
ESR1 alterations and metastasis in estrogen receptor positive breast cancer

Abstract
Endocrine therapy is essential for the treatment of patients with estrogen receptor positive (ER+) breast cancer, however, resistance and the development of metastatic disease is common. Understanding how ER+ breast cancer metastasizes is critical since the major cause of death in breast cancer is metastasis to distant organs. Results from many studies suggest dysregulation of the estrogen receptor alpha gene (ESR1) contributes to therapeutic resistance and metastatic biology. This review covers both pre-clinical and clinical evidence on the spectrum of ESR1 alterations including amplification, point mutations, and genomic rearrangement events driving treatment resistance and metastatic potential of ER+ breast cancer. Importantly, we describe how these ESR1 alterations may provide therapeutic opportunities to improve outcomes in patients with lethal, metastatic breast cancer.
Keywords
Introduction
Breast cancer is one of the leading cancer-related causes of death worldwide with more than one million new cases and more than 450,000 deaths per year according to the World Health Organization. About 70% of diagnosed cases express estrogen receptor alpha (ER)[1], where ER signaling is the defining and driving event contributing to tumor growth and disease progression in these ER+ breast tumors.
ER is a transcription factor consisting of various functional domains encoded by ESR1 located on chromosome 6 [Figure 1A]. ESR1 transcripts are generated by 2 non-coding and 8 exons that specifies protein-coding domains. The N-terminal activation function 1 (AF1) domain functions in a hormone-independent manner and is post-translationally modified by phosphorylation events that increase transcriptional and pathogenic activity[2-5] The DNA-binding domain (DBD) contains two zinc finger motifs responsible for binding to estrogen response element (ERE) DNA sequences within the enhancers and promoters of ER target genes. The C-terminal domains include the ligand-binding domain (LBD) and ligand-dependent activation function 2 (AF2) domain required for dimerization and transactivation. The LBD is required not only for estrogenic ligands but is also the domain that controls responses to anti-estrogen antagonists. The hinge domain contains the nuclear localization sequence and connects the activity from the ligand-independent AF1 and ligand-dependent AF2 together to fully promote activation of ER[6].
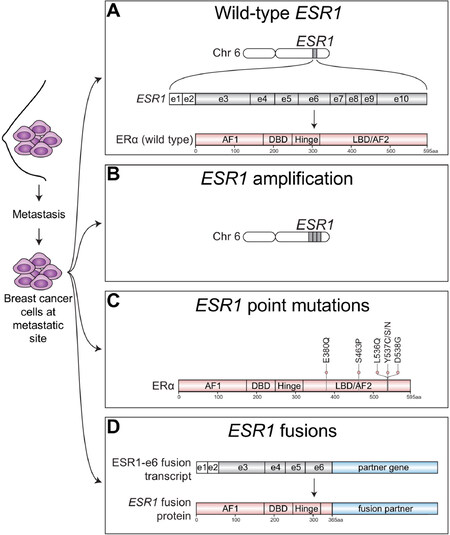
Figure 1. Spectrum of ESR1 alterations found in metastatic ER+ breast cancer. ER+ breast cancer cells that have spread beyond the breast to metastatic sties have been found to express wild-type ESR1 or harbor a variety of ESR1 alterations. A: Metastatic tumors can express wild-type estrogen receptor alpha protein (ERa), which is encoded by the estrogen receptor alpha gene (ESR1) located on chromosome (chr) 6. ESR1 transcripts are generated from 2 non-coding exons (e) depicted by white boxes and 8 coding exons depicted by gray shaded boxes; B: Metastatic ER+ tumors may also harbor amplification of ESR1 resulting in multiple copies of ESR1 and increased ER protein expression; C: Point mutations that cluster within the ligand-binding domain (LBD) of ESR1 that confer constitutive ligand-independent activation of ESR1 mutants have also been well-described in metastatic ER+ breast tumors, especially those which had been extensively pretreated with Als; D: Emerging studies have now identified structural rearrangements involving ESR1 that generate in-frame ESR1 fusion transcripts. In-frame fusion transcripts that retain the first 6 exons of ESR1 (ESR1-e6) produce stable ESR1 fusion proteins have been shown to be transcriptionally active and drive endocrine therapy resistance and metastasis in ER+ breast cancer. AF1: activation function 1 domain; DBD: DNA-binding domain; AF2: activation function 2 domain; aa: amino acid
Standard-of-care endocrine therapies that target ER itself include selective estrogen receptor modulators (SERMs), such as tamoxifen, and selective estrogen receptor degraders (SERDs), such as fulvestrant, that bind to the LBD. Aromatase inhibitors (AIs), such as letrozole, anastrozole, and exemestane, block the production of estrogens from androgens resulting in lower levels of circulating estrogen in the body. Despite the success of these agents in reducing relapse rates when given prophylactically after breast surgery and chemotherapy (adjuvant treatment), endocrine therapy resistance and the development of lethal metastatic disease is common and a major clinical problem. A major clinical feature of the disease is the long-term persistence of disseminated tumor cells despite endocrine therapy, with relapse risk continuing for decades after diagnosis[7]. The etiology of endocrine therapy resistance is complex and tremendous efforts have been made to uncover diverse mechanisms[8].
