
NSAID celecoxib: a potent mitochondrial pro-oxidant cytotoxic agent sensitizing metastatic cancers and cancer stem cells to chemotherapy


Abstract
Intermittent hypoxia within tumor microenvironments causes pro-oxidative stress impairing oxidative phosphorylation (OxPhos) and increases mitochondrial production of reactive oxygen species (ROS). In primary tumors this provokes metabolic reprogramming of both tumor cells and cancer stem cells and emergence of highly metastatic cancer cells. Tumor reprogramming is initiated by activating nuclear respiratory factors and hypoxia-inducible factors in response to changes in oxygen and ROS levels. Hence, hypoxia-induced pro-oxidative stress drives invasion and metastasis. However, it is also the Achilles’ heel of metastatic cancer cells because pro-oxidative agents further overload the mitochondria and intracellular milieu with excessive ROS to trigger apoptosis, whereas antioxidant agents promote their survival and tumor progression. Herein lies the metastatic tumor cell sensitivity to non-steroidal anti-inflammatory drugs (NSAIDs) and we and others have shown that the NSAID celecoxib exerts powerful pro-oxidative anticancer effects by directly targeting mitochondria to increase ROS production and trigger cancer cell death, including metastatic cancer cells and cancer stem cells. This review highlights the considerable benefits from appropriate NSAID use in humans against post-diagnosis metastatic tumors and the need to further develop their use as adjuvant therapy for advanced stage metastatic disease where they are already showing significantly improved clinical outcomes.
Keywords
Introduction
Cancer cells and their mitochondria adapt to higher levels of oxidative stress as they emerge from the primary tumor to become circulating tumor cells and migrate into the metastatic distant tissues[1-5]. It is clear that emerging metastatic cancer cells have undergone not only significant genetic but also metabolic changes including activation of their antioxidant systems which promote their survival by helping to detoxify heightened reactive oxygen species (ROS) levels to enable eventual metastatic outgrowth into diverse sites. In the first part of this review, the evidence for these changes in redox homeostatic mechanisms identified for reprogramming into highly metastatic cells and cancer stem cells are discussed. The second part of the review is focused on how drugs such as non-steroidal anti-inflammatory drugs (NSAIDs) like celecoxib are able to take advantage of and target the pro-oxidative state of highly metastatic cancer cells and cancer stem cells to force them into terminal states of ROS excess thereby activating cell death.
The metastatic potential of cancer cells is regulated by their redox status and ROS levels
Several lines of independent evidence have established that the metastatic potential of tumor cells and cancer stem cells are directly related to their heightened redox status and greater intrinsic capacity for ROS production[1,5,6] which becomes particularly important for metastasis[2,3][Figure 1]. The conditions that cause this development are the culmination of hypoxia generated in expanding primary tumors and resulting oxidative stress upregulating the expression and activation levels of two essential transcription factor families which allow cancer cells to cope with heightened ROS levels. These are the nuclear E2-related factors [a.k.a. nuclear respiratory factors (NRFs) as key regulators of the antioxidant and cytoprotective genes][7], that in turn increase expression of hypoxia-inducible factors (HIF’s)[8]. Both the NRF and HIF families of factors act as crucial rheostat regulators of the redox state, affected by ROS levels in cells, and have both been shown to combine together and perform key roles in tumor survival and progression under hypoxia[8]. The question is whether it is best to increase or decrease ROS as an anticancer therapeutic strategy[6]. However, before addressing the question of anticancer therapeutic strategy, the next sections of the review focus on how increased ROS levels reprogram to sustain a heightened state of ROS production and greater metastatic potential.
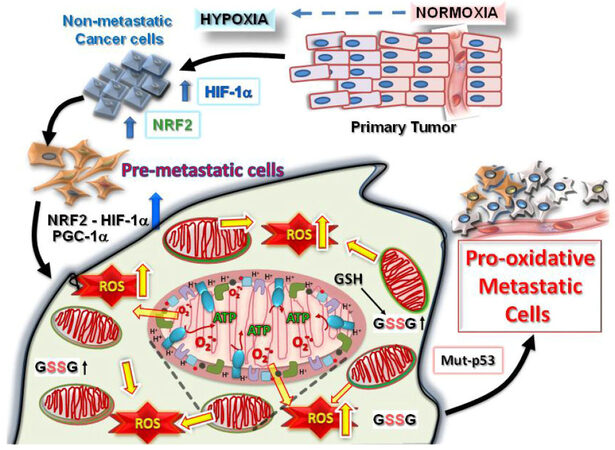
Figure 1. Tumorigenesis requires hypoxic driven metabolic reprogramming for metastatic progression. The progression of malignant states during carcinogenesis involves changes occurring within the tumor microenvironment, including regions of hypoxia, hypoglycemia and acidosis. These regions are where cancer cells evolve into pre-metastatic states after selection by the harmful conditions, resulting in the altered capacity for increased ROS production and protection from the greater oxidative stress. Eventually, other mutations (such as in cell cycle regulatory proteins, p53 or ARF) occur which allow the cells to adapt by further metabolic reprogramming to emerge as highly pro-oxidative metastatic cells. HIF: hypoxia-induced factor; PGC1α: peroxisome proliferator-activated receptor gamma coactivator 1-alpha; NRF: nuclear respiratory factor; ROS: reactive oxygen species
These events are the consequence of major changes occurring at the level of gene expression during this adaptation process and reprogramming which results when the master transcriptional regulator, nuclear respiratory factor 2 (NRF2) becomes activated and is released from the mitochondrial outer membrane to the cytosol [Figure 2]. Upon its release NRF2 transits to the nucleus to form heterodimers with other basic leucine zipper (BZIP) family members (such as Maf), binding to antioxidant response elements (AREs) in the promoter regions activating the NRF2 target genes[8]. Amongst the over 500 NRF2 target genes are many encoding proteins that collectively promote malignant cancer cell survival, such as detoxifying enzymes, antioxidant enzymes [including several key proteins of both the reduced glutathione (GSH) and thioredoxin (Trx) systems], receptors, transcription factors, metabolic enzymes, p-Akt, proteases, and many more (reviewed in[7]). NRF2 activation can cause increased mitochondrial mass[4,9,10] and induction of peroxisome proliferator-activated receptor gamma coactivator 1-alpha (PGC1α) and PGC1β; PGC1α together with NRF2 stimulate the expression of the related gene NRF1 (reviewed in[11]). PGC1α has been shown to form complexes with NRF2 as a transcriptional coactivator and promotes NRF2 and NRF1 binding to the manganese superoxide dismutase, SOD2 gene promoter[12]. Consequently, NRF1 activates nuclear genes that encode mitochondrial proteins, including mitochondrial transcription factor A (TFAM), promoting mitochondrial biogenesis[4,13] such that the tumor cells are modified to adopt increased pro-oxidative states with greater malignant potential[2,14][Figure 3].
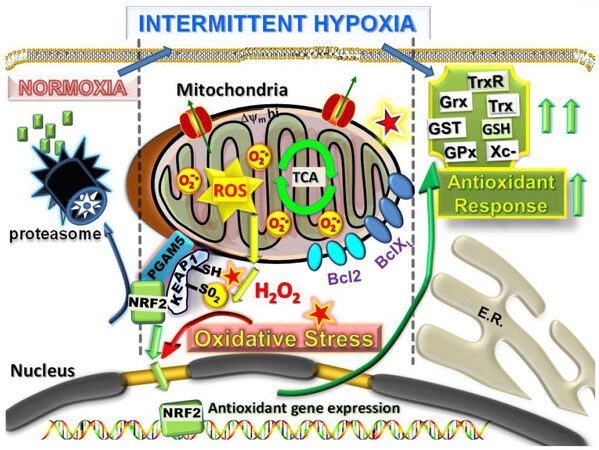
Figure 2. Intermittent hypoxia within the primary tumor microenvironment as a driver of mitochondrial ROS production and metastatic reprogramming. Mitochondrial ROS is produced extensively in cells undergoing rounds of intermittent hypoxia. In a similar manner to ischaemia/reperfusion in normal cells, the intermittent hypoxia of early primary cancer cells causes readjustments in redox homeostasis by increasing ROS activated NRF2 release from the outer redox hub (KEAP1/PGAM5) on the mitochondria, NRF2 transport to the nucleus and transcriptional activation of a large number of antioxidant defense response, including the GSH and Trx systems to counteract and detoxify the ROS. In addition, Bcl-2 and Bcl-XL are stabilized to promote cell survival under the conditions of pro-oxidative stress. NRF: nuclear respiratory factor; ROS: reactive oxygen species; Trx: thioredoxin; GSH: reduced glutathione
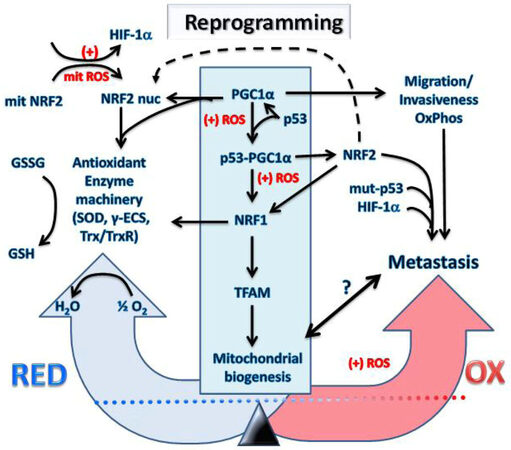
Figure 3. Transcriptional reprogramming of metabolism in cancer cells undergoing hypoxia and oxidative stress causes metastasis. The increased mitochondrial and cellular ROS (+) induced by intermittent hypoxia in cancer cells causes metabolic reprogramming. Expression levels and activation of key redox regulatory proteins: p53, PGC1α, HIF-1α, KEAP1, NRF2 and ARF leads to increased antioxidant gene expression and restoration of redox homeostasis. However, the outcome will depend on factors such as the mutational status of the genes encoding these redox regulators and resulting imbalances in their activity with potential to promote increased invasiveness, migration, OxPhos and metastasis. It is uncertain what levels of mitochondria will exist inside metastatic cells and will depend on the level of the PGC1α/NRF1/TFAM axis represented by the symbol “?” and the bidirectional arrow. The reprogramming into metastatic cancer cells is driven by NRF2, mut-p53 and HIF-1α which are critical metastatic biomarkers. HIF: hypoxia-induced factor; NRF: nuclear respiratory factor; GSH: reduced glutathione; ROS: reactive oxygen species; TFAM: mitochondrial transcription factor A; PGC1α: peroxisome proliferator-activated receptor gamma coactivator 1-alpha; KEAP1: Kelch-like ECH associated protein1; NRF: nuclear respiratory factor; γ-ECS: gamma-glutamylcysteine synthetase; Trx: thioredoxin; TrxR: thioredoxin reductase
Early studies showed PGC1α to be a potent stimulator of mitochondrial respiration and gene transcription in liver, heart, and skeletal muscle, activated under oxidative stress[15]. It is proposed that mitochondria, as an energy center important for cellular homeostasis, undergo biogenesis as an endogenous protective response mechanism designed to cope with ischemic/hypoxic insults and to counteract their detrimental effects. In either normal cells, SH-SY5Y neuroblastoma or immortalized mouse myoblast C2C12 cells undergoing oxidative stress, wild type p53 levels were shown to increase within several hours to form a complex with coactivator PGC1α and activate genes such as NRF1 and NRF2 but without affecting proliferation[16][Figure 3]. Ischemia in the brain has been shown to increase mitochondrial DNA, total mitochondrial number and expression of the mitochondrial transcription factors downstream of PGC1α (including NRF1 and TFAM), whereas the ensuing reperfusion increases oxidative stress and mitochondrial biogenesis[17]. PGC1α is a powerful controller of cell metabolism and maintains a balance between production and scavenging of pro-oxidant molecules by coordinating mitochondrial biogenesis, promoting oxidative phosphorylation [OxPhos, i.e., mitochondrial adenosine triphosphate (ATP) synthesis] and the expression of antioxidants like GSH, although the exact role of PGC1α in cancer is unclear with no consistent relationship[18].
In a study of breast cancer cells, PGC1α expression and activation were shown to significantly increase mitochondrial biogenesis and OxPhos to promote metastasis[19], and increased PGC1α levels were detected in the circulating tumor cells and metastases from a range of different murine cancer models (4T1, B16F10 and MDA-MB-231) compared to levels in the corresponding primary tumors. PGC1α was linked with greater levels of migratory/invasive cancer cells, increased mitochondrial copy number, respiration and OxPhos [Figure 3]. Silencing PGC1α in the breast cancer cells severely decreased copy number of mitochondrial DNA and visible mitochondria within the cells, suspended their invasive potential and attenuated metastasis without affecting proliferation, primary tumor growth or the epithelial-to-mesenchymal transition[19]. Unfortunately, this study did not compare mitochondrial ROS production or oxidative stress between primary and metastatic tumors. However, in another study of renal carcinoma, overexpressing PGC1α in Von Hippel-Lindau (VHL) gene defective, constitutive HIF expressing clear cell renal cell carcinoma (ccRCC) impaired cancer cell growth and upregulated expression of antioxidant enzymes, but also showed greater ROS levels and oxidative stress[20]. The HIFs were shown to directly inhibit PGC1α activity or its expression, reducing oxygen consumption and increased stabilization of HIF1α protein caused a switch in metabolism away from PGC1α driven OxPhos to increased glycolysis[20].
It would appear that the reasons for differences in the PGC1α relationship amongst different cancers may depend upon their levels of other factors such as expression of the HIF’s as inhibitors vs. other PGC1α coactivators such as p53, a transcriptional activator and interactive binding partner of PGC1α [Figure 3]. For example, PGC1α mRNA levels were substantially higher in wild-type p53 lung cancer cell lines compared to cell lines with p53 loss or missense mutations and siRNA knockdown of PGC1α inhibited cell proliferation in wild-type p53 lung cancer cell lines[21]. These results are consistent with p53 binding the PGC1α gene promoter, increasing expression[16], thereby protecting the cells after promoting ROS detoxification capacities to enable cancer cell survival under states of oxidative stress[22], particularly stress from mitochondrial ROS[23]. The increased PGC1α complexes with p53, modifying transactivating function [Figure 3] to cause cancer cell cycle arrest and activation of metabolic target genes, promoting ROS clearance in response to metabolic stress, such as from low glucose[24]. However, loss of PGC1α expression prevents the p53-mediated ROS clearance, instead enhancing p53-dependent cancer cell apoptosis[24]. Hence, when GSH levels are depleted by the gamma-glutamylcysteine synthetase (γ-ECS) inhibitor, buthionine sulfoximine (BSO) or other metabolic oxidative stress, increased p53 binds to the PGC1α gene promoter to increase its expression and together the complex then promotes cellular antioxidant defenses via NRF2-mediated expression of antioxidant enzymes such as SOD2 and c-GlutamylCysteine Ligase [a.k.a. γ-ECS, Catalytic subunit of the γ-ECS (a.k.a. γ-GCL) enzyme required in the first step of GSH synthesis], increasing GSH synthesis to restore redox homeostasis[16]. Knocking down either p53 or PGC1α prevented induction of SOD2 or γ-ECS[16]. Hence, loss of p53 function would restrict the ability of cells to defend against oxidative stress. In the latter study, it should be noted that under conditions where NRF2 was activated by pro-oxidative stress using BSO to scavenge GSH, no change was detected in mitochondrial biogenesis and neither NRF1 nor TFAM was altered at the protein or mRNA levels[16]. Based on the results of this study, it follows that cancer cells with lower GSH/GSSG ratios and a more pro-oxidative status such as that commonly found in metastatic tumor cells[1-3,5,6], particularly where p53 is either lost or mutated, will not show a strong PGC1α response. This is supported by studies of vascular smooth muscle cell responses to oxidative stress (1 mmol/L diethylenetriamine/nitric oxide adduct (DETA/NO) as a nitric oxide donor for 24 h) comparing p53 wild type to p53 null cell responses where higher ROS (2.7 fold greater increase) was produced in cells from the wild type p53 mice[25]. The question then is how does the metastatic cancer cell cope with the heightened level of oxidative stress, and whether this is related to changes in p53 function.
