
Immunotherapy in colorectal cancer treatment: actual landscape and future perspectives


Abstract
Colorectal cancer (CRC) represents the second most common cancer in Europe with marked differences in prognosis and response to treatments. In the past years research showed emerging interest in genomic and immunologic fields. The clinical heterogeneity, that occurs during the pathogenesis of CRC, is driven by chromosomal alterations and defective function of DNA mismatch repair genes. CRC is classified in four consensus molecular subtypes (CMS) with different immunogenic characteristics and prognosis. CMS1 microsatellite instable (MSI)-like and CMS4, both characterized by high levels of immune infiltration, are recognized as the most immunogenic subtypes, even though functional characteristic leading to different prognosis are reported. In particular, MSI tumors have been identified as the best candidates for immunotherapy treatment and a number of studies have evaluated the efficacy of anti-programmed cell death ligand-1 (PDL-1) and anti-cytotoxic T-lymphocyte-associated protein 4 (CTLA4) in this setting. However, literature data show that the majority of patients with CRC have microsatellite stable (MSS) tumors and this status seems related to lower response to PDL-1/programmed cell death-1 or CTLA4 blockade. The aim of this paper is to investigate the role of immunotherapy in MSI and MSS CRC.
Keywords
Introduction
Colorectal cancer (CRC) represents the second most common cancer in Europe with significant heterogeneity in prognosis and response to treatment. Prognostic factors include stage of disease, site of metastasis, and type of treatment given. Tumor genetic mutations gained a pivotal role as the prognostic factor. To date, the median overall survival (OS) for patients with metastatic CRC is about 30 months[1]. In 2015, 70% of new cases underwent potentially curative resection thanks to screening programs[2].
In the past years, research on CRC has shown an emerging interest in genomic and immunologic fields. The clinical heterogeneity that occurs during the pathogenesis of CRC is driven by chromosomal alterations and defective function of DNA mismatch repair (MMR) systems[3]. In particular, about 15%-20% of CRC show deficient mismatch repair (dMMR) systems, while chromosomal instability with functioning DNA MMR, a status defined as microsatellite stable (pMMR), is found in 80%-85% of CRC[4].
Microsatellites are defined as areas within the DNA sequence where a single nucleotide (mononucleotide) or units of two or more nucleotides are repeated in genome. They are usually located in the introns of genes and the number of repeats contained in every microsatellite is usually preserved in every single cell of the body[5]. Microsatellite instability (MSI) is defined as a clonal change in the number of repeated DNA nucleotide units in microsatellites and it arises in tumors with dMMR due to the inactivation of one of the four MMR genes: MSH2, MLH1, MSH6, and PMS2. Considering that a minority of tumors display instability in fewer than 20% of the markers studied, a classification has been proposed that identifies MSI-low (with just one unstable marker in out of the five-marker Bethesda panel) and MSI-high (with two or more unstable marker)[6].
Clinical and biologicals differences between dMMR and pMMR are well established. Specifically, dMMR causes genetic instability (aneuploidy, allelic losses, amplifications, translocations, and chromosomal gains) that influences the expression of genes leading to CRC carcinogenesis[7,8]. On the other hand, dMMR CRCs have shown better prognosis compared to pMMR tumors[9-11]. The increased mutation rate of dMMR tumors triggers an increased production of potentially immunogenic peptides or epitopes establishing a rationale for immunotherapy in this CRC subtype while few data regarding immunotherapy efficacy in pMMR tumors are available in literature[12]. In this review we analyzed the role of immunotherapy and target agents in dMMR and pMMR.
Role of the immune system in CRC
Conventionally, clinical and pathological features, along with tumor characteristics, are known to define cancer aggressiveness. Nevertheless, in the past years, tumor microenvironment (TME) has shown to play an important role in tumor growth and metastatic potential. TME is composed of epithelial cells, blood and lymphatic vessels, stromal cells, and infiltrating immune cells, including T lymphocytes, B cells, natural killer (NK) cells, dendritic cells (DCs), macrophages, and granulocytes. Each tumor displays a specific composition of TME and CRC shows a high degree of immune cell infiltration and high presence of mesenchymal stromal cells[13].
Studies in this field highlighted that different constituents of TME may influence tumor proliferation, infiltration and metastatic spread in different ways. Cancer growth or inhibition represents the result of the interplay between tumor cells and TME. Immune system has been demonstrated to be a key-mechanism of tumor regulation.
Immune system recruits, in cancer surveillance, the coordinated and balanced activation of both innate immune cells [such as macrophages, neutrophils, myeloid derived suppressor cells (MDSC), mast cells, eosinophils, and antigen-presenting cells (APCs)] and adaptive immune cells (NK cells, T and B lymphocytes cells)[14].
At first, innate immune system is recruited by abnormal cells without specific antigen recognition and inflammatory response is activated promoting angiogenesis and tumor cells proliferation. Later, adaptive immune response is triggered by interaction and recognition between non-self-antigens and peptides presented by the major histocompatibility complexes (MHC) of APCs and T cells[15].
Immune system cells play different roles during tumor immune response. CD4+ cells sustain inflammatory response by secreting a variety of cytokines such as interferon γ, tumor necrosis factor α, interleukin-2 (IL-2), and IL-17. CD4+ cell activation promotes proliferation and function of a specific subgroup of CD8+ cells called cytotoxic T lymphocytes, that are capable of direct lysis of tumor cells. CD8+ cells can also secrete cytokines causing cytotoxic response. NK cells are involved in antibody-dependent cell-mediated cytotoxicity and natural cytolytic activity against tumor cells. Macrophages destroy cancer cells through phagocytosis and release matrix-degrading substances (metalloproteinases and cysteine cathepsin proteases). Consequently, high levels of metalloproteinase represent an important factor to predict CRC prognosis and metastasis[16].
Part of the cells described above make up tumor-infiltrating lymphocytes (TILs) that showed to have a prognostic role in cancer treatment and appeared often to be associated with better clinical outcomes[17].
Mesenchymal stem cells (MSC) are non-hematopoietic stromal cells with proliferative potential, immunosuppressive properties, and ability to differentiate into several cell types. Their immunosuppressive function is releasing of proinflammatory factors, inhibiting lymphocyte proliferation and DCs maturation, promoting the production of macrophages, and regulating T cells (Treg). MSC are also involved in tumor initiation, angiogenesis, resistance to chemotherapy, invasion and metastatic process.
Criteria such as composition, density and location of TILs have shown to correlate with different prognosis indicators. Notably, in CRC the number and location of cytotoxic and memory T lymphocytes can predict tumor recurrence and prognosis in early-stage CRC[18]. Mlecnik et al.[19] observed that CRC presenting low CD8+ cytotoxic T-lymphocyte (CTL) infiltration were associated with higher tumor growth and metastatic spread. Conversely, patients whose tumor showed high density CD8+ CTL were more likely to have early-stage exordium. Moreover, among patients relapsed, CD8+ CTL infiltrate appeared to be low independently to TNM stage. These findings support the hypothesis that lymphocyte infiltration represents a strong and independent prognostic factor in CRC.
