
PIM-1 inhibition with AZD1208 to prevent osimertinib-induced resistance in EGFR-mutation positive non-small cell lung cancer


Abstract
Aim: The progression free survival of non-small cell lung cancer (NSCLC) patients has been doubled over the last years, but still single epidermal growth factor receptor (EGFR) tyrosine kinase inhibitors (TKIs) lead to incomplete responses. Compensatory signaling pathways are activated upon single EGFR TKIs. We have shown that compounds, which inhibit these pathways, are synergistic with EGFR TKIs. Proviral integration site for Moloney murine leukemia virus (PIM) has been connected to cancer therapy resistance, being involved in receptor tyrosine kinase, signal transducer and activator of transcription 3 (STAT3) and interleukin-6 signaling. We hypothesized that combined PIM and EGFR inhibition may improve the upfront therapy of EGFR-mutation positive NSCLC.
Methods: We reviewed the literature on PIM kinases, and performed cell viability assays, gene expression analyses, and immunoblotting experiments to reveal the mechanisms of action of the PIM inhibitor (AZD1208) alone and combined with osimertinib in five EGFR-mutation positive NSCLC cell lines.
Results: Osimertinib alone induced the activation of signal transducer and activator of transcription 3 (STAT3) as well as other signaling nodes. Combined osimertinib and AZD1208 yielded moderate synergistic effects in all NSCLC cell lines investigated. Among the EGFR-mutation positive cell lines examined, the H1975 and PC9 cell lines had the highest PIM1 and STAT3 mRNA expression. The combination decreased the osimertinib-induced STAT3 phosphorylation.
Conclusion: This study provides a short review on PIM kinases, and shows our results of combined PIM and EGFR inhibition in EGFR-mutation positive NSCLC cell lines. The combination was moderately synergistic but decreased STAT3 phosphorylation, an important signaling node in therapy resistance.
Keywords
Introduction
Activation of the epidermal growth factor receptor (EGFR) leads to downstream induction of signaling pathways, including phosphatidylinositol 3-kinase/AKT, Ras/mitogen-activated protein kinase and Janus kinase 2 (JAK2)/signal transducer and activator of transcription 3 (STAT3) [Figure 1]. Consequently, these pathways stimulate cell proliferation, cell motility, cell cycle progression and cell survival[1,2]. EGFR mutations were discovered in 2004[1]. This finding has radically changed the therapy of EGFR-mutation positive non-small cell lung cancer (NSCLC) patients, replacing standard chemotherapy with EGFR tyrosine kinase inhibitors (TKIs)[1,3]. Treatment with first-, second-, and third-generation EGFR TKIs has significantly improved the progression free survival of NSCLC patients, harboring EGFR mutations[4]. The third-generation EGFR TKI, osimertinib is now the standard first-line therapy of these patients[5]. However, although initially effective, monotherapy with EGFR TKIs, including osimertinib, triggers intrinsic or acquired resistance pathways[6-8], and the patients eventually succumb to their disease.
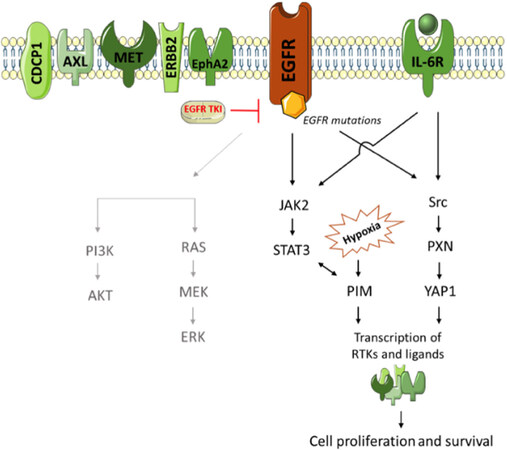
Figure 1. We present our initial literature-based model of EGFR-mutation positive NSCLC tumor cells, treated with single TKIs, and the parallel compensatory pathways that are being activated. Upon TKI treatment the downstream PI3K/AKT and RAS/MAPK pathways - in light grey - are inhibited, limiting the ability of the cells for proliferation and survival. However, our previous work has shown that this inhibition is the direct cause of the activation of the JAK2/STAT3 and Src-YAP1 pathways, and cannot abrogate coexpressed RTKs (e.g., MET, CDCP1) on the cell membrane. This in turn causes increased cell proliferation and survival, and thus leads to therapy resistance. PIM is an important signaling node in this network, potentially activated through IL-6/STAT3 and/or hypoxia. PIM was shown to be activated in multiple tumor types, and may therefore provide a druggable target for various tumor types. EGFR: epidermal growth factor receptor; NSCLC: non-small cell lung cancer; TKI: tyrosine kinase inhibitor; PI3K: phosphatidylinositol 3-kinase; MAPK: mitogen-activated protein kinase; JAK2: Janus kinase 2; STAT3: signal transducer and activator of transcription 3; YAP1: Yes-associated protein 1; RTK: receptor tyrosine kinase; PIM: proviral integration site for Moloney murine leukemia virus; IL-6R: interleukin 6 receptor; CDCP1: CUB domain-containing protein 1; EphA2: EPH receptor A2; PXN: Paxillin
Signaling nodes, including signal transducer and activator of transcription 3 (STAT3) and Src/Yes-associated protein 1 (YAP1), are not abrogated or even activated upon EGFR blockade. Moreover, besides EGFR, other receptor tyrosine kinases (RTKs) are present and induce downstream signaling, independent of EGFR inhibition[9-13]. We have demonstrated that simultaneous inhibition of EGFR, STAT3 and Src-YAP1 was highly synergistic in vitro and in vivo. Moreover, multiple RTKs and non-RTKs were up regulated at baseline or after treatment with EGFR TKIs, limiting their efficacy[6-8]. Proviral integration site for Moloney murine leukemia virus-1 (PIM-1) is a serine/threonine kinase, involved in cell cycle progression, cell growth, cell survival and therapy resistance. PIM-1 is activated in various types of cancer, amongst which prostate[14-16], colorectal[15,17-19], triple negative breast cancer (TNBC)[20,21], hematologic malignancies[22-24], neuroblastoma[25] and lung cancer[26]. For this reason, PIM kinases could provide a common target for the treatment of diverse malignancies.
