
Metabolic rewiring of stem cells and differentiated cells in cancer: the hypothetical consequences of a GABA deficiency in endocrine pancreas
 0
0Abstract
The carcinogenic mechanism proposed considers that stem cells committed to repair tissues and differentiated cells, acquire different metabolic properties, if there is an associated GABA deficiency suppressing a control system of the endocrine pancreas. This control system mediated by GABA, released with insulin, normally turns off glucagon and somatostatin release when insulin is released. A consequence of the GABA deficiency in pancreas and adrenals is a hybrid insulin-glucagon-somatostatin message, received by new mitotic stem cells displaying then a hybrid metabolic rewiring. This gives them a selective metabolic advantage over differentiated cells that become insulin resistant and only receive the glucagon- somatostatin part of the hormonal message. Indeed, their insulin receptors are desensitized by the persistent leakage of insulin resulting from the GABA deficiency that fails to close the insulin release mechanism. Thus differentiated cells are simply rewired to be plundered by stem cells. The metabolic advantage gained by stem cells blocks their own differentiation and maintains their mitotic capacity. Inevitable mutations of mitotic cells follow, the immune system is unable to eliminate a geometrically increasing number of altered stem cells, a selection of the most aggressive but metabolically successful population takes place when cancer is declared.
Keywords
Introduction
Since the early observations of Warburg on cancer metabolism showing the lactic acid increase even in the presence of oxygen (Warburg effect)[1,2], until the seminal works of Mazurek and Eigenbrodt[3] and Eigenbrodt et al.[4] on pyruvate kinase (PK) M2 blockade in tumors, much progress has been made on metabolic rewiring processes in cancer[5,6]. An increasing number of works are interestingly gathered in special issues on cancer metabolism[7]. However, they deserve to be included in a more general frame proposing a mechanistic explanation for metabolic rewiring processes in cancer. In parallel, there is great interest for diets, natural products and life styles that would decrease cancer incidence. Presumably, the idea is that a diet might influence metabolism and the epigenetic control of genes that have been implicated in cancer incidence. It is for example shown that a simple dietetic mixture: curcuma and poly unsaturated fatty acid, gives protection against a carcinogen (Dimethyl Benz Anthracene) in animal models[8]. There are several observations linking nutrition and cancer, reminding that nutrition has evidently metabolic consequences that may change the incidence of diseases, including cancer.
Paradoxically, even viral or cellular oncogenes that initially pointed toward a viral or genetic cause for cancer, reinforced the metabolic cancer model, because the genes that were up-regulated by the viral or cellular oncogenes encoded for proteins supporting the different steps of signaling pathways similar to the one activated by insulin, via tyrosine kinase receptors. With insulin signaling, glycolytic metabolism evidently came back on the stage. However, a general frame that would explain the metabolic rewiring process in cancer, its effects on stem cells and differentiated cells remained to be proposed; this was started in previous works[9-13] and is continued in this presentation.
A surprisingly low number of metabolic “switch compounds” control the direction of major metabolic pathways in a network of enzymes that support the different metabolic finalities reached by a cell in response to its receptors. The cascades of signals that follow coordinate the enzymes and the pathways direction towards a specific metabolic finality responding to a physiological situation. Schematically, the finalities to reach are: (1) the catabolic production of nutrients (glucose and ketone bodies) when food gets scarce; (2) the production of energy, with a possible selection of the energetic source (glucose, fatty acids, eventually ketone bodies); (3) the anabolic synthesis of new constituents for new mitotic cells, which requires energy and the substrates for making these constituents. In addition, anabolic hormones increase glycogen and lipid tissue stores when food is available. These metabolic finalities are controlled by the ON or OFF status of major enzymes, activated or inhibited by phosphorylation via their respective protein kinases or phosphatases, themselves controlled by “switchboard” kinases and phosphatases activated by specific receptors. In previous works we have analyzed the phosphorylation status of key enzymes in given physiological situations covering the finalities considered. A coherent set of phosphorylation of these enzymes was associated to the action of catabolic hormones (glucagon, epinephrine and cortisol). The phosphorylation status was opposite for anabolic hormones such as insulin or IGF. The conclusion was that in cancer, enzymes such as PK and pyruvate dehydrogenase (PDH) remained phosphorylated and blocked in their catabolic configuration, even if the cell had switched from catabolism to anabolism; as if the action of a switch board phosphatase had failed. In such condition, the cell had to rewire its metabolic pathways in order to by-pass the PK and PDH “bottle necks”, the resulting metabolism was favorable to its development at the detriment of other differentiated cells that become their nutritional reservoir; this perverted finality seems to be a starter for cancer. We shall first consider the status of major control switches, for the three major metabolic finalities and then analyze the unique hybrid configuration of these switches in cancer, showing how pathways are reconnected in mitotic stem cells, or in differentiated cells. It will become apparent that a non-coherent mixed hormonal signal is received by new mitotic cells with new receptors, while differentiated cells with desensitized insulin receptors, resistant to insulin, will respond preferentially to catabolic hormones and to growth hormone (GH). We have in earlier works considered that a failure of the endocrine pancreas, would explain the aberrant hybrid hormonal signaling that supports cancer metabolism. Indeed, insulin secreting beta cells in the pancreas possess a major GABA control system that normally turns off glucagon releasing alpha cells and somatostatin releasing delta cells, when beta cells release insulin and GABA. The released GABA also closes via auto receptors of beta cells, the release of insulin. Thus a GABA deficiency would not only fail to turn off glucagon release when insulin is released, sending a dual hybrid glucagon-insulin message, but also maintain a steady leakage of insulin that desensitizes with time, insulin receptors. New mitotic stem cells with new insulin receptors, not yet desensitized for insulin will then respond to both insulin and glucagon, while other differentiated cells constantly submitted to an insulin leakage become resistant to insulin. The stem cells will have to rewire their metabolic pathways in response to the hybrid message; they gain a special metabolic advantage over differentiated cells that are simply plundered, since they only respond to the catabolic component of the hybrid message.