Tumors cells are well known to develop strategies of immune escape. Indeed, they may show genetic alterations that enhance the expression of mesenchimal transition or immunosuppressive genes along with chemokines responsible for immune suppressive cells recruitment, conferring to cancer cells innate resistance to anti-programmed cell death-1 (PD-1) drugs. Different mutations might be responsible for resistance acquired after an initial benefit out of immunotherapy; during clonal expansion a resistant clone develops high proliferation potential and drives resistance advance.
For example, loss-of-function mutations in Janus Kinases 1/2 (JAK 1/2) might be responsible both for primary and adaptive resistance to immunotherapy. These inactivating mutations affect interferon gamma signaling rendering cancer cells unable to respond to interferon gamma by expressing programmed cell death ligand-1 (PDL-1) and other interferon-stimulated genes, and patients with such tumors became unlikely to respond to PD-1 blockade therapy. This mechanism has already been described in melanoma patients. Zaretsky et al.[20] analyzed biopsy samples from paired baseline and relapsing lesions in four metastatic melanoma patients who experienced disease progression after an initial objective tumor regression and found resistance-associated loss-of-function mutations in the genes encoding JAK1 or JAK2, concurrent with deletion of the wild-type allele. Shin et al.[21] described the case of one patient with dMMR colon cancer who did not respond to anti-PD-1 therapy despite a high mutational load, thus identifying JAK1/2 mutations also as potential mediators of primary resistance to PD-1 blockade.
Another mechanism that has been accounted for acquired resistance to immunotherapy in melanoma is inactivation of beta-2-microglobulin (B2M), a fundamental component of the antigen-presenting MHC I. Le et al.[22] (which included in their study 40 patients with CRC and 46 patients with 11 other histologies) identified mutations of the B2M gene in four of five tumors with acquired resistance to anti-PD-1 therapy. However, no B2M mutations were identified in primarily resistant tumors. The recognition that the above-mentioned mutations would lead to primary or acquired resistance to PD-1 therapy might be useful to building oncogenic sequencing panels used to select patients for treatments.
The tight interaction between tumor and immune system has driven to the hypothesis of cancer immunoediting. This concept reinvented tumor immunosurveillance taking into account the dual role played by immune responses as host-protective and tumor-promoting. According to immunoediting cancer growth is structured in three different phases: elimination, equilibrium and escape. In the elimination phase immune system engages both innate and adaptive response to eliminate developing tumors before they become clinically evident. If this phase is satisfactorily fulfilled and the tumor results fully eradicated, the whole process might be considered completed. However if a single cancer cell variant escapes the elimination phase it proceeds to the equilibrium phase. During the second phase clonal growth of selected cell variant is prevented by immune system, but those cells still survive in a state of dormancy. Notably, adaptive responses are engaged in the equilibrium phase which is also the time of cancer immunoediting. Also equilibrium might be the end of the entire process whether the immune system keeps under control the “survivor cells” for the lifetime of the host. Nevertheless, the continuous immune pressure on tumor cells may lead them to enter the escape phase. In this third phase tumor variants elude immune system with different mechanisms and they outgrow to clinically apparent cancer[23,24].
CMS 1-4 and immune classification
As previously reported CRC clinical pathological characteristics and tumor TMN stage largely affect CRC prognosis and drive treatment choices along with mutation in RAS and BRAF genes. Nevertheless, patients sharing same TNM stage and therapies end up with different outcomes suggesting that key factors are still missing to our knowledge and approach. To attempt a more inclusive classification, different criteria were proposed that take into account also composition, density and location of tumor immune infiltrate[25]. An example of these efforts is the “Immunoscore” for tumor immune classification promoted by Galon’s group. This immune-based classification demonstrated to have a good and independent prognostic value[26].
Furthermore, an international consortium of experts has introduced a gene expression and immune -based classification system: the “consensus molecular subtypes” of CRC, providing new prognostic and predictive tools[27]. CRC is classified in four CMS and a fifth unclassified group. CMS1 group, also called MSI-like, includes tumors with instability of microsatellite due to mutations in MMR proteins and BRAF oncogene mutations. This subtype is also characterized by a diffuse immune infiltrate, composed of T helper cells and cytotoxic T cells. CMS2 subtype, also called canonical, encopasses tumors with chromosomal instability and upregulation of c-MYC and Wnt proto-oncogene pathways. CMS3 subtype, also known as metabolic, encompasses tumors with mutated KRAS and tumors presenting metabolic dysregulation. CMS4, also known as mesenchymal subtypes includes tumors with mesenchymal phenotype, high expression of mesenchymal genes, stromal infiltration, angiogenesis and transforming growth factor beta (TGF-β) activation. The four subtypes have different frequency, immunogenic characteristics and prognosis with CMS1 and CMS4 recognized as the most immunogenic subtypes, both characterized by high levels of immune infiltration[13,17].
Becht et al.[28] in a retrospective analyses demonstrated that high levels of TILs reported in CMS1 and CMS4 have different functional characteristics leading to different prognoses. Although both subtypes have high CD8+ T lymphocytes and macrophage infiltrate, CMS1 patients show a Th1 polarization, with favorable prognosis, while CMS4 subgroup have high density of endothelial, myeloid cells and fibroblasts with higher production chemokines and cytokines that support tumor-associated inflammation, stromal invasion and, angiogenesis, resulting in worse prognosis. These findings stress the role of TME functional orientation beyond TILs composition.
Regarding the others subgroups, CMS2 and CMS3 that occur approximately in 50% of CRC, have low immune and inflammatory infiltration and, intermediate prognosis[27,28].
Also tumor genetic signature has a strong prognostic value. It is reported that stromal composition might strongly affect tumor transcriptional profile hiding tumor cell intrinsic transcriptional traits, especially in those tumors whose gene expression is largely sustained by stromal cells. Using patient-derived xenografts, Isella et al.[29] developed an approach to unmask CRC cell specific transcriptional features. Based on these findings, five CRC intrinsic subtypes (CRIS) were identified. CRIS-A includes MSI-like, BRAF- or KRAS-mutated tumors with mucinous expression and glycolytic, pro-inflammatory features. CRIS-B encompasses poorly differentiated tumors characterized by high TGF-β driven activation and stressed epithelial-mesenchymal transition traits. CRIS-C groups KRAS wild-type tumor with chromosomal instability expressing elevated levels of epidermal growth factor receptor (EGFR). CRIS-D clusters stem phenotype tumors with active Wnt pathway and insulin-like growth factor-2 amplification and overexpression. In CRIS-E subtype Wnt signaling is again observed but it is associated with Paneth-like phenotype and mutations in TP53.