PIM activation is induced by changes in the tumor microenvironment, such as hypoxia, and causes resistance to antiangiogenic drugs[15]. The combination of PIM inhibitors with antiangiogenic compounds, down regulates the expression of hypoxia inducible factor 1 (HIF1) and decreases tumor cell proliferation, the formation of tumor vasculature and the ability of the cells to metastasize[15]. A second study has also shown the involvement of PIM kinases in therapy resistance through hypoxia. Although the authors in this study propose a HIF1-independent, nuclear factor-erythroid 2 p45-related factor 2 induced hypoxia, PIM kinase inhibitors are again the solution to prevent therapy resistance and induce cell death in different tumor types[27].
PIM-1 is involved in RTKs, STAT3 and interleukin-6 (IL-6) signaling [Figure 1]. Specifically, STAT proteins bind to the promoter of the PIM-1 gene, and increase PIM-1 expression. A feedback loop, in turn, regulates the expression of STAT proteins[14,18,21,28,29]. Considering the fact that there are no approved STAT3 inhibitors, the combination of EGFR TKIs with a PIM-1 inhibitor could be of interest to block STAT3 activation. In prostate and gastric cancer, the efficacy of AKT inhibitors is attenuated by PIM-1 signaling, which upregulates the expression of MET, ERBB2 and other RTKs. Combined AKT and PIM-1 inhibition is synergistic in preclinical models[16,30]. Similarly, in the activated B cell-like (ABC) subtype of diffuse large B cell lymphoma (DLBCL), the efficacy of the Bruton’s tyrosine kinase inhibitor ibrutinib, was attenuated in the presence of PIM-1 mutations. In addition, chronic exposure of an ABC-DLBCL cell line to ibrutinib increased PIM-1 expression, and the combination of ibrutinib with the pan-PIM inhibitor (AZD1208) showed synergistic effects[24]. Finally, in MET amplified NSCLC and gastric cancer cell lines, PIM-1 was identified as a mechanism of resistance to MET inhibitors. Treatment of MET inhibitor-resistant cells with PIM inhibitors restored sensitivity to MET inhibition, both in culture and in xenograft models[26].
Several PIM inhibitors have been developed, and can be categorized into first- and next generation inhibitors. AZD1208 falls in the latter category, and inhibits all isoforms of the PIM kinase (1, 2 and 3). AZD1208 has been shown to improve outcome to radiation therapy in castrate-resistant prostate cancer cells[14]. Moreover, PIM-1 levels were correlated to MET expression in both cell lines and patient-derived tumor samples, driving migration and invasion of cancer cells. Patient-derived tissues from a phase 1 clinical trial (ClinicalTrials.gov Identifier: NCT01489722), exploring the safety, tolerability, pharmacokinetics and efficacy of AZD1208 in acute myeloid leukemia, confirmed the mechanism of action of PIM-1 through MET regulation[31].
The ability of AZD1208 to induce cell cycle arrest and apoptosis by inhibition of all PIM kinase isoforms indicates its potential as an anticancer treatment. Previous publications have shown that PIM inhibition augments the efficacy of targeted therapies in diverse tumor types. Therefore, dual PIM-1 and EGFR inhibition might modulate cell survival pathways and block progression and growth of cancer cells, by preventing the EGFR TKI-induced STAT3 and RTKs activation in EGFR-mutation positive NSCLC cell lines[6-8]. This research aims to evaluate whether the pan-PIM inhibitor AZD1208 improves the efficacy of osimertinib in EGFR-mutation positive cell lines.
Methods
Chemicals and reagents
Human lung adenocarcinoma PC-9 cells, harboring EGFR exon 19 deletion and 11-18 cells, harboring EGFR exon 21 L858R mutation were provided by F. Hoffmann-La Roche Ltd. (Basel, Switzerland), and by Dr. Mayumi Ono, (Kyushu University, Fukuoka, Japan), respectively. EGFR exon 19 deletion positive HCC4006 and HCC827 cells were purchased from the American Type Culture Collection (ATCC). The H1975 cell line, harboring both EGFR exon 21 L858R and resistant T790M mutation as well as the TNBC MBA-MB-231 cell line were purchased from ATCC. All cell lines were maintained in RPMI (Roswell Park Memorial Institute medium) 1640 supplemented with 1% penicillin/streptomycin/glutamine (Gibco) and 10% fetal bovine serum (FBS; Gibco) in a 5% CO2 37 °C cell culture incubator and routinely evaluated for mycoplasma contamination.