Downstream signaling events from aberrantly activated growth factor receptor tyrosine kinases (RTKs) such as epidermal growth factor receptor (EGFR) and HER2 (ERBB2) have been shown to phosphorylate and increase ER transcriptional activity in a hormone-independent manner[9]. ER+ tumors that exhibit ERBB2 amplification have reduced ER expression, reduced sensitivity to ER targeted therapies, and poor outcomes[10]. Nonetheless, co-targeting ER+/HER2+ breast cancer has been clinically successful. Experimental models have extended these ideas to other RTKs that are expressed by ER+ breast cancer. Interestingly, these investigations revealed a non-genomic or transcription-independent function of ER in association with EGFR[11] and insulin-like growth factor receptor (IGF1-R)[12] at the plasma membrane. However, clinical trials testing the use of EGFR inhibitors in endocrine treatment resistant ER+ breast cancer have produced modest or negative results[13] suggesting that further insight into underlying mechanisms for RTKs and ER interactions are required for successful translation of this aspect of ER function.
Since PIK3CA is the most frequently mutated gene in ER+ breast cancer[14], targeting components of the PI3K-AKT-mTOR pathway has also been described to treat endocrine refractory disease. Preclinical models demonstrated enhanced activation of the PI3K pathway in long-term estrogen deprived (LTED) ER+ breast cancer cells and a negative feedback system by which PI3K inhibition increases ER activity, potentially explaining the effectiveness of combinatorial mTOR and ER inhibition[15]. The use of an mTOR inhibitor, everolimus, in combination with endocrine therapy, significantly improves progression-free survival (PFS) for patients failing previous endocrine therapies[16], although side effects are severe and stratification of patients for this treatment is essential. Treatment with a pan-PI3K inhibitor, buparlisib, in combination with fulvestrant increased PFS with compared to fulvestrant alone in patients with ER+ locally advanced or metastatic breast cancer (BELLE-3 clinical trial)[17]. Greater benefit was observed in patients treated with buparlisib harboring PIK3CA mutations. However, significant toxicities in buparlisib treated patients have stopped further clinical trials of the drug in this setting. In contrast to pan-PI3K inhibitors, alpelisib, an agent that specifically targets the alpha isoform of PIK3CA, has been shown to overcome the toxicities associated with pan-PI3K treatment. Apelisib greatly improved PFS in patients when given in combination with fulvestrant to patients with endocrine-refractory, advanced ER+ breast cancer harboring PIK3CA mutations[18]. There was no significant benefit to PFS in patients with non-PIK3CA mutant tumors suggesting that PIK3CA status is a potential biomarker to predict response to PI3K inhibition. Results from studies also further suggest that targeting specific mutant isoforms of PI3K reduces toxicities leading to increased tolerability and therefore can be given for a longer duration compared to other pan-PI3K agents such as buparlisib.