The carcinogenic mechanism proposed considers that stem cells committed to repair tissues after a variety of injuries and differentiated cells develop different metabolic features, if there is an associated GABA failure in the endocrine pancreas[9,10]. The pancreatic GABA deficiency hypothesis that fully explained the observed metabolic rewiring in cancer was initially published in reference 9; the hypothesis was confirmed and strengthened by a set of epidemiological observations that were gathered in reference 10. These observations also included the more controversial role of some pesticides that may affect GABA, a point that was not particularly appreciated. The metabolic advantage given to stem cells by the pancreatic deficiency blocks their differentiation and maintains their mitotic capacity; while differentiated cells are plundered. Inevitable mutations follow, while the immune system becomes unable to eliminate a geometrically increasing number of altered stem cells, a selection of the most aggressive and metabolically successful population starts cancer. Presumably, this metabolic trigger for carcinogenesis starts years before cancer appears, correcting the GABA pancreatic failure, or neutralizing eventual auto-antibodies against glutamate decarboxylase (GAD), or suppressing its inhibition, might delay or avoid cancer. When cancer is declared present therapies might be backed-up by a metabolic treatment aiming to reverse the rewiring process that gave to tumor cells their metabolic advantage.
Metabolic switches orient pathways towards different metabolic finalities
We represented in Figure 1, the endocrine pancreas, with its beta, alpha and delta cells that secrete respectively insulin, glucagon and somatostatin (STH). The GABA release from beta cells inhibits alpha and delta cells (via GABA A receptors) when insulin is released; and puts an end to insulin release (via GABA B auto receptors). GABA also inhibits epinephrine release from adrenals. We describe with different colors the pathways that operate for the different metabolic finalities that a cell is susceptible to reach, and give in the corresponding colored boxes the status (increase +, decrease -, or 0 not relevant ) of the selected switches. First finality: The production of nutrients; triggered by catabolic hormones, glucagon, epinephrine, and cortisol (yellow pathways and yellow box Figure 1). When food gets scarce, the pancreas senses the drop of blood glucose, hyperpolarized beta cells retain GABA, no longer spilled over alpha cells and their relative depolarization triggers the release of glucagon. The action of glucagon on liver glucagon Gs coupled receptors, or that of epinephrine on beta adrenergic receptors in muscles, stimulates adenylate cyclase and the synthesis of cAMP. The latter, activates PKA serine kinase then Src tyrosine kinase, eliciting in fine, via specific protein kinases, the phosphorylation of a set of enzymes that are ON or OFF after phosphorylation. Glycogen phosphorylase a, is ON, supporting glycogenolysis, while PK and PDH are OFF closing the entry of the citric acid cycle, this spares oxaloacetate (OAA) at the start of the neoglucogenic pathway; pyruvate carboxylase (Pcarb) and phosphoenolpyruvate carboxykinase (PEPCK) are both activated. The blockade of PK by phosphorylation, avoids a reconversion of phosphoenolpyruvate (PEP) into pyruvate (PYR). Muscle proteolysis provides amino acids, the transamination of alanine feeds with PYR the neoglucogenic route. Since cAMP inhibits Fructose 2-6 bis phosphate (Fruc 2-6 bisP), it cancels the inhibition of neoglucogenesis and blocks glycolysis; glucose is now produced. We have to add that hypoglycemia decreases glycolysis in erythrocytes, which form less 2-3 DPG. Thus, oxygen is retained by hemoglobin and less is delivered and reduced by other cells, their NADH/NAD ratio increases, closing their citrate condensation that starts the Krebs cycle, more OAA goes to neoglucogenesis. Glycogenolysis is activated as well (phosphorylated phosphorylase a, is ON), while phosphorylated glycogen synthase is OFF. The other nutrient are ketone bodies; we know that PKA and Src activate a protein LKB1 by phosphorylation, it will stimulate AMP kinase, which inhibits acetyl CoA carboxylase (ACC) at the start of the fatty acid synthesis pathway. The inhibition of ACC decreases the product malonyl CoA, which is an inhibitor of the mitochondria carnityl transporter of fatty acids, allowing then fatty acid to enter in mitochondria. There, beta oxidation forms acetyl CoA, then ketone bodies, acetoacetate, particularly in liver. The malonyl CoA inhibition of the fatty acid transporter blocks the degradation of fatty acid when their synthesis is activated and vice versa. The ketone bodies are delivered to other cells, where they give back acetyl CoA by the reverse pathway (thiolase operating in both directions).