The pan-PIM inhibitor, AZD1208, and osimertinib were bought from Selleck Chemicals (Houston, TX, USA). Drugs were prepared in dimethylsulfoxide (DMSO) at a concentration of 10-100 mmol/L stock solutions and stored at -20 °C. Further dilutions were made in culture medium to final concentration before use. All antibodies used in our study, including dilution and company catalog number can be found in Supplementary Table 1. All primers used can be found in Supplementary Table 2.
Cell viability assay
Cells were seeded in 96-well plates at the following densities: 1.5 × 103 (PC9), 2.0 × 103 (H1975) 3.0 × 103 (HCC4006, HCC827 and 11-18), and incubated for 24 h, as previously described[6]. For the combined treatment cells were seeded in 96-well plates at the following densities: 1 × 103 (PC9 and H1975), 1.5 × 103 (HCC827, HCC4006 and 11-18), and incubated for 24 h. Cell viability was assessed using the 3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyltetrazolium bromide (MTT) assay (Sigma Aldrich, St Louis, MO, USA). Cells were treated with serial dilutions of the drugs. For the half maximal inhibitory concentration (IC50) determination, AZD1208 doses ranged from 0-200 µmol/L and treatment lasted 72 h. For the MTT viability assays with a combination of AZD1208 and osimertinib (IC50s were previously obtained) drug doses were as follows: PC9, H1975, HCC4006, HCC827 cells were treated with osimertinib ranging from 0-100 nmol/L, and AZD1208 ranging from 0-100 µmol/L, or with the combination of both. The 11-18 cells were treated with osimertinib ranging from 0-200 nmol/L, and AZD1208 ranging from 0-100 µmol/L, or with the combination of both. The combined treatment lasted 96 h. After treatment incubation, 0.5 mg/mL of MTT reagent was added to the medium in the wells for 2 h at 37 °C and formazan crystals in viable cells were solubilized with 100 µL DMSO and spectrophotometrically quantified using a microplate reader (Varioskan Flash; Thermo Fisher Scientific, Waltham, MA, USA) at 565 nm of absorbance. Fractional survival was then calculated as percentage to control cells. Data of combined drug effects were subsequently analyzed by the Chou and Talalay method[32,33]. Combination index (CoI) values < 1, = 1 and > 1 indicated synergism, additive effect and antagonism, respectively.
Western blotting
For immunoblotting experiments, 1.5 million cells were seeded in T75 flasks (Starstedt, Newton, USA). The next day, PC9 and H1975 cells were treated with 24 nmol/L osimertinib, 5 µmol/L AZD1208 and a combination of both. Untreated cells received an equivalent dose of vehicle (DMSO). After 24 h cells were washed with cold PBS and re-suspended in ice-cold radioimmunoprecipitation assay buffer (50 mmol/L Tris- hydrochloric acid in pH 7.4, 1% Nonidet P-40, 0.5% sodium deoxycholate, 0.1% sodium dodecyl sulfate (SDS), 150 mmol/L sodium chloride, 1 mmol/L ethylenediaminetetraacetic acid, 1 mmol/L sodium vanadate and 50 mmol/L sodium fluoride) containing protease inhibitor mixture (Roche Applied Science, Penzberg, Germany). Following cell lysis by sonication and centrifugation at 18620 × g for 10 min at 4 °C, the resulting supernatant was collected as the total cell lysate. Briefly, the lysates containing 45 µg proteins were electrophoresed on 10% SDS-polyacrylamide gels (Life Technologies, Carlsbad, CA, USA) and transferred to polyvinylidene difluoride membranes (Bio-Rad laboratories Inc., Hercules, CA, USA). Membranes were blocked in Odyssey blocking buffer (Li-Cor Biosciences, Lincoln, NE, USA). All target proteins were immunoblotted with appropriate primary and horseradish peroxidase (HRP)-conjugated secondary antibodies. Chemiluminescent (HRP-conjugated) bands were detected in a ChemiDoc MP Imaging System (Bio-Rad laboratories Inc.). β-actin was used as an internal control to confirm equal gel loading.