Dysregulation of cell cycle components is common in ER+ breast cancer, particularly the Cyclin D-CDK4/6-Rb axis in the luminal B subtype[19]. This includes amplification of Cyclin D1 (CCND1), gene copy gain of CDK4 and loss of negative regulators such as p16 and p18 (CDKN2A and CDKN2C)[19]. Together with downstream activity from tyrosine kinase growth factor signaling described earlier, these events promote phosphorylation of Rb and resistance to endocrine therapy[20]. CDK4/6 inhibitors such as palbociclib and ribociclib, are now FDA approved for use in combination with endocrine therapy to treat advanced stage ER+ disease. Other studies are now examining the use of such inhibitors to treat early stage ER+ disease in both neoadjuvant and adjuvant settings (ClinicalTrials.gov identifiers for PALLET NCT02296801 and PALLAS NCT02513394, respectively). Some trials have already reported promising results in the neoadjuvant setting[21].
In addition to metastatic breast tumors expressing wild-type ER [Figure 1A], alterations in ESR1 itself, such as ESR1 amplifications have been identified in metastatic ER+ disease[22][Figure 1B]. Other ESR1 alterations found in endocrine therapy resistant breast tumors include point mutations in the ligand-binding domain (LBD)[23][Figure 1C] that confer constitutive hormone-independent activation of ER and are now a well-described mutational mechanism identified in up to 40% of metastatic breast cancer cases[24]. These are especially enriched in tumors pretreated with aromatase inhibitors[25]. Emerging evidence now suggests that chromosomal rearrangement events involving ESR1 are yet another ESR1 mutational mechanism driving endocrine therapy resistance and metastatic disease progression [Figure 1D]. Hereon, we focus on the spectrum of ESR1 aberrations underlying treatment resistance and metastasis in ER+ breast cancer.
ESR1 amplification
The copy number increase of a confined area of a chromosome is defined as gene amplification/gain [Figure 1B] which may result in protein overexpression of the amplified gene therefore driving tumor biology. For example, ERBB2 amplification[26] and fibroblast growth factor receptor 1 gene (FGFR1) amplification[27] are drivers of therapeutic resistance and poor prognosis in ER+ breast cancer. The discovery of ESR1 gene amplifications in 1990[28] sparked intense interest in investigating the role of this mutational event to be a potential driver of endocrine therapy resistance and recurrent disease in ER+ breast tumors.
Incidence of ESR1 amplifications in ER+ breast cancer
ESR1 amplification is found in up to 30% of ER+ breast tumors[22,28-37] depending on the detection method and scoring systems[38]. A study by Holst et al.[29] that analyzed over 2,000 breast tumors, showed that 20.6% of tumors harbored ESR1 amplifications and 14% showed ESR1 copy number gain by using fluorescence in situ hybridization (FISH) method and validated by quantitative PCR[29]. Nearly all ESR1 amplified tumors in these samples also expressed high levels of ER protein by immunohistochemistry. Additional analysis from precancerous ductal and lobular carcinoma in situ (DCIS and LCIS) breast tumors showed over one-third of these samples also harbored ESR1 amplifications suggesting that ESR1 amplifications present in early-stage breast cancer may drive disease progression. Two other independent studies that also used FISH, both showed that ESR1 amplification frequency is between 20%-22%[34-35], consistent with Holst et al.[29]. In contrast, other studies by Brown et al.[30], Horlings et al.[31], Reis-Filho et al.[32], and Vincent-Salomon et al.[33], have shown a much lower frequency of ESR1 amplifications, in which ESR1 amplification or gain was less than 5% by using array comparative genomic hybridization (aCGH) and validated by FISH by the majority of these studies. Another study which used a multiplex ligation-dependent probe amplification (MLPA) approach to analyze 104 invasive breast cancers identified 16% of samples harbored ESR1 amplifications consisting of low level gains[36]. A variation in the frequency of ESR1 amplification found among metastatic breast samples has also been reported. A seminal study from Jeselsohn et al.[37] examined ESR1 amplification in the metastatic setting using next generation sequencing approaches. They reported the frequency of ESR1 amplification in ER+ tumors at less than 2% in both the primary and metastatic setting[37]. Using NanoString sequencing approaches, a recent study reported that 13% of ER+ metastatic breast tumors harbored ESR1 amplifications. Interestingly, the authors found an enrichment of ESR1 amplifications in bone metastatic samples, suggesting that ESR1 amplification may underlie organ-specific metastatic behavior of ER+ breast cancers[39].
