
Targeting autophagy with small molecules for cancer therapy


Abstract
Autophagy is a conserved lysosomal-dependent catabolic process that maintains the cellular homeostasis by recycling misfolded proteins and damaged organelles. It involves a series of ordered events (initiation, nucleation, elongation, lysosomal fusion and degradation) that are tightly regulated/controlled by diverse cell signals and stress. It is like a double-edged sword that can play either a protective or destructive role in cancer, by pro-survival or apoptotic cues. Recently, modulating autophagy by pharmacological agents has become an attractive strategy to treat cancer. Currently, a number of small molecules that inhibit autophagy initiation (e.g., ULK kinase inhibitors), nucleation (e.g., Vps34 inhibitors), elongation (e.g., ATG4 inhibitors) and lysosome fusion (e.g., chloroquine, hydroxyl chloroquine, etc.) are reported in pre-clinical and clinical study. Also a number of small molecules reported to induce autophagy by targeting mammalian target of rapamycin (e.g., rapamycin analogs) or adenosine 5’-monophosphate-activated protein kinase (e.g., sulforaphane). The study results suggest that many potential “druggable” targets exist in the autophagy pathway that could be harnessed for developing new cancer therapeutics. In this review, we discuss the reported autophagy modulators (inhibitors and inducers), their molecular mode of action and their applications in cancer therapy.
Keywords
Introduction
Autophagy is a natural cellular process that occurs for the maintenance of cellular homeostasis. During autophagy, catabolic degradation occurs to recycle unnecessary, dysfunctional cellular components, damaged organelles and protein aggregates[1]. It also removes intrusive pathogens to protect our body from various infectious diseases and other disorders including cancer[2,3]. The autophagic process occurs when the cells are in stress conditions such as starvation or hypoxia. Based on these cues, the cytoplasmic contents (cargo) get sequestered within the autophagosome and then fuse with lysosome for cargo degradation. Three forms of autophagy have been reported: (1) chaperon mediated autophagy; (2) microautophagy; and (3) macroautophagy[4][Table 1].
Characteristics of three types of autophagy
| Sl. No. | Type | Mechanism | Biological effects | Markers |
|---|---|---|---|---|
| 1 | Macroautophagy(commonly referred as autophagy) | Autophagosome formation | Cell killing and survival | Beclin1, Atg5, Atg12, LC3-I and LC3-II |
| 2 | Microautophagy | Vacuole invagination process | Cell killing and survival | Hsc70 multi-vesicular bodies and multi-vesicular lysosomes |
| 3 | Chaperon mediated autophagy | Receptor mediated process | Selective cell killing, T-cell activation | LAMP-2A, Hsc70 |
The type and mode of action differs from one another based on how and what target cargoes are subjected to lysosomal degradation. In chaperone mediated autophagy, the substrate with the targeting motif KFERQ is recognized by the chaperone Hsc70, and moves the individual protein substrate to lysosome degradation[5]. While, microautophagy is responsible for basal degradation of the cytoplasmic content by the direct invagination into lysosome[6]. On the other hand, macroautophagy, an ubiquitous pathway in eukaryotic cells, starts with the formation of double membrane structures called autophagosomes in which cargo sequestration occurs[3]. The early autophagosome is formed from components derived from endoplasmic reticulum, which acquires V-ATPase and LAMP to become late autophagosome[7,8]. Finally, the late autophagosome fuses with the pre-existing lysosomes to become the autolysosome[9]. The autolysosome contains unrecognizable cytoplasmic materials as they are in degradation and recycling process[10]. The multi-step process of macroautophagy (here onwards referred to as autophagy; Figure 1) is regulated by autophagy related genes, which were originally found in autophagy defective yeast mutants.
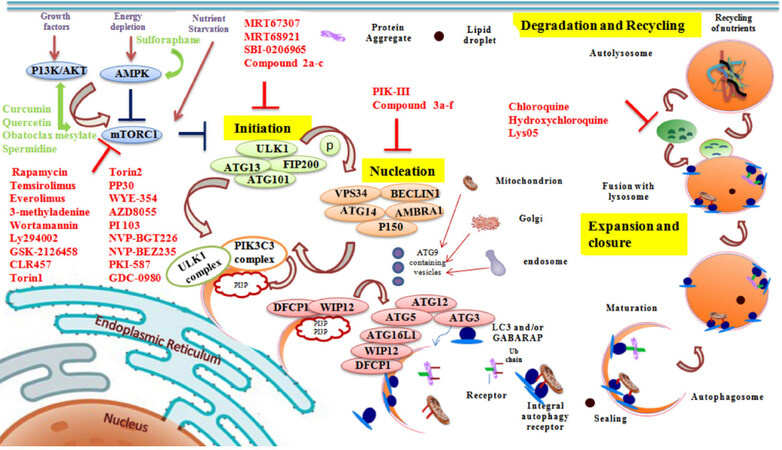
Figure 1. Cartoon representation of pathways involved in autophagy and the chemical inhibitors/inducers of the process. Autophagy involves 4 steps: (1) initiation; (2) nucleation; (3) expansion and closure; and (4) degradation and recycling. Small molecule modulators that affect different steps of autophagy are shown in red color (inhibitors) and green color (inducers)
Autophagy is initiated through diverse signaling pathways in response to major stress response and plays a pro-survival role by nutrient recycling. The stress factors include low cellular energy levels, amino acid deprivation, growth factor withdrawal, hypoxia conditions, ER stress, oxidative stress, organelle damage and infection[11]. Over a period of stress, the cells employ autophagy process either to move contents or to degrade harmful components such as damaged mitochondria or invading pathogens through the process of lysosomal degradation[12]. Aberration in autophagy process has been implicated in a wide range of diseases including neurodegenerative disorders that involve the accumulation of pathogenic proteins, inflammatory disorders and cancer[4,13]. The following sections discuss various signaling pathways involved in the autophagy process, their role in cancer and other diseases; also small molecule inhibitors that target the autophagy process that are useful for cancer therapy are detailed along with the mode of interaction with their targets, if known.
Autophagy process
Initiation step
Autophagy is a multi-step process involving, initiation, nucleation, elongation/expansion and closure steps[14]. The initiation of phagophore formation is governed by multi-protein complex known as ULK complex (Unc-51-like kinase 1, FIP200, ATG101 and ATG13), that integrates upstream nutrient and energy status and thereby initiates the process of autophagy [Figure 2]. Each protein of the ULK complex has a unique role; ULK1, a serine-threonine protein kinase, plays a key role in the scaffold formation of ULK1-ATG13-FIP200 complex[15]; ATG13, an adaptor protein mediates interaction between ULK1 and FIP200, and directly binds to LC3 as well. Another protein, ATG101 which is a subunit of ULK complex recruits downstream Atg proteins that are essential for autophagy[16,17].
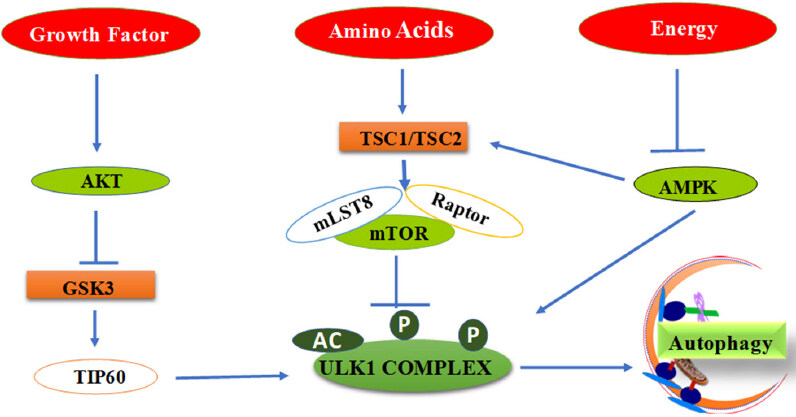
Figure 2. Diagrammatic representation of various signals leading to the formation of autophagy initiation - ULK complex
Initiation step of autophagy process in normal nutrient conditions (e.g., sensed by the levels of growth factors, amino acids and glucose) is regulated by a negative autophagy regulator mammalian target of rapamycin (mTOR) that phosphorylates two subunits, ULK1 and Atg13 of ULK complex. The complex of mTOR that consists of mTORC1, Raptor and MLST8 as subunits directly binds ULK1 protein and thereby leads to ULK complex dissociation[18]. Upon nutrient starvation, mTORC1 dissociates from ULK complex and hence ULK1 gets activated and phosphorylates ATG13, ATG101 and FIP200 in order to initiate the phagophore formation [Figure 3]. Investigations suggest that phosphorylation of ULK1 plays a vital role in the regulation of autophagy initiation process and at least 30 phosphorylation sites have been reported. However, the molecular basis and the molecules involved in ULK1 phosphorylation are yet to be completely unraveled[18].
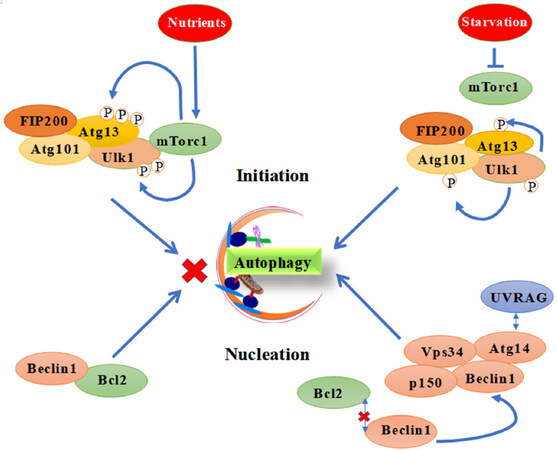
Figure 3. Schematic representation of molecules involved in the (1) initiation; and (2) nucleation process leading to the autophagophore formation during autophagy
In contrast to mTORC1, adenosine 5’-monophosphate (AMP)-activated protein kinase (AMPK) indirectly activates ULK complex by phosphorylating TSC2 and raptor. Recently, AMPK has also been shown to directly interact with and phosphorylate ULK1 in a nutrient-dependent manner. Several phosphorylation sites, including Ser555, Ser637 and Ser757 were reported. AMPK phosphorylation site Ser555 is thought to recruit phospho-binding protein to the ULK complex[19]. During glucose starvation, AMPK targeting phosphorylation sites on ULK1 are trigged and contributes to ULK1 activation [Figure 2]. AKT is a serine/threonine kinase that acts as a sensor of growth factor levels in the cell and is activated in nutrient rich condition. Upon activation, AKT phosphorylates Ser9 of GSK3, which acts as inhibitory cue for GSK3. Dephosphorylation of Ser9 activates GSK3 and eventually the activated GSK3 phosphorylates TIP60 at Ser86[20]. Further, TIP60 acetylates ULK1 and thereby increases the kinase activity of ULK1 [Figure 2]. Finally, accumulation of activated ULK complex initiates the phagophore formation[20].
Nucleation step
Phagophore formation starts with the nucleation step of autophagy process, where protein subunits including Vps34, Beclin1, Ambra1, ATG14 and p150 coordinate with each other and form a nucleation complex[21]. During nucleation, vesicular sorting protein 34 and its enzymatic product phosphatidylinositol-3-phosphate, an essential component play a vital role in recruiting other autophagy protein subunits such as WIPI-1, DFCP1, ATG5 and LC3. The effectors WIPI-1, WIPI-2 and DFCP1, binds to PI3P via WD repeats and FYVE domains, respectively[21,22]. In nucleation process, Vps34 associates with the phagophore membrane via p150 [Figure 3] that is anchored by myristic acid. Beclin-1 is the third important component for phospholipid kinase activity. Activity of Beclin-1 is affected by many different binding partners. Beclin1 dissociates from the anti-apoptotic factor Bcl2, which leads to the activation of Vps34. The association of beclin1 with Vps34 is stabilized by the other two components - UV radiation-associated resistance gene and beclin-1-associated autophagy related key regulator[23].
Elongation step
In elongation step, the maturation of the autophagosome takes place with the help of two ubiquitin like conjugation system [Figure 4]. In the early step of autophagosome maturation, Atg12 is activated by Atg7 and transferred to Atg10 thereby forming a covalent linkage with Atg5[24]. This Atg12-Atg5 conjugate complexes with Atg16L and forms autophagy elongation complex. Carboxy terminal Gly residue of Atg12 is involved in the formation of thioester linkage with the active site Cys residue of Atg5 and Atg10, and also involved in amide linkage with Lys residue of Atg5[25]. Elongation complex forms a dimer which provides a site for LC3 lipidation, a process required for association of LC3 with autophagosome membrane[26]. Although, Atg12 does not possess similarity with ubiquitin, it forms an ubiquitin-like fold and is involved in autophagy elongation step. For the autophagosome maturation, LC3 lipidation is very essential and acts as a second ubiquitin-like conjugation. This conjugation occurs in a series of reactions including proLC3-I cleavage by Atg4B, LC3-I activation by Atg7, transfer to Atg3 and finally conjugation with PE. Like Atg12, carboxy terminal Gly of LC3 is involved in thioester linkage with Cys residues of Atg7 and Atg3, and an amide linkage with PE[27]. These reactions are similar in LC3 homologues GABARAP, GATE-16 and mAtg8L. Completion of these maturation steps leads to autophagosome-lysosome fusion[28] and then degradation of the cargo.
