
Targeting histone lysine-specific demethylase KDM1A/LSD1 to control epithelial-mesenchymal transition program in breast cancers
 0
0
Abstract
Epithelial-mesenchymal transition (EMT) is a plastic and reversible process, essential for development and tissue homeostasis. Under pathological conditions, EMT causes induction of tumor growth, angiogenesis and metastasis. According to its reversible nature, the EMT program is associated with vast epigenetic changes. Targeting the epigenetic network that controls the EMT pathway in disease progression is a novel promising strategy to fight cancer metastasis. The impact of alterations in histone methylation in cancer has led to the identification of histone methyltransferases and demethylases as promising novel targets for therapy. Specifically, the lysine specific demethylase 1 (LSD1, also known as KDM1A) plays a pivotal role in the regulation of EMT. Here we present an overview of the causative role of LSD1 in the EMT process, summarizing recent findings on its emerging functions in cell migration and invasion in breast cancer.
Keywords
Introduction
Breast cancer is the most frequent cancer in women worldwide. It has a predictable incidence of 246,660 new cases (29% of all sites cancers) and 40,450 estimated deaths (14% of all sites) in 2016 in the United States[1]. The major causes of death in breast cancer patients are the result of metastasis in distant organs[2]. Metastasis is a complex biological process, which occurs through a series of steps: (1) rising of a locally invasive and migratory behavior, (2) arriving to the blood vessel and into the circulation via the blood flow, (3) halting in the distant organ, (4) surviving the initial stress, and (5) reinitiating outgrowth in distant stroma[2,3]. Metastasis involves a multitude of molecular signals[3]. Single cells leaving the primary tumor undergo epithelial to mesenchymal transition (EMT), consisting in the loss of epithelial polarity and the achievement of a mesenchymal morphology[4]. The opposite process, the mesenchymal to epithelial transition, takes place when metastatic cells switch back to an epithelial state in order to colonize secondary sites[5].
The mechanism of EMT is currently a major focus in metastasis research. The knowledge of its mechanisms is still fragmentary and more in-depth studies are needed in order to improve therapeutic approaches and influence, in the long-term, control of breast cancer outcome[6,7].
According to the reversible nature of EMT-associated processes, epigenetic mechanisms, such as DNA methylation and histone modifications, exert a great influence on the EMT program[8-11]. Particularly, wide ranges of studies demonstrate that the lysine specific demethylase 1 LSD1, also known as KDM1A, plays a pivotal role in the EMT regulation[12-15].
High levels of LSD1 expression in various tumors, including breast cancer, are correlated with poor prognosis[16-19]; several works reported that the ablation of LSD1 in breast cancer cells inhibits the invasion capabilities and suppresses their metastatic potential[20-22].
LSD1 is classified as an amine oxidase that uses FAD as the co-factor for its enzymatic action[23]; LSD1 is composed by three major domains: an N-terminal SWIRM (small alpha-helical domain), for protein stability; a central protruding Tower domain and a C-terminal amine oxidase like domain [Figure 1A]; LSD1 is able to demethylate mono and di-methylated lysine 4 and 9 of the histone H3 (H3K4, H3K9)[23-27]. Exerting its activity LSD1 can function as co-repressor, removing H3K4 methylation on gene promoters and enhancers, or co-activator, by removing H3K9 methylation, indicating that substrate specificity and the binding to different interactors define its biological outcome [Figure 1B][12]. Furthermore, LSD1 regulates methylation dynamics of non-histone proteins[16,18-23].
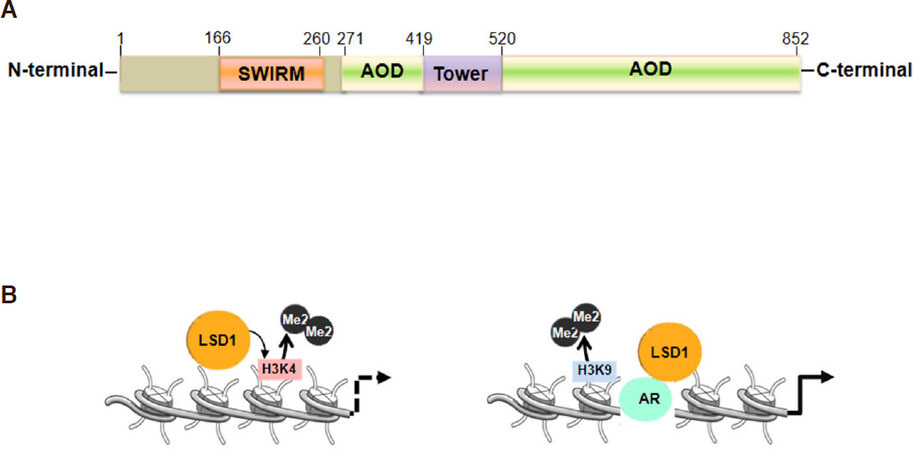
Figure 1. Lysine specific demethylase 1 (LSD1) protein domains and structure. A: In orange the SWIRM domain, in green the AOD domains and in yellow the Tower Domain; B: LSD1 selective substrate specificities: LSD1 represses promoter and enhancer activities through the demethylation of histones lysines, H3K4 or in cooperation with the androgen receptor (AR), activates transcription through Histones H3K9 demethylation
This review summarizes recent advantages in understanding the mechanisms through which LSD1 controls EMT in breast cancer, and discusses how these findings could be used to establish a new approach for therapeutic intervention in breast cancer.