Many of these traits differ from those reported in other transcriptional classification, confirming the strong influence of stromal contexture. CRIS grouping may be applied both to primary and metastatic CRC with low overlap on previous transcriptional classifications. Interestingly, CRIS subtypes were demonstrated to have new prognostic and predictive potentials[29].
Immunotherapy in MSI CRCs
In the past years, research on immunology and molecular biology fields has clarified the role of the immune system in cancer growing and metastatic potential of tumors. Interestingly, MSI tumors show a marked predisposition to express a wide variety of neoantigens reflecting a significantly high mutational burden [20 fold higher compared to microsatellite stable (MSS)], due to dMMR. The load of neoantigens and the pronounced expression of T-cell recruiting chemokines cooperate to sustain an active immune TME characterized by diffuse immune infiltrate. This explains why CMS1 subtype is recognized as highly immunogenic. This consideration builds up a strong rationale for the use of immunotherapy in MSI CRC. Furthermore, Llosa et al.[30] proposed an interaction between tumor gene expression and immune microenvironment in CRC. Not only did they report an association between MSI tumors and Th1/CTL rich infiltrate, but they also observed that MSI tumors showed enhanced expression of several immune checkpoints, as to balance such an active immune microenvironment. This might explain both the natural development and growth of tumors that should be easily eliminated by the immune system and the possible efficacy of checkpoint inhibitors in this setting.
Immune system defends our bodies from non-self antigens activating immune response. However, it is pivotal that immune defenses arise at the appropriate time and are limited when they are no more requested in order to prevent chronical inflammation and autoimmune disease. A variety of co-inhibitory checkpoints are engaged to balance activation signals.
One of the most important immune checkpoints is represented by PD-1 and PDL-1. PD-1 is expressed on activated T-cells while PDL-1 is usually expressed on APCs’ surface and their interaction mediates a co-inhibitory stimulus that limits excessive immune responses in peripheral tissues ensuring the maintenance of peripheral tolerance [Figure 1]. Another immune checkpoint is a cytotoxic T-lymphocyte-associated protein 4 (CTLA4), expressed on T-cell surface, which counteracts CD28 T-cell activation signal thus downregulating the amplitude of early stages T-cell activation[31,32].
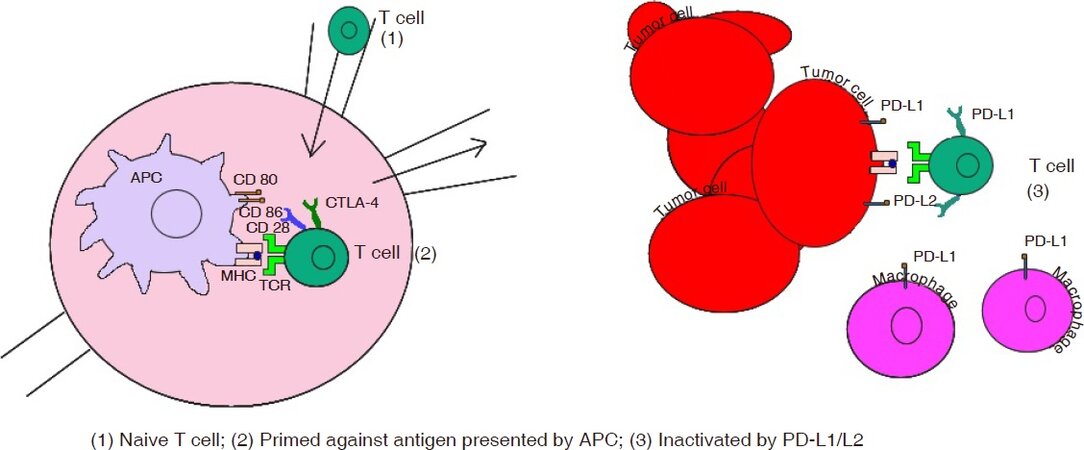
Figure 1. Interactions between cancer cells and T-cells and the role of PD-1/PDL-1 and CTLA4. PD-1: programmed cell death-1; PDL-1: programmed cell death ligand-1; CTLA4: cytotoxic T-lymphocyte-associated protein 4; APC: antigen-presenting cell; MHC: major histocompatibility complexes
The biological significance of PD-1/PDL-1 and CTLA4 suggests a therapeutic role of blockade of these pathways in different types of cancer, including CRC[33,34]. Monoclonal antibodies that block PD-1/PDL-1 or CTLA4 are currently approved in melanoma, kidney, and lung cancer treatment and are under study in other neoplastic diseases including CRC cancer. In particular, the efficacy of anti-PD-1 treatment in metastatic CRC was evaluated in a phase 2 clinical trial where pembrolizumab was administered in patients with pMMR and dMMR. Pembrolizumab was administered at the dose of 10 mg/kg every 14 days. The results of this study showed that MMR status predicted clinical benefit of immune checkpoint blockade with pembrolizumab.
In particular, the immune-related objective response rate (ORR) and immune-related 6-month progression-free survival (PFS) were 40% and 78% respectively, for dMMR CRC patients (cohort A) and 0% and 11% for pMMR CRC patients (cohort B). The median PFS was 2.2 months (95% CI, 1.4-2.8) and OS was 5 months (95% CI, 3.0 to not estimable) in the cohort with pMMR CRC. The median PFS and OS were not reached in the cohort with dMMR CRC. Indeed, the authors revealed 1782 somatic mutations per tumor in dMMR compared with 73 in pMMR tumors (P = 0.007), and high somatic mutation loads were associated with prolonged PFS (P = 0.02). In conclusion Le et al.[35] underlined that dMMR CRC is more responsive to PD-1 blockade than pMMR [Table 1].