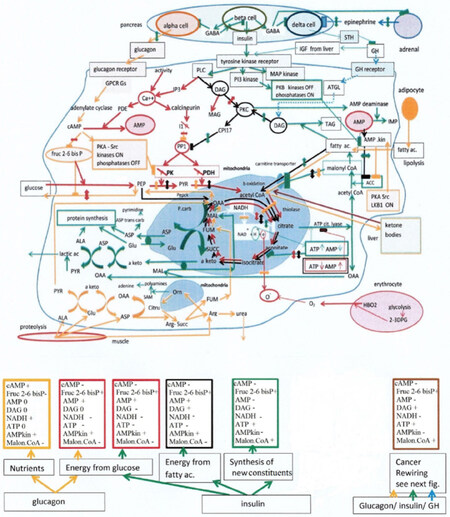
Figure 1. Top: endocrine pancreas, the alpha cell releases glucagon; it is turned off by GABA co-released with insulin from beta cells. In this way when anabolic insulin is ON catabolic hormones are OFF, GABA also inhibits epinephrine release from adrenals. Moreover, GABA turns off somatostatin release from delta cells, which stimulates growth hormone and IGF. The cell below the pancreas displays different metabolic finalities that can be reached; these are controlled by a few “switch compounds” themselves controlled by the endocrine pancreas. These compounds are listed in the boxes below the cell. The yellow pathways represent the nutrition finality forming glucose and ketone bodies; it is activated by catabolic hormones that control the switch compounds in the yellow box (+ is increase, - decrease, 0 no change or not relevant). The second and third metabolic finalities represented by the red pathways is the production of energy from glucose, in the case of glucagon or insulin, note in the corresponding red boxes the difference for diacylglycerol DAG. The next finality is the production of energy from fatty acids it is represented by the black pathways and the black box for switch compounds. The last finality represented by the green pathways and green box for the switches, is the anabolic synthesis of new constituents, it is essential for mitotic cells. The last box in brown shows for comparison the switches for mitotic tumor cells, specially studied in Figure 2. DAG: diacyl glycerol; PK: pyruvate kinase; PDH: pyruvate dehydrogenase; GH: growth hormone
Second finality: the production of energy, and selection of glycolytic or fatty acid sources of acetyl CoA. Even when glucagon, epinephrine and cortisol are released for making glucose and nutrients, muscles can reverse the situation and keep their glycolytic pathway operational, with little or no neoglucogenesis; on the other hand, glycogenolysis is still possible (Figure 1 red pathways and red box). This muscle exception is presumably linked to the fact that muscles have to be ready for escaping predators. How are the effects of cAMP on neoglucogenesis cancelled? Muscle activity is associated to the movement of charges along T tubules until reticulum sacs with ryanodine receptors in the depth of the muscle, the depolarization releases calcium in the cytosol initiating muscle contraction. Calcium will also stimulate a phosphodiesterase (PDE) converting cAMP into AMP, which restores the synthesis of Fruc2-6 bisP, the allosteric activator of glycolysis, and inhibitor of neoglucogenesis. In parallel, calcium stimulates calcineurin phosphatase, which dephosphorylates an inhibitor (I1) of a major phosphatase PP1 that gets activated, leading in fine to the dephosphorylation and activation of PK and PDH, which opens the glycolytic supply of acetyl CoA to the Krebs cycle. The citrate condensation starts, (the NADH/NAD ratio decreases via oxygen reduction). Then, since ATP is low and AMP elevated, the inhibition isocitrate dehydrogenase by ATP is cancelled and the citric acid cycle ”turns”, which decreases the citrate efflux from mitochondria and closes the fatty acid synthesis route (via ATP citrate lyase and ACC). The elevated AMP activates AMP kinase, which inhibits ACC; a low level of malonyl CoA may now open the beta oxidation of fatty acid and the fatty acid source of acetyl CoA, when glucose is missing. Well, exactly the same glycolytic energetic finality (red pathway Figure 1) will eventually operate in anabolism when insulin is released; because insulin elicits the activation of a phospholipase that splits phosphatidyl inositol (4-5) Bis P (PIP2) into diacyl glycerol (DAG) and inositol 1,4,5 Phosphate (IP3). The latter, will act on IP3 receptors of the reticulum, releasing calcium in the cytosol. Calcium stimulates as described above, a PDE neutralizing the inhibitory effect of cAMP on Fruc 2-6 bisP synthesis, and activates calcineurin, then PP1, PK and PDH, which stimulates glycolysis and opens the glycolytic entry of the Krebs cycle. But here is a new regulator to consider: DAG. If the level of DAG is low, presumably when DAG is converted into monoacyl glycerol, the glycolytic route forms acetyl CoA as above (Figure 1 second red box). However, if DAG increases, it will activate protein kinase C (PKC) and the latter elicits the synthesis of a new inhibitor of PP1 known as CPI 17, which keeps PK and PDH phosphorylated and inhibited, and closes the glycolytic supply of acetyl CoA. The fatty acid source of acetyl CoA will have to be opened. Since AMP is elevated, it stimulates AMP kinase, which inhibits ACC and the fatty acid synthesis pathway. With malonyl CoA being low, fatty acid beta oxidation is now permitted, opening the fatty acid supply of acetyl CoA, (Figure 1 black pathway and black box). The third finality: the anabolic synthesis of new constituents depends essentially on Insulin and IGF. It is represented by the green pathways, plus the glycolytic red pathway, and green box for switches. The effect of insulin on its tyrosine kinase receptor elicits anabolism: The synthesis of glycogen, of fatty acids and triglycerides (TAG), of proteins. Insulin also inhibits the opposite effects: glycogenolysis, neoglucogenesis, lipolysis and proteolysis mediated by catabolic hormones. Insulin stimulates glucose uptake, facilitating the incorporation of the glucose transporter. The influx of glucose in cells is also regulated by different affinities of liver glucokinase lower than the equivalent brain enzyme hexokinase; which serves the brain first with glucose. In parallel, insulin activates the MAP kinase mitotic and PI3kinase routes, supporting mitosis and cell survival (recall that each step of these pathways was up-regulated by viral or cellular oncogenes). We have mentioned above, the downstream stimulation by insulin, of a phospholipase C that generates IP3 and DAG. We recall that IP3 elicits the release of internal calcium, activating a PDE hydrolyzing cAMP, which cancels the inhibition of Fruc 2-6 bisP synthesis, leading to an increased glycolysis. Moreover, calcium helps the incorporation of glucose transporters in cells that have no constitutive transporter in their membrane. But the stimulation of PKC by DAG will then stimulate AMP deaminase that converts AMP into IMP; which decreases AMP and the stimulation of AMP kinase, cancelling its inhibitory effect on ACC and fatty acid synthesis that gets activated. Moreover, insulin-mediated PKB effects are opposite to those of PKA that stimulated AMP kinase. One may then expect that the inhibition of AMP kinase (low AMP and inhibition by PKB) will open the fatty acid synthesis pathway; the malonyl CoA intermediate increases, closing the fatty acid carnityl transporter and the beta oxidation source of acetyl CoA. It is thus necessary to open back the glycolytic source of acetyl CoA that had been closed via DAG PKC stimulation forming the CPI17 inhibitor of PP1, which blocked PK and PDH. Now that the synthesis of fatty acids and triglycerides is activated by insulin, DAG will be converted into triglycerides and decrease, suppressing the stimulation of PKC by DAG and the CPI17 inhibition of PP1, which opens back PK and PDH and the glycolytic source of acetyl CoA (red pathway Figure 1), while the full anabolic effects of insulin take place. Note that the efflux of citrate from mitochondria is facilitated by the blockade of isocitrate dehydrogenase by ATP (the ATP/AMP ratio is elevated). When energy is missing, this ratio decreases, the citrate efflux from mitochondria declines, closing the citric supply to the fatty acid synthesis route, while the citric Krebs cycle starts turning again; the acetyl CoA may come again from fatty acid beta oxidation or from glucose, depending on the level DAG, as indicated in the corresponding black or red boxes.