LSD1 modulate the EMT process by its participation in different complexes
In human cancers, EMT contributes to tumor progression and invasion of surrounding tissues and confers chemo-resistance. Morphological and phenotypic changes are orchestrated by transcriptional reprogramming in which several transcription factors, known as EMT-TF[34], have a role in silencing of epithelial genes (e.g., E-cadherin, Occludin) and in the activation of mesenchymal genes (e.g., N-cadherin, Vimentin, Fibronectin)[35]. EMT is characterized by reprogramming of epigenetic marks: following the induction of the EMT by the transforming growth factor beta (TGF-β), a global increase of trimethylation in H3K4 and H3K36 histones and a decrease in the dimethylation of H3K9 are observed[34]. Mcdonald et al.[35] demonstrated that these epigenetic modifications depend largely on LSD1, and that loss of LSD1 functions affects EMT-driven cell migration and chemo-resistance.
Taking in consideration different studies, it is clear that LSD1 may function as enhancer or inhibitor of EMT functioning in a cell type-specific fashion, probably due to the cells genetic background and depending on its interacting partners[36]. A number of studies have described LSD1 as a critical player in epigenetic reprogramming during EMT. Two independent reports demonstrated that LSD1 physically associates with SNAIL1 (snail family transcriptional repressor 1) in breast cancer cells[15,34,37]. The members of the SNAIL family of zinc finger transcription factors (Snail, Slug and Smuc) control the invasive phenotype and metastasis in several types of cancers, included breast cancer. In the EMT pathway, SNAIL family proteins repress the expression of epithelial genes, such as E-cadherin (CDH1), through an LSD1-dependent molecular mechanism. It has been shown that LSD1, interacting with Snail, leads to CDH1 repression[15,34][Figure 2A]. In particular, it has been defined that the N-terminal SNAG domain of Snail is essential and responsible for the interaction and recruitment of LSD1 to gene promoters. Notably, SNAG domain of Snail1 resembles a histone H3-like structure, thus, it acts as a pseudo-substrate “hook” for LSD1 to recruit it together with CoREST to the Snail1 target gene promoters [Figure 2A]. The inhibition of the Snail/Slug-LSD1 interaction, using pharmacological LSD1 inhibitors or a cell-permeable peptide corresponding to the SNAG domain of Slug, suppresses motility and invasiveness of breast cancer cells[34].
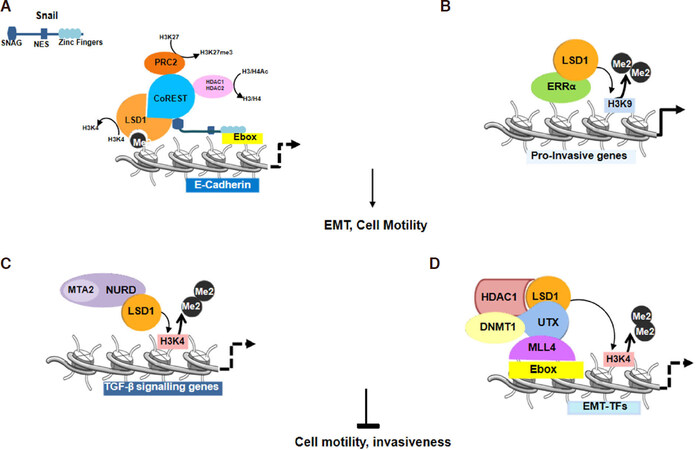
Figure 2. Lysine specific demethylase 1 (LSD1) regulates cell motility and epithelial to mesenchymal transition (EMT). A: Snail is stabilized through the formation of a ternary Snail1-LSD1-CoREST complex. SNAG domain of Snail binds LSD1 and recruits it together with CoREST to E-boxes of Snail target gene promoters. LSD1 binds the histones H3 tail and removes the activation marks on H3K4. HDAC1 and 2 deacetylate histones 3 and 4 (H3/H4), subsequently PRC2 directs the trimethylation of H3K27; B: H3K9 demethylation activity by LSD1 in complex with ERRα leads to transcriptional activation of pro-invasive genes; C: LSD1 inhibits EMT process through binding to NuRD impairing TGF-β signaling genes expression. D LSD1 with UTX functions as epigenetic silencer of EMT TFs
Notably, high expression of LSD1 or ERRα associates with breast cancer poor prognosis, decreased survival, cancer progression and metastasis, and LSD1 has been found to protect the ERRα protein from proteasome degradation[38]. The ERRα-LSD1 complex regulates a subset of genes involved in migration and invasiveness in breast cancer. The LSD1-ERRα complex binds the TSSs of common target genes and this interaction induces LSD1 to demethylate H3K9 leading to transcriptional activation[39][Figure 2B].