Clinical trials of immunotherapy in colorectal cancer
| Trial | Phase | Drugs | n (%) | Setting | PFS months | OS months | ORR | Number of identifier |
|---|---|---|---|---|---|---|---|---|
| PD-1 Blockade in Tumors with Mismatch-Repair Deficiency | ||||||||
| dMMR CRC | II | Pembrolizumab 10 mg/kg each 14 days | 21 | NR | 40 | |||
| pMMR CRC | II | Pembrolizumab 10 mg/kg each 14 days | 11 | 2.2 (95% CI, 1.4 to 2.8) | 5.0 (95% CI, 3.0 to not estimable) | 0 | ||
| Other dMMR non-CRC | II | Pembrolizumab 10 mg/kg each 14 days | 9 | 5.4 (95% CI, 3 to not estimable) | NR | 71 | ||
| CheckMate 142 Ongoing (recruiting) Estimated primary completion date December 3, 2018 | II | Nivolumab 3 mg/Kg with Ipilimumab 1 mg/Kg every 3 weeks for 4 doses followed by Nivolumab 3 mg/Kg every 2 weeks until progression | Pre-treated | NE* 9-mo rate 87%* | 55%*° | NCT02060188 | ||
| A Phase 2 Study of Pembrolizumab (MK-3475) in Combination With Azacitidine in Subjects With Chemo-refractory Metastatic Colorectal Cancer AAM 2017 n 3054 Ongoing (not recruiting) Actual primary completion date March 2016 Estimated study completation date November 2020 | II | Pembrolizumab 200 mg on day 1 of every 21 day cycle PLUS Azacitidine 100 mg daily on days 1-5 every 21 days. Treatment continued for 9 cycles or until evidence of progression of disease or unacceptable toxicity Subjects with chemo-rfractorym CRC without any further standard treatment option | 31 | Pre-treated | 2.1 m (1.8 to 2.8) | 6.2 m (3.5 to 8.7) | 3% CI (1-17) | NCT02260440 |
CheckMate142 investigated efficacy of both nivolumab monotherapy and nivolumab plus ipilimumab combination therapy in MSI CRC. In the monotherapy cohort, seventy-four pretreated dMMR/MSI-H metastatic CRC patients were treated with nivolumab 3 mg/kg every 14 days. Nivolumab provided evidence of benefit in previously treated patients with dMMR CRC, with an ORR of 34% (95% CI, 23.2-45.7) with a disease control rate (DCR) of 62% (95% CI, 50.1-73.2). Interestingly, durable responses were observed and 64% of patients had response lasting more than 12 months. Median PFS was 6.6 months and 12 months OS was 72% (95% CI, 60.0-80.9) with a median follow up of 21 months[36].
CheckMate 142 combination cohort evaluated nivolumab plus ipilimumab, an anti-CTLA4 antibody. Indeed, nivolumab and ipilimumab can act synergistically to promote T cell antitumor activity. In this cohort, one hundred ninety-nine previously treated patients with metastatic or recurrent dMMR CRC were treated with 4 doses of combination immunotherapy with nivolumab and ipilimumab followed by nivolumab. At median follow-up of 13.4 months, primary endpoint ORR was 55% (95% CI, 45.2-63.8) and DCR for 12 weeks or more was 80%. PFS rates were 76% at 9 months and 71% at 12 months while OS rates were 87% and 85%, respectively. Responses were observed irrespective of PDL-1 expression, BRAF or KRAS mutational status or history of Lynch syndrome. Regarding toxicity, no new safety signals were reported and no treatment related deaths were reported. Incidence of treatment related adverse events (73%) was comparable to monotherapy while grade 3-4 adverse events were 32% compared to 20% for monotherapy cohort. Common adverse events included fatigue, diarrhea, pruritus, fever, increase of aspartate aminotransferase and hypothyroidism.
Although the comparison is only indirect, these results suggest that a double-blockade might improve clinical outcomes, thus becoming a promising treatment option for MSI CRC[37]. Nevertheless, data from melanoma clinical trials have shown that combination of anti-PD-1 and anti-CTLA4 treatment may result in significant toxicity with 55% grade 3-4 adverse events[38]. In particular, diarrhea and colitis represented adverse events leading to discontinuation of treatment in a significant proportion of patients. On this basis, more studies about safety of this combination in treatment of CRC patients are warranted. Future investigations may further clarify the role of immunotherapy in pMMR CRC, in particular regarding the role of combination therapy compared to single agent anti-PD-1 treatment and the predictive value of PDL-1 expression [Table 2].
Ongoing clinical trials of immunotherapy in colorectal cancer
| Trial | Phase | Drugs | Setting | Number of identifier |
|---|---|---|---|---|
| MK-3475-177/KEYNOTE-177 Ongoing (not recruiting) Estimated primary completion date August 15, 2019 | III | Pembrolizumab 200 mg each 21 days for up to 35 treatments vs. chemotherapy | 1st line | NCT02563002 |
| A Study to Investigate Efficacy and Safety of Cobimetinib Plus Atezolizumab and Atezolizumab Monotherapy vs. Regorafenib in Participants With Metastatic Colorectal Adenocarcinoma (COTEZO IMblaze370) Ongoing, (not recruiting) Estimated primary completion date February 2019 | III | Regorafenib (160 mg days 1-21 every 28 days) vs. Cobimetinib plus atezolizumab (cobimetinib 60 mg days 1 to 21 plus atezolizumab 840 mg IV on day 1 and day 15 in a 28-day cycle) and atezolizumab monotherapy (atezolizumab monotherapy 1,200 milligrams (mg) on day 1 in a 21-day cycle) | 3rd line | NCT02788279 |
| A Phase 2 Study With Safety Lead-in, Evaluating TAS-102 Plus Nivolumab in Patients With Microsatellite Stable Refractory Metastatic Colorectal Cancer Ongoing (not recruit) Estimated completation date March 2018 | II | TAS102 plus Nivolumab | 3rd line | NCT02860546 |
| MK-3475-158/KEYNOTE-158 Ongoing (recruiting) Estimated primary completion date August 28, 2023 | II | Pembrolizumab 200 mg every 3 weeks for up to 35 administrations | Pre-treated | NCT02628067 |
| Phase 2 Study of MK-3475 in Patients With Microsatellite Unstable (MSI) Tumors Ongoing (recruiting) Estimated primary completion date June 2021 | II | -MSI Negative Colorectal Cancer: Pembroluzumab 10 mg/kg every 14 days -MSI Negative with Mutator Phenotype: Pembrolizumab 200 mg flat dose every 21 days | Pre-treated | NCT01876511 |
| A Phase I, Open-Label, Multi-Centre Study to Assess the Safety, Tolerability and Preliminary Anti-tumour Activity of Ascending Doses of Selumetinib (AZD6244 Hyd-sulfate) in Combination With MEDI4736 and Selumetinib in Combination With MEDI4736 and Tremelimumab in Patients With Advanced Solid Tumours Ongoing (recruiting) Estimated primary completion date July 10, 2018 | I | Selumetinib + MEDI4736 Patients with known MSI-high status will be excluded; patients with MSS, MSI-low, or unknown MSI status may be enrolled | Pre-treated | NCT02586987 |
| An Open-label, Phase II Basket Study of a hypoMEThylating Agent Oral Azacitidine and DURvalumab (MEDI4736) (Anti-PDL1) in Advanced Solid Tumors (METADUR) Ongoing (recruiting) Estimated primary completion date July 2021 | II | Azacitidine 300 mg daily for 14 consecutive days of every 28 days cycle for 3 cycles. PLUS Durvalumab 1,500 mg on Day 1 of every 28 days cycle for 12 months or until disease progression Only Microsatellite Stable Colorectal Carcinoma | Pre-treated | NCT02811497 |
| Evaluate the Efficacy of MEDI4736 in Immunological Subsets of Advanced Colorectal Cancer Ongoing (recruiting) Estimated primary completion date July 2019 | II | subjects will receive MEDI4736 for 12 months, or until PD, initiation of alternative cancer therapy, unacceptable toxicity. Following the 12-month treatment period, subjects without evidence for PD or other reason to discontinue treatment will be monitored without further treatment. Upon evidence of PD during the monitoring period, administration of MEDI4736 may resume at the Q2W schedule, for up to another 12 months Locally advanced or metastatic MSI-H CRC | 3rd line | NCT02227667 |
Immunotherapy in other CRCs subtypes
Albeit dMMR tumors proved to be responsive to immune-checkpoint inhibition, the majority of patients with CRC have pMMR tumors and this status was related to lower response to PDL-1/PD-1 or CTLA4 blockade. Hence, other molecular subtypes require different strategies. Theoretically, immunotherapy could be useful for all CRC if it was possible to convert the tumor towards a “CMS1-like” immune phenotype. CMS4 tumors (which showed the worse prognosis in terms of overall and relapse-free survival), for example, are characterized by an unfavorable, inflammed immune phenotype. They revealed high expression of mesenchymal genes, stromal cell infiltration and an angiogenic microenvironment.