Well, one of the keys for understanding the rewiring process taking place in cancer is the fact that tumor cells do maintain the synthesis of fatty acids and triglycerides, which automatically closes the fatty acid source of acetyl CoA, but tumor cells are unable to open back the glycolytic source of acetyl CoA; as if they were unable to decrease its DAG blocker, in spite of its conversion into triglycerides. A new supply of DAG kept PKC and CPI 17 stimulated, inhibiting PP1, then PK and PDH, which closes the glycolytic source of acetyl CoA. With these two sources of acetyl CoA blocked, the only possible way for tumor cells to get acetyl CoA and survive, comes from ketone bodies provided by liver cells. It is then essential to find out what keeps DAG elevated, in spite of its consummation by the active lipid synthesis pathway? Remember that carcinogenic phorbol-esters act similarly to DAG on PKC; this is discussed in the next section.
A carcinogenic metabolic rewiring process described in stem cells and differentiated cells
We have again represented in Figure 2, the endocrine pancreas but here is a blockade of GABA release, which might be the consequence of auto-antibodies against GAD that synthesizes GABA; remember that diabetes Type 1 is provoked by such antibodies that kill beta cells[14,15]. The GABA deficiency may have other causes, vitamin B6 deficiency (it is the co-factor of GAD) or a GAD mutation as in Stiff person syndrome, or result from pesticides and compounds that affect GABA uptake (betel nut addictions) and metabolism[10]. The most compelling fact in support of the hypothesis is that smoking (a major risk factor for numerous cancers, including pancreatic) impairs the GABA system. In brain, the increase of glutamate and decrease of GABA prolongs the activation of the dopaminergic rewarding sensation elicited by nicotine. In the periphery, the effects of smoking increase the systemic levels of epinephrine and norepinephrine. Whatever is the cause of the pancreatic GABA deficiency, it leads to an abnormal release of glucagon even when insulin is released, sending to cells a hybrid catabolic - anabolic message. There is also an effect on beta cells that become unable to terminate insulin release, a constant leakage of insulin will occur and desensitize insulin receptors, recalling much aspects of type 2 diabetes or metabolic syndrome. The GABA deficiency should increase somatostatin release from delta cell, but this is counteracted by an increased epinephrine release from adrenals due to the GABA deficit; thus, epinephrine strongly inhibits the delta cell and STH release, increasing the action of GH. It is not the same for alpha cells that are stimulated by epinephrine.

Figure 2. Top: endocrine pancreas with its GABA ergic controls OFF. This leads to a leakage of insulin rendering differentiated cells resistant to insulin; in addition, GABA fails to block glucagon release when insulin is released, sending a hybrid catabolic-anabolic hormonal message. The GABA deficiency increases epinephrine release from adrenals, which inhibits delta cells and somatostatin, activating growth hormone. The first cell is a new mitotic stem cell with new insulin receptors it receives the dual catabolic - anabolic message. PKA, Src are activated closing pyruvate kinase and pyruvate dehydrogenase and the glycolytic supply of acetyl CoA; but the increase of IP3 and calcium mediated by insulin, boosts glycolysis until the pyruvate kinase bottle neck, by increasing the fructose 2-6 bis Phosphate activator. Indeed, calcium stimulates a phosphodiesterase that decreases cAMP, cancelling its inhibitory effect on fructose 2-6 bis Phosphate synthesis. Note that growth hormone activates the production of DAG via ATGL, which stimulates PKC and CPI 17, inhibiting PP1 phosphatase, which maintains the glycolytic bottle neck, in spite of the conversion of DAG into triglycerides. Insulin elicits fatty acid synthesis by inhibiting via PKB, AMP kinase, which suppresses the inhibition of acetyl CoA carboxylase; the automatic increase of malonyl CoA closes the fatty acid transporter and the fatty acid source of acetyl CoA. Since the glycolytic source of acetyl CoA was off as well, the stem cell becomes dependent of ketone bodies reconverted into acetyl CoA to feed their citrate condensation, and support the synthesis of fatty acid and lipids for new mitotic cells. Note the supply of amino acids the synthesis of proteins, the transamination, and the release of lactic acid. The next cell is a liver cell resistant to insulin; it receives the catabolic glucagon part of the pancreatic message and is programmed for making glucose and ketone bodies. The fatty acid synthesis is OFF (ACC inhibition by AMP kinase) while fatty acid supplied by adipocytes, form acetyl CoA by beta oxidation, the decreased citrate condensation inhibited by NADH, leaves more acetyl CoA for the ketogenic pathway. The increased NADH comes from a lower oxygen supply and reduction. PKC-CPI17 is stimulated by DAG coming from ATGL stimulated by growth hormone. Muscle, provides amino acids supporting neoglucogenesis, note the transamination the urea cycle. The bottom cell is a muscle cell, also insulin resistant, while receiving the catabolic message (through epinephrine). Muscle activity releases calcium in the cytosol stimulating a phosphodiesterase hydrolyzing cAMP, which cancels the inhibition of fructose 2-6 bis Phosphate formation, activating glycolysis. The glycolytic bottle neck, is maintained because the inhibitor CPI17 of PP1 is again formed via PKC stimulation by DAG, produced by ATGL activated by growth hormone. The citrate condensation is not inhibited by NADH (the latter decreases since lactic acid and myoglobin pull more oxygen). The citric acid cycle turns, activity lowers the ATP/AMP ratio. Cortisol activates proteolysis providing amino acids to the liver. Doted lines are interrupted pathways. Blocks adjacent to each cell indicate the switch compound levels. DAG: diacyl glycerol; PK: pyruvate kinase; PDH: pyruvate dehydrogenase; GH: growth hormone; ATGL: adipose triglyceride lipase
Below the GABA deficient pancreas, three cells are represented: the top one is a stem cell rewiring its metabolic pathways into a carcinogenic mode after receiving the hybrid hormonal message from the deficient pancreas, the cell below is a liver differentiated cell, rewiring its metabolism while receiving the hybrid hormonal message after becoming insulin resistant. The bottom cell is of a muscle fiber also receiving the hybrid hormonal message with insulin resistance. The full lines are operational pathways; the dotted lines are pathways that have been closed. Each block adjacent to the cell gives the expected switch status in cancer: Figure 2.