Recently, mutant p53 (mut-p53) was shown to interact with NRF2, increasing p53/NRF2 complexes on select antioxidant response element (ARE) containing gene promoters to activate transcription of a specific set of genes, whilst repressing most others[26-28]. In particular, the Trx gene (TXN) is unusual along with the thioredoxin reductase (TXNRD1) as mut-p53 activated NRF2 target genes enhancing the Trx system with pro-survival and pro-migratory functions in breast cancer cells under oxidative stress, while heme oxygenase 1 (HMOX1) is a mut-p53 repressed target displaying opposite effects[26]. Mut-p53 appears to sequester NRF2 preventing its activity on most of the NRF2 regulated genes impairing its canonical antioxidant activities, directly promoting greater ROS accumulation in cancer cells by inhibiting expression of the glutamate/cystine antiporter solute carrier family 7 member 11 (SLC7A11, also called xCT), a component of the cystine/glutamate antiporter as part of the Xc-system, diminishing cytosolic production of GSH[27,28]. Analysis across a cancer cell panel for accumulation of mut-p53 protein showed a significant association with increased basal mitochondrial and cytosolic ROS levels, and decreased endogenous GSH reserves. Also, consistent with this observation, by overexpressing mut-p53 in cancer cells, system Xc- activity and GSH levels were diminished resulting in a heightened level of ROS stress. In contrast, knockout of mut-p53 decreased basal ROS levels and conferred protection against H2O2[27,28]. Hence, highly metastatic cancer cells with a mutant form of p53 or as p53 null cells which commonly occurs in advanced stages of malignancy would account for many of the cell phenotypes with greater mitochondrial ROS production [Figures 3 and 4], reflected by their lower or higher PGC1α levels and related changes in mitochondrial mass.
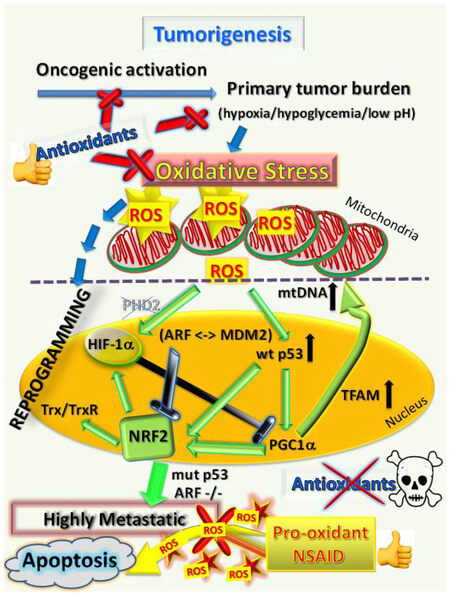
Figure 4. Antioxidants are effective cancer chemopreventatives until cancer becomes established, when they promote metastatic progression, whereas NSAIDs as pro-oxidants are effective by overloading cancer cells with excessive ROS to eliminate metastases. Once tumors are established, antioxidants will support and promote the further progression of cancer cells to metastasize. However, the opposing pro-oxidants such as the NSAID celecoxib cause excessive ROS overload and induce mass cell death in metastases or sensitize metastatic cells to enhanced chemotherapeutic killing. ROS: reactive oxygen species; HIF hypoxia-induced factor; NSAID: non-steroidal anti-inflammatory drug; Trx: thioredoxin; TrxR: thioredoxin reductase
ARF, the regulator of p53 protein levels in cells by binding the mouse double minute 2 (MDM2) homolog to prevent p53 turnover, also exerts its influence in regulating the NRF2 antioxidant system, also binding NRF2 to inhibit its activation, wherein ARF/NRF2 association prevents cAMP-response-element-binding protein (CREB) dependent acetylation of NRF2 and binding to target DNA[29]. Hence, ARF/NRF2 significantly represses NRF2 transcriptional activity and expression of SLC7A11 component of the cystine/glutamate antiporter Xc-system is dramatically suppressed when ARF is activated, again affecting GSH production and causing increased cellular ROS levels in cancer cells, much as mut-p53 does as outlined above. Hence, depending on the status of ARF and p53 mutations or deletions in cancer cells, NRF2 activation would be expected to be heightened in their absence, particularly in cellular states of elevated pro-oxidative stress via Kelch-like ECH associated protein1 (KEAP1) inactivation, potentially setting up a self-perpetuating scenario maintaining higher endemic intracellular ROS levels. Even in cells with wild type p53, activated NRF2 binds to an ARE in the MDM2 gene increasing expression[30] that in turn, promotes p53 ubiquitination and proteasomal degradation. Transcription of the SQSTM1 gene encoding the p62 autophagy regulatory protein is also stimulated by NRF2[31]. In turn, p62 sequesters KEAP1, thereby increasing NRF2 abundance[31] in another self-promoting cycle. Moreover, depending on the levels of oxidative stress, NRF2 together with mechanistic target of rapamycin (mTOR) serine/threonine kinase and adenosine-monophosphate-activated-protein-kinase (AMPK) coordinate alternative autophagy dependent pathways, with lower stress levels promoting survival whereas higher stress results in cell death[32].
Chemoprevention of carcinogenesis mediated via the KEAP1-NRF2 redox sensory hub
When cells undergo hypoxia, mitochondrial ROS production is promoted in the short term as a by-product from the respiratory chain[33,34]. Consequently, a number of events ensue ultimately resulting in greater activation of NRFs and HIFs. One of the main cell sensors of the oxidative stress resides on the outer mitochondrial membrane, comprising a ternary protein complex anchored via PGAM5 (histidine mediated serine/threonine phosphatase)[35]. This complex act as a critical redox sensory hub consisting of the bound protein, KEAP1 with many redox regulated thiol-cysteine residues in its structure available for modification by electrophilic agents, ROS mediated oxidation or other xenobiotic compounds. The PGAM5-KEAP1 master redox controller on mitochondria is a pivotal regulatory complex involved in the actions of chemopreventive agents that inhibit the development of chemically-induced carcinogenesis [Figure 2]. The role of the KEAP1 complex in cancer has already been previously extensively reviewed[36-40] and it will not be reviewed further except in relation to mechanisms of action by anticancer chemopreventative agents.
The KEAP1 cysteine thiol residues with low pKa values are especially reactive with chemopreventatives such as the isothiocyanates[41]. At physiological pH, these cysteines exist as thiolate anions that are primed for nucleophilic attack by such electrophilic agents (termed inducers) (reviewed in[42]). These thiol modifications disrupt the function of KEAP1 as an anchor or tether holding KEAP1 binding proteins such as NRF2, Bcl2 or Bcl-XL in a complex on the outer membrane with PGAM5, as a histidine phosphatase involved in regulating these interactions [Figure 2]. KEAP1 is a redox-regulated substrate adaptor protein for the Cul3 E3 ubiquitin ligase and together this complex responds to oxidative stress by controlling availability of NRF2, as well as Bcl-2 and Bcl-XL pro-survival proteins. Hence, under normal redox conditions, KEAP1 targets NRF2, Bcl-2 and Bcl-XL for ubiquitination and proteasomal degradation (reviewed in[43]). Upon cellular/mitochondrial oxidative stress, KEAP1 thiols are oxidized and inactivated preventing substrate ubiquitination, allowing NRF2, Bcl2 and Bcl-XL stabilization and release [Figure 2]. The pro-survival proteins Bcl-2 and Bcl-XL then protect the mitochondria by preventing apoptosis while NRF2 migrates to the nucleus where it activates cellular antioxidant defense genes to restore redox homeostasis[29]. Therefore, the KEAP1 hub is a major regulator of the normal host cell defense responses against oxidative stress involved in maintaining the cellular redox balance.
In this regard, KEAP1 functions as a tumor suppressor protein to prevent tumor progression by negatively regulating substrates NRF2, IKKβ and Bcl-2/Bcl-XL, consistent with KEAP1 function as a guardian against cancer[40,44]. However, when subjected to increased levels of pro-oxidative stress and enhanced ROS levels, irreversible modification of the KEAP1 master regulator either via successively more severe chemical oxidation reactions (such as the irreversible transformation of the thiol-cysteine derived sulfenic acid into sulfinic or sulfonic acids, Figure 2) or genetic mutations or other chemical modification will occur. The end result is blocking of KEAP1 function, constitutive NRF2 activity and increased Bcl-2/Bcl-XL availability which together with adaption to heightened ROS levels is commonly found inside highly metastatic tumor cells[1-3,5,6]. At this point, use of chemopreventive agents targeting the KEAP1-NRF2 master complex will be either ineffectual or could even promote more rapid tumor progression to increase metastatic disease, as outlined below.
Constitutive NRF2 mediated HIF activation and reprogrammed state of emerging metastatic cancer cells
Mutation and dysregulation of the NRF2-KEAP1 pathway are common events in cancer (reviewed in[40,44]) and KEAP1 inactivating somatic mutations have been detected in numerous cancer cells (reviewed in[40]). A study by the Cancer Genome Atlas Research Network reported that > 30% of squamous lung carcinomas have alterations in the NRF2-KEAP1-CUL3 pathway that result in high, constitutive expression of NRF2 and that somatic mutations in NRF2 will disrupt its interaction with KEAP1[45]. Epigenetic silencing of KEAP1 by hypermethylation of its promoter is found in 53% of colorectal cancers[46]. Thus, the constitutive activation of NRF2 common to metastatic cancer cells will result in many major modifications including altering the mitochondrial and cytosolic metabolism critical for tumor survival and metastasis[7]. NRF2 activation prevents cancer initiation[47,48] but in tumor cells promotes cancer progression as invasion, migration[49-51] and metastasis[52-54], and also induces multidrug resistance to chemotherapy by upregulating expression of the multidrug resistance proteins (MDR)/P-glycoprotein/ABC drug transporters[55,56]. In this regard, NRF2 functions in a similar manner during ischemia-reperfusion of normal tissues in the body[57] as well as in the metabolic shift that occurs during induced pluripotent stem cell colony formation with reprogramming after an initial burst of OxPhos and increased ROS production which increases NRF2 before a temporal peak in HIF-1 mediated glycolysis and shuttling via the pentose phosphate pathway[58]. In a related fashion a recent study showed that NRF2 activation also promotes the Warburg effect and stemness-associated properties of cancer-initiating cells[59].
Although it is well known that NRF2 and HIF-1 signaling are both regulated in response to intermittent hypoxia and ROS accumulation [Figures 2 and 3], the evidence suggests that these two signaling pathways are not simply linked by cellular context but interact to promote metastasis and play complementary roles. For example, in the state of chemoresistance induced by hypoxia they both increase expression of the multidrug resistance MDR/P-glycoproteins (reviewed in[8]). The HIF-1 promoter contains two AREs and has been shown to be negatively regulated by NRF1 but is probably also activated by NRF2 during intermittent hypoxia[60]. However, during constant hypoxia, only HIF-1 is increased but not NRF2[60]. Hence, when limiting O2 levels required for OxPhos are available, a build-up in tricarboxylic acid cycle intermediate metabolites (such as fumarate, succinate or oxaloacetate) can occur, which inhibits prolyl hydroxylase (PHD) activity (reviewed in[61]). The ensuing still relatively low ROS level (i.e., moderate oxidative stress) produced by the dysfunctional mitochondria in cells under moderate hypoxia also helps inactivate the PHDs[62]. The net effect is to prevent the enzymes such as PHD2 from targeting HIF for ubiquination and proteasomal degradation [Figure 4]. Thus, hypoxia results in stabilization and activation of HIF-1 as part of the homeostatic mechanism to mediate adaptive responses via altered gene expression (reviewed in[63]).
One critical cell-autonomous adaptive response to hypoxia controlled by HIF-1 is to act as a feedback regulator to lower mitochondrial mass (by inhibiting PGC1α, as outlined above) and alter mitochondrial and cytosolic metabolism (reviewed in[61,64]). Thus, HIF-1 is one of the main factors that mediates the adaptive metabolic responses to hypoxia, increasing glycolytic pathway flux and decreasing flux through the tricarboxylic acid cycle, in order to return mitochondrial ROS production to more normal low levels[65-67]. HIF-1 and NRF2 also help mediate increased flux through the serine synthesis pathway and mitochondrial one-carbon (folate cycle) metabolism to increase mitochondrial production of antioxidants (nicotinamide adenine dinucleotide phosphate (NADPH) and GSH). In this manner, HIF and NRF mediated reprogramming functions to protect cells from excessive oxidative stress and levels of mitochondrial ROS production, albeit promoting survival of cancer cells with heightened redox and greater oxidative status.
The evidence above provides clear support for mut-p53/NRF2 and HIF-1 in reprogramming metastatic cancer cell metabolism by blocking GSH production while increasing the pentose phosphate shunt in order to provide the NADPH required for Trx production to compensate for GSH loss and to buffer the resulting increased ROS levels in these cells to within a range that is beneficial for tumor progression. In 2015, studies were undertaken that specifically analyzed the importance of these antioxidant pathways and their role in cancer initiation vs. progression[68]. A range of murine cancer models deficient in Gclm (encoding the modifier or regulatory subunit of γ-ECS) showed that the inherent decreased GSH production caused delayed tumor initiation, invasiveness and progression, consistent with observations across a range of human cancers[68]. These studies also examined several drugs such as BSO to inhibit γ-ECS either used alone or combined with sulfasalazine (SSA) to block the Xc- glutamate/cystine antiporter and reduce cystine uptake or auranofin (AUR) to inhibit the enzyme TrxR. Early BSO (20 mmol/L) treatment in the drinking water of young animals dramatically reduced breast cancer burdens in the Gclm-/- mouse models, increasing oxidative stress damage that was proposed to hinder tumor growth. However, if BSO treatment was delayed until after the onset of tumors, then no differences in tumor development were noted. Primary breast epithelial cells isolated from the Gclm-/- mice were resistant to BSO because of a compensatory increase in the NRF2-mediated Trx antioxidant pathway, higher CD44, cystine and glutamate levels, as well as increased NADPH, but decreased GSH levels[68]. BSO (150 μmol/L) treating human MDA-MB-231 metastatic breast cancer cells or a range of other cancer lines in culture similarly increased cystine uptake whereas combining BSO with SSA (250 μmol/L) or AUR (250 nmol/L) to simultaneously diminish both GSH and Trx induced striking levels of ROS production [detected with 2’,7’ -dichlorodihydrofluorescein diacetate (DCFDA)] and apoptotic cell death. However, antioxidants N-acetyl-L-cysteine (NAC; 1 mmol/L) or Trolox (250 μmol/L) prevented the cell death[68].