Vascular endothelial growth factor-A (VEGF-A), a proangiogenic molecule produced by the tumors, has a crucial role in the development of the immunosuppressive microenvironment[39]. Given the immune-adjuvant effect that has been suggested for metastatic CRC patients treated with the anti-VEGF antibody bevacizumab[40] when combined with conventional chemotherapy, researchers are trying to further enhance the effect on the immune system by coupling anti-angiogenic treatment with immunotherapy and this strategy might be particularly relevant for CMS4 tumors. Several clinical trials are investing whether combination of bevacizumab with either immunotherapy alone or combined with targeted therapies and conventional chemotherapy might show activity in this setting (NCT02873195, NCT02291289, NCT02876224).
Another key aspect of the TME of CMS4 tumors is represented by activation of TGF-β signaling. Using a preclinical model of CT26 colon carcinoma cells, Triplett et al.[41] showed that combining aOX40 antibodies with an inhibitor of the TGF-β receptor (SM16) had a synergic action and elicited complete regression of tumors. Targeting the TGF-β pathway with galunisertinib as monotherapy and in combination with anti-PD-1 agents, induced anti-tumor immunity and tumor shrinkage also in a mouse model of mesenchymal CRC[42]. Based on these evidences, multiple TGF-β targeted therapies are currently in clinical trials.
CMS2 and CMS3 are considered as “cold” tumors, meaning that they lack immune cell infiltration. The level of expression of immunosuppressive genes is low, thus suggesting different mechanisms of immune escape. For example, the downregulation of MHC class I observed in these tumors, results in reduced presentation of tumor-associated antigens[43]. CMS3 tumors are frequently RAS mutated. In a recent study by Lal et al.[44] who used The Cancer Genome Atlas RNA-seq, KRAS-mutant CMS2 samples had reduced infiltration of cytotoxic cells and neutrophils relative to CMS1 and CMS4 and to KRAS wild-type CMS2 samples. Deregulation of mitogen-activated extracellular signal-regulated kinase (MEK) pathway is involved in carcinogenesis and maintenance of cancers. This pathway is physiologically activated by growth factors, but in pathological conditions mutations of oncogenic proteins (such as RAS and RAF) can cause the systematic activation of the MEK cascade. MEK inhibition with cobimetinib upregulates tumor major histocompatibility complex-I expression, promotes intratumoral T-cell accumulation and enhances anti-PDL-1 responses[45]. In a recent phase Ib study presented at Gastrointestinal Cancer Symposium American Society of Clinical Oncology (ASCO) 2018, sixty-six patients were enrolled to receive atezolizumab in combination with Cobimetinib in metastatic or locally advanced CRC refractory to chemotherapy. Preliminary data showed interesting results: OS was 10 months with durable responses in patients with MSS or microsatellites instable-low tumors[46]. Conversely, CMS2 tumors are usually characterized by EGFR activation without mutations in downstream pathways (e.g., KRAS mutations). Cetuximab, an anti EGFR monoclonal antibody, revealed a potential synergistic effect with monoclonal antibodies targeted to CTLA4 and PD-1 antigens and in vivo studies, especially in patients with head and neck tumors and lung cancer, are promising[47].
Other approaches that are being tested to improve immunotherapy response among CMS subtypes are represented by cytokine treatment, cancer vaccination and passive immunotherapy with adoptive T cell transfer or monoclonal antibody targeting tumor-associated antigens. Klein et al.[48] recently evaluated carcinoembryonic antigen (CEA)-IL2v (RG7813), an engineered IL-2 variant (IL-2v) with abolished IL-2Rα (CD25) binding fused to an antibody targeting CEA to increase immune infiltration and activates NK and T cells both in the periphery and within tumors. In two ongoing dose-escalation phase I studies, Tabernero et al.[49] proved the antitumor activity of CEA CD3 TCB (RG7802, RO6958688), a novel T-cell bispecific antibody targeting CEA on tumor cells and CD3 on T cells, in 11% of adult patients with advanced CEA+ solid tumors who received RG7802 as monotherapy and in 50% of patients to whom the antibody was given in combination with with atezolizumab 1200 mg Q3W.
Likewise, other malignancies, combining immunotherapy with conventional chemotherapeutic strategies or with radiotherapy (RT) might represent an useful and practical means to stimulate immune cell infiltration and elicit immune response. To this purpose, clinical trials testing the combination of anti-PDL-1/PD-1 treatment with RT or modified FOLFOX are ongoing (NCT02437071, NCT02375672). In the first one is a phase II study to evaluate the safety and abscopal effect of pembrolizumab after palliative RT or ablation in pts with unresectable/recurrent pMMR metastatic colorectal cancer, who have received ≥ 2 standard therapies, with ORR in a non-targeted lesion as primary objective[50]. After enrolling 26 patients, pembrolizumab after RT or ablation resulted feasible with a tolerable safety profile, with one patient achieving a partial response (PR) in non-irradiated lesions after RT (abscopal effect). The second one is based on the hypthesis that combination of mFOLFOX6 and pembrolizumab may enhance immunogenic cell death and improve outcome in patients with untreated, advanced CRC irrespective of MMR status. After a median follow up of 24 weeks, clinical activity was seen in patients including those with proficient MMR, with a DCR rate of 100% at 8 weeks[51].
A different strategy that is currently under evaluation to improve efficacy of immunotherapy in MSS/pMMR CRCs is combination of histone deacetylase inhibitor and PD-1 inhibitors. Entinostat, an oral, class I-selective histone deacetylase inhibitor is able to enhance anti-PD-1 activity by downregulation of immunosuppressive cell types in the TME[52] in models of renal and lung cancer.
Preliminary results of a phase II study of entinostat in combination with pembrolizumab have been recently presented at ASCO 2018 annual meeting. Sixteen pretreated MSS/pMMR CRC patients were enrolled and at data cut-off 6 patients remained on study (1 PR, 6 stable disease). The treatment showed acceptable safety with common adverse events including fatigue (37.5%), arthralgia (18.8%), and increased alkaline phosphatase (18.8%). These results can be viewed as promising, considering that have been obtained in a patient population in which objective responses have not been reported with anti-PD-1 monotherapy[53].