In the top stem cell, glucagon activates via GPCR Gs receptors, adenylate cyclase and PKA-Src signaling as for the nutritional finality, but insulin elicits the formation of IP3 and DAG. The release of calcium mediated by IP3 activates the PDE hydrolyzing cAMP into AMP, the associated increase of Fruc 2-6bisP stimulates glycolysis, while calcium triggers the incorporation of glucose transporters in the membrane, rendering the cell avid for glucose. But the increased DAG stimulates PKC and the synthesis of the CPI17 inhibitor of PP1[16], which maintains the phosphorylation of PK and PDH and closes the glycolytic source of acetyl CoA. Accumulated above this bottle neck, PEP will be converted into OAA. Then, PKC will stimulate AMP deaminase and 5’ nucleotidase[17,18], which decreases AMP and the stimulation of AMP kinase; but here, AMP kinase is already directly blocked by PKB that is activated by the insulin signal. This blockade of AMP kinase cancels its stimulation by PKA-Src via LKB1. The inhibition of AMP kinase activates ACC, eliciting the synthesis of fatty acid, while the malonyl CoA intermediate increases and turns off the beta oxidation of fatty acids, which closes the fatty acid source of acetyl CoA. With the two sources of acetyl CoA closed, the only possible supply of acetyl CoA are ketone bodies provided by liver cells. Acetoacetate is taken up and reconverted back into acetyl CoA (via beta hydroxybutyrate dehydrogenase, ketoacyl CoA transferase and thiolase that are reversible). With OAA coming from PEP above the bottle neck via PEPCK, and acetyl CoA provided by ketone bodies, the citric Krebs cycle starts. The ketone bodies supply was the only way for these cells to survive. The citric condensation reaction is activated; since oxygen reduction pulls electrons from NADH converted to NAD, which removes the NADH brake over the citrate condensation reaction. Note that the release of lactic acid generates an acid gradient around the cell, pulling oxygen from erythrocytes via the Bohr Effect. The ATP/AMP ratio being elevated isocitrate dehydrogenase is inhibited favoring the citrate efflux from mitochondria, which feeds via ATP citrate lyase, and ACC, fatty acid synthase; and the synthesis of fatty acids, then triglycerides, to make new membranes for mitotic cells. The conversion of DAG into TAG should decrease DAG and put an end to the stimulation of PKC and CPI17 and to the inhibition of PP1 that closed the glycolytic source of acetyl CoA. But this does not occur in this case, because growth hormone activated by the decrease of STH mediated by epinephrine, stimulates an enzyme: adipose triglyceride lipase (ATGL) that forms more DAG, maintaining in this way the stimulation of PKC and CPI17, which closes the glycolytic acetyl CoA supply, via PP1 inhibition that fails to activate PK and PDH; (recall once more that phorbol ester carcinogens stimulate PKC as DAG). The release of epinephrine that blocked STH release from delta cells and increased GH is a consequence of the GABA deficiency that increases epinephrine release from adrenals.
Another point to mention is that the inhibition of AMP kinase discussed above, suppresses as well the inhibition of mTOR, which liberates a factor (eIF4E) from its binding protein phosphorylated by mTOR; this factor eIF4E will then initiate the synthesis of proteins. Amino acids are provided by muscle cells, and processed via the transaminases and the mitochondria shuttle, feeding also the synthesis of adenine and pyrimidine bases. The transamination chain forms pyruvate and in fine lactic acid. In addition, the avid uptake of glucose supports the pentose phosphate pathway, forming necessary nuclear components (ribose 5P) for mitotic cells. The box adjacent to the top cell gives the status of switches for this stem cell that rewires its metabolism into a carcinogenic way.