Quantitative real time polymerase chain reaction
RNA was isolated from cell lines using phenol-chloroform-isoamyl alcohol, followed by precipitation with isopropanol in the presence of glycogen and sodium acetate. RNA was re-suspended in water and treated with DNAse I to avoid DNA contamination. Complementary DNA (cDNA) was synthesized using moloney murine leukaemia virus reverse transcriptase enzyme. cDNA was added to Taqman Universal Master Mix (Applied Biosystems, Carlsbad, CA, USA) in a 12.5 µL reaction with specific primers and probe for each gene. The primer and probe sets were designed using Primer Express 3.0 Software (Applied Biosystems) according to their Ref Seq (http://www.ncbi.nlm.nih.gov/LocusLink). Gene-specific primers are provided in Chemicals and reagents. Quantification of gene expression was performed using the ABI Prism 7900HT Sequence Detection System (Applied Biosystems) and was calculated according to the comparative Ct method. Final results were determined as follows: 2^-(ΔCt sample-ΔCt calibrator), where ΔCt values of the calibrator and sample are determined by subtracting the Ct value of the target gene from the value of the endogenous gene (β-actin). Commercial RNA controls were used as calibrators (Liver and Lung; Stratagene, La Jolla, CA, USA). In all quantitative experiments, a sample was considered not evaluable when the standard deviation of the Ct values was > 0.30 in two independent analyses. As a result, the number of evaluable samples varied among the genes examined.
Results
Baseline mRNA expression of EGFR-mutation positive cells and sensitivity to PIM inhibition
We first evaluated the baseline mRNA expression of PIM, STAT3 and other relevant genes[8], in our panel of five EGFR-mutation positive NSCLC cell lines. As can be seen in Figure 2A, both PC9 and H1975 cell lines had higher PIM-1, STAT3 and JAK2 mRNA expression compared to the average gene expression of all five cell lines. Hereafter, cell viability assays were performed and the IC50s of single AZD1208 treatment were determined. The IC50s of osimertinib have been previously calculated and reported[6]. As expected, none of the cell lines was sensitive to single AZD1208 treatment, with IC50s ranging from 17.1 to 23.0 µmol/L [Figure 2B, left panel], while all of them were sensitive to osimertinib[6][Figure 2B, right panel].
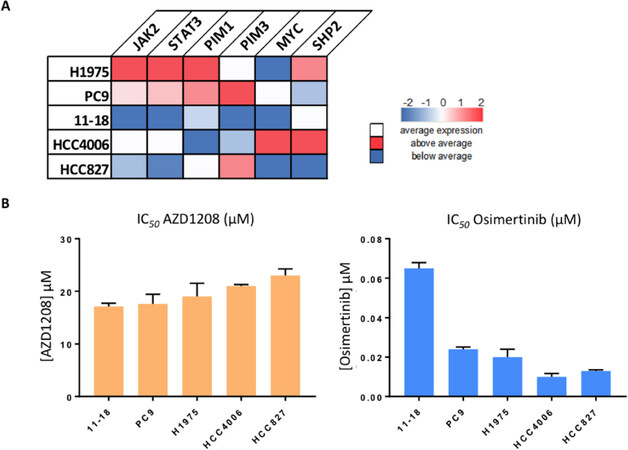
Figure 2. Baseline mRNA expression of EGFR-mutation positive cells and sensitivity to PIM inhibition. A: baseline mRNA expression of various genes in five EGFR-mutation-positive NSCLC cell lines (H1975, PC9, 11-18, HCC4006 and HCC827). Heatmap depicts gene mRNA expression (columns) in the different cell lines (rows) compared to the average mRNA expression of each gene in all cell lines. Experiments were performed in biological triplicates, and the average of these experiments are shown; B: cell viability assays were performed in all cell lines, to explore the effect of AZD1208 in vitro, and to determine the IC50 in each cell line. Results of previously performed experiments with osimertinib in the same cell lines are also shown. Experiments were performed in biological triplicates, and the average and standard deviations are shown. EGFR: epidermal growth factor receptor; PIM: proviral integration site for Moloney murine leukemia virus; NSCLC: non-small cell lung cancer; IC50: half maximal inhibitory concentration; JAK2: Janus kinase 2; STAT3: signal transducer and activator of transcription 3; SHP2 (PTPN11): tyrosine-protein phosphatase non-receptor type 11
Combination of osimertinib plus a PIM inhibitor in EGFR-mutation positive cells
Taking into consideration our previous findings, that osimertinib alone induces the activation of resistant signaling nodes[6], we then tried to evaluate if AZD1208 increases the effect of osimertinib on cell growth inhibition. Dual treatment revealed moderate synergistic effects, with a CoI between 0.79 and 0.85 [Figure 3A, left panel]. The lowest CoIs and therefore stronger synergism, were seen in the PC9 [CoI of 0.79; 95% confidence interval (CI): 0.66-0.92] and HCC827 (CoI of 0.79; 95%CI: 0.69-0.89) cell lines. A moderate synergism was seen in HCC4006 cells, with a CoI of 0.83 (95%CI: 0.79-0.87). The combination was less effective, but still moderately synergistic, in the 11-18 (CoI of 0.85; 95%CI: 0.69-1.00) and H1975 (CoI of 0.85; 95%CI: 0.74-0.96) cell lines [Figure 3A, left panel].