In summary, the results above are consistent with the NRF2-Trx mediated reprogramming of tumor cells into cancer stem cells and the emergence of highly metastatic cancer cells, inhibiting the GSH antioxidant pathway but increasing the Trx/TrxR antioxidant pathway, ROS production and oxidative stress.
The use of antioxidants in post-diagnosis cancer therapy will be harmful by promoting activation of the NRF2-HIF-1 axis, providing insight into the importance of redox status in cancer metastasis
In 2015, an elegant study showed that human patient derived melanoma cell lines transplanted subcutaneously, intravenously or into the spleen of NSG immunodeficient mice produced circulating melanoma cells and metastases with high levels of mitochondrial ROS production, transmembrane electrical potential and NADPH levels via the folate pathway but also lower mitochondrial mass and GSH/GSSG ratios when compared to that of the primary tumors[69]. As outlined in the previous section, highly metastatic tumors are reprogrammed by NRF2-HIF-1 to undergo metabolic changes allowing them to increase mitochondrial NADPH levels to help detoxify and thus attenuate or buffer against higher ROS and oxidative stress. In the 2015 study, NAC was applied in vivo with the aim of inhibiting ROS in these cells and lowering metastasis in their melanoma model, but it failed. To the contrary, systemic treatment with NAC enhanced the levels of circulating tumor cells and significantly increased their numbers of metastases[69]. Unfortunately, these investigators did not examine the metastases for their levels of NRF2 expression or ROS level. It should be noted here that several studies have reported that NAC can activate NRF2 expression in treated cells[70,71], possibly acting via miR141 to lower KEAP1 expression levels[72].
Although antioxidant therapy was predicted early on to be a potentially effective means for treating cancer patients, it became a highly controversial area of debate[3,73], similar to the role of ROS in cancer[74]. However, more recent studies like the one above have repeatedly shown that the use of antioxidants can be counter-productive and instead accelerate more malignant tumor phenotypes, particularly those associated with metastasis[52,69,75,76]. These findings are consistent with the intracellular redox status as playing a crucial role in tumor survival, progression and development of the metastatic malignant phenotype[77]. Alternative, more natural interventions to treat cancers with antioxidants like NAC or soluble vitamin E (Trolox) were originally aimed at decreasing ROS levels as the driver of malignancy. However, such interventions were only successful if applied during the early stages of carcinogenesis, as outlined above[68]. In another study of mouse models with B-RAF- and K-RAS-induced lung cancers, antioxidants were again shown to significantly increase the metastatic potential of pre-existing cancer cells by stimulating tumor progression and lowering survival rates[78]. Transcriptome analysis revealed that the structurally unrelated NAC and vitamin E produced similar changes, lowering expression of cellular antioxidant genes and increasing tumor proliferation by decreasing levels of ROS, oxidative DNA damage and p53 expression in murine and human lung tumor cells. Knockdown or inactivation of p53 similarly increased tumor growth and obviated the effects of antioxidants. This evidence implies that the use of antioxidants promotes oncogenic cancer cell growth once established by inhibiting the ROS-activated wild type p53 axis, which would otherwise act to suppress tumor initiation and development by causing cell death. However, once p53 is mutated or deleted then the cell death from greater ROS levels and oxidative stress will be avoided.
In 2015, another study[76] administering NAC also showed increased lymph node and lung metastases in the BrafCA/+Ptenfl/flTyr-Cre+/0 mouse model of spontaneous malignant melanoma, but had little impact on the number or size of the primary tumors (NAC dose 1 g/liter, ~6 mmol/L changed weekly, corresponding to 114 to 229 mg/kg body weight for an adult male mouse). Similarly, NAC or vitamin E at 200 and 20 μmol/L respectively increased the migration and invasiveness of human malignant melanoma cells in vitro but did not affect their proliferation. Either of these two antioxidants greatly increased the GSH/GSSG ratios in the melanoma cells and in lymph node metastases from the mouse model. The effects of increased tumor migration in vitro were inhibited by BSO (1 mmol/L) showing a dependency on nascent GSH synthesis. Furthermore, NAC or vitamin E did not alter the ROS levels detected in treated cells but increased the activation of the small guanosine triphosphatase (GTPase) Ras homolog gene family, member A (RHOA) involved in tumor cell migration and invasion and blocking downstream RHOA signaling abolished the antioxidant-induced migration. These results confirmed that antioxidants and the GSH system are important in enhancing metastatic cancer progression.
It is noteworthy[79] that NAC treatment has been shown to increase activation and protein levels of HIF-1α in rat brain after ischemia/reperfusion. In another study of fetal lung alveolar epithelial cells, NAC over the range 1-50 mmol/L given as a pretreatment for 24 h dose-dependently enhanced and stabilized the subsequent levels of hypoxia-induced (3% O2 for 4 h) activation of HIF-1α protein, but decreased nuclear p65 NF-κB and DNA binding activity[80]. Analyses of changes in GSH homeostasis with increasing O2 + NAC levels revealed correspondingly increased GSH/GSSG ratios in cultured cells. These results indicate that the effects of NAC as an antioxidant will depend on the amount of the agent applied and available levels of oxygen and can activate HIF-1 in cancer cells[81].
It should be noted that NAC reacts differently with the various oxyradicals found in cells undergoing pro-oxidative stress. For example, NAC rapidly reacts with hypochlorous acid or hydroxyl radicals with a rate constant of 1.36 × 1010 M-1 s-1 whereas reaction with superoxide (O2-, 65 M-1 s-1) and H2O2 (0.16-0.85 M-1 s-1) is much slower[82,83]. In mitochondria, NAC becomes desulfurated to form H2S, which is subsequently oxidized to sulfane sulfur (protein-SH + H2S (from NAC) + 1/2 O2 → protein-SSH + H2O) as a key mediator of the antioxidative and cytoprotective effects of NAC[84]. However, NAC also undergoes direct interactions with proteins containing reactive cysteine thiol groups, such as Raf-1, MEK and ERK via thiol-disulfide exchange[85] and hence, could react with KEAP1 to allow NRF2 activation. Furthermore, NAC directly reacts with many other small molecules (isothiocyanates, diallyl sulfides or triterpenoids), which themselves have the ability to interact with thiol-containing proteins. Thus, great caution must be exercised when interpreting results where NAC has been used primarily for its antioxidant effect or to demonstrate the involvement of ROS in drug-induced cancer cell death, as NAC effects may vary depending on the concentration applied, the oxygen level and other reactive drugs in the system. Furthermore, agents like NAC could work independently of KEAP1 to directly activate NRF2 by acetylating critical lysine residues[86] enabling NRF2 to go to the nucleus of cells[87].
The NAC mediated activation of HIF-1 protein stabilization is more likely to occur under long-term NAC treatment due to modifying redox homeostasis via PHD inhibition and NRF2 activation, whereas over the short-term, high levels of NAC directly inhibit HIF-1 activity and the hypoxic responses taking place inside cancer cells in vitro and in vivo[88-90]. Thus, treating either PyMT or EO771 breast cancer cell lines with 25 mmol/L NAC prevented HIF-1α stabilization (over 2 h) under either hypoxia or normoxia in vitro[88]. NAC treatment had no effect on HIF-2 expression. Over 8 h, NAC treatment (10-25 mmol/L) prevented stabilization of HIF-1α and decreased vascular endothelial growth factor (VEGF) secretion in response to hypoxia in breast tumor cells in vitro, but did not alter the hypoxia-induced increase in mRNA expression for VEGF and lysyl oxidase (LOX). In vivo, mice supplemented with NAC (40 mmol/L fresh daily) in drinking water showed significantly increased GSH levels in their blood within 48 h and maintained these elevated levels for ensuing weeks of continued treatment. NAC (40 mmol/L/daily in drinking water) did not translate to a difference in the primary tumor growth or the hypoxic state of primary tumors (by either HIF-1 expression or hypoxia level detected with pimonidazole) which remained similar to that seen in primary tumors in the untreated control mice[88]. However, NAC treatment given in vivo did significantly increase the lung metastatic burden in the EO771 experimental breast cancer metastasis model, consistent with NAC antioxidant as not advisable for post-diagnosis cancer therapy. Again, it would have been of interest to examine the NRF2-HIF-1 levels inside the metastatic tumor cell population.
In a related study using several different models of tumorigenesis including human P493 B lymphoma cells with conditional MYC or PC3 prostate carcinoma cells with 10 mmol/L NAC diminished HIF-1α protein stabilization and activity over 8-24 h and VEGF secretion under hypoxic conditions (1% O2)[89], it was identified that NAC treatment also lowered the MYC induced ROS production and g-H2AX level in the cancer cells but there were no other signs of genomic instability. In an inducible MYC transgenic murine hepatocarcinoma model, providing NAC (40 mmol/L/daily in drinking water) to pregnant females prevented offspring from subsequently developing liver cancers compared to untreated mice. Hence, NAC or vitamin C used at very high levels to remove cellular ROS caused PHD2 reactivation and HIF-1α degradation in a VHL-dependent manner with loss of HIF-1 over the short term, even in hypoxia[89]. A study of epithelial to mesenchymal transition 6 (EMT6) triple negative breast cancer cells in mice undergoing metastatic colonization over several days during extravasation into the lungs showed during this time that metastasizing cells increased their HIF-1 activity in a manner that was hypoxia-independent but ROS-dependent[90]. This activation of HIF-1 most likely correlates with ROS mediated inactivation of KEAP1 and/or stabilization of active NRF2 to upregulate HIF-1 expression. The increased HIF-1 level was confirmed by correlating with induced expression of lactate dehydrogenase A and phosphorylation of the E1a subunit of pyruvate dehydrogenase, consistent with HIF-1 reprogramming of energy metabolism from a predominant oxidative (OxPhos-dependent) state to a non-oxidative anaerobic glycolysis-dependent state.
The bolus administration of very high doses of NAC (1 g/kg/administration, 2 injections/day from 1 to 6 days after i.v. transplantation of tumor cells) or the use of the HIF-1 inhibitor, YC-1, impeded the metabolic reprogramming of cancer cells, eventually suppressing the formation of metastatic lung tumors[90]. These results are consistent with an earlier study of B16F10 metastasis to the lung after subcutaneous injection where increasing NAC to very high dosage (up to 4 g/kg dose, with the latter having no metastases at all) showed dose related inhibition of both primary tumor size and corresponding numbers of lung metastases after 4 weeks[91]. This situation would be consistent with NAC at very high levels inhibiting the ROS-NRF2-HIF-1-mediated metabolic reprogramming responsible for migration, invasion and survival of metastatic cancers during their metastatic colonization in the lungs. Very high NAC levels will help adapt the redox homeostasis in cancer cells back to a lower level by lowering ROS and pro-oxidative stress, increasing GSH/GSSG ratios and preventing tumor growth.
Further evidence for the role of HIF-1 in tumor metastasis was shown by treating B16F10 cells with intermittent hypoxia, in which case significantly increased levels of ROS generation and HIF-1 protein levels were obtained[92]. Mice were subjected to whole body intermittent hypoxia after implantation of B16F10 melanoma cells, which increased the number and weight of metastatic colonies growing in their lungs[92]. Examining the lungs containing tumor metastases showed greater oxidative stress assessed by increased p22phox, SOD mRNA levels and NRF2 protein levels, as well as increased inflammatory markers, TNF-α and IL-6 mRNA levels and NF-κB p65 protein levels. In these studies, mice were treated with Tempol (4-hydroxy-2,2,6,6-tetramethylpiperidine-N-oxyl), a broadly effective agent for detoxifying ROS as a cell membrane-permeable nitroxide that dismutates superoxide, facilitates hydrogen peroxide metabolism by catalase-like actions, and limits formation of hydroxyl radicals when it is reduced to the amine derivative. Thus, Tempol treatment counteracted the hypoxia/ROS-induced melanoma lung metastasis in mice by decreasing the levels of oxidative stress and inflammatory responses.
A related study using intermittent hypoxia to treat breast cancer cell lines in culture showed similar findings when cells adapted after successive rounds of hypoxia were then injected intravenously into syngeneic mice[93]. Again, as in the studies above, intermittent hypoxia treatment of breast cancer cell cultures subsequently enhanced their metastatic seeding and outgrowth into the lungs when transplanted in vivo. Furthermore, exposing these mammary tumor cells to intermittent hypoxia promoted clonal diversity, upregulated metastasis-associated gene expression, induced a pro-tumorigenic secretory profile, increased stem-like cell marker expression, and gave rise to tumor-initiating cells at a relatively higher frequency[93]. Thus, the evidence from many studies is consistent that intermittent hypoxia reprograms cancer cells by inducing a number of genetic, molecular, biochemical, and cellular changes to support tumor cell survival, colonization, and the creation of a permissive microenvironment to enhance metastatic growth.
In a study repurposing common drugs used to treat human type 2 diabetes mellitus, including the hypoglycemic dipeptidyl peptidase-4 inhibitors (DPP-4i) saxagliptin and sitagliptin, as well as α-lipoic acid, it was shown that their use did not increase the frequency of primary tumor incidence[52]. However, these drugs did increase the risk of metastasis from existing tumors[52]. Specifically, the drugs induced prolonged activation of NRF2 and a cellular antioxidant response by inhibiting KEAP1-dependent ubiquitination mediated NRF2 degradation. Therefore, it was proposed that these drugs were acting as antioxidants, which is doubtful. Rather they are more likely to be reactive drugs capable of NRF2 activation. Thus, in cellular states with heightened oxidative stress, the KEAP1 cysteine sulfhydryl groups may be modified by electrophilic reactive species that disrupt KEAP1-NRF2 interactions [Figures 2 and 3]. This would cause NRF2 release and activation to upregulate expression of metastasis-associated proteins, increase cancer cell migration, promoting metastasis, as seen in xenograft mouse models[52]. Accordingly, knockdown of NRF2 expression attenuated naturally occurring or DPP-4i-induced tumor metastasis, whereas NRF2 activation accelerated metastasis. In human liver cancer tissue samples, higher NRF2 expression correlated with metastasis[52]. Hence, this is a further mechanism whereby agents that first appear to be antioxidants, when used during cancer therapy could activate greater NRF2 signaling to promote cancer metastatic progression in patients.
Another aspect to NRF2’s role in tumorigenesis relates to the host immune response. It was shown that NRF2 deficiency in the host animal but not of the cancer cells led to increased local tumor growth in the NRF2 null mice after subcutaneous injection of wild type B16F10 melanoma cells, as indicated by an increased proportion of animals with locally palpable tumor mass and time-dependent increases in tumor volume at the primary site of injection[94]. Further, the NRF2 null host mice showed a remarkable increase in lung metastasis by B16F10 melanoma cells as compared with wild-type mice[94]. Thus, factors such as a hypoxic tumor microenvironment would normally promote an anticancer immune response, but not in the absence of any capacity for expression of NRF2 in the host stroma and immune cells. Again, the results are consistent with the proposal that the usage of systemic antioxidant therapy which will act to suppress NRF2 protein levels in host cells could also be highly counterproductive due to their inhibitory potential for host immune responses.