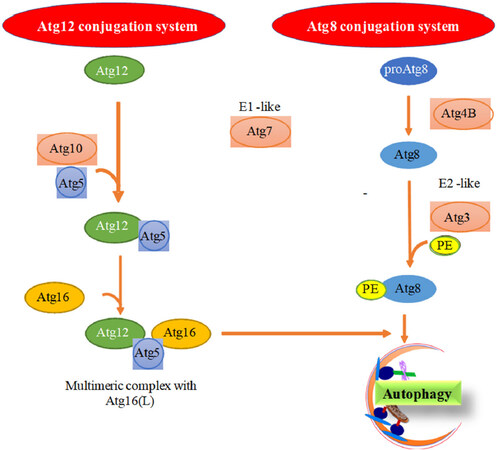
Figure 4. Two ubiquitin like conjugation system is involved in the autophagophore elongation/maturation process
Fusion and degradation step
Degradation and recycling of cellular components is a central function of all living cells to meet cell demands. In final stage of autophagy, matured autophagosome fuses with multivesicular endosomes and lysosomes. Degradation of cytosolic components is not a random process and thus several proteins such as Vps34/SKD1 and Rab11 involvement is necessary to accomplish autophagosome-lysosome fusion process[29]. A recent study has reported that components of HOPS complex (homotypic fusion and protein sorting) plays a major role in the formation of autophagosome-endosome fusion. Moreover, dysfunction or absence of subunits of the ESCRT III complex and proteins required for biogenesis of endosomes severely affects the fusion process[30]. The fusion of inner membrane of autophagosome delivers the cytosolic proteins to lysosomes, where hydrolysis takes place to complete the degradation of the cargo.
Role of autophagy in various diseases
Dysregulation in the autophagy process results in various diseases. Defects or deregulation is especially important in cancer, ageing related disease, neurodegenerative diseases and lysosomal storage diseases[2]. In aging process, the functional role of autophagy is expulsion of aggregated protein which increases the lifespan; in the case of defective autophagy process, the formation of vacuoles and improper fusion of vacuoles with the lysosomes results in impaired protein flux[31]. In infectious disease, the functional role of autophagy is to remove the bacterial and viral pathogens through sequestration in autophagic vacuoles and then degradation. This provides immunity against pathogens[32]; while a defective autophagy process provides a conducive environment for pathogens. In lysosomal storage disorders, the removal of lysosomal stock such as fatty acids, cholesterol are defective and an increased number of autophagosomes and reduced organelle turnover occur[33]. In the case of neurodegeneration disorders, the neuronal protein aggregates are removed by autophagic process; while, in the defective process the protein aggregates accumulate in neurons leading to neurodegenaration[34].
Role of autophagy in cancer
Autophagy is a complex process that responds to a variety of stressful environments such as nutrient deprivation, abnormal protein accumulation and damaged organelle, and thereby maintains the cellular homeostasis[35]. Autophagy plays a cyto-protective role by clearing the damaged organelles, misfolded proteins and ROS, thus confining the genomic instability and aberrant mutations that ultimately leads to cancer. Consequently, autophagy machinery can be defined as cell survival mechanism in normal and as death mechanism in cancer cells.
However, deregulation of autophagy has been reported in a variety of diseases including cancer. Many reports have shown that autophagy plays dual role in cancer development. In early stage of cancer, autophagy suppresses/abolishes tumor formation by clearing the damaged proteins and organelles, and thereby induces cell death; whereas in advanced cancers, the stress mediated properties of autophagy has been hijacked by tumor cells to meet their increased metabolic requirements that are indispensable for tumor survival and rapid proliferation. Hence, autophagy has been reported as a tumor promoter in advanced cancers. Additionally, the regulation of autophagy through diverse signaling mechanisms can contribute upregulation/downregulation of tumor suppressor/oncogenes, and that can lead to inhibition/induction of cancer development[36,37]. For example, negative regulation of tumor suppressor genes through different signaling mechanisms (i.e., mTOR, AMPK, etc.) could induce autophagy and suppression of cancer initiation; whereas, activation of oncogenes could lead to inhibition of autophagy and promotion of cancer development.
It has been widely reported that autophagy modulates cancer growth and development, and this depends on cancer type, stage, and genetic context. A basal level of autophagy is considered as a cancer suppressive mechanism in normal cells. However, abnormal levels of autophagy in stressful conditions (i.e., Hypoxia, ROS, etc.) lead to inhibition of break down of damaged organelles and proteins, and subsequent cancer development. Nonetheless, it has been reported that mutation in autophagy related proteins leads to tumor suppression/promotion in a variety of cancers. For example, BECN1 related proteins (e.g., BIF-1 etc.) have been found abnormal/absent in gastric and colorectal cancer[38,39]. Further, mutation in UVRAG protein reported to reduce autophagy, and consequent colorectal cancer development[40]. On the other hand, an unexpected high basal-level of autophagy has been reported in several types of RAS-activated cancers (e.g., pancreatic cancers) and inhibition of autophagy in these cancers hinders the tumor formation[41].
In order to identify alterations of different genes that are involved in autophagy signaling pathways, we have analyzed 1,087 cancer patient samples data from Cbioportal database (http://www.cbioportal.org/). Through this data analysis, we have noticed that mTOR gene shows high alterations with 12% (altered/profiled ratio = 128/1,087) and PIK3C3 shows 9% (altered/profiled ratio = 97/1,087) alterations. Further we also observed the alterations of other genes including ULK1 (5%), UVRAG (5%), beclin1 (2.7%), ATG4B (4%), ATG16L1 (2.1%), ATG5 (4%) and ATG12 (2.8%). Altogether, these studies explain that cell transformation and deregulation of many signaling pathways are connected directly or indirectly with autophagy modulation. These evidences, suggest that autophagy has a dual role in cancer and is dependent on biological factors such as the driving oncogene, tumor suppressor involved and tumor type. Hence, autophagy is considered as a double-edged sword, by both protecting from and promoting cancer[42,43].
Autophagy acts as a tumor suppressor during tumorigenesis
Autophagy is widely documented as a tumor suppressive mechanism, as its deregulation leads to genomic instability, aberrant mutations, tumor formation and metastasis[44]. Primarily, the role of beclin1 in autophagy has been studied extensively. For example, mice having monoallelic deletion of beclin1 gene induce tumor formation. As is evident, the allelic loss of beclin1 was found in 40%-75% in breast, ovarian, and prostate cancers[44,45]. It is well documented that Beclin-1 promotes autophagy by binding to Vps34 via its conserved domain that was reported essential for tumor suppression. Recently, phosphorylation of multiple tyrosine residues of beclin1 has also been observed, which leads to a decrease in the activity of beclin1/PI3KC3 complex and thereby the reduction of autophagy[46].
The reduction of beclin1 protein levels is also reported in many brain cancers. A study conducted to investigate beclin1 mRNA expression in different histotypes of brain tumors reported the expression levels vary based on the type of tumor. After examining mRNA expression in 212 primary brain tumors, the study identified low expression in most high-grade ependymal neoplasms, astrocytic and atypical meningiomas; whereas, high expression in low-grade tumors and medulloblastomas[47]. Additionally, monoallelic deletion/mutations in UVRAG protein and decreased expression levels of Bif-1 were also reported in colon, gastric, breast, prostate and bladder cancers[38]. The reported results clearly indicate that autophagy related protein Beclin-1 and its regulators (i.e., UVRAG and Bif-1) mediate tumor suppression.
Further, deregulation of several proteins of PI3K/Akt pathway were also reported to impair autophagy mechanism and can lead to tumorigenesis. For example, phosphatase and tensin homolog protein was reported to inhibit Akt survival pathway, and thereby induces autophagy mechanism. However, a mutation in PTEN gene leads to constitutive activation of Akt and inhibition of autophagy, leading to cancer formation[44,48]. Furthermore, the accumulations of p62 aggregates were reported to cause several cancers owing to impaired autophagy mechanism. Another study by Kang et.al.[49], identified the frameshift mutations with mononucleotide repeats in ATG genes in gastric and colorectal carcinomas. Further, this study suggested that these mutations are associated with cancer progression by autophagy deregulation. The study investigated the expression of BNIP3 (Bcl-2/adenovirus E1B 19 kDa-interacting protein), a key regulator of mitochondrial autophagy in breast cancer and reported that BNIP3 expression is significantly lost in invasive breast cancers and suggested that breast cancers cells shows high proliferation with low BNIP3 expression. Thus, collectively these evidences suggest that autophagy plays essential role in tumor suppression and conversely its deregulation leads to cancer.
Autophagy acts as a cell survival mechanism in cancer cells
Genome-wide screening studies show that many genes are involved in the regulation of autophagy either through suppression or enhancement of autophagy[50]. High throughput analyses also contribute to the understanding of autophagy regulation at protein level and in terms of protein-protein interactions. Further, research in yeast and animals suggests that stress-induced autophagy under nutrient-limiting condition promotes cell survival by influencing the bioenergetics of the cell. A study conducted in human prostate cancer cells PC3 and LNCaP, and breast cancer cells MCF7 shows that autophagy acts as a survival mechanism in hypoxic tumor cells. Hypoxia inducible factor1, a positive regulator of autophagy enhances tumor metabolism and metastasis[51], and thereby limits the radiation and chemotherapy. Hypoxia inducible factor1 is involved in the induction of BNIP3 and BNIP3L which disrupts beclin1-bcl2 complex, and releases beclin1 to induce autophagy. Further, BNIP3 induced autophagy acts as an adaptive survival mechanism in hypoxia tumors[52]. In vitro and in vivo studies also reported that autophagy acts as survival mechanism in squamous cell carcinoma by protecting endoG-mediated apoptosis[3].
Additionally, it has been reported that RAS-activating mutations induce high basal-level of autophagy, and consequently assist in the development of lung, colon, and pancreatic cancers; hence inhibition of autophagy in these cancers hinders the tumor growth[44,48,53]. Genetic studies carried on mice also disclosed that deletion of autophagic gene FIP200 inhibits the cell growth in mammary tumors[41]. Moreover, mutations in BRAF protein reported to induce high levels of autophagy in CNS tumor[53], melanoma[54] and thyroid cancers[45]; while, inhibition BRAF lead to impaired autophagy and decreased cell proliferation and cancer growth. All together, these studies suggest that inhibition of autophagy could be an appropriate strategy for the treatment of cancer and targeting the autophagy pathway with small molecules would be fruitful.
Autophagy modulators for cancer therapy
mTOR inhibitors
The mTOR, a member of PI3K family, is critical for serving as a primary regulator of cell growth, proliferation, metabolism and survival[55]. The catalytic subunit of both mTOR1 and mTOR2 complex is involved in many oncogenic signaling pathways. The hyperactive characteristic of mTOR in many human cancers led to target this protein kinase as therapeutic target. Therefore, inhibiting mTOR has gained much attention in anti-cancer therapy. Rapamycin [Figure 5] with two binding moieties is the first generation inhibitor of mTOR. In order to form ternary complex, one binding moiety of rapamycin binds with FKBP12 and the other binds with mTOR[56]. Initially, rapamycin was recognized as immunosuppressant which blocks T-cell activation and later on the anti-cancer activity was documented. Several rapalogs were generated by replacing C-40-O with different moieties and among them Temsirolimus is the first to get FDA approval for cancer treatment[57]. Recent studies have shown that rapamycin can also act as a cytostatic agent, slowing or arresting the growth of various cancer cell lines.
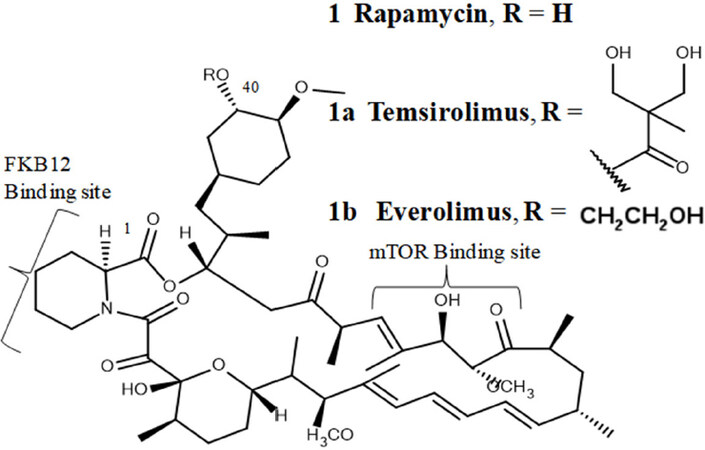
Figure 5. Mammalian target of rapamycin (mTOR) inhibitors
Pan-PI3K inhibitors
PI3K is an essential subunit of PI3K-AKT-mTOR pathway involved in cell proliferation and survival; it is a well known protein kinase for the induction of autophagy. In several cancers including diffuse intrinsic pontine glioma, glioblastoma, paediatric high-grade glioma, breast cancer and cutaneous melanoma, over activation of this pathway has been observed and hence inhibition of PI3K has become an important target in several cancers[58]. PI3K inhibitors including 3-methyladenine (3-MA) and wortmannin [Figure 6] are well characterized as autophagy inhibitors based on their inhibitory effect on the autophagy induction. Besides inhibiting PI3K with an IC50 of 60 µmol/L, 3-MA has also been reported as inhibitor of Vps34 with an IC50 25 µmol/L. Cell culture studies revealed that 3-MA suppresses cell migration and leads to cancer cell death under normal as well as starvation conditions[56]. Surprisingly, prolonged treatment (upto 9h) with 3-MA has shown autophagy flux promotion by increasing the autophagic markers levels such as LC3 protein[59].