The next cell in Figure 2 is a differentiated liver cell rendered resistant to insulin; this is due to the leakage of insulin resulting from the pancreatic GABA deficiency. However, this cell responds to the glucagon part of the hybrid pancreatic message. The Gs coupled glucagon receptor is activated, adenylate cyclase forms cAMP, and PKA-Src are operational; cAMP inhibits the formation of Fruc 2-6bis P, closing glycolysis while opening neoglucogenesis. In parallel, PKA-Src will phosphorylate and inhibit PK and PDH closing the entry of the citric acid cycle. The cell receives amino acids from muscle proteolysis; the amino acids follow the transamination chain as indicated, forming pyruvate that feeds, via Pcarb and PEPCK the neoglucogenic pathway. There is also an activation of the urea cycle. In parallel to neoglucogenesis, glucose is also produced by glycogenolysis (phosphorylase a, is ON). The transaminations are interacting as indicated in Figure 2, with the mitochondria shuttle. Note that PKA-Src activates LKB1, which stimulates AMP kinase, this closes the fatty acid synthesis pathway (AMP kinase inhibits ACC). Now fatty acids provided by adipocyte lipolysis, get transported in the liver cell mitochondria since malonyl CoA is low, their beta oxidation forms acetyl CoA. With the NADH being elevated, the citrate condensation is inhibited. Thus, acetyl CoA in excess will follow the ketogenic route giving acetoacetate. Note that growth hormone stimulates ATGL and DAG formation, which stimulates PKC and the CPI17 inhibitor of phosphatase PP1, maintaining the inhibition of PK and PDH, which closes the glycolytic entry of the citric acid cycle. The stimulation of PKC should activate AMP deaminase, decreasing AMP and the stimulation of AMP kinase, but AMP kinase is already efficiently activated by LKB1, which maintains the inhibition of the fatty acid synthesis pathway. Note also that the ATP/AMP ratio that controls the citrate efflux via the inhibition of isocitrate dehydrogenase by ATP is not operational here, since the above reaction, the citrate condensation, is already inhibited by an elevated NADH/NAD ratio. Presumably a low supply of glucose decreases 2-3DPG in erythrocytes, less oxygen is delivered and reduced by the liver cell and fewer electrons are pulled away from NADH, which inhibits the citrate condensation reaction. Probably, less lactic acid is released by liver cells, since pyruvate feeds the neoglucogenic route, rather than lactate dehydrogenase, which locally decreases the Bohr Effect and oxygen supply to the liver. This liver cell is programmed to feed the above stem cell with nutrients, glucose and particularly ketone bodies, while receiving amino acids from muscle, and fatty acids from adipose tissue.
The bottom cell in Figure 2 is a muscle cell; again, it is resistant to insulin like other differentiated cells, and receives the catabolic part of the hybrid pancreatic message. For muscles, epinephrine rather than glucagon, activates via Gs coupled beta adrenergic receptors, adenylate cyclase and PKA-Src. But here, there is a muscle specificity that has to maintain glycolysis as discussed earlier. Muscle activity releases calcium from internal stores, stimulating a PDE that hydrolyses cAMP, this cancels the inhibition by cAMP on Fruc2-6 bis P formation; the increase of this allosteric activator of glycolysis at the phospho fructokinase 1 step, boosts glycolysis, while calcium stimulates the membrane incorporation of the glucose transporter. In addition, the alpha epinephrine receptors of Go or Gi type are predominant in muscle limiting the increase cAMP, via Gs coupled beta adrenergic receptors. Calcium should also activate calcineurin phosphatase and neutralize the inhibition of PP1 by I1 in order to open back PK and PDH by de-phosphorylation, but here the activation of PKC by DAG forms the CPI 17 inhibitor of PP1 closing the glycolytic acetyl CoA supply. This DAG is formed by ATGL stimulated by GH; we explained above that the GABA deficit elicits the release of epinephrine that blocks efficiently delta cells, decreasing STH, which stimulates GH and ATGL. As occurred for the liver cell, the LKB1 stimulation of AMP kinase inhibits ACC and closes the fatty acid synthesis route, which automatically opens the beta oxidation of fatty acids into acetyl CoA. But in contrast to the liver cell, the citrate condensation is operational in muscle. It is activated by the decrease of NADH resulting from the increased reduction of oxygen. Indeed, the lactic acid released supports the Bohr Effect, which liberates more oxygen from oxy- hemoglobin in acid conditions. Moreover, muscle myoglobin pulls more oxygen into the muscle. The condensation of acetyl CoA coming from fatty acids, and OAA provided by PEP, above the PK bottle neck, forms citrate. The efflux of citrate will not feed the fatty acid synthesis, we have seen that the pathway was closed (via the activation of AMP kinase by LKB1) thus citrate will be processed by the Krebs cycle. Muscle activity consumes ATP and forms AMP; since ATP is lowered, it does not inhibit isocitrate dehydrogenase, the citric acid cycle “turns”. Another essential point is that cortisol stimulates proteolysis in muscle; indeed, glucagon increases ACTH release and the release of cortisol from adrenals. Note that the amino acids will feed the liver cell forming glucose, and the stem cell synthesizing its proteins; follow the interaction of the amino acids with the transaminases and mitochondrial shuttle, and the lactic acid release for both stem cells and muscle supporting locally a Bohr Effect. In liver cells, pyruvate will preferentially follow the neoglucogenic route, and less goes to lactate dehydrogenase.