In summary, although dietary antioxidants may be beneficial in helping prevent carcinogenesis in the initiation stages, they appear to be ill advised in the period post-cancer diagnosis where these agents promote greater malignancy and metastatic progression by helping activate the NRF2-HIF-1 axis. Hence, a different approach will be required to enhance anticancer responses post-diagnosis which will target the specific reprogrammed differences existing in the more highly advanced/metastatic tumor cells.
NSAIDs as chemopreventatives and effective anticancer agents
The NSAIDs are the most commonly used medicine for inflammatory diseases, providing effective management of pain, fever, flushing and edema. This therapeutic benefit is ascribed to their designated (purported) function as inhibitors of the cyclooxygenases (prostaglandin-endoperoxide synthases as enzymes involved in producing pro-inflammatory prostanoids, including thromboxane and prostaglandins)[95,96]. However, extensive support for NSAIDs having other functions as anticancer drugs is emerging and this evidence is reviewed here.
First evidence for anticancer activity from chemoprevention with the use of NSAIDs in familial adenomatous polyposis
Celecoxib was originally developed as a selective cyclooxygenase-2 (COX-2) NSAID used to treat the pain and inflammation of osteoarthritis, rheumatoid arthritis, ankylosing spondylitis and other acute forms of pain. Celecoxib was designed to provide analgesia similar to the earlier NSAIDs such as ibuprofen and naproxen but offering much lower gastrointestinal side effects by not targeting COX-1. Early on, NSAIDs were recognized for lowering the risk of colorectal cancers (for review, see[97]) and in 2004, Celebrex (celecoxib) was the first to gain United States Federal Drug Admininistration (USFDA) approval for the purpose of decreasing polyp formation in familial adenomatous polyposis (FAP) [Table 1]. In this situation, celecoxib acts as a chemopreventative agent, lowering the incidence of polyp formation by about 30%, thereby impeding patients’ progression to developing advanced colorectal cancer[98]. In patients post-diagnosis after treatment for sporadic polyposis, taking celecoxib (400 mg daily) was also shown to decrease by 41% the incidence of adenoma recurrence or onset of advanced adenoma detected after 5 years[99,100]. More recently the USFDA has given the combination of another NSAID, sulindac with the ornithine decarboxylase inhibitor, eflornithine, [difluoromethylornithine (DFMO)] fast-track status for use in FAP, although it has yet to be approved. When the polyp burden was assessed for the entire colorectum by endoscopy, the DFMO/sulindac treated FAP patient group showed a lower 3-year incidence of subsequent high-risk adenomas by > 90 % vs. only a 36 % decrease (P = 0.01) in the sulindac monotherapy group. However, more clinical trials are required to complete the supportive evidence before approval can be granted[101]. A similar international randomized trial comparing combined celecoxib + DFMO to celecoxib alone showed a synergy with the combination providing an average decrease for video based assessment of global polyps by 80% vs. 33% for celecoxib alone (P = 0.03)[102]. From the above outcomes (see Table 1 for summary), it is clear that chemoprevention with NSAIDs works very successfully for colorectal cancer.
Summary of NSAID prevention against metastatic cancers in clinical trials
| Type of cancer | NSAID | Treatment | Combination with | Clinical outcome | Ref. |
|---|---|---|---|---|---|
| FAP | Celecoxib | 400 mg bi-daily | None | Lowering polyp formation (30%) | [97,98] |
| CRC | Celecoxib | 400 mg daily | None | Lowering 5-year risk of advanced adenoma by 41% | [99,100] |
| CRC | Sulindac | 150 mg daily | +DFMO (750 mg daily) | 3-year high risk adenomas lower by > 90% vs. sulindac monotherapy (30%) | [101] |
| CRC | Celecoxib | 400 mg bi-daily | +DFMO (250-1250 mg daily) | Decrease global polyp assessment by 80% vs. celecoxib monotherapy (33%) | [102] |
| Proximal colon | Aspirin | 75, 100, 300 or 600 mg/day | None | 50% reduction in deaths from CRC with metastasis free at diagnosis; 50%-70% reduction in distant metastases | [103,104] |
| CRC | Aspirin vs. other NSAIDS | Any dose | None | Long term low dose decreased CRC mortality by 56% over 10-year follow-up & by 40% post-diagnosis mortality in KRAS wild type CRC | [111] |
| Distant metastasis by Br or Pr Ca | Several NSAIDS | pre- vs. post-operative NSAID use vs. non-users | None | NSAIDS decreased incidence of metastatic cancer post-cancer diagnosis by ~ 50% | [120] |
| Unretractable metastatic CRC | Celecoxib | 200 mg bi-daily 400 mg bi-daily | FOLFOX4 Capecitabine | 45% survival at 3 years, 4 CR’s 93/195 complete response rate | [168-172] |
| REACT Her2-, resected Br Ca. | Celecoxib | 400 mg daily | 48% decreased recurrence after 2 years | [173] | |
| NSCLC meta-analysis | COXIBs | Chemotherapy | 40% increase in response rates | [174] | |
| STAMPEDE prostate cancer | Celecoxib | 400 mg bi-daily | Zoledronic acid (4 mg) | 22% increased overall survival at median follow-up of 5 years | [175] |
Evidence for NSAID based chemoprevention against colorectal cancer in general
Several more recent studies have indicated that low, non-toxic doses of NSAIDs (including the low cost drug, aspirin) should be considered for approval or at least recommended for extended use across the entire population for the chemoprevention of colorectal cancers[103]. For example, it has been shown that aspirin use was more effective than either fecal occult blood testing (RR = 0.36; 95% CI: 0.22-0.59) or flexible sigmoidoscopy (RR = 0.37; CI: 0.22-0.62) in preventing death from or cancer development in the proximal colon and was equally effective to the other screening methods for lowering the colorectal cancer incidence and mortality[104]. One biomarker for responsiveness to aspirin under consideration is the tumor phosphatidylinositol-4,5-bisphosphate 3-kinase, catalytic subunit alpha (PIK3CA) mutation linked with greater effectiveness from regular aspirin use post-diagnosis by lowering total mortality from colorectal cancer by 29%-46% (RR = 0.71; CI: 0.51-0.99, P = 0.04[105] and HR = 0.54; CI: 0.31-0.94, P = 0.01[106]). Aspirin (N-acetylsalicylic acid) extensively acetylates proteins in vivo and may also react to put salicylate groups on proteins[107]. As such, aspirin related drugs can modulate KEAP1 function[108] inducing NRF2 signaling as an antioxidant chemopreventative drug[109] or as an alternative mechanism similar to other NSAIDs by increasing ROS levels in cancer cells as outlined further below. The exact effects exerted by drugs such as NAC or aspirin on cellular redox will depend on their relative concentrations, reaction rates and affinity for GSH (pro-oxidative effect) or the Cys-thiol groups on redox regulatory proteins such as the KEAP1/NRF2 hub (antioxidant effect) vs. TrXR (antioxidant effect)[110].
NSAIDs as chemopreventatives post-cancer diagnosis lower the incidence of recurrence or metastasis
In a comprehensive study of 2,419 patients with invasive colorectal cancer during 1997-2008 from registries in the USA, Canada and Australia, with a median follow-up period of 10.8 years since diagnosis, survival in the post-diagnostic non-users was compared with NSAID users[111]. The results showed significant decrease in all-cause mortality [hazard ratio (HR) = 0.75; CI: 0.59-0.95] and marked reduction in colorectal cancer specific mortality (HR = 0.44; CI: 0.47-0.86), notably with aspirin use. By comparison, the decreased mortality from any NSAID use post-diagnosis was only significantly improved in the Kirsten Rat Sarcoma (KRAS) wild-type protein expressing tumors (HR = 0.60; CI: 0.46-0.80), but not for the more malignant KRAS-mutant tumors (HR = 1.24; CI: 0.78-1.96).
Beyond FAP and general colorectal cancer, the evidence is now sufficiently substantial for recommendations that the population consider taking NSAIDs regularly over the long term in low doses as a chemoprevention against all types of cancer[112-114]. Historically, many population-based longitudinal studies with other cancer types and patients prescribed NSAIDs have been reported, including several recent meta-analyses summarizing the findings[104,111,114,115]. The outcomes from many studies have consistently outlined the benefits accrued from using NSAIDs either in the setting of pre- or postoperative use to treat cancer[116], and particularly in a manner similar to that with FAP, by lowering risks of recurrence or progression to metastatic cancer post-diagnosis[113,117-119]. Given the abundance of recent meta-analyses, such studies will not be reviewed here except for those having a direct bearing on the main point of this review - that the NSAIDs preferentially work when used as a therapy for advanced stage metastatic disease (for a summary of the clinical evidence, in Table 1).
In particular, in this regard, a recent very large retrospective meta-analysis of the decrease in cancer metastasis with NSAID use is noteworthy and reported on data from 16 previous studies of various cancer types and a total of 202,780 participants[120]. The common observation from their analysis was the significantly lower risk ratios for distant metastasis found across the majority of cancer types comparing pre- vs. postoperative NSAID use relative to non-users [overall response rate (ORR) = 0.708; CI: 0.586-0.856, and RR = 0.484; CI: 0.393-0.595, respectively]. This included prostate cancer (pre-diagnostic use: RR = 0.874; CI: 0.787-0.97; post-diagnostic use: RR = 0.482; CI: 0.359-0.647), and breast cancer (pre-diagnostic use: RR = 0.644; CI: 0.565-0.735; post-diagnostic use: RR = 0.485; CI: 0.362-0.651). These results are typical and show that the NSAIDs in general will decrease the incidence of metastatic cancer post-cancer diagnosis by about 50%.
Enhanced clinical outcomes from using NSAIDs combined with chemotherapy for advanced stage metastatic cancers
The reasons for the consistent differences observed between pre- and post-diagnostic use or pre- vs. post-operational use, with post-use showing a much lower relative risk of cancer related mortality have yet to be conclusively identified. However, we propose that one essential basis for these differences relates to the effectiveness of the drugs with the timing of treatment (post being more important and NSAIDs are much more effective in this situation) together with the extent of metastatic burden of the disease (with the NSAIDs showing activity predominantly greater effective benefit in the context of metastatic disease for the reasons outlined below). Importantly, overall in the above large scale study, comparing to the reference non-user group, those cancer patients prescribed the NSAIDs showed a significant and marked reduction in their subsequent risk from developing metastatic tumors (RR = 0.623; CI: 0.515-0.753, P < 0.001). From these studies and many others, it can be concluded that in a majority of cases the outcomes clearly demonstrate the benefits from NSAID prescriptions after cancer diagnosis, which are commonly associated with lower all-cause mortality amongst cancer patients [Table 1]. The lowering of post-diagnostic cancer with NSAID use applies not only to FAP and colorectal cancer but also to breast[121], prostate[122,123], melanoma[124], oesophageal[125], gastrointestinal[126] and endometrial[127,128] cancers. Clearly, if the NSAIDs are utilized and administered with the correct timing and for the appropriate stages of advanced disease, they should work across all types of cancers and lower the burden caused by metastatic disease.
At this point, it should be noted that a few studies have been reported which did not find associations between aspirin or NSAID use and lower cancer mortality[121,129,130] and in some cases, they have been associated with even greater mortality[131]. Importantly, considerable caution and care must be taken with such studies where patients may be using the NSAIDs to offset pain in the terminal phases of cancers. For example, when NSAID use during the last three years of patient follow-up before death was excluded, it completely reversed the findings from one of higher mortality to a much lower mortality shown for the NSAID users relative to non-users, in line with the majority of studies. Hence, including the time period up until death (i.e., overall survival) can greatly and grossly adversely affect the observations[122]. In addition, the importance of comprehensive exposure definitions (duration of use, timing, consistency and intensity/dose) and evaluation of potential effect modification, co-morbidities or other user characteristics such as gastrointestinal and cardiovascular status, blood pressure, body mass index or obesity should also be evaluated[132].
The importance of cancer staging in the clinical benefit from the NSAIDs and why their use enhances outcomes as chemopreventatives or chemosensitizing agents that induce greater pro-oxidative stress
To summarize, different cancer cell types and stages alter the efficacy of the NSAIDs considerably when tested as anticancer agents, particularly when the bulk of supportive evidence in the clinical setting of metastatic malignant disease post-cancer diagnosis is assessed. This situation has, until now, been further compounded by a lack of a precise understanding of how the NSAIDs act to kill cancer cells (for review see[133]). Current understanding of NSAID function as anticancer agents and our recent elucidation of their mitochondrial targeting (as mitocans) is reviewed below, as well as clinical data from human trials in advanced cancer. The mounting evidence is now clear that NSAIDs, and particularly celecoxib, significantly enhance advanced cancer patient responses to the existing commonly used chemotherapies and lower the burden of metastatic disease. One of the main aims of this review is then to promote increased understanding and extended clinical usage of celecoxib when treating advanced stage metastatic disease, for example in drug unresponsive tumors like triple negative breast cancers.
Based on the deregulated redox homeostasis in cancer cells and increased ROS levels promoting tumor growth and malignant progression by metabolic reprogramming in tumors associated with enhanced antioxidant ability as a common feature, it has been proposed that tumors can be sensitized to chemotherapy and other canonical antitumor treatments by disabling antioxidant defenses (NADPH and GSH) through metabolic inhibition[134,135]. Overloading cancer cells by exacerbating oxidative stress potentiates chemotherapeutic responses and can also improve responses to radiation therapy[135,136]. Such studies underscore the importance of understanding the regulatory systems operating in cancer cells to then be able to use agents like the NSAIDs appropriately for therapeutic benefit in treating disease [Figure 4]. Whereas it has been commonly reported that the mechanism of cancer therapy obtained with NSAIDs can be ascribed to their activity as potent drugs capable of inhibiting the COX’s, attempts to link RRs in cancer patients with tumor levels of COX expression have been largely unsuccessful[137,138]. While these actions may account for a fraction of the events in response to NSAID treatment occurring in vivo, the bulk of recent evidence shows that targeting such enzyme systems is inadequate and does not explain the majority of their anticancer functions, but rather, indicates that other more important off-target activities in cancer cells do exist. We propose that one of the key targets of NSAIDs is the mitochondria in cancer cells and that NSAIDs should be repurposed for post-diagnostic therapy of cancer by exploiting pro-oxidative ROS production to kill metastatic cancer cells.
NSAIDs function as pro-oxidative anticancer drugs independent of COX or other enzymatic inhibition
Several lines of evidence have convincingly shown that COX inhibition is not the main mode of action for the anticancer effectiveness of NSAIDs. First, comparing the relative anticancer activities and structure/function of the different NSAIDs revealed that their actions as anticancer agents usually involve higher or lower drug concentrations than the inhibition constant (Ki or Kd) required to inhibit the COX activities[139-141], with many working independently of their COX inhibitory potential. For instance, doses of acetylsalicylic acid used to decrease inflammation are much higher than those required to inhibit COX activity. Second, several studies have established that NSAID derivatives and homologs that do not inhibit COX function, nevertheless exhibit undiminished anticancer responses[142-145]. Third, the evidence shows COX inhibitors to be equally effective against COX-null cancer cell lines[146,147]. Thus, on multiple bases, it can be concluded that COX inhibition is not the predominant driver of the anticancer effect exhibited by this class of drugs.