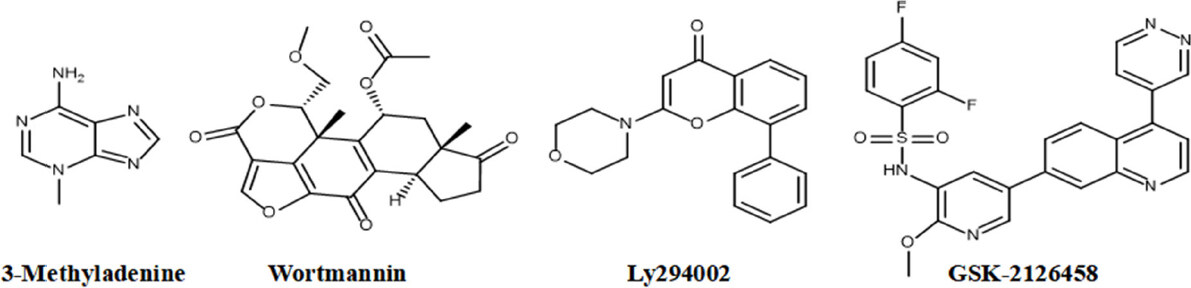
Figure 6. Pan-PI3K inhibitors
Wortmannin, a fungal metabolite is a selective, irreversible and potent inhibitor of PI3K that inhibits autophagic sequestration. It has been demonstrated that the lower concentration (nanomolar) of wortmannin potently and specifically inhibits PI3K; whereas, higher concentrations can inhibit the ataxia telangiectasia gene-related DNA-dependent protein kinase. At physiological pH (6-8.5), wortmannin compete with ATP and ATP analogs binds to PI3K, this suggests that wortmannin binds in the substrate binding site of PI3K. More importantly, site directed mutagenesis studies shows that Lys802 is essential to form nucleophilic interaction[60]. These observations of wortmannin interactions with PI3K provide the molecular basis for designing better inhibitors for PI3K kinase family proteins to treat cancer via autophagy inhibition.
Ly294002, an inhibitor of PI3K class family proteins is derived from the flavonoid quercetin. Ly294002 is not completely selective for PI3K family proteins, and additionally act on other unrelated proteins and lipid kinases[61]. Ly294002 binds at the ATP binding site and is more stable in solution than wortmannin. More importantly, this compound shows its inhibitory effect with IC50 values of 0.5, 0.97 and 0.7 µmol/L for PI3Kα, β and δ targets, respectively[61,62]. To improve the selectivity and specificity, many Ly294002 analog were synthesized; SF1126 a prodrug of Ly294002 entered into clinical trial but was recently halted.
CLR457, a potential inhibitor of all PI3K isoforms is an orally bioavailable inhibitor with antineoplastic activity. It has been extensively characterized by in vitro biochemical methods and in vivo tumor xenograft studies[63]. Dose limiting toxicity studies show that CLR457 potently inhibited PI3K isoforms including p110α (IC50 = 89 nmol/L), p110β (IC50 = 56 nmol/L), p110δ (IC50 = 39 nmol/L) and p110γ (IC50 = 230 nmol/L). However, the characteristics such as poor tolerability and limited antitumor activity of CLR457 resulted in the termination of clinical development.
Recently, omipalisib (GSK2126458) is presented as an autophagy inhibitor that specifically binds to PI3K in PI3K/mTOR signaling pathway. It directly targets Akt phosphorylation by PI3K and reverse phosphorylation of Akt by mTOR. It is an orally bioavailable dual ATP-competitive inhibitor of PI3K and mTOR with high potency[64]. The indirect inhibition of Akt by omipalisib induces cytotoxicity and promotes autophagic cell death at 0.5 μmol/L dose. Further, investigations carried out to know whether the mechanism of cell death occurs through autophagy or apoptosis reported that there is no significant difference in treated and untreated cells when apoptosis markers were used[65]. Cell culture studies reported that GSK2126458 arrests cell cycle at G1 phase and affects proliferation of several cell lines such as breast cancer cell lines T47D and BT474 with IC50 values of 3 and 2.4 nmol/L, respectively. Currently, omipalisib is in clinical trials for idiopathic pulmonary fibrosis and solid tumors. The difficulties in PI3K inhibitors such as lower solubility of wortmannin and broad spectrum inhibition of Ly294002 and limited antitumor activity of CLR457, further necessitate the identification of new inhibitors for PI3K.
Pan-mTORC inhibitors
Activation of mTORC1 and mTORC2 is important in many cancers. Compounds with ATP competitive mechanism inhibiting both mTORC1 and mTORC2 offer better alternative to rapalogs. Torin 1 [Figure 7], is a commercially available autophagy inhibitor that is selective and highly potent ATP-competitive inhibitor of both mTORC1 and mTORC2. Torin 1 shows more efficacy towards blocking the phosphorylation of mTORC1 and mTORC2, when compared with rapamycin a well-known classical mTOR allosteric inhibitor. In vitro kinase assay reveals that Torin 1 has an IC50 of 3 nmol/L, 3 µmol/L, 1.8 µmol/L and 1 µmol/L for mTOR, hVps34, PI3K-α and DNA-PK, respectively. The results show that Torin 1 is more selective for mTOR inhibition over other kinases[60]. Second generation ATP-competitive inhibitor, Torin 2 is a potent and selective inhibitor of mTOR with better pharmacokinetics profile to overcome the limitations of Torin 1. In vitro studies revealed that Torin 2 reduced cell proliferation in several cancer cell lines and exhibited combinatorial response with AZD6244, an inhibitor of MEK kinase in the molar ratio of 1:50. Torin 2 shows inhibition of several PI3KK family proteins including mTOR, ATM, ATR and DNA-PKs with an IC50 value of less than 10 nmol/L[66]; whereas, Torin 1 inhibits only mTOR, ATR and DNA-PK. Further, effects of Torin 2 in autophagosome formation were examined and found to induce autophagy[66].
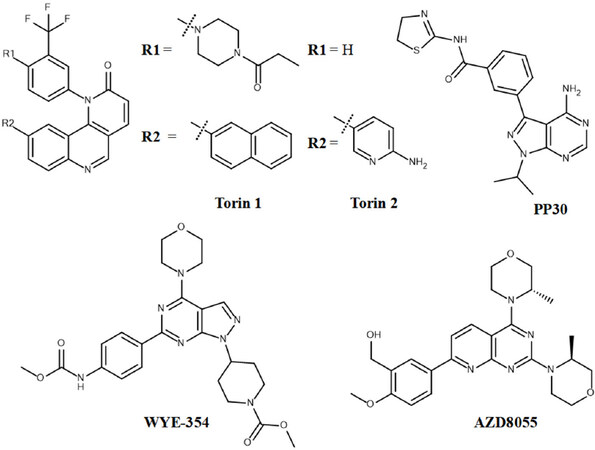
Figure 7. Pan-mTORC inhibitors
AZD8055 is another novel ATP-competitive inhibitor of mTOR kinase (IC50 of 0.8 nmol/L) that shows approximately 1000-fold selectivity against other kinases[67]. Remarkably, in xenografts studies, AZD8055 shows substantial growth inhibition and suggests that AZD8055 can be a potent therapeutic drug in many human cancer treatments[67]. AZD8055 treated acute myeloid leukemia cells have shown significant decrease in cell cycle progression and cell proliferation in blast phase. More interestingly, AZD8055 treatment results in decreased growth of leukemic progenitors but not normal immature CD34+ cells[68]. Another independent study also revealed that AZD8055 treated chronic lymphoid leukemia (CLL) cells show significant reduction in CLL cell proliferation and increase in apoptosis. Currently, this inhibitor is in phase 1 clinical trials.
WYE-354, another heterocyclic compound is a powerful dual ATP-competitive kinase inhibitor that selectively blocks mTORC1/2 activity with an IC50 value of 4.3 nmol/L. Although it displays a weak inhibition of PI3Kα, it has no inhibitory effect on other proteins/kinases. In vitro analysis revealed that WYE-354 potently blocks several cancer cell proliferation including LNCap, A498, MDA-MB-231, MDA-MB-361, MDA-MB-468 and HCT116 cell lines. Mechanistically, WYE-354 arrests cell cycle at G1 phase and induces apoptosis by activation of caspase cascade. Autophagy inhibition by shRNA and autophagy inhibitors such as bafilomycin A1 and 3-MA, notably sensitized WYE-354 mediated anti-colon cancer cell activity. In vivo studies, further witnessed that WYE-354 administration in severe combined immunodeficient mice inhibited growth of xenografts[69]. PP30 is an adenine-mimetic pyrazolopyrimidine scaffold compound that selectively inhibits mTORC1 (IC50 = 8 nmol/L) and mTORC2 (IC50 = 80 nmol/L).
PI3K/mTOR inhibitors
mTOR shares high sequence homology with the hinge region of PI3K, as they belong to the same family of phosphatidylinositol 3-kinase. Hence, several small molecules target both mTOR and PI3K simultaneously. PI-103 [Figure 8] belongs to pyridofuropyrimidine class of compounds and is a multi-target inhibitor that inhibits PI3K and mTOR. Studies using human leukemia cell lines including MV4-11, OCI-AML3 and MOLM14 clearly indicated that PI-103 treatment arrested the cell cycle at G1 phase and eventually reduced the cell proliferation in these cells. The effects of PI-103 in AML patient samples have shown that 82% reduction of AML progenitor clonogenecity. The significant increase in apoptosis is also observed in blast cells when treated with 1.0 µmol/L of PI-103. On the other hand, PI3K/Akt and mTOR inhibition has also been shown when the AML blast cells treated with RAD001 and IC87114 (RAD + IC); but the mechanism of antiproliferative effect is yet to be elucidated. At the same time, this study also reported that inhibitory effect of PI-103 is not much higher than that of RAD + IC in AML blast cells[70].
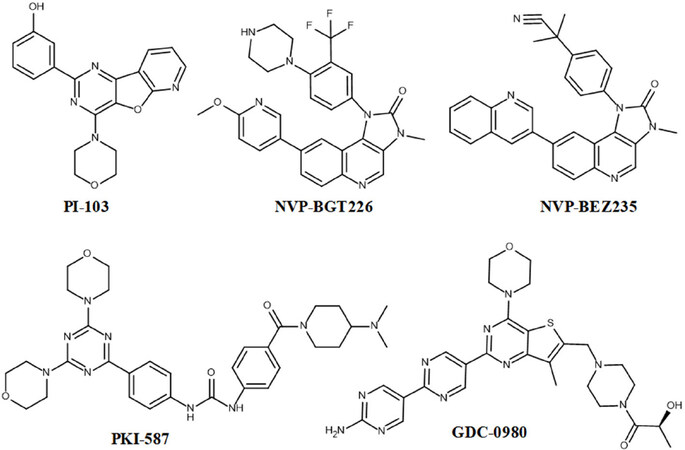
Figure 8. Dual PI3K/mTOR inhibitors
PKI-587, a dual ATP competitive inhibitor of known therapeutic targets PI3K (PI3K-α and PI3K-γ) and mTOR is an orally bioavailable inhibitor[56]. It shows potent inhibitory efficacies of PI3K-α, PI3K-γ and mTOR with IC50 values of 0.4, 5.4 and 1.6 nmol/L, respectively. PKI-587 not only inhibits wild type PI3K, but also exhibits inhibitory activity against most commonly occurring mutants including H1047R and E545K of PI3K with an IC50 value of 0.6 nmol/L for both forms. In in vitro, PKI-587 has exhibited excellent antitumor activity in over 50 human cancer cell lines[71]. In in vivo, PIK-587 showed inhibition of tumor growth in MDA-MB-261, BT474, HCT116, H1975 and U87MG xenograft models, when administrated intravenously.