Among co-regulated genes LSD1-ERRα enhances the expression of the matrix metalloproteinase 1 (MMP1), a secreted protein involved in extracellular matrix degradation and cell invasion, and this can account for the capacities of LSD1-ERRα to induce tumor progression[39].
Although the majority of work indicates that LSD1 is a key positive regulator of the EMT program, in cooperation with other proteins such as the NuRD complex, it could promote opposite effects. It has been reported that the LSD1/NuRD complex inhibits TGF-β signaling pathway, reducing breast cancer metastatic potential[20][Figure 2C].
LSD1 has been found to have a role also as epigenetic regulator of EMT-TFs transcription factors expression [SNAIL, zinc finger E-box-binding homeobox 1 (ZEB1) and ZEB2][34]. It has been reported that in breast cancer stem cells LSD1 interacts with the Ubiquitously transcribed tetratricopeptide repeat, X chromosome (UTX)[14]. UTX functions as lysine-specific demethylase on H3K27me2/H3K27me3. Choi et al.[14] demonstrated that the role of UTX in epigenetic silencing of EMT-TF is not due to its H3K27 demethylation activity but to the ability to disrupt c-Myc and MLL4 recruitment on the E-boxes of their promoters. LSD1 recruited by UTX on the E-boxes form a transcriptional repressive complex with histone deacetylase 1 (HDAC1) and DNA methyltransferase 1 (DNMT1) [Figure 2D]. LSD1 together with UTX in this case of human breast cancers functions directly as epigenetic silencer of EMT TFs and consequently acts as tumor suppressor[14].
LSD1 post-translational targeting in EMT modulation
Several studies established that LSD1 activity and stability is altered by post-translational modifications[40,41]. LSD1 is acetylated in epithelial, but not in mesenchymal cells. In particular, the acetyltransferase MOF (or KAT8, lysine acetyltransferase 8) is critically involved in LSD1-induced EMT, acting on LSD1 acetylation[21]. MOF-mediated acetylation of LSD1 on lysines 432, 433 and 436 alters the association between LSD1 and target chromatin loci, increasing H3K4me2 levels and expression of epithelial markers. Moreover, while LSD1 is ubiquitously expressed, MOF is enriched in epithelial cells, but down-regulated in mesenchymal cells[21]. Thus, MOF-mediated acetylation of LSD1 may represent a crucial regulatory switch that, modulating LSD1 control of the EMT process. Accordingly, in epithelial breast cancer cells, MOF blocks LSD1 association with epithelial gene promoters, increases H3K4me2 levels at these loci and activates E-cadherin expression[21][Figure 3A].