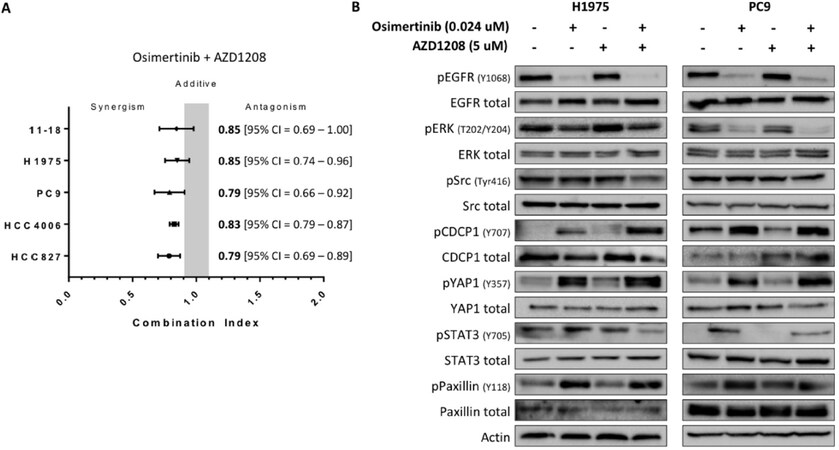
Figure 3. The effects of AZD1208 and osimertinib on RTKs and downstream components in EGFR-mutation positive NSCLC cells. A: in all cell lines, cell viability assays were performed to explore the effect of combined osimertinib and AZD1208 treatment. The obtained CoI was calculated based on the Chou and Talalay method, and values < 1, = 1 and > 1 indicate synergism, additive effect and antagonism, respectively. Experiments were performed in biological triplicates, and the average and standard deviations plus 95%CIs are shown; B: two EGFR-mutation positive NSCLC cell lines (H1975 and PC9) were treated with single osimertinib (0.024 µmol/L), single AZD1208 (5 µmol/L) or with the combination. Untreated cells received an equivalent dose of vehicle (DMSO). Cell lysates were used for immunoblotting and changes in RTKs and non-RTKs upon the different treatments were investigated in the two cell lines. Experiments were performed in biological triplicates with similar results, and a representative blot is shown. RTK: receptor tyrosine kinase; EGFR: epidermal growth factor receptor; NSCLC: non-small cell lung cancer; CoI: combination index; 95%CI: 95% confidence interval; DMSO: dimethylsulfoxide; CDCP1: CUB domain -containing protein 1; YAP1: yes-associated protein 1; STAT3: signal transducer and activator of transcription 3
We then tried to see the correlation between signaling nodes and cellular responses after combined treatment with osimertinib plus AZD1208. For this, we selected the PC9 and H1975 cell lines, which distinguished among the rest of the cell lines, having the highest PIM-1, STAT3 and JAK2 mRNA expression [Figure 2A]. Changes in the protein expression of RTKs, STAT3 and other proteins after single osimertinib, AZD1208 or combined treatment were studied, using immunoblotting experiments [Figure 3B, right panel]. The results confirmed our previous findings[6,8], as single osimertinib increased the phosphorylation of CUB-domain-containing protein 1 (CDCP1), YAP1, paxillin and STAT3 in the PC9 and H1975 cell lines. Osimertinib abrogated or diminished EGFR and ERK phosphorylation, but it had no effect on Src phosphorylation [Figure 3B, right panel]. In contrast, single AZD1208 treatment was unable to inhibit activated EGFR and ERK, but in comparison with osimertinib, it induced at a much lower level the phosphorylation of CDCP1, YAP1, STAT3 and paxillin. The combination of osimertinib plus AZD1208, abolished EGFR and ERK activation in both cell lines, but paradoxically, was unable to reverse - or even increased - the osimertinib-induced CDCP1 and YAP1 phosphorylation. To our positive surprise, the double combination reversed the osimertinib-induced STAT3 phosphorylation in both cell lines, and diminished paxillin and Src phosphorylation in PC9 and H1975 cells, respectively [Figure 3B, right panel]. Overall these data reconfirm our previous findings that, even in oncogene addicted EGFR-mutation positive cancer cells, single EGFR TKIs are insufficient[6-8,34]. Concomitant PIM-1 inhibition may reverse some of the deleterious effects of osimertinib on the activation of oncogenic signaling pathways.
Discussion
Herein, we have reviewed the available literature on PIM and we have found that this kinase is involved in multiple tumor types, including lung cancer[26]. The survival of lung cancer patients has been improved in the last decade due to the availability of new and more effective targeted therapies. EGFR-mutation positive patients are treated with EGFR TKIs, and the last years we have seen the development and approval of three generations of these agents[35]. However, ultimately resistance mechanisms take over and treatment options are not indefinite. Furthermore, about 20% of EGFR-mutation positive patients have intrinsic resistance to EGFR TKIs[8,34], indicating that molecularly targeted therapy is not a one-size-fits-all approach. Therefore, defining biomarkers of resistance and designing a truly personalized therapy is of great relevance. We have previously seen, and we confirm in the present work, that EGFR TKIs, including osimertinib, induce STAT3 and Src-YAP1 activation[6-8]. Rational double or triple combinations prevented this phenomenon and increased the efficacy of EGFR TKI monotherapy in EGFR-mutation positive models[6-8]. Since PIM was shown to be a key player in IL-6/STAT3 signaling[14,18,21,28,29], we hypothesized that by combining the PIM inhibitor, AZD1208 with osimertinib we may obtain similar results[6-8].
