives on targeting of myeloid derived suppressor cells in the treatment of cancer. Front Oncol 2013;3:49.
134. Damuzzo V, Pinton L, Desantis G, Solito S, Marigo I, Bronte V, Mandruzzato S. Complexity and challenges in defining myeloid-derived suppressor cells. Cytometry Part B: Clinical Cytometry 2015;88:77-91.
135. Grant CR, Liberal R, Mieli-Vergani G, Vergani D, Longhi MS. Regulatory T-cells in autoimmune diseases: challenges, controversies and—yet—unanswered questions. Autoimmun Rev 2015;14:105-16.
136. Miyamoto DT, Zheng Y, Wittner BS, Lee RJ, Zhu H, Broderick KT, Desai R, Fox DB, Brannigan BW, Trautwein J, Arora KS, Desai N, Dahl DM, Sequist LV, Smith MR, Kapur R, Wu CL, Shioda T, Ramaswamy S, Ting DT, Toner M, Maheswaran S, Haber DA. RNA-Seq of single prostate CTCs implicates noncanonical Wnt signaling in antiandrogen resistance. Science 2015;349:1351-6.
137. Dago AE, Stepansky A, Carlsson A, Luttgen M, Kendall J, Baslan T, Kolatkar A, Wigler M, Bethel K, Gross ME, Hicks J, Kuhn P. Rapid phenotypic and genomic change in response to therapeutic pressure in prostate cancer inferred by high content snalysis of single circulating tumor cells. PLoS One 2014;9:e101777.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Shi X, Chakraborty P, Chaudhuri A. Unmasking tumor heterogeneity and clonal evolution by single-cell analysis. J Cancer Metastasis Treat 2018;4:47. http://dx.doi.org/10.20517/2394-4722.2018.32
AMA Style
Shi X, Chakraborty P, Chaudhuri A. Unmasking tumor heterogeneity and clonal evolution by single-cell analysis. Journal of Cancer Metastasis and Treatment. 2018; 4: 47. http://dx.doi.org/10.20517/2394-4722.2018.32
Chicago/Turabian Style
Shi, Xiaoshan, Papia Chakraborty, Amitabha Chaudhuri. 2018. "Unmasking tumor heterogeneity and clonal evolution by single-cell analysis" Journal of Cancer Metastasis and Treatment. 4: 47. http://dx.doi.org/10.20517/2394-4722.2018.32
ACS Style
Shi, X.; Chakraborty P.; Chaudhuri A. Unmasking tumor heterogeneity and clonal evolution by single-cell analysis. J. Cancer. Metastasis. Treat. 2018, 4, 47. http://dx.doi.org/10.20517/2394-4722.2018.32
About This Article
Special Issue
Copyright

Data & Comments
Data


 Cite This Article 19 clicks
Cite This Article 19 clicks

 Like This Article 24
likes
Like This Article 24
likes






Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.