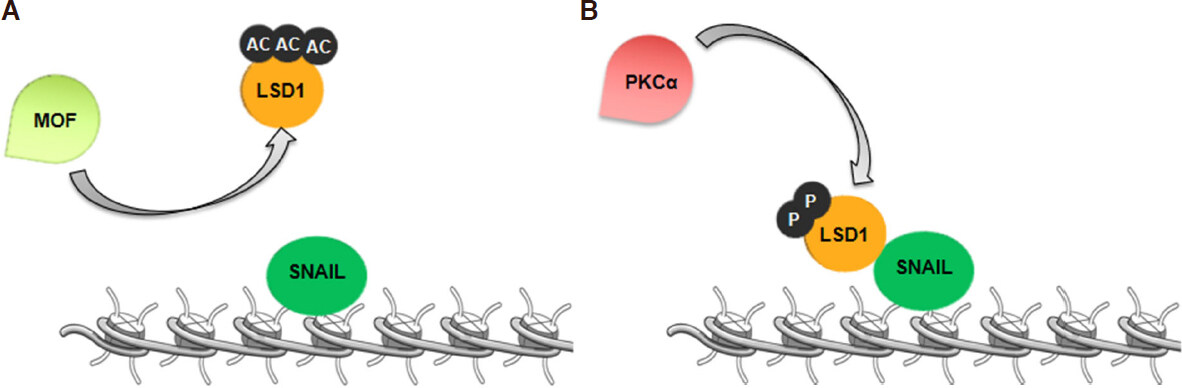
Figure 3. Lysine specific demethylase 1 (LSD1) is differently modified at post-translation level in epithelial and mesenchymal cells. A: In epithelial cells, MOF acetylates LSD1, and this modification induces the dissociation of LSD1/SNA1 complex. Then, LSD1 cannot block CDH1 transcription; B: in mesenchymal cells, PKCα phosphorylates LSD1. LSD1 phosphorylated interacts with SNA1 in order to silence CDH1 gene
In addition, recent evidences highlight that phosphorylation of LSD1 on Serine-111 (analogous to murine serine-112) is crucial for its activity in the EMT programs. It has been reported that PKCα (Protein kinase C alpha), that phosphorylate LSD1 at Serine-111, is required for LSD1 association with the E-cadherin promoter[42]. Specifically, LSD1-mediated demethylation at E-cadherin promoter is PKCα dependent and knockdown of PKCα impairs EMT induced by LSD1 overexpression in breast cancer cells [Figure 3B]. Another study reports that, upon induction of EMT, LSD1 is transiently induced and regulates epigenetic changes that result in transcriptional activation of several EMT-related genes in breast cancer cells. Phosphorylation of LSD1 at Serine-111 by a different PKC isoform, the PKC-θ (Protein kinase C theta), regulates its pro-EMT function[22]. Indeed, PKC-θ co-localizes with LSD1 in mesenchymal-like, but not in epithelial breast cancer cells and co-binding with LSD1 occurs predominantly at promoter regions of genes activated during EMT. Moreover, LSD1 phosphorylated at Ser-111 is enriched in chemo-resistant breast cancer cells with mesenchymal phenotype after therapy. In vivo studies also demonstrate that targeting LSD1 in combination with chemotherapy significantly reduces tumor growth by inhibiting the mesenchymal properties generated by chemotherapy alone[22].
Thus, targeting specific LSD1 post-translational modifications may be instrumental to modulate LSD1 functions and poses the basis to develop selective therapeutic tools.
LSD1/HIF-1α axis in breast cancers
Several studies demonstrate that LSD1 acts also on several non-histone substrates, such as lysine 370 on p53, which allows the binding of 53BP1[28,43]. Furthermore, LSD1 regulates the protein stability of the DNMT1[29], E2F1 (E2F transcription factor 1)[30], MYPT1 (myosin phosphatase)[31], STAT3 (signal transducer and activator of transcription 3)[32] and HIF-1α (hypoxia inducible factor 1 subunit alpha)[33,44] through its non-histone demethylase activity. LSD1 demethylation of non-histone substrates confirms its versatility to regulate different processes. One of the most important LSD1 non-histone substrate is HIF-1α, the master transcriptional regulator of developmental response to hypoxia[45]. High levels of HIF-1α have been strongly implicated in cancer biology, as well as a number of other pathophysiologies, vascularization and angiogenesis, metabolism, cell survival, and tumor invasion. HIF-1α protein accumulates under a hypoxic microenvironment, and it appears increased in various cancers. HIF-1α plays a crucial role in adaptive responses of the hypoxic tumor cells and it functions as a transcriptional activator of genes that regulate biological processes required for tumor survival and progression. Furthermore, high levels of HIF-1α induce up-regulation of different EMT-associated transcription factors, which in turn activate or repress EMT-associated signaling pathways and modulate EMT-associated inflammatory cytokines[46]. In breast cancer cells, EMT is prompted by ZEB1-MYB-E-cadherin signaling under hypoxic stress, which induces higher expression of ZEB1 and lower expression of MYB[47]. In particular, HIF-1α promotes EMT through the regulation of E-cadherin, SNAIL, ZEB1, TWIST (twist family bHLH transcription factor 1), and transcription factor 3 (TCF3, also known as E47)[47-49]. In this contest, LSD1 has a pivotal role in HIF-1α post-translation regulation. LSD1 demethylates lysine 32 (K32) of HIF-1α, while the same site can be methylated by SET9 inducing HIF-1α protein degradation[50][Figure 4A].