A novel inhibitor apitolisib, also known as RG7422/GDC-0980 is an orally available dual PI3K and mTOR inhibitor with excellent pharmaceutical and pharmacokinetics properties. The GDC-0980 inhibition of PI3K and mTOR overexpression has shown significant reduction in tumor cell growth by inducing apoptosis. In cell free assays, GDC-0980 treatment results in the inhibition of class 1 PI3K isoforms α, β, δ and γ at low nanomolar IC50 values of 5, 27, 7 and 14 nmol/L, respectively and mTOR inhibition with Ki value of 17 nmol/L. These preclinical data show high potency and selectivity of GDC-0980 inhibitory activity. However, GDC-0980 has less effectiveness than everolimus in metastatic renal cell carcinoma[72].
NVP-BGT226, a potent orally bioavailable dual inhibitor of PI3K and mTOR signaling pathways, blocks cell cycle at G0/G1 phase and induces autophagy and apoptosis. It is shown that NVP-BGT226 suppresses the growth of primary myeloma and common myeloma cell lines at nanomolar concentrations. Specifically, NVP-BGT226 inhibits PI3Kα, β and γ isoforms with IC50 values of 4, 63 and 38 nmol/L, respectively. The analysis of NVP-BGT226 effects in hepatocellular carcinoma (HCC) shows cell growth and proliferation inhibition with potent cytotoxic activity. Hence, the capabilities of NVP-BGT226 in targeting PI3K and mTOR may represent it as an anticancer agent in HCC[73]. NVP-BEZ235, an imidazo[4,5-c]quinoline derivative, is a dual kinase inhibitor of PI3K and mTOR that binds to the ATP binding site and halts cell cycle at G1 phase. When the compound is given orally to animal models, it displayed disease stasis of human cancers[74]. Although co-crystallization studies of this compound with targets are ongoing, docking studies revealed that NVP-BEZ235 forms H-bond with ATP binding cleft residues including Val851, Asp933, and Ser774 of PI3Kα homology model. This compound has entered phase1 clinical trials for the treatment of breast cancer, advanced solid tumors and cowden syndrome.
Unc-51-like kinases inhibitors
ULK belong to serine/threonine kinase family proteins, and play a crucial role in autophagy regulation[34]. Humans contain four ULK kinases including ULK1, ULK2, ULK3 and ULK4. Among them, ULK1 is well studied and it is utmost important for autophagy initiation. Under nutrient deprivation, ULK1 is activated by several up-stream signals (e.g., AMPK, etc.) and then initiates autophagy process by recruiting various other proteins (i.e., FIP200, ATG101 and ATG13 for ULK complex) to the on-site of autophagy initiation. Thus, ULK1 and its associated proteins (i.e., ULK complex) play essential roles in cell survival mechanism under nutrient deficiency[75]. However, disruption of ULK1 and its associated protein complex lead to autophagy inhibition and cell death. As cancer cells generate energy and nutrients through autophagy mechanism and eventually help in cell survival and tumor progression, disruption of ULK1 function by developing small molecule inhibitors has become an attractive approach to treat cancer[75,76]. As a proof of concept, few ULK1 inhibitors have been reported in the literature [Figure 9].
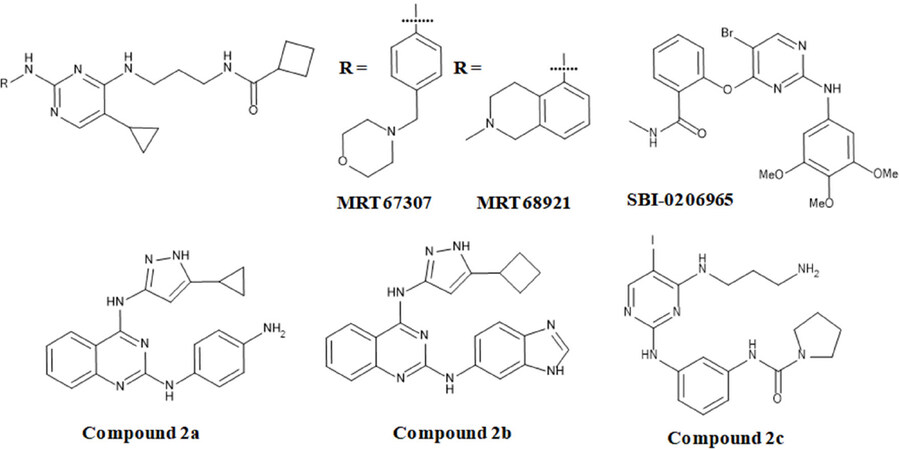
Figure 9. ULK1 inhibitors
MRT67307 and MRT68921 are two closely related derivatives with different substitution pattern on the pyrimidine ring. MRT67307 inhibits ULK1 and ULK2 with an IC50 of 45 and 38 nmol/L, respectively. This compound led to the identification of MRT68921, that has 15-fold improved inhibition of ULK1 (IC50 = 2.9 nmol/L) and 30-fold improved inhibition of ULK2 (IC50 = 1.1 nmol/L), when compared with MRT67307. Studies using MRT67307 and MRT68921 in MEFs cells show that these compounds are able to block autophagic flux. In addition, MRT68921 treated cells show significant increase in SQSTM1 level and decrease in LC3-2/LC3-1 ratio. These results suggested that MRT68921 treatment efficiently inhibits ULK-mediated autophagy[77]. However, the molecular basis of this block remains to be elucidated. SBI-0206965 is a potent and specific inhibitor of ULK1 with an IC50 of 108 nmol/L and also selectively inhibits ULK2 with IC50 of 711 nmol/L, but with less efficacy when compared with ULK1. Inhibition of ULK1 in NSCLC cells results in anti-proliferative effect[51]. Specifically, SBI-0206965 suppresses phosphorylation events in cells that are mediated by ULK1. SBI-0206965 at 10 µmol/L concentration shows high selectivity and inhibits only 10 out of 456 kinases.
In 2015, Lazarus et al.[75] reported the first crystal structure of ULK1 in complex with multiple inhibitors. The structure consists of an N-terminal kinase domain, a serine-proline rich region, and a C-terminal interacting domain. They used a standard 32P-ATP radioactive assay to screen a collection of 746 compounds against ULK1 that led to the identification of several pyrazole aminoquinazolines as ULK1 inhibitors. For example, the identified compound 2a showed dose dependent inhibition with IC50 of 160 nmol/L when re-tested in in vitro assay. Further, co-crystallization of this compound with ULK1 demonstrated that compound 2a [Figure 10] bound in the ATP binding site by making hinge region interaction with its amino pyrazole core group. Moreover, they observed that the aniline moiety made contacts with Asp165 of DFG motif, and the cyclopropyl substituent moiety fitted into the pocket close to the gate keeper residue methionine. Quinazoline ring interacts through position 6 and 7 with the backbone of kinase. In conformity with this steric obstruction, no compounds with substituents at these positions showed activity against ULK1.
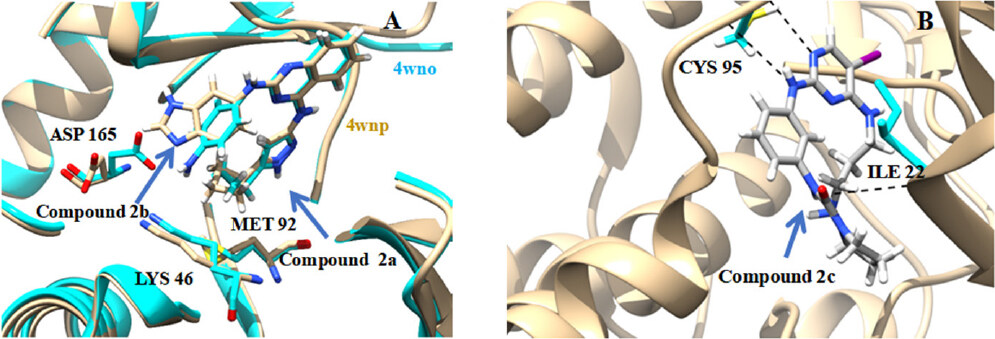
Figure 10. A: Superimposition of the binding mode of compound 2a (PDB ID: 4WNO) and compound 2b (PDB ID: 4WNP) in ULK1; B: binding mode of compound 2c (PDB ID: 5CI7) in ULK1. Hydrogen bond interactions are represented in black dotted lines. Compound 2c with a diaminopropyl group, a closely related pyrimidine analog of MRT67307, showed dose dependent inhibition with an IC50 of 120 nmol/L. Co-crystallization of compound 2c with ULK1 [Figure 10B] showed that it has similar orientation with the diaminopropyl group occupying a similar space as that of quinazoline of compound 2a. The main difference in the kinase is the movement in the β sheet in the N-terminal lobe with Gly23 towards the inhibitor, which allows Ile22 to twist away from the bulky diaminopropyl substituent on the pyrimidine. The other difference is the gatekeeper methionine moves towards iodine group, to adopt a suitable dipole-dipole interaction. A flexible region involving Ile22 was required to pack above the aminopropyl group[78]
Further, modifications made to these series of compounds led to the identification of compound 2b with improved potency with an IC50 of 8 nmol/L[56]. However, the co-crystallization of this compound in similar condition (as of compound 2a) with ULK1 produced different space groups that indicate compound 2b produced conformational changes in the kinase domain, which led to the improved activity. Detailed analysis revealed that there were major changes in the conformation of interlobe loop, the side chain of Asp165 (DFG loop residue) and methionine of gatekeeper residue[75]. Testing of compound 2b against a small panel of kinases showed non-selectivity, suggesting the need for improving the selectivity and potency against ULK1.
Vacuolar protein sorting 34 inhibitors
Vps34 is a lipid kinase that belongs to subgroup of class III PI3K family protein. The major function of this family protein is to phosphorylate the 3-hydroxyl group of inositol ring of phosphatidylinositol (PtdIns) lipid substrates to generate PtdIns3P[22,79,80]. Vps43 interacts with multiple protein subunits and forms Vps34 complex I (i.e., Vps34, Vps15, Beclin 1, and Atg14L) that precisely catalyzes the phosphorylation of PtdIns to PtdIns3P and anchors with intracellular membranes. Further, the membrane-bound PtdIns3P interacts with proteins that contains FYVE, PX, or WD40 domain and involves in vesicle trafficking and autophagy mechanism. Thus, Vps34 complex is essential for initiation/induction of autophagy during nutrient deprivation through regulation of mTOR pathway. Therefore, the disruption of Vps34 complex leads to autophagy inhibition and cancer cell death. Hence, Vps34 has become an attractive drug target for cancer therapy and a number of inhibitors have been reported in the literature, and few compounds have entered into clinical studies.
Recently, Dowdle et al.[81], identified a hit containing bisaminopyrimidine core as a potent and selective Vps34 kinase inhibitor using high-throughput screening of compound libraries. Optimization of this compound led to the identification of 4-aminopyridine-containing PIK-III inhibitor [Figure 11] with improved potency and selectivity[81]. Additional biochemical testing and profiling of this compound showed that PIK-III is at least 100-fold selective for Vps34 as compared to related kinases including PI3Kα, mTOR, and additional 44 protein kinases. Moreover, they also demonstrated that PIK-III inhibited the co-localization and distribution of PtdIns3P specific lipid binding domain (FYVE) fused with GFP with an IC50 of 55 nmol/L concentration and that is > 10,000 times more potent than the non-selective Vps34 inhibitor 3-MA.
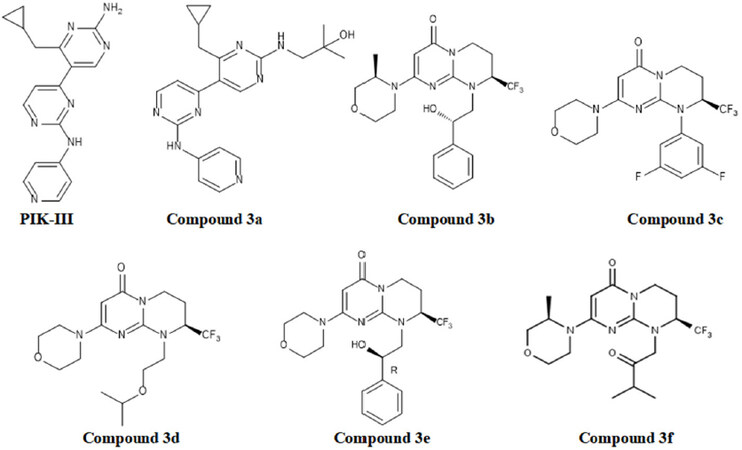
Figure 11. Vps34 inhibitors
Co-crystallization of PIK-III with human Vps34 kinase [Figure 12A] revealed that the overall structural geometry was comparable to that of PI3Kγ and Drosophila melanogaster Vps34 structures[81]. Further, analysis revealed that the structure appears like a typical lipid and protein kinase structure that has a relatively narrow active site with hydrophobic pocket appropriate for binding co-planar aromatic compounds. Binding mode of PIK-III to Vps34 structure demonstrated that the cyclopropyl group occupied the hydrophobic pocket that was formed with side chains of Phe612, Pro618 and Phe684. Two hydrogen bond interactions were observed between PIK-III acceptor/donor and backbone amide & carbonyl oxygen of Ile685. In addition, solvent mediated hydrogen bond network bridges were observed between aminopyrimidine moiety of PIK-III and side chains of Asp671 and Asp644. Superimposition of Vps34 and PI3Kα active site revealed that in both the structures the hydrophobic cavity was enclosed/covered with P-loop residues, however their relative orientations were quite different with respect to their hinge regions. In Vps34 it is relatively displaced towards hinge region, whereas in PI3Kα structure it is wider and proximal to hinge region. In Vps34 structure, the relative orientation of Phe612 has significant role as it allows cyclopropyl group to fit into the hydrophobic pocket to acquire optimal interaction with hinge region. Whereas, in PI3Kα structure the corresponding phenylalanine was replaced with methionine residue and it doesn’t allow the cyclopropyl group to fit into the pocket. Thus, this structural difference makes an ideal tool for developing selective inhibitors of Vps34 and also to explicitly measure pharmacological consequences of VPS34 inhibition in vivo.