Similar to the other NSAIDs, structurally related homologs of celecoxib exist with even greater potency as anticancer agents, but they do not bind or inhibit COX’s. For example, dimethylcelecoxib is a COX-null celecoxib derivative containing an additional methyl group compared to the prototypic 3-methylcelecoxib. Zhu et al.[148] (2002) showed that modifying the side groups and enlargement of the hydrophobic aryl moiety by adding the second methyl group (as 2,5-dimethylcelecoxib or more specifically 4-(5-(2,5-Dimethylphenyl)-3-(trifluoromethyl)-1H-pyrazol-1-yl)benzenesulfonamide) promoted apoptosis more effectively than celecoxib. This mechanism is in contrast to the action of celecoxib in COX-2 inhibition, which has stringent requirements in regard to the stereo-specific arrangement of the 3’ methyl group on celecoxib and the benzenesulphonamide moiety. Another COX-null homolog of celecoxib, E7123 or 4-(5-(2,5-dimethylphenyl)-3-(trifluoromethyl)-4,5-dihydro-1H-pyrazol-1-yl)benzenesulfonamide was also much more potent than celecoxib in killing cancer cells[149-151].
NSAIDs target mitochondrial ROS production to trigger apoptosis of metastatic cancer cells and cancer stem cells and celecoxib is a potent exemplar
Previously, we reviewed the role of NSAIDs as “hitting the bulls-eye” in cancer cells by targeting mitochondrial function to trigger cell death via the intrinsic mitochondrial pathway of apoptosis[152]. Our more recent studies examined five different NSAIDs and showed that adding them to metastatic cancer cell lines in culture resulted in a progressive increase in ROS production from mitochondria to trigger ensuing cytotoxicity, by activating the intrinsic apoptotic signaling pathway. Celecoxib showed much greater potency than the other NSAIDs tested. Similar observations applied to isolated and purified preparations of mitochondria where upon addition of celecoxib in low micromolar concentrations abruptly induced production of superoxide by disrupting the respiratory chain electron transfer and mitochondrial metabolism thereby inducing ROS production directly from the mitochondria[153]. These results applied to mitochondria isolated from both normal tissues and hepatoma cells. Thus, our data indicate that when the mitochondria are removed from their normal intracellular milieu with the cytosol full of antioxidant systems, they become very sensitive to the direct action of celecoxib on ROS production. Furthermore, we showed[153,154] that one aspect of celecoxib’s activities important for cancer cell death is that it can, at sufficiently high levels, directly inhibit mitochondrial respiration, the transmembrane electrical potential and ATP production and induces excess superoxide as a by-product from the electron transport chain, which in turn, triggers caspase activation and apoptosis of cancer cells. Thus, even at low levels, celecoxib interferes with the mitochondrial respiratory pathway of cancer cells to promote excessive ROS production[153]. Moreover, celecoxib at doses assayed to block OxPhos and cellular growth (10 μmol/L) severely decreased triple negative breast cancer cell (MDA-MB-231 and MDA-MB-468) migration (60%) and invasiveness (25%-55%) potential[154]. Celecoxib was recently shown to inhibit breast cancer stem cell self-renewal, sensitize against chemo-resistance, inhibit EMT, and attenuate metastasis and tumorigenesis[155]. Although a similar report on bladder cancer suggested that the mechanism for the actions of celecoxib on cancer stem cells is mediated by inhibiting COX-2 and prostaglandin synthesis[156], this is unlikely to be the cause given the evidence cited above for mitochondrial pro-oxidant activity.
Celecoxib (1-10 μmol/L) treatment of J774 myelomonocytic leukemia cells, vascular smooth muscle cells or human umbilical vein and aortic endothelial cells has been shown to increase mitochondrial ROS and NRF2 nuclear activation via PI3K/Akt, p-38 and p-ERK signaling[157,158] or AMPK/CREB[159]. Celecoxib together with hypoxia produced greater expression levels of heme oxygenase HO-1[157]. This activation was inhibited by pretreating cells for 30 min with 10 mmol/L NAC and was COX-independent but was not seen with rofecoxib, ibuprofen, naproxen or indomethacin[157-159]. It follows that celecoxib should uniquely activate NRF2 inside metastatic cancer cells or cancer stem cells, but this is unlikely to be sufficient to protect against the excessive mitochondrial ROS overloading the antioxidant system with ensuing cytotoxicity [Figure 4].
Celecoxib in combination with chemotherapy synergistically improves responses against advanced stage metastatic disease in pre-clinical animal models of cancer
Although many reports of pre-clinical studies with animal or human xenografted cancer cell lines treated with NSAIDs have been published, only celecoxib will be reviewed here where the focus has been on celecoxib and its exceptional ability to target metastastic cancer cells and synergize with chemotherapy. Thus, synergistic anticancer effects have been attained by combining celecoxib in murine models of colorectal cancer with either 5-fluorouracil (5-FU)[160] or with oxaliplatin[161]; in melanoma models with dacarbazine[162] or with doxorubicin for metastatic murine breast cancer[163]. Hence, the pre-clinical findings are consistent with the ability of celecoxib to chemosensitize cancer cells rendering them more susceptible to other anticancer drugs. Drugs like celecoxib have proven to offer further advantages in that they have been shown to kill cancer cells independently of MDR[164,165] or p53 or DNA mismatch repair enzymes (reviewed in[152,166]), because as we have shown, they kill by targeting mitochondrial metabolism[152-154,167].
Celecoxib in combination with chemotherapy has shown curative efficacy in clinical trials of advanced stage metastatic human cancers
An extensive analysis of NSAIDs and their use to treat human cancer is beyond the scope of this review. A basic Pubmed search restricted to celecoxib with the key words "clinical trial", "cancer" and "celecoxib" provided about 424 studies. In the site https://clinicaltrials.gov, 359 listed studies include celecoxib and cancer for either the prevention, treatment and decreased symptoms or cancer recurrence for a wide range of cancers including: breast, bladder, pancreatic, colorectal, lung, head and neck, prostate, ovarian, uterine, liver and bile duct, cervical and renal. The main message from these studies is that where the NSAIDs have been combined with the standard of care treatments in the clinical setting for advanced stage metastatic disease, they have often shown significant improvements in outcomes. The successful results of clinical trials where celecoxib has been combined with commonly used chemotherapies are summarized in Table 1 and as follows.
A “curative” efficacy was reported following combination standard of care chemotherapy (FOLFOX4) with celecoxib for advanced unresectable metastatic adenocarcinoma of the colorectal area[168]. The Activate tumor from Dormancy And Potentiate its Targeting (ADAPT) phase II trial examined capecitabine and celecoxib ± radiation following first-line chemotherapy and showed a higher complete response (CR) rate and prolonged survival with the celecoxib combination in stage IV unresectable metastatic colorectal cancer patients at the 10-year follow-up[169-172]. The Randomised EuropeAn celecoxib trial (REACT) of primary breast cancer subgroup analysis after 5-year follow-up showed that the 655 breast cancer patients who did not have subsequent adjuvant chemotherapy, nevertheless greatly benefited from being prescribed celecoxib, with a decreased recurrence (HR = 0.62; CI: 0.38-1.00)[173]. In non small cell lung cancer (NSCLC), a statistically significant improved response with COX-2 inhibitors added to first-line treatment was reported for advanced stage disease (RR = 1.39; CI: 1.19-1.63). Increased ORRs were also observed with COX-2 inhibitors added to chemotherapy (RR = 1.40; CI: 1.20-1.63)[174]. In the large Systemic Therapy in Advancing or Metastatic Prostate Cancer: Evaluation of Drug Efficacy (STAMPEDE) trial, similar to the REACT breast cancer study above, subgroup analysis of patients with metastases at baseline showed a significant improvement in both the overall survival (HR = 0.78; CI, 0.62-0.99, P = 0.04) and failure-free survival (HR = 0.77; CI: 0.63-0.93, P = 0.008) for the celecoxib/zoledronic acid group compared with the control group[175].
Conclusion
It is becoming clear from the greater understanding of differences occurring in tumor cells during the progression to advanced stages of metastatic disease that a precise basis exists for specifically targeting such tumors and eliminating them. Thus, with reversible or irreversible changes in the NRF2-HIF-1 axis, redox mediated reprogramming of gene expression occurs associated with metabolic change and greater endemic mitochondrial ROS/pro-oxidative states. Hence, we have now identified a specific cancer drug target, the mitochondria. Based on the evidence, we can conclude that use of antioxidant strategies is ill advisable after cancer diagnosis, as it is too late to prevent tumors from arising, but instead will promote their further metastatic progression. However, pro-oxidative agents like celecoxib which target mitochondrial ROS production to further tip the redox balance over and beyond the limits of cell survival by overwhelming the antioxidant defense systems in these tumor cells, will cause their mass destruction. This pro-oxidative overkill synergizes when combined with standard chemotherapeutic treatments targeting other aspects of cancer cell replication and survival, significantly improving patient responses and survival with post-diagnosis treatment, lowering recurrence rates. Such combination therapies have even shown significant curative benefits for hitherto refractory tumors. The implications from these findings are that repurposing drugs such as NSAIDs like celecoxib or other agents that work in similar fashion should be highly encouraged, as should their use in more clinical trials of metastatic disease and where biomarkers such as constitutive NRF2-HIF protein expression are well defined.
Declarations
AcknowledgmentsThe authors thank Prof. R.K. Ralph for proofreading and editing of the manuscript.
Authors’ contributionsDesign: Ralph SJ, Pritchard R
Literature research, data analysis: Ralph SJ, Nozuhur S, Moreno-Sánchez R, Rodríguez-Enríquez S, Pritchard R
Manuscript writing: Ralph SJ
Manuscript editing and revision: Ralph S J, Pritchard R, Moreno-Sánchez R, Enríquez SR
Availability of data and materialsNot applicable.
Financial support and sponsorshipThis work was partially supported by CONACyT-Mexico grants No. 239930 and 281428 to RMS and 283144 to SRE. Rhys Pritchard was supported by an Australian postgraduate research award (APRA).
Conflicts of interestAll authors declared that there are no conflicts of interest.
Ethical approval and consent to participateNot applicable.
Consent for publicationNot applicable.
Copyright© The Author(s) 2018.
REFERENCES
1. Freitas M, Baldeiras I, Proenca T, Alves V, Mota-Pinto A, Sarmento-Ribeiro A. Oxidative stress adaptation in aggressive prostate cancer may be counteracted by the reduction of glutathione reductase. FEBS Open Bio 2012;2:119-28.
3. Peiris-Pagès M, Martinez-Outschoorn UE, Sotgia F, Lisanti MP. Metastasis and oxidative stress: are antioxidants a metabolic driver of progression? Cell Metab 2015;22:956-8.
4. Piantadosi CA, Carraway MS, Babiker A, Suliman HB. Heme oxygenase-1 regulates cardiac mitochondrial biogenesis via Nrf2-mediated transcriptional control of nuclear respiratory factor-1. Circ Res 2008;103:1232-40.
5. Rodic S, Vincent MD. Reactive oxygen species (ROS) are a key determinant of cancer's metabolic phenotype. Int J Cancer 2018;142:440-8.
6. Wang J, Yi J. Cancer cell killing via ROS: to increase or decrease, that is the question. Cancer Biol Ther 2008;7:1875-84.
7. Rojo de la Vega M, Chapman E, Zhang DD. NRF2 and the hallmarks of cancer. Cancer Cell 2018;34:21-43.
8. Toth RK, Warfel NA. Strange bedfellows: nuclear factor, erythroid 2-Like 2 (Nrf2) and hypoxia-inducible factor 1 (HIF-1) in tumor hypoxia. Antioxidants (Basel) 2017;6:pii: E27.
9. Athale J, Ulrich A, MacGarvey NC, Bartz RR, Welty-Wolf KE, Suliman HB, Piantadosi CA. Nrf2 promotes alveolar mitochondrial biogenesis and resolution of lung injury in Staphylococcus aureus pneumonia in mice. Free Radic Biol Med 2012;53:1584-94.
10. MacGarvey NC, Suliman HB, Bartz RR, Fu P, Withers CM, Welty-Wolf KE, Piantadosi CA. Activation of mitochondrial biogenesis by heme oxygenase-1-mediated NF-E2-related factor-2 induction rescues mice from lethal Staphylococcus aureus sepsis. Am J Respir Crit Care Med 2012;185:851-61.
11. Dominy JE, Puigserver P. Mitochondrial biogenesis through activation of nuclear signaling proteins. Cold Spring Harb Perspect Biol 2013;5:pii: a015008.
12. Cherry AD, Suliman HB, Bartz RR, Piantadosi CA. Peroxisome proliferator-activated receptor γ co-activator 1-α as a critical co-activator of the murine hepatic oxidative stress response and mitochondrial biogenesis in Staphylococcus aureus sepsis. J Biol Chem 2014;289:41-52.
13. Scarpulla RC. Nuclear control of respiratory chain expression by nuclear respiratory factors and PGC-1-related coactivator. Ann N Y Acad Sci 2008;1147:321-34.
14. Ralph SJ, Rodríguez-Enríquez S, Neuzil J, Saavedra E, Moreno-Sánchez R. The causes of cancer revisited: "mitochondrial malignancy" and ROS-induced oncogenic transformation - why mitochondria are targets for cancer therapy. Mol Aspects Med 2010;31:145-70.
15. Handschin C, Spiegelman BM. Peroxisome proliferator-activated receptor gamma coactivator 1 coactivators, energy homeostasis, and metabolism. Endocr Rev 2006;27:728-35.
16. Aquilano K, Baldelli S, Pagliei B, Cannata SM, Rotilio G, Ciriolo MR. p53 orchestrates the PGC-1α-mediated antioxidant response upon mild redox and metabolic imbalance. Antioxid Redox Signal 2013;18:386-99.
17. Yin W, Signore AP, Iwai M, Cao G, Gao Y, Chen J. Rapidly increased neuronal mitochondrial biogenesis after hypoxic-ischemic brain injury. Stroke 2008;39:3057-63.
18. Gravel SP. Deciphering the dichotomous effects of PGC-1α on tumorigenesis and metastasis. Front Oncol 2018;8:75.
19. LeBleu VS, O'Connell JT, Gonzalez Herrera KN, Wikman H, Pantel K, Haigis MC, de Carvalho FM, Damascena A, Domingos Chinen LT, Rocha RM, Asara JM, Kalluri R. PGC-1α mediates mitochondrial biogenesis and oxidative phosphorylation in cancer cells to promote metastasis. Nat Cell Biol 2014;16:992-1003, 1-15.
20. LaGory EL, Wu C, Taniguchi CM, Ding CC, Chi JT, von Eyben R, Scott DA, Richardson AD, Giaccia AJ. Suppression of PGC-1α is critical for reprogramming oxidative metabolism in renal cell carcinoma. Cell Rep 2015;12:116-27.
21. Taguchi A, Delgado O, Celiktaş M, Katayama H, Wang H, Gazdar AF, Hanash SM. Proteomic signatures associated with p53 mutational status in lung adenocarcinoma. Proteomics 2014;14:2750-9.
22. Vazquez F, Lim JH, Chim H, Bhalla K, Girnun G, Pierce K, Clish CB, Granter SR, Widlund HR, Spiegelman BM, Puigserver P. PGC1α expression defines a subset of human melanoma tumors with increased mitochondrial capacity and resistance to oxidative stress. Cancer Cell 2013;23:287-301.