Conclusions and perspectives
An overview of the carcinogenic mechanism proposed, considers that stem cells receive an anabolic message: “divide and make the constituents for new cells” in order to replace altered cells by whatever cause. These stem cells will then have to synthesize fatty acids and lipids for making new membranes. Fatty acid synthesis automatically closes via malonyl CoA, their beta oxidation, and shuts the fatty acid source of acetyl CoA. Since there is an associated pancreatic GABA failure, there is a catabolic message sent in parallel to insulin, and glucagon cuts the glycolytic supply of acetyl CoA as well, by eliciting the phosphorylating of PK and PDH. Normally this occurs in neoglucogenesis and should be reversed by the anabolic insulin message; but here, this does not occur. Insulin did stimulate via IP3 and calcium the PDE that hydrolyses cAMP, which boosted glycolysis by increasing Fruc2-6 bisP, and calcium did stimulate glucose uptake by incorporating transporters in the membrane, but the end of glycolysis remains OFF, closing the glycolytic supply of acetyl CoA. The persistent blockade of the de-phosphorylation of PK and PDH and their inhibition, comes from an elevated DAG that maintains the stimulation of PKC and CPI 17 inhibitor of PP1, which fails to activate PK and PDH. What kept DAG elevated is again a consequence of the GABA failure. Indeed, the GABA deficiency increases epinephrine release as well, which turns off STH and thus activates GH; the latter will stimulate ATGL, which increases DAG that stimulates PKC, closing the glycolytic source of acetyl CoA. Since the two sources of acetyl CoA, from fatty acids, and from glycolysis are blocked, the mitotic stem cell will have to get acetyl CoA from ketone bodies, reconverted into acetyl CoA by the reversible ketogenic pathway. This is possible if liver cells are maintained in catabolism to form ketone bodies. Well, this is again a consequence of the GABA failure, because a persistent leakage of insulin desensitizes their insulin receptors, rendering differentiated cells resistant to insulin, while responding to glucagon. Adipocytes provide fatty acids that will form in the liver these ketone bodies. As for the muscles, which are also insulin resistant, they receive the catabolic message through epinephrine and cortisol, the beta oxidation of fatty acids operates in muscles, but since the citric acid cycle Krebs cycle “turns” in muscles, acetyl CoA condenses with OAA coming via PEPCK and forms citrate, rather than ketone bodies. In muscles an intense proteolysis takes place, they provide amino acids to stem cells for making their proteins, and to liver for neoglucogenesis. The adipocytes respond preferentially to the catabolic component of the hybrid pancreatic message (they become insulin resistant) and provide fatty acids to liver and muscle. The amino acids processed by the transamination chain form pyruvate, and lactic acid is released by stem cells and muscles. In liver, a large part of this pyruvate feeds the neoglucogenic route.
Thus, the new mitotic stem cells that have not been chronically desensitized for insulin receive the dual hybrid message and the metabolic advantage they gain over the differentiated cells that are plundered, opens the road for these stem cells to cancer. Their differentiation is blocked, their number increases geometrically, inevitable mutations select the most aggressive population, the immune system fails to eliminate such cells.
A comment of some perspectives might be useful; the finding that diabetes type 1 result from the auto- immune destruction of beta cells by anti-GAD antibodies is certainly a major discovery, particularly if we consider that GABA is an essential regulator in the endocrine pancreas. These anti-GAD antibodies were detected in most cases, and were even implicated in type 2 diabetes. An interesting attempt to prevent diabetes using a “GAD vaccination” for preserving beta cells, did not yet give the expected results. Presumably, the network of anti-GAD antibodies and anti-idiotype antibodies is an equilibrium that is difficult to displace. There is still some condition to be found and hopefully a preventive vaccination will someday become efficient. This question is particularly relevant here, since we suspect that the GABA deficiency of beta cells in the pancreas is the starting point of a metabolic rewiring process of stem cells and differentiated cells that leads to the carcinogenic transformation of stem cells. Hence, preserving the pancreatic GABA ergic system[19] may not only prevent diabetes but presumably cancer as well. It would be interesting to look for eventual anti-GAD antibodies in cancer as it was done for diabetes. One may also hope that neutralizing these antibodies would give protection. The GABA deficiency may have other causes, a vitamin B6 deficiency for example, since it is the co-factor for GAD. Indeed, conditions that inhibit GAD or neutralize vitamin B6 (pesticides, amines and others) have been found to be carcinogenic[10]; this is also the case for diseases that genetically affects GAD. In Stiff person syndrome for example, the neurological symptoms prevail, but the cancer incidence is more elevated. It would be an interesting perspective to look for anti-GAD antibodies long before cancer develops and eventually find out procedures for neutralizing them, and develop “an anticancer vaccination”. It is certainly a difficult project, particularly if we consider the difficulty encountered for the diabetes type 1 vaccination attempt. Checking GABA metabolism and preserving this control system of the pancreas that depends of vitamins (B6) and nutrition, might decrease cancer incidence, particularly if its alteration by pesticides and other compounds that affect GAD is avoided. Moreover, it might be an interesting perspective to explore the pancreatic GABA system in relation to cancer, using noninvasive imaging methods for GABA as those developed for brain.
The other more practical perspective is to try to normalize the rewiring process that was presented in detail, even though many parts were deduced, and require more experimental work. A few compounds such as isocitrate, lipoic acid and others, gave encouraging results in animal cancer models (the list of compounds is given in previous publications)[5,11,13,20]. However, only a small number of potentially useful compounds were tested, in spite of the fact that many more should be given for normalizing the complex rewiring processes associated to cancer. One would gain to suppress the metabolic advantage given to stem cells by cutting their supply of ketone bodies; or to block the effect of GH on ATGL, which increases DAG, inhibitors of ATGL have been developed[21]; or to control the cAMP and AMP ratio, and AMP kinase[22]; or the mitochondria citrate efflux[23]; or to decrease the DAG activation of PKC, there are several PKC inhibitors discussed in reference[13]; or to inhibit the fatty acid synthesis pathway. A final remark concerns the direct effects of GABA on pancreatic ductal adenocarcinoma and presumably on other cancers. Apparently, GABA B receptor activation mediates a decrease of adenylate cyclase that attenuates the progression of pancreatic cancer[24]. This does not contradict the fact that a GABA deficiency of Beta cell fails to terminate insulin release, which desensitizes insulin receptors, while glucagon is released, leading to the described metabolic rewiring process that initiates cancer.