In addition to immune strategies focused on PD-1/PDL-1 axis and CTLA4 and against cancer immunotolerance, a series of different approaches (albeit still on the side of immunotherapeutic approaches) are recently been investigated in CRC. T lymphocytes engineered to express chimeric antigen receptors (CAR-T cells) have been tested for their potential role as therapeutic agents in CRC. In a recent paper of Magee et al.[54], CAR-T cells expressing the human specific GUC2YC antigen variable fragment were able to determine an increase of cytokine production and upregulation of markers of inflammation. The cells were also able to induce a somewhat specific killing of CRC cells who did express GUC2YC, whereas GUC2YC-deficient cells were spared. This was proven in in-vitro and in mice xenografts, suggesting further development of CAR-T cells engineered to express this antigen.
However, in another paper of Huang et al.[55], it is also suggested that, albeit interesting, development of CAR-T cells therapy for CRC patients should first be complemented by the addition of some forms of treatment able to induce indoleamine 2,3-dioxygenase 1 (IDO1) downregulation. The authors have examined the effects of CAR-T cells targeting EGFR variant III on CRC cell lines and correlated the effectiveness of treatment on the basis of either IDO1 downregulation or normal expression on the basis of the expression, in cell lines, of mir-153. In particular, due to the inhibitory effect on the expression of IDO1 of mir-153, the authors were able to find a significant correlation between CAR-T cells mediated killing of CRC cells and high levels of expression of mir-153, thus suggesting that CAR-T cells treatment “per se” is not enough to induce some meaningful tumor response.
Albeit manipulation of the mutational load of CRC patients is a mere piece of science fiction, it is well-known that, for treatments that are focused on PD-1/PDL-1 axis, mutational load might represent the best way to identify those patients who could benefit from this kind of strategy (more than the simplistic way of assessment of patients as in microsatellite stable/unstable). In particular, in a recent paper of Fabrizio et al.[56], authors tested 6004 cases of CRC by matching MSI assessment (MSS or MSI-L vs. MSI-H) and estimation of tumor mutational burden (TMB high or low). Authors found that the matching was not exactly perfect, with 302 cases (5% cases) having MSI-H status and 301/302 (99.7%) MSI-H cases having TMB high status but also with 164/5702 (2.9%) MSS cases having also TMB high status. Authors were able to confirm the activity of an anti-PD1 inhibitor in patients having TMB high status, thus suggesting that screening patients on the basis of MSI-H status positivity is somehow restricting the number of patients that could ultimately benefit from anti-PD1/PDL-1 treatment.
These data suggest that, at least in the foreseeable future, more data are needed to further assess the clinical impact of these treatment approaches in everyday practice, as there are a few crucial topics still to be addressed (namely the fitness of T cells, how to increase sensitivity of the TME towards T cell mediated killing and the selection of patients that benefit best from these treatment approaches).
Conclusion
In the past few years, introduction of new therapeutic approaches and better selection of patients have significantly changed treatment strategy of CRC and definitely improved patient outcome.
Immunotherapy has been the most important revolution in cancer treatment of recent years and it continues to show impressive results in lethal malignancies such as melanoma or lung cancer. Still, results observed in CRC with checkpoint inhibitors immunotherapy are modest if compared to other tumor entities and limited to a small subset of patients with MSI. In this context, a better knowledge of tumor immune microenvironment is essential to developing effective therapeutic strategies and overcoming resistance.
Interestingly, molecular characterization of CRC has shown that CMSs are associated with specific immune infiltration profiles corresponding with characteristic mechanisms of immune escape.
In particular, CMS1 subtype presents the most favourable situation for immunotherapy efficacy with high immune infiltration rich in Th1 cells and TILs, explaining the efficacy of checkpoint inhibitors in this subtype. CMS4 also presents high immune infiltrate but with an unfavourable, inflamed molecular orientation characterized by intratumoral MDSC, M2-macrophages and B-cells associated with pro-inflammatory gene expression, including myeloid chemokines, immune suppressive molecules and complement factors. In this situation, the combination of checkpoint inhibitors with TGF pathway inhibition represents a promising strategy as well as the use of angiogenesis inhibitors or anti-MDSCs treatment. On the contrary, CMS2 and CMS3 are poorly immunogenic tumors with scarce immune infiltrate. In this context, combination of checkpoint inhibitors with MEK-inhibition or anti-EGFR monoclonal antibodies could allow to overcome resistance. In addition, monoclonal antibodies targeting tumor-associated antigens, such as CEA, engineered with IL-2 may be able to increase immune infiltration and activates NK and T cells also in tumors with poor immune infiltration. Other strategies which may be effective in the setting of CMS2 and CMS3 are the combination of chemotherapy and immune checkpoint inhibitors or passive immunotherapy treatments as cancer vaccines with primed DCs.
In conclusion, the development of new effective immunotherapeutic strategies in CRC should be driven by a better knowledge of mechanisms of resistance to current treatments and take in account differences in immune microenvironment between different molecular subtypes to find the best treatment for each patient.
Declarations
Authors’ contributionsResponsible for the paper: Berardi R
Concept, design, definition of intellectual content: Bittoni A
Literature search: Meletani T, Sotte V, Cantini L
Manuscript preparation: Meletani T, Sotte V, Cantini L, Giampieri R
Manuscript editing: Meletani T, Sotte V, Cantini L, Giampieri R, Bittoni A
Manuscript revision: Berardi R, Bittoni A, Meletani T, Sotte V, Cantini L, Giampieri R
Availability of data and materialsNot applicable.
Financial support and sponsorshipNone.
Conflicts of interestAll authors declared that there are no conflicts of interest.
Ethical approval and consent to participateNot applicable.
Consent for publicationNot applicable.
Copyright© The Author(s) 2018.
REFERENCES
1. Van Cutsem E, Cervantes A, Adam R, Sobrero A, Van Krieken JH, et al. ESMO consensus guidelines for the management of patients with metastatic colorectal cancer. Ann Oncol 2016;27:1386-422.
3. Grady WM, Carethers JM. Genomic and epigenetic instability in colorectal cancer pathogenesis. Gastroenterology 2008;135:1079-99.
4. de la Chapelle A, Hampel H. Clinical relevance of microsatellite instability in colorectal cancer. J Clin Oncol 2010;28:3380-7.
5. de Weger VA, Turksma AW, Voorham QJ, Euler Z, Bril H, et al. Clinical effects of adjuvant active specific immunotherapy differ between patients with microsatellite-stable and microsatellite-instable colon cancer. Clin Cancer Res 2012;18:882-9.
6. Boland CR, Thibodeau SN, Hamilton SR, Sidransky D, Eshleman JR, et al. A National Cancer Institute Workshop on microsatellite instability for cancer detection and familial predisposition: development of international criteria for the determination of microsatellite instability in colorectal cancer. Cancer Res 1998;58:5248-57.