Figure 4. Non-histone lysine specific demethylase 1 (LSD1) substrate: hypoxia inducible factor-1 alpha (HIF-1α) protein stability is modulated by LSD1. A: Metylation-demethylation by SET7/9 and LSD1 respectively regulates HIF-1α degradation; B: LSD1-mediated regulation of HIF-1α protein stability during hypoxia: In early stage of hypoxia, the LSD1 (FAD-dependent) demethylase activity protects HIF-1α from RACK1-mediated degradation and HIF-1α activates transcription of its target genes. In prolonged hypoxia, the absence of FAD impairs FAD-dependent LSD1 activity on HIF-1α with consequent RACK1-dependent HIF-1α protein degradation
Moreover, in breast cancer, it has been reported that LSD1 regulates indirectly HIF-1α at post- translation level influencing its stability and avoiding its degradation. In particular, LSD1 upregulates HIF-1α demethylating the RACK1 (receptor for activated C kinase 1) protein (a component of the HIF-1α ubiquitination machinery) on lysine 271 (K271). After that, the degradation of HIF-1α is suppressed. These results indicate that in breast cancer the levels of FAD in the cells and the FAD-dependent LSD1 activity on RACK1-K271me2 could be the main determinants of HIF-1α stability during prolonged hypoxia[51][Figure 4B]. Accordingly, it has been shown that LSD1 inhibitors also impair the accumulation of HIF-1α, which is implicated in the activation of EMT in breast cancer. Therefore, even if at the moment there is a lack of evidences for an in vivo link between LSD1/HIF-1α/EMT, this pathway needs to be further explored since FAD modulation could represent a potential therapeutic strategy to regulate LSD1 and HIF-1α pathway.
Conclusion
Oncogenic roles of LSD1 in the pathogenesis of different epithelial cancers, such prostate, bladder, liver, non-small cell lung cancer and neuroblastomas, have been widely reported.
In breast cancer, LSD1 expression increases with cancer progression and its overexpression is positively correlated with the estrogen receptor negative status; high levels of LSD1 are a considerate molecular marker for predictive aggressive biology in ER-negative[17,24] and basal-like[17] breast cancer. Moreover, the depletion of LSD1 reduces proliferation and invasiveness of breast cancer cells in vitro.
In breast cancer, the oncogenic role of LSD1 depends on its versatility to interact with different partners. Current clinical trials utilizing epigenetic drugs for combination therapy have been shown to be promising in treating metastatic cancers. Vasilatos et al.[52] suggest that the combination therapy of LSD1 and HDAC inhibitors leads to expression activation of genes such as E-cadherin in Triple-negative breast cancer (TNBC)[52-54]. More recently, Yang et al.[55] proposed that LSD1 interacts with SIN3A/HDAC complex, which plays a role in EMT-induced cancer stemness inhibiting a series of genes, such as TERT (Telomerase reverse transcriptase), CUL4A (Cullin4A), TGFB2 (Transforming growth factor beta 2 ), MDM2 (Mouse double minute 2 homolog ), RHOA (Ras homolog family member A) and HIF-1α[55].
Progresses have been made in drug targeting for breast cancer over the years. Current clinical treatments typically involve surgery if disease is promptly diagnosed. Moreover, depending on molecular characteristics of injury, breast cancer surgery may be followed by radiation, chemotherapy, hormone therapy and targeted therapy. Nevertheless, the major limitation of targeted anticancer therapies is the intrinsic or acquired resistance[56].
Epigenetic therapies are prime candidates for adjuvant treatments to improve cancer therapy efficacy. Thus, establishing how exactly LSD1 regulates cell proliferation and invasive capacity will potentially facilitate the development of epigenetic therapies to attenuate tumor progression and metastasis in human breast cancer.
Recent studies identify LSD1 as a potent inhibitor of anti-tumor immunity and responsiveness to immunotherapy. LSD1 inhibition leads to double-stranded RNA stress and activation of type 1 interferon, which stimulates anti-tumor T cell response and represses tumor growth[57].
Qin et al.[58] have recently shown that the inhibition of LSD1 reactivates key immune checkpoint regulator and cytotoxic T cell-attracting chemokines in TNBC to immune checkpoint blocking antibodies. In vivo, the inhibition of chemokine receptors, by combined treatment with PD-1 antibody and LSD1 inhibitors, suppresses tumor growth and metastasis[58].
In conclusion, LSD1 inhibitors represent a promising epigenetic approach to treat breast Cancer. Furthermore, for a possible therapeutic goal, developing new drugs that target LSD1 and its partners or immune modulators would carry high innovation and translational potential.
Declarations
Authors’ contributionsAll authors partecipated in drafting the article and to its critical revision and approved the final version to be published.
Availability of data and materialsNot applicable.
Financial support and sponsorshipThis work was supported by grants from AIRC (I.G. 13173).
Conflicts of interestAll authors declared that there are no conflicts of interest.
Ethical approval and consent to participateNot applicable.