As expected, single AZD1208 was not effective, but the combination of AZD1208 and osimertinib augmented the effect of each drug alone on cell viability in a moderate synergistic manner, in five EGFR-mutation positive cell lines. This synergistic effect is probably due to the AZD1208-based inhibition of resistance pathways. Our model, in Figure 4, indicates that combined EGFR and PIM inhibition does not prevent the activation of all relevant resistance signaling nodes, but it is able to decrease STAT3 phosphorylation. Since currently there are no approved STAT3 inhibitors in clinical use, PIM inhibition merits further investigation in lung cancer as well as other tumor types. AZD1208 is not in clinical development any more[22] and we plan to extend the current work using the pan-PIM inhibitor LGH447 (PIM447). LGH447 has single agent activity, and blocks resistance pathways when combined with standard of care treatments[22,36-38]. The agent is currently being tested in a phase I/II clinical trial (ClinicalTrials.gov Identifier, NCT01456689) for relapsed/refractory multiple myeloma, and safety and efficacy results are awaited.
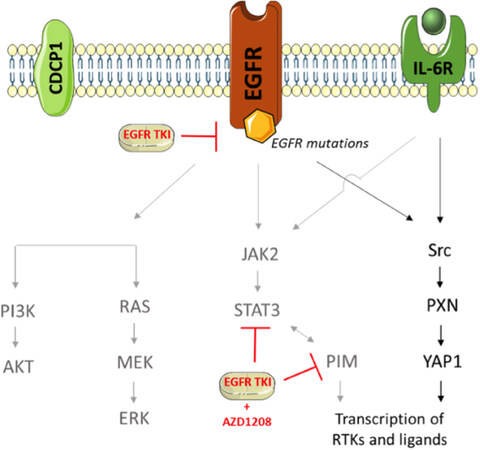
Figure 4. Obtained model of EGFR-mutation positive NSCLC tumor cells, after dual EGFR- (osimertinib) and PIM- (AZD1208) inhibition. Upon osimertinib treatment, the downstream PI3K/AKT and RAS/MAPK pathways - in light grey - are inhibited, lowering cell proliferation and survival. However, AZD1208 treatment now blocks both the activation of PIM and the activation of the JAK/STAT3 pathway. This leads to moderate synergistic effects in vitro. However, the Src-YAP1 pathway is still activated (PC9) or only slightly inhibited (H1975), and can therefore compensate for the blockade induced by osimertinib and AZD1208. EGFR: epidermal growth factor receptor; NSCLC: non-small cell lung cancer; PIM: proviral integration site for Moloney murine leukemia virus; PI3K: phosphatidylinositol 3-kinase; MAPK: mitogen-activated protein kinase; JAK2: Janus kinase 2; STAT3: signal transducer and activator of transcription 3; YAP1: Yes-associated protein 1; CDCP1: CUB domain-containing protein 1; IL-6R: Interleukin 6 receptor; PXN: Paxillin
Declarations
Authors’ contributionsMade substantial contributions to conception and design of the study and performed data analysis and interpretation: Bracht JWP, Karachaliou N, Rosell R
Performed data acquisition, as well as provided administrative, technical, and material support: Bracht JWP, Karachaliou N, Berenguer J, Fernandez-Bruno M, Filipska M, Pedraz-Valdunciel C, Codony-Servat C, Codony-Servat J, Rosell R
Availability of data and materialsNot applicable.
Financial support and sponsorshipWork in Dr. Rosell’s laboratory is partially supported by a grant from La Caixa Foundation; an Instituto de Salud Carlos III grant (RESPONSE, PIE16/00011, PI14/01678); a Marie Skłodowska-Curie Innovative Training Networks European Grant (ELBA No 765492); and a Spanish Association Against Cancer (AECC) grant (PROYE18012ROSE).
Conflicts of interestAll authors declared that there are no conflicts of interest.
Ethical approval and consent to participateNot applicable.
Consent for publicationNot applicable.
Copyright© The Author(s) 2019.
REFERENCES
1. Sordella R, Bell DW, Haber DA, Settleman J. Gefitinib-sensitizing EGFR mutations in lung cancer activate anti-apoptotic pathways. Science 2004;305:1163-7.
2. Lazzara MJ, Lane K, Chan R, Jasper PJ, Yaffe MB, et al. Impaired SHP2-mediated extracellular signal-regulated kinase activation contributes to gefitinib sensitivity of lung cancer cells with epidermal growth factor receptor-activating mutations. Cancer Res 2010;70:3843-50.
3. Karachaliou N, Fernandez-Bruno M, Bracht JWP, Rosell R. EGFR first- and second-generation TKIs-there is still place for them in EGFR-mutant NSCLC patients. Transl Cancer Res 2018; doi: 10.21037/tcr.2018.10.06.
4. Rosell R, Carcereny E, Gervais R, Vergnenegre A, Massuti B, et al. Erlotinib versus standard chemotherapy as first-line treatment for European patients with advanced EGFR mutation-positive non-small-cell lung cancer (EURTAC): a multicentre, open-label, randomised phase 3 trial. Lancet Oncol 2012;13:239-46.
5. Soria JC, Ohe Y, Vansteenkiste J, Reungwetwattana T, Chewaskulyong B, et al. Osimertinib in untreated EGFR-mutated advanced non-small-cell lung cancer. N Engl J Med 2018;378:113-25.