Correlation between ESR1 amplification, protein expression, and clinical significance
Many studies show positive correlation between ESR1 amplification and ER protein expression suggesting that amplification may lead to production of elevated levels of oncogenic ER protein[28,29,34,35]. Interestingly, studies have shown that ESR1 amplification in a subset of ER+ breast cancers were associated with tamoxifen resistance and poor prognosis[40,41]. In contrast, contradicting studies have identified ESR1 amplification as an indicator of longer disease-free survival and increased sensitivity to tamoxifen treatment[35,42]. These conflicting results suggest that more dedicated studies will be required to fully understand the clinical implications of ESR1 amplifications. Results from other studies have identified ESR1 amplification in benign and early-stage breast cancer and is associated with endocrine therapy resistance. Discovery of ESR1 amplifications in benign papillomas and early-stage breast cancer such as ductal hyperplasias suggests that ESR1 amplifications may play a role in the tumor initiation process since high expression of ER in benign breast cells is associated with higher breast cancer risk[29,43,44], but these findings still require further validation. The insignificant difference of ESR1 amplification between invasive and non-invasive breast cancers suggests that ESR1 copy number alteration might not be used as a key predictive marker for invasion and metastasis, however its enrichment in recurrent disease, especially after endocrine therapy treatment, suggests that it likely plays a role in intrinsic and/or acquired resistance to endocrine therapy and metastatic disease progression[45-48].
Although the use of endocrine agents that block estrogen production (AIs) or block ER function (SERM/SERD) are front-line therapies to treat metastatic ER+ breast cancer, the use of high-dose estrogens has also been reported to be effective. This approach was first described over 70 years ago before the discovery of anti-estrogens to treat advanced breast cancer[49]. More recently, a study reported a breast cancer patient harboring an ESR1 amplification showed tumor regression in a liver metastasis after receiving estradiol treatment as a primary therapy[50]. Another study using a patient-derived xenograft (PDX) model harboring an ESR1 amplification derived from a patient with endocrine-refractory disease demonstrated that tumor growth was suppressed with estradiol treatment[47]. These results were corroborated in an independent study using a LTED ER+ MCF7 breast cancer cell model system in which such cells acquire ESR1 amplification during long term estrogen deprivation showed an apoptotic response upon estradiol treatment[48]. Collectively, these studies suggest a role for ESR1 amplification in driving endocrine therapy resistance and metastasis and that treating ESR1 amplified tumors with intermediate doses of estradiol (6 mg daily) is an option for some patients.
The presence of ESR1 amplification in some breast cancers is undeniable. However, a clear link between the presence of ESR1 amplifications in breast tumors and endocrine therapy resistance and metastasis remains to be shown. Deeper multi-dimensional characterization of relapsed and/or metastatic breast tumors at the RNA, DNA, and protein levels may aid to better understand its prognostic value. Therefore, more studies will be required to better understand the functional and therapeutic significance of ESR1 amplifications in driving endocrine therapy resistance and metastasis.
CYP19A1 amplification
While ESR1 amplification has been an intense area of investigation underlying endocrine therapy resistance as described above, a study focusing on genomic aberrations of the drug target of AIs, aromatase (CYP19A1), has deepened our understanding of endocrine-refractory ER+ breast tumors. Copy number alterations in the gene encoding aromatase, CYP19A1, also has been shown to promote resistance to AIs in patients with metastatic ER+ breast cancer. While CYP19A1 amplification is very rare in primary untreated ER+ breast cancers, Magnani et al.[51] found that 21.5% of AI-refractory relapsed tumors to harbor CYP19A1 amplification, suggesting that CYP19A1 amplification is an acquired endocrine therapy resistance mechanism[51]. This study also revealed that both CYP19A1 and ESR1 were frequently co-amplified in AI treated patients, further suggesting that these two amplification events may function collaboratively. To better understand the role of CYP19A1 amplification and endocrine therapy resistance, a LTED MCF7 ER+ breast cancer cell model was used which was found to acquire copy number alterations around the CYP19A1 locus compared to parental cells MCF7 cells[51]. Elevated levels of both CYP19A1 mRNA and CYP19A1 protein were observed in CYP19A1 amplified LTED cells compared to parental cells. The functional consequences of CYP19A1 amplification in the LTED cells were increased aromatase activity, enhanced ER recruitment to regulatory regions on DNA of target genes and their transcriptional activation leading to reduced sensitivity to AI treatment[51]. These results suggest that CYP19A1 amplification, in addition to ESR1 amplification, could potentially represent biomarkers of endocrine therapy resistance. More studies are needed to validate these findings in more patient datasets. Furthermore, deeper studies focusing on how these amplification events contribute to the metastatic behavior of endocrine-refractory ER+ breast tumors are needed. These results highlight the possibility that response to standard-of-care endocrine therapies are not only as a consequence of ESR1 amplification but may also be critically dependent on the status of the target genes of endocrine therapies themselves.