7. Hermsen M, Postma C, Baak J, Weiss M, Rapallo A, et al. Colorectal adenoma to carcinoma progression follows multiple pathways of chromosomal instability. Gastroenterology 2002;123:1109-19.
8. Meijer GA, Hermsen MA, Baak JP, van Diest PJ, Meuwissen SG, et al. Progression from colorectal adenoma to carcinoma is associated with non-random chromosomal gains as detected by comparative genomic hybridisation. J Clin Pathol 1998;51:901-9.
9. Merok MA, Ahlquist T, Røyrvik EC, Tufteland KF, Hektoen M, et al. Microsatellite instability has a positive prognostic impact on stage II colorectal cancer after complete resection: results from a large, consecutive Norwegian series. Ann Oncol 2013;24:1274-82.
10. Samowitz WS, Curtin K, Ma KN, Schaffer D, Coleman LW, et al. Microsatellite instability in sporadic colon cancer is associated with an improved prognosis at the population level. Cancer Epidemiol Biomarkers Prev 2001;10:917-23.
11. Gelsomino F, Barbolini M, Spallanzani A, Pugliese G, Cascinu S. The evolving role of microsatellite instability in colorectal cancer: a review. Cancer Treat Rev 2016;51:19-26.
12. Segal NH, Parsons DW, Peggs KS, Velculescu V, Kinzler KW, et al. Epitope landscape in breast and colorectal cancer. Cancer Res 2008;68:889-92.
13. Roelands J, Kuppen PJK, Vermeulen L, Maccalli C, Decock J, et al. Immunogenomic classification of colorectal cancer and therapeutic implications. Int J Mol Sci 2017; doi: 10.3390/ijms18102229.
14. Voena C, Chiarle R. Advances in cancer immunology and cancer immunotherapy. Discov Med 2016;21:125-133.
15. Schiavoni G, Gabriele L, Mattei F. The tumor microenvironment: apitch for multiple players. Front Oncol 2013;3:90.
16. Herrera M, Herrera A, Domínguez G, Silva J, García V, et al. Cancer-associated fibroblast and M2 macrophage markers together predict outcome in colorectal cancer patients. Cancer Sci 2013;104:437-44.
17. Basile D, Garattini SK, Bonotto M, Ongaro E, Casagrande M, et al. Immunotherapy for colorectal cancer: where are we heading? Expert Opin Biol Ther 2017;17:709-21.
18. Pagès F, Kirilovsky A, Mlecnik B, Asslaber M, Tosolini M, et al. In situ cytotoxic and memory T cells predict outcome in patients with early-stage colorectal cancer. J Clin Oncol 2009;27:5944-51.
19. Mlecnik B, Tosolini M, Kirilovsky A, Berger A, Bindea G, et al. Histopathologic-based prognostic factors of colorectal cancers are associated with the state of the local immune reaction. J Clin Oncol 2011;29:610-8.
20. Zaretsky JM, Garcia-Diaz A, Shin DS, Escuin-Ordinas H, Hugo W, et al. Mutations associated with acquired resistance to pd-1 blockade in melanoma. N Engl J Med 2016;375:819-29.
21. Shin DS, Zaretsky JM, Escuin-Ordinas H, Garcia-Diaz A, Hu-Lieskovan S, et al. Primary resistance to PD-1 blockade mediated by JAK1/2 mutations. Cancer Discov 2017;7:188-201.
22. Le DT, Durham JN, Smith KN, Wang H, Bartlett BR, et al. Mismatch-repair deficiency predicts response of solid tumors to PD-1 blockade. Science 2017;357:409-13.
23. Khong HT, Restifo NP. Natural selection of tumor variants in the generation of “tumor escape” phenotypes. Nat Immunol 2002;3:999-1005.
24. Brzostek J, Gascoigne NR, Rybakin V. Cell type-specific regulation of immunological synapse dynamics by B7 ligand recognition. Front Immunol 2016;7:24.
25. Galon J, Pagès F, Marincola FM, Thurin M, Trinchieri G, et al. The immune score as a new possible approach for the classification of cancer. J Transl Med 2012;10:1.
26. Galon J, Pagès F, Marincola FM, Angell HK, Thurin M, et al. Cancer classification using the immunoscore: a worldwide task force. J Transl Med 2012;10:205.
27. Guinney J, Dienstmann R, Wang X, de Reyniès A, Schlicker A, et al. The consensus molecular subtypes of colorectal cancer. Nat Med 2015;21:1350-6.
28. Becht E, de Reyniès A, Giraldo NA, Pilati C, Buttard B, et al. Immune and stromal classification of colorectal cancer is associated with molecular subtypes and relevant for precision immunotherapy. Clin Cancer Res 2016;22:4057-66.
29. Isella C, Brundu F, Bellomo SE, Galimi F, Zanella E, et al. Selective analysis of cancer-cell intrinsic transcriptional traits defines novel clinically relevant subtypes of colorectal cancer. Nat Commun 2017;8:15107.
30. Llosa NJ, Cruise M, Tam A, Wicks EC, Hechenbleikner EM, et al. The vigorous immune microenvironment of microsatellite instable colon cancer is balanced by multiple counter-inhibitory checkpoints. Cancer Discov 2015;5:43-51.
31. Verhagen J, Sabatos CA, Wraith DC. The role of CTLA-4 in immune regulation. Immunol Lett 2008;115:73-4.
32. Buchbinder EI, Desai A. CTLA-4 and PD-1 pathways: similarities, differences, and implications of their inhibition. Am J ClinOncol 2016;39:98-106.
33. Nishimura H, Okazaki T, Tanaka Y, Nakatani K, Hara M, et al. Autoimmune dilated cardiomyopathy in PD-1 receptor-deficient mice. Science 2001;291:319-22.
34. Chamoto K, Al-Habsi M, Honjo T. Role of PD-1 in immunity and diseases. Curr Top Microbiol Immunol 2017;410:75-97.
35. Le DT, Uram JN, Wang H, Bartlett BR, Kemberling H, et al. PD-1 blockade in tumors with mismatch-repair deficiency. N Engl J Med 2015;372:2509-20.
36. Andre T, Lonardi S, Wong M, Lenz HJ, Gelsomino F, et al. Nivolumab + ipilimumab combination in patients with DNA mismatch repair-deficient/microsatellite instability-high (dMMR/MSI-H) metastatic colorectal cancer (mCRC): first report of the full cohort from CheckMate-142. J Clin Oncol 2018.
37. Overman MJ, Lonardi S, Wong KYM, Lenz HJ, Gelsomino F, et al. Durable clinical benefit with nivolumab plus ipilimumab in DNA mismatch repair-deficient/microsatellite instability-high metastatic colorectal cancer. J Clin Oncol 2018;36:773-9.