6. Chaib I, Karachaliou N, Pilotto S, Codony Servat J, Cai X, et al. Co-activation of STAT3 and YES-associated protein 1 (YAP1) pathway in EGFR-mutant NSCLC. J Natl Cancer Inst 2017;109.
7. Codony-Servat C, Codony-Servat J, Karachaliou N, Molina MA, Chaib I, et al. Activation of signal transducer and activator of transcription 3 (STAT3) signaling in EGFR mutant non-small-cell lung cancer (NSCLC). Oncotarget 2017;8:47305-16.
8. Karachaliou N, Chaib I, Cardona AF, Berenguer J, Bracht JWP, et al. Common Co-activation of AXL and CDCP1 in EGFR-mutation-positive Non-smallcell lung cancer associated with poor prognosis. EBioMedicine 2018;29:112-27.
9. Gao SP, Mark KG, Leslie K, Pao W, Motoi N, et al. Mutations in the EGFR kinase domain mediate STAT3 activation via IL-6 production in human lung adenocarcinomas. J Clin Invest 2007;117:3846-56.
10. Fan W, Tang Z, Yin L, Morrison B, Hafez-Khayyata S, et al. MET-independent lung cancer cells evading EGFR kinase inhibitors are therapeutically susceptible to BH3 mimetic agents. Cancer Res 2011;71:4494-505.
11. Lee HJ, Zhuang G, Cao Y, Du P, Kim HJ, et al. Drug resistance via feedback activation of Stat3 in oncogene-addicted cancer cells. Cancer Cell 2014;26:207-21.
12. Zhong Z, Wen Z, Darnell JE Jr. Stat3: a STAT family member activated by tyrosine phosphorylation in response to epidermal growth factor and interleukin-6. Science 1994;264:95-8.
13. Nan J, Du Y, Chen X, Bai Q, Wang Y, et al. TPCA-1 is a direct dual inhibitor of STAT3 and NF-kappaB and regresses mutant EGFR-associated human non-small cell lung cancers. Mol Cancer Ther 2014;13:617-29.
14. Tursynbay Y, Zhang J, Li Z, Tokay T, Zhumadilov Z, et al. Pim-1 kinase as cancer drug target: An update. Biomed Rep 2016;4:140-6.
15. Casillas AL, Toth RK, Sainz AG, Singh N, Desai AA, et al. Hypoxia-Inducible PIM kinase expression promotes resistance to antiangiogenic agents. Clin Cancer Res 2018;24:169-80.
16. Cen B, Mahajan S, Wang W, Kraft AS. Elevation of receptor tyrosine kinases by small molecule AKT inhibitors in prostate cancer is mediated by Pim-1. Cancer Res 2013;73:3402-11.
17. Zhang M, Liu T, Sun H, Weng W, Zhang Q, et al. Pim1 supports human colorectal cancer growth during glucose deprivation by enhancing the Warburg effect. Cancer Science 2018;109:1468-79.
18. Weirauch U, Beckmann N, Thomas M, Grunweller A, Huber K, et al. Functional role and therapeutic potential of the Pim-1 kinase in colon carcinoma. Neoplasia 2013;15:783-94.
19. Liu K, Gao H, Wang Q, Wang L, Zhang B, et al. Hispidulin suppresses cell growth and metastasis by targeting PIM1 through JAK2/STAT3 signaling in colorectal cancer. Cancer Sci 2018;109:1369-81.
20. Braso-Maristany F, Filosto S, Catchpole S, Marlow R, Quist J, et al. PIM1 kinase regulates cell death, tumor growth and chemotherapy response in triple-negative breast cancer. Nat Med 2016;22:1303-13.
21. Zhao W, Qiu R, Li P, Yang J. PIM1: a promising target in patients with triple-negative breast cancer. Med Oncol 2017;34:142.
22. Keane NA, Reidy M, Natoni A, Raab MS, O’Dwyer M. Targeting the Pim kinases in multiple myeloma. Blood Cancer J 2015;5:e325.
23. Koblish H, Li YL, Shin N, Hall L, Wang Q, et al. Preclinical characterization of INCB053914, a novel pan-PIM kinase inhibitor, alone and in combination with anticancer agents, in models of hematologic malignancies. PLoS One 2018;13:e0199108.
24. Kuo HP, Ezell SA, Hsieh S, Schweighofer KJ, Cheung LW, et al. The role of PIM1 in the ibrutinib-resistant ABC subtype of diffuse large B-cell lymphoma. Am J Cancer Res 2016;6:2489-501.
25. Brunen D, de Vries RC, Lieftink C, Beijersbergen RL, Bernards R. PIM kinases are a potential prognostic biomarker and therapeutic target in neuroblastoma. Mol Cancer Ther 2018;17:849-57.
26. An N, Xiong Y, LaRue AC, Kraft AS, Cen B. Activation of Pim kinases is sufficient to promote resistance to MET small-molecule inhibitors. Cancer Res 2015;75:5318-28.