23. Choi HI, Kim HJ, Park JS, Kim IJ, Bae EH, Ma SK, Kim SW. PGC-1α attenuates hydrogen peroxide-induced apoptotic cell death by upregulating Nrf-2 via GSK3β inactivation mediated by activated p38 in HK-2 Cells. Sci Rep 2017;7:4319.
24. Sen N, Satija YK, Das S. PGC-1α, a key modulator of p53, promotes cell survival upon metabolic stress. Mol Cell 2011;44:621-34.
25. Popowich DA, Vavra AK, Walsh CP, Bhikhapurwala HA, Rossi NB, Jiang Q, Aalami OO, Kibbe MR. Regulation of reactive oxygen species by p53: implications for nitric oxide-mediated apoptosis. Am J Physiol Heart Circ Physiol 2010;298:H2192-200.
26. Lisek K, Campaner E, Ciani Y, Walerych D, Del Sal G. Mutant p53 tunes the NRF2-dependent antioxidant response to support survival of cancer cells. Oncotarget 2018;9:20508-23.
27. Clemons NJ, Liu DS, Duong CP, Phillips WA. Inhibiting system xC- and glutathione biosynthesis - a potential Achilles' heel in mutant-p53 cancers. Mol Cell Oncol 2017;4:e1344757.
28. Liu DS, Duong CP, Haupt S, Montgomery KG, House CM, Azar WJ, Pearson HB, Fisher OM, Read M, Guerra GR, Haupt Y, Cullinane C, Wiman KG, Abrahmsen L, Phillips WA, Clemons NJ. Inhibiting the system xC-/glutathione axis selectively targets cancers with mutant-p53 accumulation. Nat Commun 2017;8:14844.
29. Chen D, Tavana O, Chu B, Erber L, Chen Y, Baer R, Gu W. NRF2 is a major target of ARF in p53-independent tumor suppression. Mol Cell 2017;68:224-32.
30. Todoric J, Antonucci L, Di Caro G, Li N, Wu X, Lytle NK, Dhar D, Banerjee S, Fagman JB, Browne CD, Umemura A, Valasek MA, Kessler H, Tarin D, Goggins M, Reya T, Diaz-Meco M, Moscat J, Karin M. Stress-activated NRF2-MDM2 cascade controls neoplastic progression in pancreas. Cancer Cell 2017;32:824-39.
31. Komatsu M, Kurokawa H, Waguri S, Taguchi K, Kobayashi A, Ichimura Y, Sou YS, Ueno I, Sakamoto A, Tong KI, Kim M, Nishito Y, Iemura S, Natsume T, Ueno T, Kominami E, Motohashi H, Tanaka K, Yamamoto M. The selective autophagy substrate p62 activates the stress responsive transcription factor Nrf2 through inactivation of Keap1. Nat Cell Biol 2010;12:213-23.
32. Kapuy O, Papp D, Vellai T, Bánhegyi G, Korcsmáros T. Systems-level feedbacks of NRF2 controlling autophagy upon oxidative stress response. Antioxidants (Basel) 2018;7:pii: E39.
33. Li P, Zhang D, Shen L, Dong K, Wu M, Ou Z, Shi D. Redox homeostasis protects mitochondria through accelerating ROS conversion to enhance hypoxia resistance in cancer cells. Sci Rep 2016;6:22831.
34. Kim B, Song YS. Mitochondrial dynamics altered by oxidative stress in cancer. Free Radic Res 2016;50:1065-70.
35. Lo SC, Hannink M. PGAM5 tethers a ternary complex containing Keap1 and Nrf2 to mitochondria. Exp Cell Res 2008;314:1789-803.
36. Tian H, Zhang B, Di J, Jiang G, Chen F, Li H, Li L, Pei D, Zheng J. Keap1: one stone kills three birds Nrf2, IKKβ and Bcl-2/Bcl-xL. Cancer Lett 2012;325:26-34.
37. Giudice A, Arra C, Turco MC. Review of molecular mechanisms involved in the activation of the Nrf2-ARE signaling pathway by chemopreventive agents. Methods Mol Biol 2010;647:37-74.
38. Dinkova-Kostova AT, Fahey JW, Kostov RV, Kensler TW. KEAP1 and done? Targeting the NRF2 pathway with sulforaphane. Trends Food Sci Technol 2017;69:257-69.
39. Sihvola V, Levonen AL. Keap1 as the redox sensor of the antioxidant response. Arch Biochem Biophys 2017;617:94-100.
40. Jaramillo MC, Zhang DD. The emerging role of the Nrf2-Keap1 signaling pathway in cancer. Genes Dev 2013;27:2179-91.
41. Dinkova-Kostova AT, Holtzclaw WD, Cole RN, Itoh K, Wakabayashi N, Katoh Y, Yamamoto M, Talalay P. Direct evidence that sulfhydryl groups of Keap1 are the sensors regulating induction of phase 2 enzymes that protect against carcinogens and oxidants. Proc Natl Acad Sci U S A 2002;99:11908-13.
42. Dinkova-Kostova AT, Kostov RV, Canning P. Keap1, the cysteine-based mammalian intracellular sensor for electrophiles and oxidants. Arch Biochem Biophys 2017;617:84-93.
43. Canning P, Bullock AN. New strategies to inhibit KEAP1 and the Cul3-based E3 ubiquitin ligases. Biochem Soc Trans 2014;42:103-7.
44. Leinonen HM, Kansanen E, Pölönen P, Heinäniemi M, Levonen AL. Dysregulation of the Keap1-Nrf2 pathway in cancer. Biochem Soc Trans 2015;43:645-9.
45. Cancer Genome Atlas Research Network. Comprehensive genomic characterization of squamous cell lung cancers. Nature 2012;489:519-25.
46. Hanada N, Takahata T, Zhou Q, Ye X, Sun R, Itoh J, Ishiguro A, Kijima H, Mimura J, Itoh K, Fukuda S, Saijo Y. Methylation of the KEAP1 gene promoter region in human colorectal cancer. BMC Cancer 2012;12:66.
47. Satoh H, Moriguchi T, Takai J, Ebina M, Yamamoto M. Nrf2 prevents initiation but accelerates progression through the Kras signaling pathway during lung carcinogenesis. Cancer Res 2013;73:4158-68.
48. Tao S, Rojo de la Vega M, Chapman E, Ooi A, Zhang DD. The effects of NRF2 modulation on the initiation and progression of chemically and genetically induced lung cancer. Mol Carcinog 2018;57:182-92.
49. Tochhawng L, Deng S, Pervaiz S, Yap CT. Redox regulation of cancer cell migration and invasion. Mitochondrion 2013;13:246-53.
50. Jeddi F, Soozangar N, Sadeghi MR, Somi MH, Samadi N. Contradictory roles of Nrf2/Keap1 signaling pathway in cancer prevention/promotion and chemoresistance. DNA Repair (Amst) 2017;54:13-21.
51. Sadeghi MR, Jeddi F, Soozangar N, Somi MH, Samadi N. The role of Nrf2-Keap1 axis in colorectal cancer, progression, and chemoresistance. Tumour Biol 2017;39:1010428317705510.
52. Wang H, Liu X, Long M, Huang Y, Zhang L, Zhang R, Zheng Y, Liao X, Wang Y, Liao Q, Li W, Tang Z, Tong Q, Wang X, Fang F, Rojo de la Vega M, Ouyang Q, Zhang DD, Yu S, Zheng H. NRF2 activation by antioxidant antidiabetic agents accelerates tumor metastasis. Sci Transl Med 2016;8:334ra51.
53. Zhang C, Wang HJ, Bao QC, Wang L, Guo TK, Chen WL, Xu LL, Zhou HS, Bian JL, Yang YR, Sun HP, Xu XL, You QD. NRF2 promotes breast cancer cell proliferation and metastasis by increasing RhoA/ROCK pathway signal transduction. Oncotarget 2016;7:73593-606.
54. DeNicola GM, Karreth FA, Humpton TJ, Gopinathan A, Wei C, Frese K, Mangal D, Yu KH, Yeo CJ, Calhoun ES, Scrimieri F, Winter JM, Hruban RH, Iacobuzio-Donahue C, Kern SE, Blair IA, Tuveson DA. Oncogene-induced Nrf2 transcription promotes ROS detoxification and tumorigenesis. Nature 2011;475:106-9.
55. Ji L, Li H, Gao P, Shang G, Zhang DD, Zhang N, Jiang T. Nrf2 pathway regulates multidrug-resistance-associated protein 1 in small cell lung cancer. PLoS One 2013;8:e63404.
56. Jeddi F, Soozangar N, Sadeghi MR, Somi MH, Shirmohamadi M, Eftekhar-Sadat AT, Samadi N. Nrf2 overexpression is associated with P-glycoprotein upregulation in gastric cancer. Biomed Pharmacother 2018;97:286-92.
57. Xu B, Zhang J, Strom J, Lee S, Chen QM. Myocardial ischemic reperfusion induces de novo Nrf2 protein translation. Biochim Biophys Acta 2014;1842:1638-47.
58. Hawkins KE, Joy S, Delhove JM, Kotiadis VN, Fernandez E, Fitzpatrick LM, Whiteford JR, King PJ, Bolanos JP, Duchen MR, Waddington SN, McKay TR. NRF2 orchestrates the metabolic shift during induced pluripotent stem cell reprogramming. Cell Rep 2016;14:1883-91.
59. Chang CW, Chen YS, Tsay YG, Han CL, Chen YJ, Yang CC, Hung KF, Lin CH, Huang TY, Kao SY, Lee TC, Lo JF. ROS-independent ER stress-mediated NRF2 activation promotes warburg effect to maintain stemness-associated properties of cancer-initiating cells. Cell Death Dis 2018;9:194.
60. Malec V, Gottschald OR, Li S, Rose F, Seeger W, Hänze J. HIF-1 alpha signaling is augmented during intermittent hypoxia by induction of the Nrf2 pathway in NOX1-expressing adenocarcinoma A549 cells. Free Radic Biol Med 2010;48:1626-35.
61. Philip B, Ito K, Moreno-Sánchez R, Ralph SJ. HIF expression and the role of hypoxic microenvironments within primary tumours as protective sites driving cancer stem cell renewal and metastatic progression. Carcinogenesis 2013;34:1699-707.
62. Niecknig H, Tug S, Reyes BD, Kirsch M, Fandrey J, Berchner-Pfannschmidt U. Role of reactive oxygen species in the regulation of HIF-1 by prolyl hydroxylase 2 under mild hypoxia. Free Radic Res 2012;46:705-17.
63. Semenza GL. Hypoxia-inducible factors: coupling glucose metabolism and redox regulation with induction of the breast cancer stem cell phenotype. EMBO J 2017;36:252-9.
64. Marín-Hernández A, Gallardo-Pérez JC, Ralph SJ, Rodríguez-Enríquez S, Moreno-Sánchez R. HIF-1alpha modulates energy metabolism in cancer cells by inducing over-expression of specific glycolytic isoforms. Mini Rev Med Chem 2009;9:1084-101.
65. Okamoto A, Sumi C, Tanaka H, Kusunoki M, Iwai T, Nishi K, Matsuo Y, Harada H, Takenaga K, Bono H, Hirota K. HIF-1-mediated suppression of mitochondria electron transport chain function confers resistance to lidocaine-induced cell death. Sci Rep 2017;7:3816.
66. Zhang H, Gao P, Fukuda R, Kumar G, Krishnamachary B, Zeller KI, Dang CV, Semenza GL. HIF-1 inhibits mitochondrial biogenesis and cellular respiration in VHL-deficient renal cell carcinoma by repression of C-MYC activity. Cancer Cell 2007;11:407-20.
67. Fukuda R, Zhang H, Kim JW, Shimoda L, Dang CV, Semenza GL. HIF-1 regulates cytochrome oxidase subunits to optimize efficiency of respiration in hypoxic cells. Cell 2007;129:111-22.
68. Harris IS, Treloar AE, Inoue S, Sasaki M, Gorrini C, Lee KC, Yung KY, Brenner D, Knobbe-Thomsen CB, Cox MA, Elia A, Berger T, Cescon DW, Adeoye A, Brüstle A, Molyneux SD, Mason JM, Li WY, Yamamoto K, Wakeham A, Berman HK, Khokha R, Done SJ, Kavanagh TJ, Lam CW, Mak TW. Glutathione and thioredoxin antioxidant pathways synergize to drive cancer initiation and progression. Cancer Cell 2015;27:211-22.
69. Piskounova E, Agathocleous M, Murphy MM, Hu Z, Huddlestun SE, Zhao Z, Leitch AM, Johnson TM, DeBerardinis RJ, Morrison SJ. Oxidative stress inhibits distant metastasis by human melanoma cells. Nature 2015;527:186-91.
70. Zhang L, Zhu Z, Liu J, Zhu Z, Hu Z. Protective effect of N-acetylcysteine (NAC) on renal ischemia/reperfusion injury through Nrf2 signaling pathway. J Recept Signal Transduct Res 2014;34:396-400.
71. Ji L, Liu R, Zhang XD, Chen HL, Bai H, Wang X, Zhao HL, Liang X, Hai CX. N-acetylcysteine attenuates phosgene-induced acute lung injury via up-regulation of Nrf2 expression. Inhal Toxicol 2010;22:535-42.
72. Wang LL, Huang YH, Yan CY, Wei XD, Hou JQ, Pu JX, Lv JX. N-acetylcysteine ameliorates prostatitis via miR-141 regulating Keap1/Nrf2 signaling. Inflammation 2016;39:938-47.
73. Moss RW. Should patients undergoing chemotherapy and radiotherapy be prescribed antioxidants? Integr Cancer Ther 2006;5:63-82.
75. Hiramoto K, Satoh H, Suzuki T, Moriguchi T, Pi J, Shimosegawa T, Yamamoto M. Myeloid lineage-specific deletion of antioxidant system enhances tumor metastasis. Cancer Prev Res (Phila) 2014;7:835-44.
76. Le Gal K, Ibrahim MX, Wiel C, Sayin VI, Akula MK, Karlsson C, Dalin MG, Akyürek LM, Lindahl P, Nilsson J, Bergo MO. Antioxidants can increase melanoma metastasis in mice. Sci Transl Med 2015;7:308re8.
77. Cairns RA, Harris IS, Mak TW. Regulation of cancer cell metabolism. Nat Rev Cancer 2011;11:85-95.
78. Sayin VI, Ibrahim MX, Larsson E, Nilsson JA, Lindahl P, Bergo MO. Antioxidants accelerate lung cancer progression in mice. Sci Transl Med 2014;6:221ra15.
79. Zhang Z, Yan J, Taheri S, Liu KJ, Shi H. Hypoxia-inducible factor 1 contributes to N-acetylcysteine's protection in stroke. Free Radic Biol Med 2014;68:8-21.
80. Haddad JJ, Olver RE, Land SC. Antioxidant/pro-oxidant equilibrium regulates HIF-1alpha and NF-kappa B redox sensitivity. Evidence for inhibition by glutathione oxidation in alveolar epithelial cells. J Biol Chem 2000;275:21130-9.
81. Jin WS, Kong ZL, Shen ZF, Jin YZ, Zhang WK, Chen GF. Regulation of hypoxia inducible factor-1α expression by the alteration of redox status in HepG2 cells. J Exp Clin Cancer Res 2011;30:61.