The best mixtures that will reverse the rewiring process still remain to be compared, in animal cancer models, then in properly built clinical trials. It is also essential to find an adequate diet and control the amino acid supplied, in order to attenuate the effects of the metabolic rewiring process in cancer; and then develop a metabolic treatment with non-toxic mixtures that would normalize cancer metabolism. In our opinion, this would be useful for backing up, if necessary, present cancer therapies.
Declarations
Authors’ contributionsIsraël M solely contributed to this study.
Availability of data and materialsNot applicable.
Financial support and sponsorshipNone.
Conflicts of interestThe author declared that there are no conflicts of interest.
Ethical approval and consent to participateNot applicable.
Consent for publicationNot applicable.
Copyright© The Author(s) 2019.
REFERENCES
4. Eigenbrodt E, Gerbracht U, Mazurek S, Presek P, Friis R. Carbohydrate metabolism and neoplasia: new perspectives for diagnosis and therapy. In: Prestlow TG, Prestlow TP, editors. Biochemical and molecular aspects of selected cancers. Academic Press Inc.; 1994. pp. 311-85.
6. Icard P, Lincet H. Global view of the biochemical pathways involved in the regulation of metabolism of cancer cells. Biochimica et Biophysica Acta 2012;1826:423-33.
7. Newberne PM, Newberne JW. Lipotropes and cancer. Biomedical Research 2012;23:225-9.
8. Siddiqui RA, Harvey KA, Walker C, Altenburg J, Xu Z, et al. Characterization of synergistic anti-cancer effects of docosahexaenoic acid and curcumin on DMBA-induced mammary tumorigenesis in mice. BMC Cancer 2013;13:418-34.
9. Israël M. A possible primary cause of cancer: deficient cellular interactions in endocrine pancreas. Molecular Cancer 2012;11:63-8.
10. Israël M. A primary cause of cancer: GABA deficiency in endocrine pancreas. Cancer Therapy 2012;8:171-83.
11. Israël M. Signaling and metabolism in cancer: endocrine pancreas deficiency and hybrid anabolism-catabolism, drugs that undo the process. Cancer Therapy 2014;10:1-12.
12. Israël M. Comment on cancer metabolism and on the role of the endocrine pancreas. J Clin Med Res 2014;6:490-1.
13. Israël M. Altered controls transforming normal metabolism into carcinogenic. J Cancer Prev Curr Res 2017;7:00228-33.
14. Solimena M, Folli F, Denis-Donni S, Comi GC, De Camilli P, et al. Autoantibodies to glutamic acid decarboxylase in a patient with stiff-man syndrome, epilepsy, and type I diabetes. N Engl J Med 1988;318:1012-20.
15. Beam CA, MacCallum C, Herold KC, Wherrett DK, Palmer J, et al. GAD vaccine reduces insulin loss in recently diagnosed type 1 diabetes: findings from a Bayesian meta-analysis. Diabetologia 2017;60:43-9.
16. Eto M. Regulation of cellular protein phosphatase 1 (PP1) by phosphorylation of the CPI-17 family, C-kinase- activated PP1 inhibitors. J Biol Chem 2009;284:35273-7.
17. Hohl CM. AMP deaminase in piglet cardiac myocytes: effect on nucleotide metabolism during ischemia. Am J Physiol 1999;276:H1502-10.
18. Node K, Kitakaze M, Minamino T, Tada M, Inoue M, et al. Activation of ecto-5‘-nucleotidase by protein kinase C, and its role in ischaemic tolerance in canine heart. Br J Pharmacol 1977;120:273-81.
19. Wan Y, Wang Q, Prud’homme GJ. Gabaergic system in the endocrine pancreas: a new target for diabetes treatment. Diabetes Metab Syndr Obes 2015;8:79-87.
20. Schwartz L, Abolhassani M, Guais A, Sanders E, Steyaert JM, et al. A combination of alpha lipoic acid and calcium hydroxycitrate is efficient against mouse cancer models: preliminary results. Oncol Rep 2010;23:1407-16.
21. Mayer N, Schweiger M, Romauch M, Grabner GF, Eichmann TO, et al. Development of small-molecule inhibitors targeting adipose triglyceride lipase. Nat Chem Biol 2013;9:785-7.
22. Ouyang J, Parakhia RA, Ochs RJ. Metformin activates AMP kinase through inhibition of AMP deaminase. J Biol Chem 2011;286:1-11.
23. Bucay AH. The biological significance of cancer: mitochondria as a cause of cancer and the inhibition of glycolysis with citrate as a cancer treatment. Medical Hypotheses 2007;69:826-8.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Israël M. Metabolic rewiring of stem cells and differentiated cells in cancer: the hypothetical consequences of a GABA deficiency in endocrine pancreas. J Cancer Metastasis Treat 2019;5:12. http://dx.doi.org/10.20517/2394-4722.2018.78
AMA Style
Israël M. Metabolic rewiring of stem cells and differentiated cells in cancer: the hypothetical consequences of a GABA deficiency in endocrine pancreas. Journal of Cancer Metastasis and Treatment. 2019; 5: 12. http://dx.doi.org/10.20517/2394-4722.2018.78
Chicago/Turabian Style
Israël, Maurice. 2019. "Metabolic rewiring of stem cells and differentiated cells in cancer: the hypothetical consequences of a GABA deficiency in endocrine pancreas" Journal of Cancer Metastasis and Treatment. 5: 12. http://dx.doi.org/10.20517/2394-4722.2018.78
ACS Style
Israël, M. Metabolic rewiring of stem cells and differentiated cells in cancer: the hypothetical consequences of a GABA deficiency in endocrine pancreas. J. Cancer. Metastasis. Treat. 2019, 5, 12. http://dx.doi.org/10.20517/2394-4722.2018.78
About This Article
Copyright

Data & Comments
Data
0

 Cite This Article 16 clicks
Cite This Article 16 clicks

 Like This Article 30
likes
Like This Article 30
likes






Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.