Consent for publicationNot applicable.
Copyright© The Author(s) 2019.
REFERENCES
3. Valastyan S, Weinberg RA. Tumor metastasis: molecular insights and evolving paradigms. Cell 2011;147:275-92.
4. Wang Y, Zhou BP. Epithelial-mesenchymal transition---a hallmark of breast cancer metastasis. Cancer Hallm 2013;1:38-49.
5. Li X, Pei D, Zheng H. Transitions between epithelial and mesenchymal states during cell fate conversions. Protein Cell 2014;5:580-91.
6. Unger-Saldaña K. Challenges to the early diagnosis and treatment of breast cancer in developing countries. World J Clin Oncol 2014;5:465-77.
7. Sun L, Fang J. Epigenetic regulation of epithelial-mesenchymal transition. Cell Mol Life Sci 2016;73:4493-515.
8. Hübner MR, Spector DL. Role of H3K27 demethylases Jmjd3 and UTX in transcriptional regulation. Cold Spring Harb Symp Quant Biol 2010;75:43-9.
9. Ramadoss S, Chen X, Wang CY. Histone demethylase KDM6B promotes epithelial-mesenchymal transition. J Biol Chem 2012;287:44508-17.
10. Enkhbaatar Z, Terashima M, Oktyabri D, Tange S, Ishimura A, et al. KDM5B histone demethylase controls epithelial-mesenchymal transition of cancer cells by regulating the expression of the microRNA-200 family. Cell Cycle 2013;12:2100-12.
11. Jiang GM, Wang HS, Zhang F, Zhang KS, Liu ZC, et al. Histone deacetylase inhibitor induction of epithelial-mesenchymal transitions via up-regulation of Snail facilitates cancer progression. Biochim Biophys Acta 2013;1833:663-71.
12. Ambrosio S, Saccà CD, Majello B. Epigenetic regulation of epithelial to mesenchymal transition by the Lysine-specific demethylase LSD1/KDM1A. Biochim Biophys Acta Gene Regul Mech 2017;1860:905-10.
13. Ambrosio S, Amente S, Saccà CD, Capasso M, Calogero RA, et al. LSD1 mediates MYCN control of epithelial-mesenchymal transition through silencing of metastatic suppressor NDRG1 gene. Oncotarget 2017;8:3854-69.
14. Choi HJ, Park JH, Park M, Won HY, Joo HS, et al. UTX inhibits EMT-induced breast CSC properties by epigenetic repression of EMT genes in cooperation with LSD1 and HDAC1. EMBO Rep 2015;16:1288-98.
15. Lin T, Ponn A, Hu X, Law BK, Lu J. Requirement of the histone demethylase LSD1 in Snai1-mediated transcriptional repression during epithelial-mesenchymal transition. Oncogene 2010;29:4896-904.
16. Amente S, Lania L, Majello B. The histone LSD1 demethylase in stemness and cancer transcription programs. Biochim Biophys Acta 2013;1829:981-6.
17. Nagasawa S, Sedukhina AS, Nakagawa Y, Maeda I, Kubota M, et al. LSD1 overexpression is associated with poor prognosis in basal-like breast cancer, and sensitivity to PARP inhibition. PLoS One 2015;10:e0118002.
18. Lim S, Janzer A, Becker A, Zimmer A, Schüle R, et al. Lysine-specific demethylase 1 (LSD1) is highly expressed in ER-negative breast cancers and a biomarker predicting aggressive biology. Carcinogenesis 2010;31:512-20.
19. Schulte JH, Lim S, Schramm A, Friedrichs N, Koster J, et al. Lysine-specific demethylase 1 is strongly expressed in poorly differentiated neuroblastoma: implications for therapy. Cancer Res 2009;69:2065-71.
20. Wang Y, Zhang H, Chen Y, Sun Y, Yang F, et al. LSD1 is a subunit of the NuRD complex and targets the metastasis programs in breast cancer. Cell 2009;138:660-72.
21. Luo H, Shenoy AK, Li X, Jin Y, Jin L, et al. MOF acetylates the histone demethylase LSD1 to suppress epithelial-to-mesenchymal transition. Cell Rep 2016;15:2665-78.
22. Boulding T, McCuaig RD, Tan A, Hardy K, Wu F, et al. LSD1 activation promotes inducible EMT programs and modulates the tumour microenvironment in breast cancer. Sci Rep 2018;8:73.
23. Shi Y, Lan F, Matson C, Mulligan P, Whetstine JR, et al. Histone demethylation mediated by the nuclear amine oxidase homolog LSD1. Cell 2004;119:941-53.