82. Aruoma OI, Halliwell B, Hoey BM, Butler J. The antioxidant action of N-acetylcysteine: its reaction with hydrogen peroxide, hydroxyl radical, superoxide, and hypochlorous acid. Free Radic Biol Med 1989;6:593-7.
83. Samuni Y, Goldstein S, Dean OM, Berk M. The chemistry and biological activities of N-acetylcysteine. Biochim Biophys Acta 2013;1830:4117-29.
84. Ezeriņa D, Takano Y, Hanaoka K, Urano Y, Dick TP. N-Acetyl cysteine functions as a fast-acting antioxidant by triggering intracellular H2S and sulfane sulfur production. Cell Chem Biol 2018;25:447-59.
85. Kim KY, Rhim T, Choi I, Kim SS. N-acetylcysteine induces cell cycle arrest in hepatic stellate cells through its reducing activity. J Biol Chem 2001;276:40591-8.
86. Hayes JD, Dinkova-Kostova AT. Epigenetic control of NRF2-directed cellular antioxidant status in dictating life-death decisions. Mol Cell 2017;68:5-7.
87. Pehar M, Ball LE, Sharma DR, Harlan BA, Comte-Walters S, Neely BA, Vargas MR. Changes in protein expression and lysine acetylation induced by decreased glutathione levels in astrocytes. Mol Cell Proteomics 2016;15:493-505.
88. Sceneay J, Liu MC, Chen A, Wong CS, Bowtell DD, Möller A. The antioxidant N-acetylcysteine prevents HIF-1 stabilization under hypoxia in vitro but does not affect tumorigenesis in multiple breast cancer models in vivo. PLoS One 2013;8:e66388.
89. Gao P, Zhang H, Dinavahi R, Li F, Xiang Y, Raman V, Bhujwalla ZM, Felsher DW, Cheng L, Pevsner J, Lee LA, Semenza GL, Dang CV. HIF-dependent antitumorigenic effect of antioxidants in vivo. Cancer Cell 2007;12:230-8.
90. Zhao T, Zhu Y, Morinibu A, Kobayashi M, Shinomiya K, Itasaka S, Yoshimura M, Guo G, Hiraoka M, Harada H. HIF-1-mediated metabolic reprogramming reduces ROS levels and facilitates the metastatic colonization of cancers in lungs. Sci Rep 2014;4:3793.
91. Albini A, D'Agostini F, Giunciuglio D, Paglieri I, Balansky R, De Flora S. Inhibition of invasion, gelatinase activity, tumor take and metastasis of malignant cells by N-acetylcysteine. Int J Cancer 1995;61:121-9.
92. Li L, Ren F, Qi C, Xu L, Fang Y, Liang M, Feng J, Chen B, Ning W, Cao J. Intermittent hypoxia promotes melanoma lung metastasis via oxidative stress and inflammation responses in a mouse model of obstructive sleep apnea. Respir Res 2018;19:28.
93. Chen A, Sceneay J, Gödde N, Kinwel T, Ham S, Thompson EW, Humbert PO, Möller A. Intermittent hypoxia induces a metastatic phenotype in breast cancer. Oncogene 2018;37:4214-25.
94. Zhu H, Jia Z, Trush MA, Li YR. Nrf2 deficiency promotes melanoma growth and lung metastasis. React Oxyg Species (Apex) 2016;2:308-14.
95. Süleyman H, Demircan B, Karagöz Y. Anti-inflammatory and side effects of cyclooxygenase inhibitors. Pharmacol Rep 2007;59:247-58.
96. Patrignani P, Patrono C. Cyclooxygenase inhibitors: from pharmacology to clinical read-outs. Biochim Biophys Acta 2015;1851:422-32.
97. Tsioulias GJ, Go MF, Rigas B. NSAIDs and colorectal cancer control: promise and challenges. Curr Pharmacol Rep 2015;1:295-301.
98. Steinbach G, Lynch PM, Phillips RK, Wallace MH, Hawk E, Gordon GB, Wakabayashi N, Saunders B, Shen Y, Fujimura T, Su LK, Levin B, Godio L, Patterson S, Rodriguez-Bigas MA, Jester SL, King KL, Schumacher M, Abbruzzese J, DuBois RN, Hittelman WN, Zimmerman S, Sherman JW, Kelloff G. The effect of celecoxib, a cyclooxygenase-2 inhibitor, in familial adenomatous polyposis. N Engl J Med 2000;342:1946-52.
99. Arber N, Eagle CJ, Spicak J, Rácz I, Dite P, Hajer J, Zavoral M, Lechuga MJ, Gerletti P, Tang J, Rosenstein RB, Macdonald K, Bhadra P, Fowler R, Wittes J, Zauber AG, Solomon SD, Levin B; PreSAP Trial Investigators. Celecoxib for the prevention of colorectal adenomatous polyps. N Engl J Med 2006;355:885-95.
100. Arber N, Spicak J, Rácz I, Zavoral M, Breazna A, Gerletti P, Lechuga MJ, Collins N, Rosenstein RB, Eagle CJ, Levin B. Five-year analysis of the prevention of colorectal sporadic adenomatous polyps trial. Am J Gastroenterol 2011;106:1135-46.
101. Burke CA, Dekker E, Samadder NJ, Stoffel E, Cohen A. Efficacy and safety of eflornithine (CPP-1X)/sulindac combination therapy versus each as monotherapy in patients with familial adenomatous polyposis (FAP): design and rationale of a randomized, double-blind, Phase III trial. BMC Gastroenterol 2016;16:87.
102. Lynch PM, Burke CA, Phillips R, Morris JS, Slack R, Wang X, Liu J, Patterson S, Sinicrope FA, Rodriguez-Bigas MA, Half E, Bulow S, Latchford A, Clark S, Ross WA, Malone B, Hasson H, Richmond E, Hawk E. An international randomised trial of celecoxib versus celecoxib plus difluoromethylornithine in patients with familial adenomatous polyposis. Gut 2016;65:286-95.
103. Burn J, Sheth H. The role of aspirin in preventing colorectal cancer. Br Med Bull 2016;119:17-24.
104. Emilsson L, Holme Ø, Bretthauer M, Cook NR, Buring JE, Løberg M, Adami HO, Sesso HD, Gaziano MJ, Kalager M. Systematic review with meta-analysis: the comparative effectiveness of aspirin vs. screening for colorectal cancer prevention. Aliment Pharmacol Ther 2017;45:193-204.
105. Paleari L, Puntoni M, Clavarezza M, DeCensi M, Cuzick J, DeCensi A. PIK3CA mutation, aspirin use after diagnosis and survival of colorectal cancer. A systematic review and meta-analysis of epidemiological studies. Clin Oncol (R Coll Radiol) 2016;28:317-26.
106. Liao X, Lochhead P, Nishihara R, Morikawa T, Kuchiba A, Yamauchi M, Imamura Y, Qian ZR, Baba Y, Shima K, Sun R, Nosho K, Meyerhardt JA, Giovannucci E, Fuchs CS, Chan AT, Ogino S. Aspirin use, tumor PIK3CA mutation, and colorectal-cancer survival. N Engl J Med 2012;367:1596-606.
107. Alfonso L, Ai G, Spitale RC, Bhat GJ. Molecular targets of aspirin and cancer prevention. Br J Cancer 2014;111:61-7.
108. Dunlap T, Piyankarage SC, Wijewickrama GT, Abdul-Hay S, Vanni M, Litosh V, Luo J, Thatcher GR. Quinone-induced activation of Keap1/Nrf2 signaling by aspirin prodrugs masquerading as nitric oxide. Chem Res Toxicol 2012;25:2725-36.
109. Jian Z, Tang L, Yi X, Liu B, Zhang Q, Zhu G, Wang G, Gao T, Li C. Aspirin induces Nrf2-mediated transcriptional activation of haem oxygenase-1 in protection of human melanocytes from H2 O2 -induced oxidative stress. J Cell Mol Med 2016;20:1307-18.
110. Satoh T, Lipton S. Recent advances in understanding NRF2 as a druggable target: development of pro-electrophilic and non-covalent NRF2 activators to overcome systemic side effects of electrophilic drugs like dimethyl fumarate. F1000Res 2017;6:2138.
111. Hua X, Phipps AI, Burnett-Hartman AN, Adams SV, Hardikar S, Cohen SA, Kocarnik JM, Ahnen DJ, Lindor NM, Baron JA, Newcomb PA. Timing of aspirin and other nonsteroidal anti-inflammatory drug use among patients with colorectal cancer in relation to tumor markers and survival. J Clin Oncol 2017;35:2806-13.
112. Kuo CN, Pan JJ, Huang YW, Tsai HJ, Chang WC. Association between nonsteroidal anti-inflammatory drugs and colorectal cancer: a population-based case-control study. Cancer Epidemiol Biomarkers Prev 2018;27:737-45.
113. Elwood PC, Morgan G, Pickering JE, Galante J, Weightman AL, Morris D, Kelson M, Dolwani S. Aspirin in the treatment of cancer: reductions in metastatic spread and in mortality: a systematic review and meta-analyses of published studies. PLoS One 2016;11:e0152402.
114. Chubak J, Whitlock EP, Williams SB, Kamineni A, Burda BU, Buist DS, Anderson ML. Aspirin for the prevention of cancer incidence and mortality: systematic evidence reviews for the U.S. preventive services task force. Ann Intern Med 2016;164:814-25.
115. Huang XZ, Chen Y, Wu J, Zhang X, Wu CC, Zhang CY, Sun SS, Chen WJ. Aspirin and non-steroidal anti-inflammatory drugs use reduce gastric cancer risk: a dose-response meta-analysis. Oncotarget 2017;8:4781-95.
116. Li P, Wu H, Zhang H, Shi Y, Xu J, Ye Y, Xia D, Yang J, Cai J, Wu Y. Aspirin use after diagnosis but not prediagnosis improves established colorectal cancer survival: a meta-analysis. Gut 2015;64:1419-25.
117. Usman MW, Luo F, Cheng H, Zhao JJ, Liu P. Chemopreventive effects of aspirin at a glance. Biochim Biophys Acta 2015;1855:254-63.
118. Veettil SK, Lim KG, Ching SM, Saokaew S, Phisalprapa P, Chaiyakunapruk N. Effects of aspirin and non-aspirin nonsteroidal anti-inflammatory drugs on the incidence of recurrent colorectal adenomas: a systematic review with meta-analysis and trial sequential analysis of randomized clinical trials. BMC Cancer 2017;17:763.
119. Johnson CC, Jankowski M, Rolnick S, Yood MU, Alford SH. Influence of NSAID use among colorectal cancer survivors on cancer outcomes. Am J Clin Oncol 2017;40:370-4.
120. Zhao X, Xu Z, Li H. NSAIDs use and reduced metastasis in cancer patients: results from a meta-analysis. Sci Rep 2017;7:1875.
121. Moris D, Kontos M, Spartalis E, Fentiman IS. The role of NSAIDs in breast cancer prevention and relapse: current evidence and future perspectives. Breast Care (Basel) 2016;11:339-44.
122. Veitonmäki T, Murtola TJ, Talala K, Taari K, Tammela T, Auvinen A. Non-steroidal anti-inflammatory drugs and cancer death in the finnish prostate cancer screening trial. PLoS One 2016;11:e0153413.
123. Zhou CK, Daugherty SE, Liao LM, Freedman ND, Abnet CC, Pfeiffer R, Cook MB. Do aspirin and other NSAIDs confer a survival benefit in men diagnosed with prostate cancer? A pooled analysis of NIH-AARP and PLCO cohorts. Cancer Prev Res (Phila) 2017;10:410-20.
124. Rachidi S, Wallace K, Li H, Lautenschlaeger T, Li Z. Postdiagnosis aspirin use and overall survival in patients with melanoma. J Am Acad Dermatol 2018;78:949-56.
125. van Staalduinen J, Frouws M, Reimers M, Bastiaannet E, van Herk-Sukel MP, Lemmens V, de Steur WO, Hartgrink HH, van de Velde CJ, Liefers GJ. The effect of aspirin and nonsteroidal anti-inflammatory drug use after diagnosis on survival of oesophageal cancer patients. Br J Cancer 2016;114:1053-9.
126. Frouws MA, Bastiaannet E, Langley RE, Chia WK, van Herk-Sukel MP, Lemmens VE, Putter H, Hartgrink HH, Bonsing BA, Van de Velde CJ, Portielje JE, Liefers GJ. Effect of low-dose aspirin use on survival of patients with gastrointestinal malignancies; an observational study. Br J Cancer 2017;116:405-13.
127. Matsuo K, Cahoon SS, Yoshihara K, Shida M, Kakuda M, Adachi S, Moeini A, Machida H, Garcia-Sayre J, Ueda Y, Enomoto T, Mikami M, Roman LD, Sood AK. Association of low-dose aspirin and survival of women with endometrial cancer. Obstet Gynecol 2016;128:127-37.
128. Zhang D, Bai B, Xi Y, Zhao Y. Can aspirin reduce the risk of endometrial cancer? A systematic review and meta-analysis of observational studies. Int J Gynecol Cancer 2016;26:1111-20.
129. Spence AD, Busby J, Johnston BT, Baron JA, Hughes CM, Coleman HG, Cardwell CR. Low-dose aspirin use does not increase survival in 2 independent population-based cohorts of patients with esophageal or gastric cancer. Gastroenterology 2018;154:849-60.
130. Dixon SC, Nagle CM, Wentzensen N, Trabert B, Beeghly-Fadiel A, Schildkraut JM, Moysich KB, deFazio A, Risch HA, Rossing MA, Doherty JA, Wicklund KG, Goodman MT, Modugno F, Ness RB, Edwards RP, Jensen A, Kjær SK, Høgdall E, Berchuck A, Cramer DW, Terry KL, Poole EM, Bandera EV, Paddock LE, Anton-Culver H, Ziogas A, Menon U, Gayther SA, Ramus SJ, Gentry-Maharaj A, Pearce CL, Wu AH, Pike MC, Webb PM; Australian Ovarian Cancer Study Group. Use of common analgesic medications and ovarian cancer survival: results from a pooled analysis in the Ovarian Cancer Association Consortium. Br J Cancer 2017;116:1223-8.
131. Brasky TM, Felix AS, Cohn DE, McMeekin DS, Mutch DG, Creasman WT, Thaker PH, Walker JL, Moore RG, Lele SB, Guntupalli SR, Downs LS, Nagel CI, Boggess JF, Pearl ML, Ioffe OB, Park KJ, Ali S, Brinton LA. Nonsteroidal anti-inflammatory drugs and endometrial carcinoma mortality and recurrence. J Natl Cancer Inst 2017;109:1-10.
132. Verdoodt F, Kjaer SK, Friis S. Influence of aspirin and non-aspirin NSAID use on ovarian and endometrial cancer: summary of epidemiologic evidence of cancer risk and prognosis. Maturitas 2017;100:1-7.
133. Gurpinar E, Grizzle WE, Piazza GA. NSAIDs inhibit tumorigenesis, but how? Clin Cancer Res 2014;20:1104-13.
134. Panieri E, Santoro MM. ROS homeostasis and metabolism: a dangerous liason in cancer cells. Cell Death Dis 2016;7:e2253.
135. Vander Heiden MG, DeBerardinis RJ. Understanding the intersections between metabolism and cancer biology. Cell 2017;168:657-69.