ESR1 point mutations
When patients with ER+ breast cancer relapse, up to 15% have lost ER expression and therefore targeting ER in this population is likely to be ineffective, although false negative ER results are a concern if the ER analysis was conducted on bone biopsies exposed to acid formalin, or if the analysis was conducted on samples prone to degradation such as cells detected in pleural fluid. The remaining 85% of patients may initially benefit from first-line endocrine therapy, but metastatic disease progression due to acquired resistance is inevitable. One well-established mechanism explaining this relentless pattern of acquired endocrine therapy resistance is the acquisition of activating point mutations that cluster within the ligand-binding domain (LBD) of ESR1[Figure 1C]. Substitution of tyrosine at position 537 to serine (Y537S) in the LBD of ESR1 was first reported to confer constitutive, ligand-independent activity of ER in experimental breast cancer models[52]. However, such mutations were not known to occur in human tumors until Fuqua et al.[23] reported that estrogen-independent activation could be driven by another Y537 substitution, Y537N, that was identified in a metastatic sample from a breast cancer patient who experienced disease progression on hormonal therapy. This study also showed that Y537N was able to drive resistance to tamoxifen in experimental models.
Frequent ESR1 point mutations in endocrine-refractory, metastatic ER+ breast cancer
Advances in sequencing technologies have allowed more sensitive detection and thus insights into the landscape of ESR1 LBD point mutations in both primary and metastatic ER+ breast tumors. Three ESR1 mutations, Y537S, Y537N, and D538G were identified by next-generation sequencing in 14 out of 80 patient samples with endocrine-refractory, metastatic ER+ breast cancer[53]. Notably, all breast tumors from patients that were found to harbor ESR1 LBD point mutations were treated with AIs. Interestingly, these alterations were not detected in matched primary samples and were also not detected in separate large sets of treatment naïve patients. Analysis of an independent ER negative (ER-) cohort also failed to detect any ESR1 point mutations in the LBD[53]. Although ESR1 mutations were found in 3% of primary samples in this population, alterations in Y537 and D538 residues of ESR1 were enriched in patients treated extensively with AIs[53]. These results suggest that these ESR1 LBD mutations are acquired, or detected, in patients after treatment with endocrine therapy.
In addition to Y537 alterations, frequent amino acid substitution of aspartate 538 to glutamate (D538G) was identified in liver metastases from 5 out of 13 metastatic ER+ breast samples[54]. Another study which enrolled 11 metastatic ER+ breast cancer patients with exposure to serial endocrine therapies, identified that over half of these patient’s metastatic samples harbored ESR1 mutations localized in the LBD, that included Y537S, Y537C, Y537N, D538G, and L536Q mutations[55]. Further evidence for the recurrent presence of Y537 and D538 mutations in the LBD of ESR1 was shown in 9 out of 76 metastatic samples from patients with ER+ disease[37]. One patient from this study acquired a tyrosine substitution to cysteine mutation (Y537C) at the metastatic site, which was not detected prior to treatment[37]. Taken together, these studies indicate the most frequent ESR1 LBD point mutations are those affecting Y537 and D538 residues. Furthermore, the presence of ESR1 point mutations predominately appear in late-stage breast cancer patients that have been treated with multiple lines of endocrine therapies but rarely in treatment naïve cases. This strongly suggests a role for ESR1 point mutations in acquired endocrine resistance and metastasis.