24. Forneris F, Binda C, Vanoni MA, Mattevi A, Battaglioli E. Histone demethylation catalysed by LSD1 is a flavin-dependent oxidative process. FEBS Lett 2005;579:2203-7.
25. Forneris F, Binda C, Battaglioli E, Mattevi A. LSD1: oxidative chemistry for multifaceted functions in chromatin regulation. Trends Biochem Sci 2008;33:181-9.
26. Lan F, Nottke AC, Shi Y. Mechanisms involved in the regulation of histone lysine demethylases. Curr Opin Cell Biol 2008;20:316-25.
27. Forneris F, Binda C, Vanoni MA, Battaglioli E, Mattevi A. Human histone demethylase LSD1 reads the histone code. J Biol Chem 2005;280:41360-5.
28. Huang J, Sengupta R, Espejo AB, Lee MG, Dorsey JA, et al. p53 is regulated by the lysine demethylase LSD1. Nature 2007;449:105-8.
29. Wang J, Hevi S, Kurash JK, Lei H, Gay F, et al. The lysine demethylase LSD1 (KDM1) is required for maintenance of global DNA methylation. Nat Genet 2009;41:125-9.
30. Kontaki H, Talianidis I. Lysine methylation regulates E2F1-induced cell death. Mol Cell 2010;39:152-60.
31. Cho H-S, Suzuki T, Dohmae N, Hayami S, Unoki M, et al. Demethylation of RB regulator MYPT1 by histone demethylase LSD1 promotes cell cycle progression in cancer cells. Cancer Res 2011;71:655-60.
32. Yang J, Huang J, Dasgupta M, Sears N, Miyagi M, et al. Reversible methylation of promoter-bound STAT3 by histone-modifying enzymes. Proc Natl Acad Sci USA 2010;107:21499-504.
33. Kim Y, Nam HJ, Lee J, Park DY, Kim C, et al. Methylation-dependent regulation of HIF-1α stability restricts retinal and tumour angiogenesis. Nat Commun 2016;7:10347.
34. Ferrari-Amorotti G, Chiodoni C, Shen F, Cattelani S, Soliera AR, et al. Suppression of invasion and metastasis of triple-negative breast cancer lines by pharmacological or genetic inhibition of slug activity. Neoplasia 2014;16:1047-58.
35. McDonald OG, Wu H, Timp W, Doi A, Feinberg AP. Genome-scale epigenetic reprogramming during epithelial-to-mesenchymal transition. Nat Struct Mol Biol 2011;18:867-74.
36. Stemmler MP, Eccles RL, Brabletz S, Brabletz T. Non-redundant functions of EMT transcription factors. Nat Cell Biol 2019;21:102.
37. Serrano-Gomez SJ, Maziveyi M, Alahari SK. Regulation of epithelial-mesenchymal transition through epigenetic and post-translational modifications. Mol Cancer 2016;15:18.
38. Carnesecchi J, Cerutti C, Vanacker JM, Forcet C. ERRα protein is stabilized by LSD1 in a demethylation-independent manner. PLoS One 2017;12:e0188871.
39. Carnesecchi J, Forcet C, Zhang L, Tribollet V, Barenton B, et al. ERRα induces H3K9 demethylation by LSD1 to promote cell invasion. Proc Natl Acad Sci U S A 2017;114:3909-14.
40. Han X, Gui B, Xiong C, Zhao L, Liang J, et al. Destabilizing LSD1 by Jade-2 promotes neurogenesis: an antibraking system in neural development. Mol Cell 2014;55:482-94.
41. Peng B, Shi R, Jiang W, Ding YH, Dong MQ, et al. Phosphorylation of LSD1 by PLK1 promotes its chromatin release during mitosis. Cell Biosci 2017;7:15.
42. Feng J, Xu G, Liu J, Zhang N, Li L, et al. Phosphorylation of LSD1 at Ser112 is crucial for its function in induction of EMT and metastasis in breast cancer. Breast Cancer Res Treat 2016;159:443-56.
43. Scoumanne A, Chen X. The lysine-specific demethylase 1 is required for cell proliferation in both p53-dependent and -independent manners. J Biol Chem 2007;282:15471-5.
44. Liu X, Chen Z, Xu C, Leng X, Cao H, et al. Repression of hypoxia-inducible factor α signaling by Set7-mediated methylation. Nucleic Acids Res 2015;43:5081-98.
45. Semenza GL, Agani F, Booth G, Forsythe J, Iyer N, et al. Structural and functional analysis of hypoxia-inducible factor 1. Kidney Int 1997;51:553-5.
46. Bao B, Azmi AS, Ali S, Ahmad A, Li Y, et al. The biological kinship of hypoxia with CSC and EMT and their relationship with deregulated expression of miRNAs and tumor aggressiveness. Biochim Biophys Acta 2012;1826:272-96.