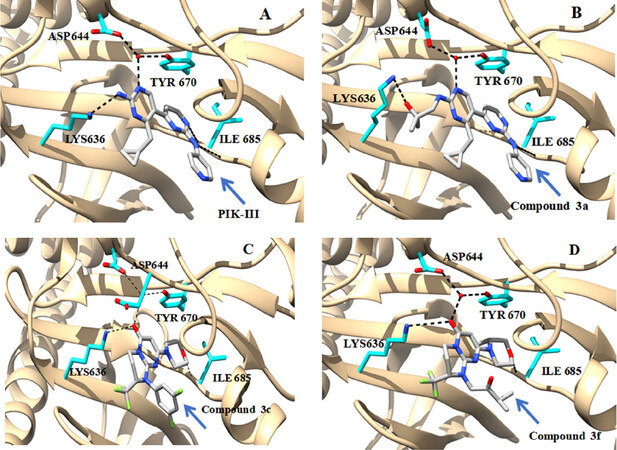
Figure 12. 3D binding mode and interaction of PIK-III (PDB ID: 4PH4) (A), compound 3a (PDB ID: 5ENN) (B), compound 3c (PDB ID: 4UWF) (C) and compound 3f (PDB ID: 4UWL) (D) with Vps34. Hydrogen and water mediated hydrogen bonds are represented in black dotted lines
Further, docking and structure-activity relationship (SAR) study led to the identification of compound 3a with greater potency and improved metabolic stability providing an excellent candidate for in vivo pharmacokinetics evaluation[80]. Compound 3a showed exceptionally selective activity over other lipid and protein kinases (> 100-fold against more than 280 kinases evaluated, except TAK1 and PI3Kδ). Co-crystallization of compound 3a with Vps34 [Figure 12] revealed that its binding mode is similar to that of PIK-III. Moreover, compound 3a prevented the degradation of various autophagy substrates (p62, NCOA4, NBR1, NDPS2 and FTH1) as that of PIK-III. Pharmacokinetics profile of compound 3a revealed that it is rapidly absorbed and showed moderate mean systemic clearance (30 mL/min/kg) with good oral bioavailability (F% = 47). Oral administration of compound 3a to RKO colon cancer bearing mice at 50 mg/kg twice a day for 7 days showed time-dependent accumulation of LC3-II with reduced autophagy capacity without reduction of tumor volume. Hence, additional studies with long term drug administration need to be evaluated.
Pasquier et al.[82], reported a series of tetrahydropyrimidopyrimidinone derivatives [Figure 11] as Vps34 inhibitors using a cell based high-throughput phenotype screening campaigning. The reported compounds comprise/contain a pyrimidinone moiety with a morpholine group as hinge binder and shown to have Vps43 inhibition in kinase profiling assay panel. However, these compounds also showed cross-reactivity with class I PIKs (isoformsα, β, δ, and γ) and at a lower concentration level, with mTOR. Further, evaluation of these compounds activity against Vps34 using GFP-FYVE cell based assay revealed that compound 3b shows higher cellular potency compared to other compounds[82]. Hence, compound 3b was selected as advanced hit for back screening. Additional screening of tetrahydropyrimidopyrimidone derivatives and SAR studies focused on potency and selectivity over lipid kinases. Substitution at 1-position of the pyrimidinone scaffold led to the identification of compounds 3c, 3d and 3e [Figure 11] with enhanced Vps34 enzymatic potency, substantial GFP-FYVE cellular potency, attractive ligand efficiency (LE) and ligand lipophilicity efficiency (LLE) values. Moreover, these compounds also showed favorable in vitro ADME properties and sensible microsomal stability.
X-ray co-crystal structure of compound 3c [Figure 12C], 3d and 3e with human Vps34 demonstrated that all the compounds adapted a DFG-in conformation and were involved in hinge region interaction via oxygen atom of the morpholine ring. Moreover, this moiety was also involved in making promising vander Waals interactions with adjacent residues. The aromatic ring of pyrimidinone moiety was stacked in between Ile634 and Ile760 side chains, whereas carbonyl function of this moiety was involved in making hydrogen bond interaction with catalytic Ly636 side chain, and water mediated H-bond interactions with Asp644 and Tyr670 side chains. The (2S)-trifluoromethyl group of tetrahydropyrimidine ring was pointed towards the hydrophobic pocket under the P-loop residues Phe612 to Ala619. Compounds 3c-e show difference only at their N-substituent that points towards the exit of the ATP binding site. Further, modification of hinge binder moiety led to improved selectivity, and replacing the trifluoromethyl moiety on tetrahydropyrimidinone ring gained Vps34 potency and lost selectivity against other lipid kinases. From the final step of optimization, compound 3f was selected as Vps34 inhibitor for in vivo pharmacological study.
In vitro studies revealed that compound 3f displayed IC50 values of 2 and 82 nmol/L on Vps34 enzymatic assay and GFP-FYVE cellular assay, respectively. Biophysical characterization using surface plasmon resonance and isothermal titration calorimetry demonstrated that the compound 3f has a binding affinity (KD) of 2.59 and 2.7 nmol/L, respectively. Moreover, compound 3f showed good physiochemical/drug-like properties (such as LE = 0.41, and LLE = 6.22). Further, co-crystallization of compound 3f with Vps34 [Figure 12D] revealed that methyl group of morpholine moiety points towards Met682. Like compound 3e, water mediated hydrogen bonds were observed between carbonyl oxygen atom and Asp761 of Vps43. Thus, this binding orientation provided enhanced Vps34 selectivity and potency. In vivo pharmacokinetics (PK) profiles of compound 3f disclosed a good oral bioavailability (F% = 85) with maximal plasma concentration observed at 0.5 h and reasonable systemic clearance. Moreover, PK/PD experiments using GFP-FYVE H1299 tumors xenografted in SCID mice revealed that compound 3f had sustainable inhibition (> 80%) of granular staining, and a dose dependent target modulation.
Lysomotrophic agents
Chloroquine [Figure 13], a widely used inhibitor of autophagy which inhibits last stage of autophagy is initially discovered to treat malaria and inflammatory diseases. Although, the mode of action of bafilomycin A1 and lysosomal protease inhibitors were well characterized, mode of action of chloroquine still remains unclear. However, it is believed that chloroquine inhibits autophagic flux through rising pH and thereby inactivates lysosomal hydrolases[83]. Currently, chloroquine (CQ) and hydroxychloroquine (HCQ) are being investigated as autophagy modulator in Phase II/III trials for cancer therapy[84]. Recent in vitro and in vivo studies revealed that chloroquine affects the endo-lysosomal system and golgi complex thereby modulates autophagic flux through decreasing autophagosome-lysosome fusion[85]. This study invalidated lysosomal degradation function of chloroquine.
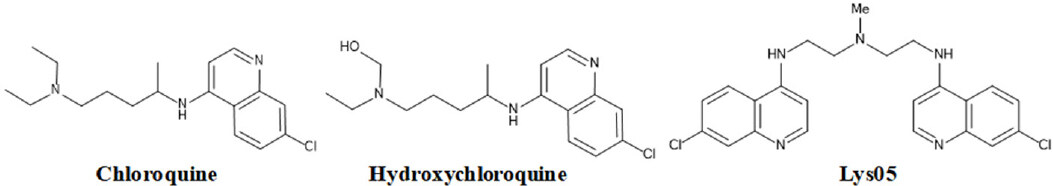
Figure 13. Lysomotrophic inhibitors
HCQ, a derivative of CQ is a 4-aminoquinoline that has antimalarial and anti-inflammatory activities; it is currently being investigated as the inhibitor for autophagy. Several clinical trials of HCQ in combination with other anti-cancer drugs (e.g., temozolomide, bortezomib, temsirolimus, vorinostat, doxorubicin, etc.) showed partial response and stable disease outcome for various cancers (melanoma, colorectal cancer, myeloma and renal cell carcinoma)[86]. As a drug, this basic compound alkalinizes acidic environment of lysosomes and thereby prevents autophagosome-lysosome fusion. HCQ is proved to be threefold less toxic than CQ and can augment the cytotoxicity of a number of chemotherapies and targeted therapies. A recent meta-analysis of clinical trials of CQ and HCQ concluded that their use in cancer patients has better treatment response, when used in combination with existing anti-cancer therapy[87].
Based on the 4-aminoquinoline core structure of CQ and HCQ, Lys05 was designed; it was more potent in in vitro and in vivo as a single agent. The increased activity of Lys05 was due to the bivalent aminoquinoline rings, C7-chlorine and a short triamine linker. Lys05 trihydrochloride is water soluble and shows potent anti-tumor activity in several human cancer cell lines as a single agent. Intermittent high dose or chronic daily dosing of Lys05 at lower doses have shown early blockage of autophagy in melanoma and colon cancer xenograft models[88]. Comparative study in cancer cells also revealed that HCQ at 100 µmol/L cannot show complete deacidification of endovascular compartment; whereas 50 µmol/L of Lys05 has shown complete deacidification. Further, in mice models Lys05 at high dose (80 mg/kg, i.p.) causes Paneth cell dysfunction with loss of lysozyme biosynthesis and bowel pseudo-obstruction[88,89]. Studies evidenced that high dose of Lys05 is associated with intestinal toxicity and it has been also observed that high dose of HCQ also causes low grade nausea and constipation in patients. Lys05 is a new lysosomal inhibitor that has a potential to be further developed as a drug for cancer treatment.
Autophagy inducers
In addition to the above agents/compounds that were designed to modulate the autophagy process by specifically interacting with targets in autophagy pathways, there are other agents that induce autophagy. Particularly, naturally occurring compounds have multiple modes of action and targets different pathways. Some of them induce autophagy by targeting autophagy pathway signaling molecules and are discussed in this section [Figure 14].
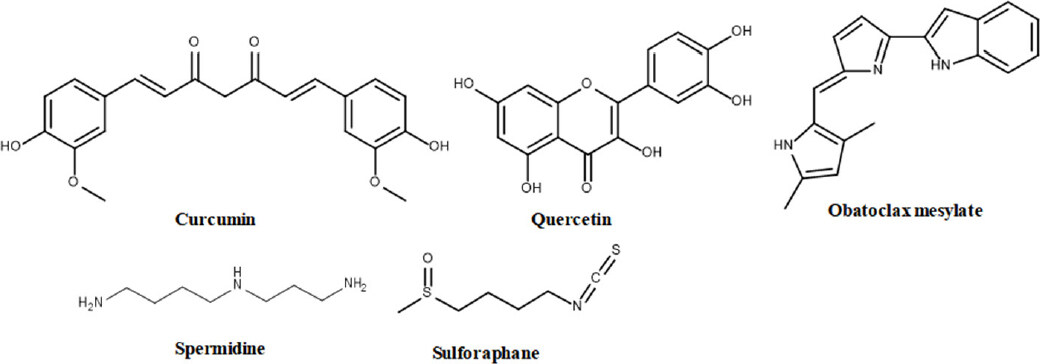
Figure 14. Autophagy inducers
Curcumin
Curcumin, a natural compound of golden spice turmeric shows numerous activities including anti-inflammatory, antimicrobial, antioxidant, hypoglycemic and wound healing activities. Considering these activities, curcumin has been investigated in many clinical conditions such as multiple myeloma, breast cancer and non small cell lung cancer. Although, it has proven efficacy in several clinical aspects, curcumin has therapeutic limitations due to rapid systemic elimination, rapid metabolism and poor absorption. Curcumin showed anticancer effects by sensitizing chemotherapy and radiation therapy[90]. In gastric cancer cell lines SGC-7901 and BGC-823, curcumin significantly inhibited cell proliferation by exhibiting autophagy induction. The studies have reported that curcumin induces autophagy by its dual functionality in up-regulation of p53 and p21, and down-regulation of PI3K/Akt/mTOR signaling pathways[91]. Another recent investigation also reported that curcumin induces autophagy in human pancreatic cancer cell lines[91]. Curcumin treatment of human lung adenocanrcinoma cell line A549 showed autophagy induction by means of increased AMPK phosphorylation[92].