38. Larkin J, Chiarion-Sileni V, Gonzalez R, Grob JJ, Cowey CL, et al. Combined nivolumab and ipilimumab or monotherapy in untreated melanoma. N Engl J Med 2015;373:23-34.
39. Voron T, Colussi O, Marcheteau E, Pernot S, Nizard M, et al. VEGF-A modulates expression of inhibitory checkpoints on CD8+ T cells in tumors. J Exp Med 2015;212:139-48.
40. Limagne E, Euvrard R, Thibaudin M, Rébé C, Derangère V, et al. Accumulation of MDSC and Th17 cells in patients with metastatic colorectal cancer predicts the efficacy of a FOLFOX-bevacizumab drug treatment regimen. Cancer Res 2016;76:5241-52.
41. Triplett TA, Tucker CG, Triplett KC, Alderman Z, Sun L, et al. STAT3 signaling is required for optimal regression of large established tumors in mice treated with anti-OX40 and TGFβ receptor blockade. Cancer Immunol Res 2015;3:526-35.
42. Schaer D, Li Y, Castaneda S, Inigo I, Surguladze D, et al. Targeting the TGF pathway with galunisertib, a TGF-RI SMI, promotes anti-tumor immunity leading to durable, complete responses, as monotherapy and in combination with checkpoint inhibition. J Immunother Cancer 2015;3:P402.
43. Watson NF, Ramage JM, Madjd Z, Spendlove I, Ellis IO, et al. Immunosurveillance is active in colorectal cancer as downregulation but not complete loss of MHC class I expression correlates with a poor prognosis. Int J Cancer 2006;118:6-10.
44. Lal N, White BS, Goussous G, Pickles O, Mason MJ, et al. KRAS mutation and consensus molecular subtypes 2 and 3 are independently associated with reduced immune infiltration and reactivity in colorectal cancer. Clin Cancer Res 2018;24:224-33.
45. Ebert PJR, Cheung J, Yang Y, McNamara E, Hong R, et al. MAP kinase inhibition promotes T cell and anti-tumor activity in combination with PD-L1 checkpoint blockade. Immunity 2016;44:609-21.
46. Bendell JC, Bang YJ, Chee CE, Ryan DP, McRee AJ, et al. A phase Ib study of safety and clinical activity of atezolizumab (A) and cobimetinib (C) in patients (pts) with metastatic colorectal cancer (mCRC). J Clin Oncol 2018.
47. Chen N, Fang W, Zhan J, Hong S, Tang Y, et al. Upregulation of PD-L1 by EGFR activation mediates the immune escape in EGFR-driven NSCLC: implication for optional immune targeted therapy for NSCLC patients with EGFR mutation. J Thorac Oncol 2015;10:910-23.
48. Klein C, Waldhauer I, Nicolini VG, Freimoser-Grundschober A, Nayak T, et al. Cergutuzumab amunaleukin (CEA-IL2v), a CEA-targeted IL-2 variant-based immunocytokine for combination cancer immunotherapy: overcoming limitations of aldesleukin and conventional IL-2-based immunocytokines. Oncoimmunology 2017;6:e1277306.
49. Tabernero J, Melero I, Ros W, Argiles G, Marabelle A, et al. Phase Ia and Ib studies of the novel carcinoembryonic antigen (CEA) T-cell bispecific (CEA CD3 TCB) antibody as a single agent and in combination with atezolizumab: Preliminary efficacy and safety in patients with metastatic colorectal cancer (mCRC). J Clin Oncol 2017; doi: 10.1200/JCO.2017.35.15_suppl.3002.
50. Segal NH, Kemeny NE, Cercek A, Reidy DL, Raasch PJ, et al. Non-randomized phase II study to assess the efficacy of pembrolizumab (Pem) plus radiotherapy (RT) or ablation in mismatch repair proficient (pMMR) metastatic colorectal cancer (mCRC) patients. J Clin Oncol 2016; doi: 10.1200/JCO.2016.34.15_suppl.3539.
51. Shahda S, Noonan AM, Bekaii-Saab TS, O’Neil BH, Sehdev A, et al. A phase II study of pembrolizumab in combination with mFOLFOX6 for patients with advanced colorectal cancer. J Clin Oncol 2017; doi: 10.1200/JCO.2017.35.15_suppl.3541.
52. Orillion A, Hashimoto A, Damayanti N, Shen L, Adelaiye-Ogala R, et al. Entinostat neutralizes myeloid-derived suppressor cells and enhances the antitumor effect of PD-1 inhibition in murine models of lung and renal cell carcinoma. Clin Cancer Res 2017;23:5187-201.
53. Azad NS, Shirai K, McRee AJ, Opyrchal M, Johnson DB, et al. ENCORE 601: a phase 2 study of entinostat in combination with pembrolizumab in patients with microsatellite stable metastatic colorectal cancer. J Clin Oncol 2018; doi: 10.1200/JCO.2018.36.15_suppl.3557.
54. Magee MS, Abraham TS, Baybutt TR, Flickinger JC Jr, Ridge NA, et al. Human GUCY2C-targeted chimeric antigen receptor (CAR)-expressing T cells eliminate colorectal cancer metastases. Cancer Immunol Res 2018;6:509-16.
55. Huang Q, Xia J, Wang L, Wang X, Ma X, et al. miR-153 suppresses IDO1 expression and enhances CAR T cell immunotherapy. J Hematol Oncol 2018;11:58.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Bittoni A, Sotte V, Meletani T, Cantini L, Giampieri R, Berardi R. Immunotherapy in colorectal cancer treatment: actual landscape and future perspectives. J Cancer Metastasis Treat 2018;4:55. http://dx.doi.org/10.20517/2394-4722.2018.37
AMA Style
Bittoni A, Sotte V, Meletani T, Cantini L, Giampieri R, Berardi R. Immunotherapy in colorectal cancer treatment: actual landscape and future perspectives. Journal of Cancer Metastasis and Treatment. 2018; 4: 55. http://dx.doi.org/10.20517/2394-4722.2018.37
Chicago/Turabian Style
Bittoni, Alessandro, Valeria Sotte, Tania Meletani, Luca Cantini, Riccardo Giampieri, Rossana Berardi. 2018. "Immunotherapy in colorectal cancer treatment: actual landscape and future perspectives" Journal of Cancer Metastasis and Treatment. 4: 55. http://dx.doi.org/10.20517/2394-4722.2018.37
ACS Style
Bittoni, A.; Sotte V.; Meletani T.; Cantini L.; Giampieri R.; Berardi R. Immunotherapy in colorectal cancer treatment: actual landscape and future perspectives. J. Cancer. Metastasis. Treat. 2018, 4, 55. http://dx.doi.org/10.20517/2394-4722.2018.37
About This Article
Special Issue
Copyright

Data & Comments
Data


 Cite This Article 23 clicks
Cite This Article 23 clicks

 Like This Article 27
likes
Like This Article 27
likes






Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.