47. Hugo HJ, Pereira L, Suryadinata R, Drabsch Y, Gonda TJ, et al. Direct repression of MYB by ZEB1 suppresses proliferation and epithelial gene expression during epithelial-to-mesenchymal transition of breast cancer cells. Breast Cancer Res 2013;15:R113.
48. Krishnamachary B, Zagzag D, Nagasawa H, Rainey K, Okuyama H, et al. Hypoxia-inducible factor-1-dependent repression of E-cadherin in von Hippel-Lindau tumor suppressor-null renal cell carcinoma mediated by TCF3, ZFHX1A, and ZFHX1B. Cancer Res 2006;66:2725-31.
49. Moreno-Bueno G, Portillo F, Cano A. Transcriptional regulation of cell polarity in EMT and cancer. Oncogene 2008;27:6958-69.
50. Lee JY, Park JH, Choi HJ, Won HY, Joo HS, et al. LSD1 demethylates HIF1α to inhibit hydroxylation and ubiquitin-mediated degradation in tumor angiogenesis. Oncogene 2017;36:5512-21.
51. Yang SJ, Park YS, Cho JH, Moon B, An HJ, et al. Regulation of hypoxia responses by flavin adenine dinucleotide-dependent modulation of HIF-1α protein stability. EMBO J 2017;36:1011-28.
52. Vasilatos SN, Katz TA, Oesterreich S, Wan Y, Davidson NE, et al. Crosstalk between lysine-specific demethylase 1 (LSD1) and histone deacetylases mediates antineoplastic efficacy of HDAC inhibitors in human breast cancer cells. Carcinogenesis 2013;34:1196-207.
53. Cao C, Wu H, Vasilatos SN, Chandran U, Qin Y, et al. HDAC5-LSD1 axis regulates antineoplastic effect of natural HDAC inhibitor sulforaphane in human breast cancer cells. Int J Cancer 2018;143:1388-401.
54. Cao C, Vasilatos SN, Bhargava R, Fine JL, Oesterreich S, et al. Functional interaction of histone deacetylase 5 (HDAC5) and lysine-specific demethylase 1 (LSD1) promotes breast cancer progression. Oncogene 2017;36:133-45.
55. Yang Y, Huang W, Qiu R, Liu R, Zeng Y, et al. LSD1 coordinates with the SIN3A/HDAC complex and maintains sensitivity to chemotherapy in breast cancer. J Mol Cell Biol 2018;10:285-301.
56. Ellis LM, Hicklin DJ. Resistance to targeted therapies: refining anticancer therapy in the Era of molecular oncology. Clin Cancer Res 2009;15:7471-8.
57. Sheng W, LaFleur MW, Nguyen TH, Chen S, Chakravarthy A, et al. LSD1 ablation stimulates anti-tumor immunity and enables checkpoint blockade. Cell 2018;174:549-63.e19.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Saccà CD, Gorini F, Ambrosio S, Amente S, Majello B. Targeting histone lysine-specific demethylase KDM1A/LSD1 to control epithelial-mesenchymal transition program in breast cancers. J Cancer Metastasis Treat 2019;5:15. http://dx.doi.org/10.20517/2394-4722.2018.95
AMA Style
Saccà CD, Gorini F, Ambrosio S, Amente S, Majello B. Targeting histone lysine-specific demethylase KDM1A/LSD1 to control epithelial-mesenchymal transition program in breast cancers. Journal of Cancer Metastasis and Treatment. 2019; 5: 15. http://dx.doi.org/10.20517/2394-4722.2018.95
Chicago/Turabian Style
Saccà, Carmen D., Francesca Gorini, Susanna Ambrosio, Stefano Amente, Barbara Majello. 2019. "Targeting histone lysine-specific demethylase KDM1A/LSD1 to control epithelial-mesenchymal transition program in breast cancers" Journal of Cancer Metastasis and Treatment. 5: 15. http://dx.doi.org/10.20517/2394-4722.2018.95
ACS Style
Saccà, CD.; Gorini F.; Ambrosio S.; Amente S.; Majello B. Targeting histone lysine-specific demethylase KDM1A/LSD1 to control epithelial-mesenchymal transition program in breast cancers. J. Cancer. Metastasis. Treat. 2019, 5, 15. http://dx.doi.org/10.20517/2394-4722.2018.95
About This Article
Special Issue
Copyright

Data & Comments
Data
0

 Cite This Article 27 clicks
Cite This Article 27 clicks

 Like This Article 36
likes
Like This Article 36
likes






Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.