Quercetin
Quercetin, a well-known cancer therapeutic agent and autophagy modulator is abundantly present in vegetables and fruits, and suppress tumor proliferation[93]. It has been shown that quercetin suppresses Akt/mTOR and Bcl-2 signaling pathways and activates LC3, ERK and caspase. Studies in human neuroglioma cells U87 and in vivo studies revealed that quercetin nanoparticles significantly inhibit neuroglioma cell proliferation by inducing autophagy and apoptosis. Cell proliferation assay of neuroglioma cells showed that quercetin nanoparticles treatment with 10-50 µg/mL gradually reduced cell viability[94]. A recent study reports that quercetin modulate p-STAT3/Bcl2 pathway and there by induces protective autophagy in ovarian cancer[95].
Obatoclax mesylate
Obatoclax, a small molecule indole bipyrrole compound is an inhibitor of Bcl2 family proteins and exhibited anticancer activity in acute lymphoblastic leukemia[96]. In addition to its involvement in apoptosis, obatoclax has also been involved in triggering cell death via autophagy induction[97]. Primarily, this compound plays a key role in stimulation of necrosome assembly on autophagosome membrane to induce autophagy. Co-immunoprecipitation studies indicated that obatoclax enhances the interaction between Atg5 and necrosome components including FAD, RIP and RIP3. The same study investigated the role of obatoclax with 200 nmol/L concentration in rhabdomyosarcoma cells to unravel whether cell death occurs via apoptosis or autophagy and noticed little DNA fragmentation when the most of cell lost their viability.
Spermidine
Spermidine is a polyamine present in most of the mammalian cells and has important roles in several cellular activities under physiological conditions. It is well known for its longevity-promoting activity in association with autophagy enhancement. A study conducted in differentiated rat pheochromocytoma PC12 and human embryonic kidney 293 cells, revealed the possible role of spermidine in autophagy. After treating the cells with spermidine, the cell death analysis and caspase activity assays indicated that spermidine restricts neuronal injury via inhibiting caspase-3 dependent Beclin1 cleavage. Eventually, this mechanism elevates dysregulated autophagy flux levels[98]. Very recent immunoprecipitation and immunohistochemistry studies in human chondrocytes and in vivo study conducted in mice reported that the cells treated with spermidine (100 nmol/L) showed autophagy activation and promoted chondrogenesis[98]. However, spermidne role in cancer is still controversial as some studies reported that spermidine do not induce tumorigenesis[99], whereas some studies established that spermidine synthase inhibitors show little cell growth reduction in cancer cell lines[100].
Sulforaphane
Sulforaphane, an organic isothiocyanate compound derived from glucosinolates shows both cytoprotective and cytotoxic activities. It activates nuclear factor E2-related factor 2 signaling that elevates the expression of antioxidant response proteins in oxidative stress. A study in murine osteosarcoma cells reported that sulforaphane induces apoptosis through cell cycle arrest and inhibits tumor cell growth[101]. It has also been reported that the treatment of sulforaphane initiates various cellular processes in human prostate cancer[102]. To investigate whether sulforaphane is reducing the cell growth or encourages cell death, a study was conducted in human lens cell line and reported that sulforaphane reduced the cell viability in human lens cell line and promotes cell death. The study also revealed that sulforaphane promotes ER stress and autophagy induction via MAPK signaling[103], and longer treatment with sulforaphane has shown significant decrease in AMPK phosphorylation at thr-172[104] in human prostate cancer cells.
FDA approved drugs with autophagy modulation activity
In addition to the above discussed autophagy modulators, few FDA approved drugs (for different indications) are reported to have additional autophagy modulator activities [Table 2]. This section is intended to list out these drugs and provide brief information and mode of autophagy modulation by these drugs. Some of these drugs are already used as anticancer agents, while some are used for other indications. These data suggests that some of the approved anticancer drug may be partly working through autophagy modulation. Moreover, understanding the exact molecular mode of action of these drugs could help to repurpose these drugs for cancer therapy in the future.
FDA approved drugs for various human diseases that are reported to have autophagy modulating activity
| Sl. No. | Drug | Mode of action | FDA approved use | Nature of the modulator |
|---|---|---|---|---|
| 1 | Temozolomide | Alkylating agent | Glioblastoma multiforme | Autophagy inducer |
| 2 | Rapamycin | Inhibits mTOR | Transplant rejection | Autophagy inducer |
| 3 | Metformin | Activates AMPK | Type 2 Diabetes | Autophagy inducer |
| 4 | Gefitinib | PI3K/AKT/mTOR pathway | Metastatic non-small cell lung cancer (NSCLC) | Autophagy inducer |
| 5 | Bortezomib | Activates AMPK | Multiple myeloma | Autophagy inhibitor |
| 6 | Sodium Phenylbutyrate | Improves cathepsin D and B activities and lysosomal-autophagic function | Urea cycle disorders and acute promyelocytic leukemia | Autophagy inhibitor |
| 7 | Carbamazepine | Reduces inositol and Ins (1,4,5)P3 levels | Epilepsy | Autophagy inducer |
| 8 | Verapamil | Inhibits Ca2+ channel lowers intracytosolic Ca2+ levels | Hypertension | Autophagy inducer |
| 9 | Rilmenidine | Reduces cAMP levels | Hypertension | Autophagy inducer |
| 10 | Choloroquine and Hydroxycholoroquine | Inhibits lysosomal function | Malaria, Lupus and rheumatoid arthritis | Autophagy inhibitor |
| 11 | Pantoprazole | Proton pump inhibitor, increased endosomal pH | Erosive esophagitis | Autophagy inhibitor |
| 12 | Celecoxib | Inhibits cyclooxygenase 2 | Rheumatoid arthritis and osteoarthritis | Autophagy inhibitor |
Temozolamide, an alkylating agent is a FDA approved drug for the treatment of glioblastoma multiforme in combination with radiotherapy[105]. Recent studies have investigated the role of temozolamide in autophagy modulation and reported that it induces autophagy in glioblastoma cancer cells through EGFR independent manner[106]. Temozolamide also showed cytotoxicity in PI3K/AKT/mTOR pathway inhibited adenoma cells[107]. Gefitinib which targets tyrosine kinase activity of EGFR and binds competitively to the ATP binding site is a FDA approved drug for treating the patients with non-small cell lung cancer (NSCLC)[108]. Gefitinib induces autophagy in lung cancer cells through blocking of PI3K/AKT/mTOR pathway[109] and also exhibited autophagy induction in combination with clarithromycine in NSCLC cells[110].
Metformin, a biguanide antihyperglycemic agent is FDA approved for treating non-insulin-dependent diabetes mellitus. It controls glucose level by means of decreasing hepatic glucose production and also by increasing insulin-mediated glucose uptake. It is reported that metformin is involved in autophagy induction by AMPK dependent manner. Several cancer models have shown significant growth inhibition upon metformin treatment[111]. A study also reported that metformin promotes autophagy and selectively inhibits esophageal squamous cell carcinoma cell growth by down regulating STAT3 signaling[112]. Studies on human multiple myeloma cells show that metformin inhibits the cell proliferation by promoting autophagy and cell cycle arrest and this study suggested metformin dual repression of mTORC1 and mTORC2 via AMPK activation[113].
Bortezomib is a proteosome inhibitor that specifically inhibits nuclear factor kappaB and is a FDA approved drug to treat multiple myeloma[114]. Bortezomib has shown anticancer activities in several human cancers including prostate cancer, colon cancer, ovarian cancer and breast cancer. Studies have explored the possible role of Bortezomib in autophagy and found that it promotes cancer cell death through blockage of autophagic flux in ERK phosphorylation dependent manner[115]. Sodium phenylbutyrate is a chemical chaperon that inhibits histone deacetylase and is FDA approved drug to treat urea cycle disorders. Sodium phenylbutyrate is also under clinical investigation in several human diseases including hemoglobinopathies, motor neuron diseases, cancer and cystic fibrosis[115]. Sodium phenylbutyrate has been shown to reduce dithiothreitol or tunicamycin induced autophagy[116].
Carbamazepine is a FDA approved anticonvulsant drug for the treatment of epilepsy. Evidences indicate that carbamazepine diminishes hepatocellular death in autophagy dependent manner. A study on SW480 colon cancer cell lines revealed that carbamazepine decreases β-Catenin and VEGF levels, resulting in antitumor activity[117]. Verapamil and Rilmenidine are well known drugs for the treatment of hypertension and now these drugs are under investigation to unravel their role as autophagy modulators in cancer. Autophagy induction has been observed upon treatment with verapamil in COLO 205 cells with cytoprotective activity[118]. It has been observed that Rilmenidine promotes autophagy in an mTOR independent manner, however the reduction in disease progression has not been observed[119].
Pantoprazole is a protein pump inhibitor used to treat certain esophagus and stomach related problems. Various studies reported that pantoprazole sensitized cancer cells to anticancer drugs by suppressing autophagy induction in time- and dose-dependent manner[120-122]. Celecoxib is a nonsteroidal anti-inflammatory drug that is FDA approved for Rheumatoid arthritis and osteoarthritis treatment. Recent studies have evaluated celecoxib activities in cancer and reported its antitumor effects in solid tumors. Also autophagy reduced significantly upon treatment with celecoxib in imatinib resistant chronic myeloid leukemia cells[123]. All together, although these drugs have proven to be useful in various human diseases, currently many studies are investigating their impact in cancer though autophagy modulation.
Conclusion
Autophagy is a conserved cellular process that is essential for the cells to cope-up with adverse conditions, such as the lack of nutrients or bacterial/viral infections. In such conditions, autophagy plays a pro-survival role by recycling the cellular components by lysosomal degradation and also helps in the removal of pathogenic organism by sequesterization and degradation. Because of its central role in maintaining cellular homeostasis, alterations in the autophagic process through aberrant signaling has been linked to several disease states such as cancer and neurodegeneration, among others. In cancer, autophagy has the role of a double edge sword, with tumor suppressing activity during the initial stages of cancer development and pro-survival effects in the later stages of cancer development.
The relevance of autophagy modulation in cancer is increasingly appreciated and many therapeutic targets involved in autophagy process have been identified. In this review, we highlighted the role of promising autophagy signaling pathway/biomarkers that could be modulated for cancer therapy. Further, we also reviewed and analyzed the available autophagy inducers/inhibitors. Except for the mTOR targeting rapalogs, no other autophagy targeting agents are approved in the clinics for cancer treatment. Currently, chloroquine and hydroxychloroquine are being clinically tested as autophagy modulators either alone or in combination with other anti-cancer therapeutics (e.g., temozolomide, bortezomib, temsirolimus, vorinostat, doxorubicin, etc.). Eventhough hydroxychloroquine shows partial response and improves treatment outcomes in various cancers such as melanoma, colorectal cancer, myeloma and renal cell carcinoma, dose related toxicity may preclude its widespread use as autophagy modulator. However, this may change soon as several groups are developing molecules to various drug targets such as ULK and Vps34 within the autophagy signaling pathway. Developing selective autophagy modulators with minimal cross-talk with other targets will be challenging and crucial for making autophagy modulation as a successful strategy for cancer therapy. Moreover, patient selection based on the alteration in the autophagy signaling pathways could pave way for better treatment outcomes for autophagy modulators.
The importance of autophagy related research can be fathomed from the ever increasing number of publications (90 articles in the year 2000, 2050 in 2010 and 6700 articles in 2018; scopus data) and the fact that 2016 Nobel prize in Physiology or Medicine was awarded to Prof. Yoshinori Ohsumi for his research on Autophagy in yeast. Eventhough, we have made big strides in understanding the autophagy process, the complete molecular machinery of autophagy, the signaling process involved and their roles in various disease conditions are not yet completely elucidated in humans. Future research should hold promise as well as provide insights in these directions for identifying better treatment for cancer and other diseases.
Declarations
Authors’ contributionsCollected the literature and wrote the article: Kondapuram SK, Sarvagalla S
Collected the literature, corrected the article and approved: Coumar MS
Availability of data and materialsNot applicable.
Financial support and sponsorshipNone.
Conflicts of interestAll authors declared that there are no conflicts of interest.
Ethical approval and consent to participateNot applicable.
Consent for publicationNot applicable.
Copyright© The Author(s) 2019.
REFERENCES
1. Morel E, Mehrpour M, Botti J, Dupont N, Hamaï A, et al. Autophagy: a druggable process. Annu Rev Pharmacol Toxicol 2017;57:375-98.
2. Saha S, Panigrahi DP, Patil S, Bhutia SK. Autophagy in health and disease: a comprehensive review. Biomed Pharmacother 2018;104:485-95.
3. Masui A, Hamada M, Kameyama H, Wakabayashi K, Takasu A, et al. Autophagy as a survival mechanism for squamous cell carcinoma cells in endonuclease g-mediated apoptosis. PLoS One 2016;11:1-19.
5. Li W, Yang Q, Mao Z. Chaperone-mediated autophagy: machinery, regulation and biological consequences. Cell Mol Life Sci 2011;68:749-63.