136. Liu J, Wang Z. Increased oxidative stress as a selective anticancer therapy. Oxid Med Cell Longev 2015;2015:294303.
137. Edelman MJ, Wang X, Hodgson L, Cheney RT, Baggstrom MQ, Thomas SP, Gajra A, Bertino E, Reckamp KL, Molina J, Schiller JH, Mitchell-Richards K, Friedman PN, Ritter J, Milne G, Hahn OM, Stinchcombe TE, Vokes EE; Alliance for Clinical Trials in Oncology. Phase III randomized, placebo-controlled, double-blind trial of celecoxib in addition to standard chemotherapy for advanced non-small-cell lung cancer with cyclooxygenase-2 overexpression: CALGB 30801 (Alliance). J Clin Oncol 2017;35:2184-92.
138. Groen HJ, Sietsma H, Vincent A, Hochstenbag MM, van Putten JW, van den Berg A, Dalesio O, Biesma B, Smit HJ, Termeer A, Hiltermann TJ, van den Borne BE, Schramel FM. Randomized, placebo-controlled phase III study of docetaxel plus carboplatin with celecoxib and cyclooxygenase-2 expression as a biomarker for patients with advanced non-small-cell lung cancer: the NVALT-4 study. J Clin Oncol 2011;29:4320-6.
139. Patel MI, Subbaramaiah K, Du B, Chang M, Yang P, Newman RA, Cordon-Cardo C, Thaler HT, Dannenberg AJ. Celecoxib inhibits prostate cancer growth: evidence of a cyclooxygenase-2-independent mechanism. Clin Cancer Res 2005;11:1999-2007.
140. Williams CS, Watson AJ, Sheng H, Helou R, Shao J, DuBois RN. Celecoxib prevents tumor growth in vivo without toxicity to normal gut: lack of correlation between in vitro and in vivo models. Cancer Res 2000;60:6045-51.
141. Xu B, Wang Y, Yang J, Zhang Z, Zhang Y, Du H. Celecoxib induces apoptosis but up-regulates VEGF via endoplasmic reticulum stress in human colorectal cancer in vitro and in vivo. Cancer Chemother Pharmacol 2016;77:797-806.
142. Sobolewski C, Rhim J, Legrand N, Muller F, Cerella C, Mack F, Chateauvieux S, Kim JG, Yoon AY, Kim KW, Dicato M, Diederich M. 2,5-Dimethyl-celecoxib inhibits cell cycle progression and induces apoptosis in human leukemia cells. J Pharmacol Exp Ther 2015;355:308-28.
143. Schönthal AH. Antitumor properties of dimethyl-celecoxib, a derivative of celecoxib that does not inhibit cyclooxygenase-2: implications for glioma therapy. Neurosurg Focus 2006;20:E21.
144. Pyrko P, Kardosh A, Liu YT, Soriano N, Xiong W, Chow RH, Uddin J, Petasis NA, Mircheff AK, Farley RA, Louie SG, Chen TC, Schönthal AH. Calcium-activated endoplasmic reticulum stress as a major component of tumor cell death induced by 2,5-dimethyl-celecoxib, a non-coxib analogue of celecoxib. Mol Cancer Ther 2007;6:1262-75.
145. Schiffmann S, Maier TJ, Wobst I, Janssen A, Corban-Wilhelm H, Angioni C, Geisslinger G, Grösch S. The anti-proliferative potency of celecoxib is not a class effect of coxibs. Biochem Pharmacol 2008;76:179-87.
146. Waskewich C, Blumenthal RD, Li H, Stein R, Goldenberg DM, Burton J. Celecoxib exhibits the greatest potency amongst cyclooxygenase (COX) inhibitors for growth inhibition of COX-2-negative hematopoietic and epithelial cell lines. Cancer Res 2002;62:2029-33.
147. Jendrossek V, Handrick R, Belka C. Celecoxib activates a novel mitochondrial apoptosis signaling pathway. FASEB J 2003;17:1547-9.
148. Zhu J, Song X, Lin HP, Young DC, Yan S, Marquez VE, Chen CS. Using cyclooxygenase-2 inhibitors as molecular platforms to develop a new class of apoptosis-inducing agents. J Natl Cancer Inst 2002;94:1745-57.
149. Bosch R, Dieguez-Gonzalez R, Céspedes MV, Parre-o M, Pavón Má, Gra-ena A, Sierra J, Mangues R, Casanova I. A novel inhibitor of focal adhesion signaling induces caspase-independent cell death in diffuse large B-cell lymphoma. Blood 2011;118:4411-20.
150. Casanova I, Bosch R, Lasa A, Parre-o M, Céspedes MV, Brunet S, Nomdedéu JF, Mangues MA, Sierra J, Mangues R. A celecoxib derivative inhibits focal adhesion signaling and induces caspase-8-dependent apoptosis in human acute myeloid leukemia cells. Int J Cancer 2008;123:217-26.
151. Bosch R, Moreno MJ, Dieguez-Gonzalez R, Céspedes MV, Gallardo A, Trias M, Gra-ena A, Sierra J, Casanova I, Mangues R. A novel orally available inhibitor of focal adhesion signaling increases survival in a xenograft model of diffuse large B-cell lymphoma with central nervous system involvement. Haematologica 2013;98:1242-9.
152. Ralph SJ, Pritchard R, Rodríguez-Enríquez S, Moreno-Sánchez R, Ralph RK. Hitting the bull's-eye in metastatic cancers-NSAIDs elevate ROS in mitochondria, inducing malignant cell death. Pharmaceuticals (Basel) 2015;8:62-106.
153. Pritchard R, Rodríguez-Enríquez S, Pacheco-Velázquez SC, Bortnik V, Moreno-Sánchez R, Ralph S. Celecoxib inhibits mitochondrial O2 consumption, promoting ROS dependent death of murine and human metastatic cancer cells via the apoptotic signalling pathway. Biochem Pharmacol 2018;154:318-34.
154. Pacheco-Velázquez SC, Robledo-Cadena DX, Hernández-Reséndiz I, Gallardo-Pérez JC, Moreno-Sánchez R, Rodríguez-Enríquez S. Energy metabolism drugs block triple negative breast metastatic cancer cell phenotype. Mol Pharm 2018;15:2151-64.
155. Huang C, Chen Y, Liu H, Yang J, Song X, Zhao J, He N, Zhou CJ, Wang Y, Huang C, Dong Q. Celecoxib targets breast cancer stem cells by inhibiting the synthesis of prostaglandin E2 and down-regulating the Wnt pathway activity. Oncotarget 2017;8:115254-69.
156. Kurtova AV, Xiao J, Mo Q, Pazhanisamy S, Krasnow R, Lerner SP, Chen F, Roh TT, Lay E, Ho PL, Chan KS. Blocking PGE2-induced tumour repopulation abrogates bladder cancer chemoresistance. Nature 2015;517:209-13.
157. Wang JS, Ho FM, Kang HC, Lin WW, Huang KC. Celecoxib induces heme oxygenase-1 expression in macrophages and vascular smooth muscle cells via ROS-dependent signaling pathway. Naunyn Schmiedebergs Arch Pharmacol 2011;383:159-68.
158. Hamdulay SS, Wang B, Birdsey GM, Ali F, Dumont O, Evans PC, Haskard DO, Wheeler-Jones CP, Mason JC. Celecoxib activates PI-3K/Akt and mitochondrial redox signaling to enhance heme oxygenase-1-mediated anti-inflammatory activity in vascular endothelium. Free Radic Biol Med 2010;48:1013-23.
159. Al-Rashed F, Calay D, Lang M, Thornton CC, Bauer A, Kiprianos A, Haskard DO, Seneviratne A, Boyle JJ, Schönthal AH, Wheeler-Jones CP, Mason JC. Celecoxib exerts protective effects in the vascular endothelium via COX-2-independent activation of AMPK-CREB-Nrf2 signalling. Sci Rep 2018;8:6271.
160. Irie T, Tsujii M, Tsuji S, Yoshio T, Ishii S, Shinzaki S, Egawa S, Kakiuchi Y, Nishida T, Yasumaru M, Iijima H, Murata H, Takehara T, Kawano S, Hayashi N. Synergistic antitumor effects of celecoxib with 5-fluorouracil depend on IFN-gamma. Int J Cancer 2007;121:878-83.
161. Zhao S, Cai J, Bian H, Gui L, Zhao F. Synergistic inhibition effect of tumor growth by using celecoxib in combination with oxaliplatin. Cancer Invest 2009;27:636-40.
162. Sadhu SS, Wang S, Averineni RK, Seefeldt T, Yang Y, Guan X. In-vitro and in-vivo inhibition of melanoma growth and metastasis by the drug combination of celecoxib and dacarbazine. Melanoma Res 2016;26:572-9.
163. van Wijngaarden J, van Beek E, van Rossum G, van der Bent C, Hoekman K, van der Pluijm G, van der Pol MA, Broxterman HJ, van Hinsbergh VW, Löwik CW. Celecoxib enhances doxorubicin-induced cytotoxicity in MDA-MB231 cells by NF-kappaB-mediated increase of intracellular doxorubicin accumulation. Eur J Cancer 2007;43:433-42.
164. Angelini A, Iezzi M, Di Febbo C, Di Ilio C, Cuccurullo F, Porreca E. Reversal of P-glycoprotein-mediated multidrug resistance in human sarcoma MES-SA/Dx-5 cells by nonsteroidal anti-inflammatory drugs. Oncol Rep 2008;20:731-5.
165. Fantappiè O, Solazzo M, Lasagna N, Platini F, Tessitore L, Mazzanti R. P-glycoprotein mediates celecoxib-induced apoptosis in multiple drug-resistant cell lines. Cancer Res 2007;67:4915-23.
166. Hiľovská L, Jendželovský R, Fedoročko P. Potency of non-steroidal anti-inflammatory drugs in chemotherapy. Mol Clin Oncol 2015;3:3-12.
167. Moreno-Sánchez R, Bravo C, Vásquez C, Ayala G, Silveira LH, Martínez-Lavín M. Inhibition and uncoupling of oxidative phosphorylation by nonsteroidal anti-inflammatory drugs: study in mitochondria, submitochondrial particles, cells, and whole heart. Biochem Pharmacol 1999;57:743-52.
168. Jin CH, Wang AH, Chen JM, Li RX, Liu XM, Wang GP, Xing LQ. Observation of curative efficacy and prognosis following combination chemotherapy with celecoxib in the treatment of advanced colorectal cancer. J Int Med Res 2011;39:2129-40.
169. Debucquoy A, Roels S, Goethals L, Libbrecht L, Van Cutsem E, Geboes K, Penninckx F, D'Hoore A, McBride WH, Haustermans K. Double blind randomized phase II study with radiation+5-fluorouracil+/-celecoxib for resectable rectal cancer. Radiother Oncol 2009;93:273-8.
170. Lin EH, Curley SA, Crane CC, Feig B, Skibber J, Delcos M, Vadhan SR, Morris J, Ayers GD, Ross A, Brown T, Rodriguez-Bigas MA, Janjan N. Retrospective study of capecitabine and celecoxib in metastatic colorectal cancer: potential benefits and COX-2 as the common mediator in pain, toxicities and survival? Am J Clin Oncol 2006;29:232-9.
171. Lin EH, Kim EY, Wang L, Fong C, Shankaran V, Wu X. ADAPT therapy vs capecitabine bevacizumab in stage IV colorectal cancer: pooled 10-year survival experience and a phase II study update. Available from: http://ascopubs.org/doi/abs/10.1200/JCO.2016.34.15_suppl.e15046. [Last accessed on 12 Sep 2018].
172. Lin EH, Patel SA, Chou J, Kim EY, Shankaran V, Coveler AL, Harris WP, Park JO, Fichera A, Mann GN, Chiorean EG, Pritchard CC, Sinanan M, Upton M, Storer B, Yeung RS, Li L. A phase II trial of maintenance ADAPT therapy targeting colon cancer stem cells in patients with metastatic colorectal cancer. Available from: http://ascopubs.org/doi/abs/10.1200/jco.2014.32.15_suppl.tps3650. [Last accessed on 12 Sep 2018].
173. Coombes RC, Tovey H, Kilburn L, Mansi J, Palmieri C, Bartlett J, Hicks J, Makris A, Evans A, Loibl S, Denkert C, Murray E, Grieve R, Coleman R, Schmidt M, Klare P, Rezai M, Rautenberg B, Klutinus N, Rhein U, Mousa K, Ricardo-Vitorino S, von Minckwitz G, Bliss J. A phase III multicentre double blind randomised trial of celecoxib versus placebo in primary breast cancer patients (REACT – Randomised EuropeAn celecoxib trial). Available from: http://cancerres.aacrjournals.org/content/78/4_Supplement/GS3-03. [Last accessed on 12 Sep 2018].
174. Zhou YY, Hu ZG, Zeng FJ, Han J. Clinical Profile of cyclooxygenase-2 inhibitors in treating non-small cell lung cancer: a meta-analysis of nine randomized clinical trials. PLoS One 2016;11:e0151939.
175. James ND, Sydes MR, Clarke NW, Mason MD, Dearnaley DP, Spears MR, Ritchie AWS, Russell JM, Parker CC, Rentsch CA, Wallace J, Barber J, Lydon A, Parmar MKB. Celecoxib with or without zoledronic acid for hormone-naïve prostate cancer: survival results from STAMPEDE (NCT00268476). Clin Adv Hematol Oncol 2016;14:11-3.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Ralph SJ, Nozuhur S, Moreno-Sánchez R, Rodríguez-Enríquez S, Pritchard R. NSAID celecoxib: a potent mitochondrial pro-oxidant cytotoxic agent sensitizing metastatic cancers and cancer stem cells to chemotherapy. J Cancer Metastasis Treat 2018;4:49. http://dx.doi.org/10.20517/2394-4722.2018.42
AMA Style
Ralph SJ, Nozuhur S, Moreno-Sánchez R, Rodríguez-Enríquez S, Pritchard R. NSAID celecoxib: a potent mitochondrial pro-oxidant cytotoxic agent sensitizing metastatic cancers and cancer stem cells to chemotherapy. Journal of Cancer Metastasis and Treatment. 2018; 4: 49. http://dx.doi.org/10.20517/2394-4722.2018.42
Chicago/Turabian Style
Ralph, Stephen John, Sam Nozuhur, Rafael Moreno-Sánchez, Sara Rodríguez-Enríquez, Rhys Pritchard. 2018. "NSAID celecoxib: a potent mitochondrial pro-oxidant cytotoxic agent sensitizing metastatic cancers and cancer stem cells to chemotherapy" Journal of Cancer Metastasis and Treatment. 4: 49. http://dx.doi.org/10.20517/2394-4722.2018.42
ACS Style
Ralph, SJ.; Nozuhur S.; Moreno-Sánchez R.; Rodríguez-Enríquez S.; Pritchard R. NSAID celecoxib: a potent mitochondrial pro-oxidant cytotoxic agent sensitizing metastatic cancers and cancer stem cells to chemotherapy. J. Cancer. Metastasis. Treat. 2018, 4, 49. http://dx.doi.org/10.20517/2394-4722.2018.42
About This Article
Copyright

Data & Comments
Data


 Cite This Article 26 clicks
Cite This Article 26 clicks

 Like This Article 44
likes
Like This Article 44
likes






Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.