27. Warfel NA, Sainz AG, Song JH, Kraft AS. PIM kinase inhibitors kill hypoxic tumor cells by reducing Nrf2 signaling and increasing reactive oxygen species. Mol Cancer Ther 2016;15:1637-47.
28. Jin B, Wang Y, Wu CL, Liu KY, Chen H, et al. PIM-1 modulates cellular senescence and links IL-6 signaling to heterochromatin formation. Aging Cell 2014;13:879-89.
29. Liu J, Qu X, Shao L, Hu Y, Yu X, et al. Pim-3 enhances melanoma cell migration and invasion by promoting STAT3 phosphorylation. Cancer Biol Ther 2018;19:160-8.
30. Lee M, Lee KH, Min A, Kim J, Kim S, et al. Pan-Pim kinase inhibitor AZD1208 suppresses tumor growth and synergistically interacts with akt inhibition in gastric cancer cells. Cancer Res Treat 2018; doi: 10.4143/crt.2017.341.
31. Cen B, Xiong Y, Song JH, Mahajan S, DuPont R, et al. The Pim-1 protein kinase is an important regulator of MET receptor tyrosine kinase levels and signaling. Mol Cell Biol 2014;34:2517-32.
32. Chou TC. Drug combination studies and their synergy quantification using the Chou-Talalay method. Cancer Res 2010;70:440-6.
33. Narayan RS, Fedrigo CA, Brands E, Dik R, Stalpers LJ, et al. The allosteric AKT inhibitor MK2206 shows a synergistic interaction with chemotherapy and radiotherapy in glioblastoma spheroid cultures. BMC Cancer 2017;17:204.
34. Karachaliou N, Cardona AF, Bracht JWP, Aldeguer E, Drozdowskyj A, et al. Integrin-linked kinase (ILK) and src homology 2 domain-containing phosphatase 2 (SHP2): novel targets in EGFR-mutation positive non-small cell lung cancer (NSCLC). EBioMedicine 2019;39:207-14.
35. Ettinger DS, Aisner DL, Wood DE, Akerley W, Bauman J, et al. NCCN guidelines insights: non-small cell lung cancer, version 5.2018. J Natl Compr Canc Netw 2018;16:807-21.
36. Raab MS, Ocio EM, Thomas SK, Günther A, Goh YT, et al. Phase 1 study update of the novel pan-pim kinase inhibitor LGH447 in patients with relapsed/refractory multiple myeloma. Blood 2014.
37. Paino T, Garcia-Gomez A, Gonzalez-Mendez L, San-Segundo L, Hernandez-Garcia S, et al. The novel Pan-PIM kinase inhibitor, PIM447, displays dual antimyeloma and bone-protective effects, and potently synergizes with current standards of care. Clin Cancer Res 2017;23:225-38.
38. Langowski JL, Holash J, Burger M, Zang R, Zavorotinskaya T, et al. The Pan-PIM kinase inhibitor LGH447 shows activity in PIM2-dependent multiple myeloma and in AML models. Blood 2013;122:1666.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Bracht JWP, Karachaliou N, Berenguer J, Fernandez-Bruno M, Filipska M, Pedraz-Valdunciel C, Codony-Servat C, Codony-Servat J, Rosell R. PIM-1 inhibition with AZD1208 to prevent osimertinib-induced resistance in EGFR-mutation positive non-small cell lung cancer. J Cancer Metastasis Treat 2019;5:22. http://dx.doi.org/10.20517/2394-4722.2018.111
AMA Style
Bracht JWP, Karachaliou N, Berenguer J, Fernandez-Bruno M, Filipska M, Pedraz-Valdunciel C, Codony-Servat C, Codony-Servat J, Rosell R. PIM-1 inhibition with AZD1208 to prevent osimertinib-induced resistance in EGFR-mutation positive non-small cell lung cancer. Journal of Cancer Metastasis and Treatment. 2019; 5: 22. http://dx.doi.org/10.20517/2394-4722.2018.111
Chicago/Turabian Style
Bracht, Jillian Wilhelmina Paulina, Niki Karachaliou, Jordi Berenguer, Manuel Fernandez-Bruno, Martyna Filipska, Carlos Pedraz-Valdunciel, Carles Codony-Servat, Jordi Codony-Servat, Rafael Rosell. 2019. "PIM-1 inhibition with AZD1208 to prevent osimertinib-induced resistance in EGFR-mutation positive non-small cell lung cancer" Journal of Cancer Metastasis and Treatment. 5: 22. http://dx.doi.org/10.20517/2394-4722.2018.111
ACS Style
Bracht, JWP.; Karachaliou N.; Berenguer J.; Fernandez-Bruno M.; Filipska M.; Pedraz-Valdunciel C.; Codony-Servat C.; Codony-Servat J.; Rosell R. PIM-1 inhibition with AZD1208 to prevent osimertinib-induced resistance in EGFR-mutation positive non-small cell lung cancer. J. Cancer. Metastasis. Treat. 2019, 5, 22. http://dx.doi.org/10.20517/2394-4722.2018.111
About This Article
Copyright

Data & Comments
Data


 Cite This Article 34 clicks
Cite This Article 34 clicks

 Like This Article 24
likes
Like This Article 24
likes






Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.