6. Li WW, Li J, Bao JK. Microautophagy: lesser-known self-eating. Cell Mol Life Sci 2012;69:1125-36.
8. Nascimbeni AC, Giordano F, Dupont N, Grasso D, Vaccaro MI, et al. ER-plasma membrane contact sites contribute to autophagosome biogenesis by regulation of local PI3P synthesis. EMBO J 2017;36:2018-33.
9. Nakamura S, Yoshimori T. New insights into autophagosome-lysosome fusion. J Cell Sci 2017;130:1209-16.
10. Klionsky DJ, Eskelinen EL, Deretic V. Autophagosomes, phagosomes, autolysosomes, phagolysosomes, autophagolysosomes... wait, I’m confused. Autophagy 2014;10:549-51.
11. Fang Y, Tan J, Zhang Q. Signaling pathways and mechanisms of hypoxia-induced autophagy in the animal cells. Cell Biol Int 2015;39:891-8.
13. Eskelinen EL, Saftig P. Autophagy: a lysosomal degradation pathway with a central role in health and disease. Biochim Biophys Acta - Mol Cell Res 2009;1793:664-73.
14. Dikic I, Elazar Z. Mechanism and medical implications of mammalian autophagy. Nat Rev Mol Cell Biol 2018;19:349-64.
15. Amaravadi R, Kimmelman AC, White E. Recent insights into the function of autophagy in cancer. Genes Dev 2016;30:1913-30.
16. Zachari M, Ganley IG. The mammalian ULK1 complex and autophagy initiation. Essays Biochem 2017;61:585-96.
18. Dunlop EA, Hunt DK, Acosta-Jaquez HA, Fingar DC, Tee AR. ULK1 inhibits mTORC1 signaling, promotes multisite raptor phosphorylation and hinders substrate binding. Autophagy 2011;7:737-47.
20. Lin S-Y, Li TY, Liu Q, Zhang C, Li X, et al. GSK3-TIP60-ULK1 signaling pathway links growth factor deprivation to autophagy. Science 2012;336:477-81.
22. Mercer TJ, Gubas A, Tooze SA. A molecular perspective of mammalian autophagosome biogenesis. J Biol Chem 2018;293:5386-95.
23. Kang R, Zeh HJ, Lotze MT, Tang D. The beclin 1 network regulates autophagy and apoptosis. Cell Death Differ 2011;18:571-80.
24. Kim JH, Hong SB, Lee JK, Han S, Roh KH, et al. Insights into autophagosome maturation revealed by the structures of ATG5 with its interacting partners. Autophagy 2015;11:75-87.
25. Sakoh-Nakatogawa M, Matoba K, Asai E, Kirisako H, Ishii J, et al. Atg12-Atg5 conjugate enhances E2 activity of Atg3 by rearranging its catalytic site. Nat Struct Mol Biol 2013;20:433-9.
26. Lao Y, Xu N. Autophagy in cancer chemoprevention: identifcation of novel autophagy modulators with anticancer potential. Compr Toxicol Second Ed 2016;1379:151-63.
27. Manni D. Oxidation-dependent regulation of the selective autophagy receptor SQSTM1/p62. Available from: https://theses.ncl.ac.uk/dspace/handle/10443/3675 [Last accessed on 3 Apr 2019].
28. Weidberg H, Shvets E, Shpilka T, Shimron F, Shinder V, et al. LC3 and GATE-16/GABARAP subfamilies are both essential yet act differently in autophagosome biogenesis. EMBO J 2010;29:1792-802.
29. Fader CM, Sánchez D, Furlán M, Colombo MI. Induction of autophagy promotes fusion of multivesicular bodies with autophagic vacuoles in K562 cells. Traffic 2008;9:230-50.
30. Lee JA, Beigneux A, Ahmad ST, Young SG, Gao FB. Escrt-III dysfunction causes autophagosome accumulation and neurodegeneration. Curr Biol 2007;17:1561-7.
31. Li C, White SH, Warren LK, Wohlgemuth SE. Skeletal muscle from aged American Quarter Horses shows impairments in mitochondrial biogenesis and expression of autophagy markers. Exp Gerontol 2018;102:19-27.
32. Mawatwal S, Behura A, Ghosh A, Kidwai S, Mishra A, et al. Calcimycin mediates mycobacterial killing by inducing intracellular calcium-regulated autophagy in a P2RX7 dependent manner. Biochim Biophys Acta - Gen Subj 2017;1861:3190-200.
33. Settembre C, Fraldi A, Rubinsztein DC, Ballabio A. Lysosomal storage diseases as disorders of autophagy. Autophagy 2008;4:113-4.
34. Cai Y, Arikkath J, Yang L, Guo ML, Periyasamy P, et al. Interplay of endoplasmic reticulum stress and autophagy in neurodegenerative disorders. Autophagy 2016;12:225-44.
35. Singh SS, Vats S, Chia AYQ, Tan TZ, Deng S, et al. Dual role of autophagy in hallmarks of cancer. Oncogene 2018;37:1142-58.
36. Comel A, Sorrentino G, Capaci V, Del Sal G. The cytoplasmic side of p53’s oncosuppressive activities. FEBS Lett 2014;588:2600-9.
37. Choi AM, Ryter SW, Levine B. Autophagy in human health and disease. N Engl J Med 2013;368:651-62.
38. Takahashi Y, Coppola D, Matsushita N, Cualing HD, Sun M, et al. Bif-1 interacts with Beclin 1 through UVRAG and regulates autophagy and tumorigenesis. Nat Cell Biol 2007;9:1142-51.
39. Coppola D, Khalil F, Eschrich SA, Boulware D, Yeatman T, et al. Down-regulation of bax-interacting factor-1 in colorectal adenocarcinoma. Cancer 2008;113:2665-70.
40. He S, Zhao Z, Yang Y, O’Connell D, Zhang X, et al. Truncating mutation in the autophagy gene UVRAG confers oncogenic properties and chemosensitivity in colorectal cancers. Nat Commun 2015;6:7839.
41. Perera RM, Stoykova S, Nicolay BN, Ross KN, Fitamant J, et al. Transcriptional control of autophagy-lysosome function drives pancreatic cancer metabolism. Nature 2015;524:361-5.
43. Thorburn A. Autophagy and its effects: making sense of double-edged swords. PLoS Biol 2014;12:e1001967.
45. Koustas E, Papavassiliou AG, Karamouzis MV. The role of autophagy in the treatment of BRAF mutant colorectal carcinomas differs based on microsatellite instability status. PLoS One 2018;13:e0207227.
46. Boutouja F, Brinkmeier R, Mastalski T, Magraoui F, Platta HW. Regulation of the tumor-suppressor beclin 1 by distinct ubiquitination cascades. Int J Mol Sci 2017;18:E2541.
47. Miracco C, Cosci E, Oliveri G, Luzi P, Pacenti L, et al. Protein and mRNA expression of autophagy gene beclin 1 in human brain tumours. Int J Oncol 2007;30:429-36.
48. Yang ZJ, Chee CE, Huang S, Sinicrope FA. The role of autophagy in cancer: therapeutic implications. Mol Cancer Ther 2011;10:1533-41.
49. Kang MR, Kim MS, Oh JE, Kim YR, Song SY, et al. Frameshift mutations of autophagy-related genes ATG2B, ATG5, ATG9B and ATGI2 in gastric and colorectal cancers with microsatellite instability. J Pathol 2009;217:702-6.
50. Lipinski MM, Hoffman G, Ng A, Zhou W, Py BF, et al. A genome-wide siRNA screen reveals multiple mTORC1 independent signaling pathways regulating autophagy under normal nutritional conditions. Dev Cell 2010;18:1041-52.
51. Tang F, Hu P, Yang Z, Xue C, Gong J, et al. SBI0206965, a novel inhibitor of Ulk1, suppresses non-small cell lung cancer cell growth by modulating both autophagy and apoptosis pathways. Oncol Rep 2017;37:3449-58.
52. Bellot G, Garcia-Medina R, Gounon P, Chiche J, Roux D, et al. Hypoxia-induced autophagy is mediated through hypoxia-inducible factor induction of BNIP3 and BNIP3L via their BH3 domains. Mol Cell Biol 2009;29:2570-81.
53. Karnoub AE, Weinberg RA. Ras oncogenes: split personalities. Nat Rev Mol Cell Biol 2008;9:517-31.
54. Ahn JH, Lee M. Autophagy-dependent survival of mutant B-Raf melanoma cells selected for resistance to apoptosis induced by inhibitors against oncogenic B-Raf. Biomol Ther 2013;21:114-20.
56. He S, Li Q, Jiang X, Lu X, Feng F, et al. Design of small molecule autophagy modulators: a promising druggable strategy. J Med Chem 2018;61:4656-87.
57. Kwitkowski VE, Prowell TM, Ibrahim A, Farrell AT, Justice R, et al. FDA approval summary: temsirolimus as treatment for advanced renal cell carcinoma. Oncologist 2010;15:428-35.
58. Koschmann C, Farooqui Z, Kasaian K, Cao X, Zamler D, et al. Multi-focal sequencing of a diffuse intrinsic pontine glioma establishes PTEN loss as an early event. NPJ Precis Oncol 2017;1:32.
59. Klionsky DJ, Eskelinen EL, Deretic V. Autophagosomes, phagosomes, autolysosomes, phagolysosomes, autophagolysosomes… wait, I’m confused. Autophagy 2014;10:549-51.
60. Wymann MP, Bulgarelli-Leva G, Zvelebil MJ, Pirola L, Vanhaesebroeck B. Wortmannin inactivates phosphoinositide 3-Kinase by covalent modification of Lys-802, a residue involved in the phosphate transfer reaction. Mol Cell Biol 1996;16:1722-33.
61. McNamara CR, Degterev A. Small-molecule inhibitors of the PI3K signaling network. Future Med Chem 2011;3:549-65.
62. Gharbi SI, Zvelebil MJ, Shuttleworth SJ, Hancox T, Saghir N, et al. Exploring the specificity of the PI3K family inhibitor LY294002. Biochem J 2007;404:15-21.
63. Harding JJ, Bauer TM, Tan DSW, Bedard PL, Rodon J, et al. Characterization and phase I study of CLR457, an orally bioavailable pan-class I PI3-kinase inhibitor. Invest New Drugs 2019;37:271-81.
64. Knight SD, Adams ND, Burgess JL, Chaudhari AM, Darcy MG, et al. Discovery of GSK2126458, a highly potent inhibitor of PI3K and the mammalian target of rapamycin. ACS Med Chem Lett 2010;1:39-43.
65. Basu D, Salgado CM, Bauer B, Khakoo Y, Patel JR, et al. The dual PI3K/mToR inhibitor omipalisib/GSK2126458 inhibits clonogenic growth in oncogenically-transformed cells from neurocutaneous melanocytosis. Cancer Genomics and Proteomics 2018;15:239-48.
66. Liu Q, Xu C, Kirubakaran S, Zhang X, Hur W, et al. Characterization of torin2, an ATP-competitive inhibitor of mTOR, ATM, and ATR. Cancer Res 2013;73:2574-86.
67. Nelson EE, Guyer AE. The development of the ventral prefrontal cortex and social flexibility. Dev Cogn Neurosci 2011;1:233-45.
68. Chresta CM, Davies BR, Hickson I, Harding T, Cosulich S, et al. AZD8055 is a potent, selective, and orally bioavailable ATP-competitive mammalian target of rapamycin kinase inhibitor with in vitro and in vivo antitumor activity. Cancer Res 2010;70:288-98.
69. Wang L, Zhu YR, Wang S, Zhao S. Autophagy inhibition sensitizes WYE-354-induced anti-colon cancer activity in vitro and in vivo. Tumor Biol 2016;37:11743-52.
70. Park S, Chapuis N, Bardet V, Tamburini J, Gallay N, et al. PI-103, a dual inhibitor of class IA phosphatidylinositide 3-kinase and mTOR, has antileukemic activity in AML. Leukemia 2008;22:1698-706.
71. Mallon R, Feldberg LR, Lucas J, Chaudhary I, Dehnhardt C, et al. Antitumor efficacy of PKI-587, a highly potent dual PI3K/mTOR kinase inhibitor. Clin Cancer Res 2011;17:3193-203.
72. Powles T, Lackner MR, Oudard S, Escudier B, Ralph C, et al. Randomized open-label phase II trial of apitolisib (GDC-0980), a novel inhibitor of the PI3K/mammalian target of rapamycin pathway, versus everolimus in patients with metastatic renal cell carcinoma. J Clin Oncol 2016;34:1660-8.
73. Simioni C, Cani A, Martelli AM, Zauli G, Alameen AA, et al. The novel dual PI3K/mTOR inhibitor NVP-BGT226 displays cytotoxic activity in both normoxic and hypoxic hepatocarcinoma cells. Oncotarget 2015;6:17147-60.
74. Li H, Jin X, Zhang Z, Xing Y, Kong X. Inhibition of autophagy enhances apoptosis induced by the PI3K/AKT/mTor inhibitor NVP-BEZ235 in renal cell carcinoma cells. Cell Biochem Funct 2013;31:427-33.
75. Lazarus MB, Novotny CJ, Shokat KM. Structure of the human autophagy initiating kinase ULK1 in complex with potent inhibitors. ACS Chem Biol 2015;10:257-61.
76. Martin KR, Celano SL, Solitro AR, Gunaydin H, Scott M, et al. A potent and selective ULK1 inhibitor suppresses autophagy and sensitizes cancer cells to nutrient stress. iScience 2018;8:74-84.
77. Limpert AS, Lambert LJ, Bakas NA, Bata N, Brun SN, et al. Autophagy in cancer: regulation by small molecules. Trends Pharmacol Sci 2018;39:53-63.
78. Lazarus MB, Shokat KM. Discovery and structure of a new inhibitor scaffold of the autophagy initiating kinase ULK1. Bioorganic Med Chem 2015;23:5483-8.
79. Matsunaga K, Morita E, Saitoh T, Akira S, Ktistakis NT, et al. Autophagy requires endoplasmic reticulum targeting of the PI3-kinase complex via Atg14L. J Cell Biol 2010;190:511-21.
80. Honda A, Harrington E, Cornella-Taracido I, Furet P, Knapp MS, et al. Potent, selective, and orally bioavailable inhibitors of VPS34 provide chemical tools to modulate autophagy in vivo. ACS Med Chem Lett 2016;7:72-6.
81. Dowdle WE, Nyfeler B, Nagel J, Elling RA, Liu S, et al. Selective VPS34 inhibitor blocks autophagy and uncovers a role for NCOA4 in ferritin degradation and iron homeostasis in vivo. Nat Cell Biol 2014;16:1069-79.
82. Pasquier B, El-Ahmad Y, Filoche-Rommé B, Dureuil C, Fassy F, et al. Discovery of (2 S)-8-[(3 R)-3-methylmorpholin-4-yl]-1-(3-methyl-2-oxobutyl)-2-(trifluoromethyl)-3,4-dihydro-2 H -pyrimido[1,2- a ]pyrimidin-6-one: a novel potent and selective inhibitor of Vps34 for the treatment of solid tumors. J Med Chem 2015;58:376-400.
83. Manic G, Obrist F, Kroemer G, Vitale I, Galluzzi L. Chloroquine and hydroxychloroquine for cancer therapy. Mol Cell Oncol 2014;1:e29911.
84. Shi TT, Yu XX, Yan LJ, Xiao HT. Research progress of hydroxychloroquine and autophagy inhibitors on cancer. Cancer Chemother Pharmacol 2017;79:287-94.
85. Mauthe M, Orhon I, Rocchi C, Zhou X, Luhr M, et al. Chloroquine inhibits autophagic flux by decreasing autophagosome-lysosome fusion. Autophagy 2018;14:1435-55.
86. Chude CI, Amaravadi RK. Targeting autophagy in cancer: update on clinical trials and novel inhibitors. Int J Mol Sci 2017;18:E1279.
87. Xu R, Ji Z, Xu C, Zhu J. The clinical value of using chloroquine or hydroxychloroquine as autophagy inhibitors in the treatment of cancers: a systematic review and meta-analysis. Medicine (Baltimore) 2018;97:e12912.
89. McAfee Q, Zhang Z, Samanta A, Levi SM, Ma XH, et al. Autophagy inhibitor Lys05 has single-agent antitumor activity and reproduces the phenotype of a genetic autophagy deficiency. Proc Natl Acad Sci 2012;109:8253-8.
90. Kasi PD, Tamilselvam R, Skalicka-Woźniak K, Nabavi SF, Daglia M, et al. Molecular targets of curcumin for cancer therapy: an updated review. Tumor Biol 2016;37:13017-28.
91. Zhu Y, Bu S. Curcumin induces autophagy, apoptosis, and cell cycle arrest in human pancreatic cancer cells. Evidence-Based Complement Altern Med 2017;2017:5787218.
92. Xiao K, Jiang J, Guan C, Dong C, Wang G, et al. Curcumin induces autophagy via activating the AMPK signaling pathway in lung adenocarcinoma cells. J Pharmacol Sci 2013;123:102-9.
93. Kobori M, Takahashi Y, Sakurai M, Akimoto Y, Tsushida T, et al. Quercetin suppresses immune cell accumulation and improves mitochondrial gene expression in adipose tissue of diet-induced obese mice. Mol Nutr Food Res 2016;60:300-12.
94. Lou M, Zhang LN, Ji PG, Feng FQ, Liu JH, et al. Quercetin nanoparticles induced autophagy and apoptosis through AKT/ERK/Caspase-3 signaling pathway in human neuroglioma cells: In vitro and in vivo. Biomed Pharmacother 2016;84:1-9.
95. Liu Y, Gong W, Yang ZY, Zhou XS, Gong C, et al. Quercetin induces protective autophagy and apoptosis through ER stress via the p-STAT3/Bcl-2 axis in ovarian cancer. Apoptosis 2017;22:544-57.
96. Hwang JJ, Kuruvilla J, Mendelson D, Pishvaian MJ, Deeken JF, et al. Phase I dose finding studies of obatoclax (GX15-070), a small molecule Pan-BCL-2 family antagonist, in patients with advanced solid tumors or lymphoma. Clin Cancer Res 2010;16:4038-45.
97. Basit F, Cristofanon S, Fulda S. Obatoclax (GX15-070) triggers necroptosis by promoting the assembly of the necrosome on autophagosomal membranes. Cell Death Differ 2013;20:1161-73.
98. Yang Y, Chen S, Zhang Y, Lin X, Song Y, et al. Induction of autophagy by spermidine is neuroprotective via inhibition of caspase 3-mediated Beclin 1 cleavage. Cell Death Dis 2017;8:e2738.
99. Sacitharan PK, Lwin S, Gharios GB, Edwards JR. Spermidine restores dysregulated autophagy and polyamine synthesis in aged and osteoarthritic chondrocytes via EP300. Exp Mol Med 2018;50:123.
100. Welsh PA, Sass-Kuhn S, Prakashagowda C, McCloskey D, Feith D. Spermine synthase overexpression in vivo does not increase susceptibility to DMBA/TPA skin carcinogenesis or Min-Apc intestinal tumorigenesis. Cancer Biol Ther 2012;13:358-68.
101. Matsui TA, Murata H, Sakabe T, Sowa Y, Horie N, et al. Sulforaphane induces cell cycle arrest and apoptosis in murine osteosarcoma cells in vitro and inhibits tumor growth in vivo. Oncol Rep 2007;18:1263-8.
102. Xiao D, Powolny AA, Antosiewicz J, Hahm ER, Bommareddy A, et al. Cellular responses to cancer chemopreventive agent D,L-sulforaphane in human prostate cancer cells are initiated by mitochondrial reactive oxygen species. Pharm Res 2009;26:1729-38.
103. Liu H, Smith AJ, Ball SS, Bao Y, Bowater RP, et al. Sulforaphane promotes ER stress, autophagy, and cell death: implications for cataract surgery. J Mol Med 2017;95:553-64.
104. Wiczk A, Hofman D, Konopa G, Herman-Antosiewicz A. Sulforaphane, a cruciferous vegetable-derived isothiocyanate, inhibits protein synthesis in human prostate cancer cells. Biochim Biophys Acta - Mol Cell Res 2012;1823:1295-305.
105. Cohen MH, Johnson JR, Pazdur R. Food and drug administration drug approval summary: temozolomide plus radiation therapy for the treatment of newly diagnosed glioblastoma multiforme. Clin Cancer Res 2005;11:6767-71.
106. Würstle S, Schneider F, Ringel F, Gempt J, Lämmer F, et al. Temozolomide induces autophagy in primary and established glioblastoma cells in an EGFR independent manner. Oncol Lett 2017;14:322-8.
107. Dai C, Zhang B, Liu X, Ma S, Yang Y, et al. Inhibition of PI3K/AKT/mTOR pathway enhances temozolomide-induced cytotoxicity in pituitary adenoma cell lines in vitro and xenografted pituitary adenoma in female nude mice. Endocrinology 2013;154:1247-59.
108. Chen B, Xiao F, Li B, Xie B, Zhou J, et al. The role of epithelial-mesenchymal transition and IGF-1R expression in prediction of gefitinib activity as the second-line treatment for advanced nonsmall-cell lung cancer. Cancer Invest 2013;31:454-60.
109. Zhao ZQ, Yu ZY, Li J, Ouyang XN. Gefitinib induces lung cancer cell autophagy and apoptosis via blockade of the PI3K/AKT/mTOR pathway. Oncol Lett 2016;12:63-8.
110. Sugita S, Ito K, Yamashiro Y, Moriya S, Che XF, et al. EGFR-independent autophagy induction with gefitinib and enhancement of its cytotoxic effect by targeting autophagy with clarithromycin in non-small cell lung cancer cells. Biochem Biophys Res Commun 2015;461:28-34.
111. Dowling RJO, Goodwin PJ, Stambolic V. Understanding the benefit of metformin use in cancer treatment. BMC Med 2011;9:33.
112. Feng Y, Ke C, Tang Q, Dong H, Zheng X, et al. Metformin promotes autophagy and apoptosis in esophageal squamous cell carcinoma by downregulating Stat3 signaling. Cell Death Dis 2014;5:1-12.
113. Wang Y, Xu W, Yan Z, Zhao W, Mi J, et al. Metformin induces autophagy and G0/G1 phase cell cycle arrest in myeloma by targeting the AMPK/mTORC1 and mTORC2 pathways. J Exp Clin Cancer Res 2018;37:1-12.
114. Field-smith A, Morgan GJ, Davies FE. Bortezomib (velcadetrade mark) in the treatment of multiple myeloma. Ther Clin Risk Manag 2006;2:271-9.
115. Kao C, Chao A, Tsai CL, Chuang WC, Huang WP, et al. Bortezomib enhances cancer cell death by blocking the autophagic flux through stimulating ERK phosphorylation. Cell Death Dis 2014;5:e1510.
116. Yang X, Srivastava R, Howell SH, Bassham DC. Activation of autophagy by unfolded proteins during endoplasmic reticulum stress. Plant J 2016;85:83-95.
117. Akbarzadeh L, Moini Zanjani T, Sabetkasaei M. Comparison of anticancer effects of carbamazepine and valproic acid. Iran Red Crescent Med J 2016;18:e37230.
118. Kania E, Pająk B, O’Prey J, Sierra Gonzalez P, Litwiniuk A, et al. Verapamil treatment induces cytoprotective autophagy by modulating cellular metabolism. FEBS J 2017;284:1370-87.
119. Shimizu T, Ono T, Yoshida T, Cho F, Goto N. Morphometrical study of physical growth in infant cynomolgus monkeys using multivariate analysis. Jikken Dobutsu 1988;37:145-51.
120. Tan Q, Joshua AM, Wang M, Bristow RG, Wouters BG, et al. Up-regulation of autophagy is a mechanism of resistance to chemotherapy and can be inhibited by pantoprazole to increase drug sensitivity. Cancer Chemother Pharmacol 2017;79:959-69.
121. Tan Q, Joshua AM, Saggar JK, Yu M, Wang M, et al. Effect of pantoprazole to enhance activity of docetaxel against human tumour xenografts by inhibiting autophagy. Br J Cancer 2015;112:832-40.
122. Cao Y, Chen M, Tang D, Yan H, Ding X, et al. The proton pump inhibitor pantoprazole disrupts protein degradation systems and sensitizes cancer cells to death under various stresses article. Cell Death Dis 2018;9:604.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Kondapuram SK, Sarvagalla S, Coumar MS. Targeting autophagy with small molecules for cancer therapy. J Cancer Metastasis Treat 2019;5:32. http://dx.doi.org/10.20517/2394-4722.2018.105
AMA Style
Kondapuram SK, Sarvagalla S, Coumar MS. Targeting autophagy with small molecules for cancer therapy. Journal of Cancer Metastasis and Treatment. 2019; 5: 32. http://dx.doi.org/10.20517/2394-4722.2018.105
Chicago/Turabian Style
Kondapuram, Sree Karani, Sailu Sarvagalla, Mohane Selvaraj Coumar. 2019. "Targeting autophagy with small molecules for cancer therapy" Journal of Cancer Metastasis and Treatment. 5: 32. http://dx.doi.org/10.20517/2394-4722.2018.105
ACS Style
Kondapuram, SK.; Sarvagalla S.; Coumar MS. Targeting autophagy with small molecules for cancer therapy. J. Cancer. Metastasis. Treat. 2019, 5, 32. http://dx.doi.org/10.20517/2394-4722.2018.105
About This Article
Special Issue
Copyright

Data & Comments
Data


 Cite This Article 41 clicks
Cite This Article 41 clicks

 Like This Article 19
likes
Like This Article 19
likes






Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.