Although formalin-fixed paraffin-embedded tumor specimens are widely used for next generation sequencing to capture ESR1 mutations used by studies as described above[37,53,54], collection of plasma circulating DNA to detect ESR1 mutations by droplet digital PCR (ddPCR) have now been implemented in several clinical trials[56-59]. Such “liquid biopsies” have shown that collecting circulating DNA samples maintains the genomic landscape of the primary tumor suggesting that less invasive detection methods may efficiently identify ESR1 point mutations once the disease has become resistant to treatment and/or has become metastatic. Interestingly, Y537 and D538 substitutions were identified in 7% of ER+ primary tumors using ddPCR, which may lead us to review the conclusion that ESR1 point mutations rarely exist in primary tumor, towards the idea that rare ESR1 mutant sub-clones exist in primary breast tumors that become selected for over time[60].
Experimental models of ESR1 point mutations
Several preclinical breast cancer models harboring ESR1 LBD point mutations have been generated, providing research platforms to characterize the functional, transcriptional, and pharmacological properties of these mutations. ER point mutant proteins have been overexpressed by transfecting[37,53,54] or transducing lentiviral vectors[55,61] encoding ESR1 mutant constructs into various ER+ breast cancer cell line models. The growth promoting properties of ESR1 mutant expressing cell line models have shown that ESR1 LBD mutants drive hormone-independent proliferation that is resistant to tamoxifen treatment[23,37,47,53,54]. Although fulvestrant efficiently inhibited the growth of point mutation bearing cells in a dose-dependent manner, growth was not reversed to levels of wild-type ESR1 expressing cells[37,47].
Since the expression of exogenous ESR1 variant transcripts encoded by expression vectors is often initiated from non-endogenous human promoters that drive very high expression of constructs, it is unlikely to mimic the expression levels in human breast tumors harboring ESR1 point mutations. To more accurately recapitulate tumor-related ESR1 mutational events, CRISPR/Cas9 approaches have been utilized to knock in ESR1 mutated sequences into ER+ breast cancer cells[62,63]. Both heterozygous and homozygous knock-in models have been shown to mediate resistance to endocrine therapies[62,63].
Transcriptional properties of ESR1 mutations in the LBD include their ability to drive constitutive hormone-independent transcriptional activation and enhance cell proliferation[23,37,47,53-55]. Human embryonic kidney 293T cells transfected with Y537C, Y537N, and D538G mutant constructs strongly activate an ERE-luciferase reporter in a ligand-independent manner compared to wild-type ER. Luciferase activity was unaffected by clinically relevant doses of tamoxifen and fulvestrant, however, high doses of these agents blocked ESR1 mutant driven ERE-luciferase reporter activity[37,53-55]. These ESR1 point mutations have also been shown to drive estrogen-independent activation of ER target genes in ER+ breast cancer cells[37,53,54]. The recruitment of ESR1-Y537S mutant to ER target genes and their expression driven by the mutant were further validated by ChIP-seq and RNA-seq[62].
ESR1 mutant-driven estrogen-independent tumor growth was also validated in both ER+ cell xenografts and patient-derived xenograft (PDX) models[47,53]. A PDX harboring ESR1-Y537S, WHIM20, has been generated from a patient with endocrine-refractory metastatic ER+ breast cancer that retains genomic features of the human counterpart[47]. This WHIM20 PDX model demonstrated estrogen-independent tumor growth[47].
Despite such in-depth studies of transcriptional and growth-promoting properties endowed by ESR1 LBD point mutations, the role of such mutations in driving cell invasion and tumor metastasis is underexplored. A scratch wound assay was performed on Y537S and D538G mutant expressing MCF7 cells to examine cell motility which showed enhanced cell migration under hormone-deprived conditions driven by these ESR1 mutants[54,61]. A recent study sheds light on ER mutant-driven metastatic biology, showing a remarkable enrichment of metastasis-associated gene sets in ESR1 mutant cells[64]. Consequently, Y537S and D538G mutant expressing MCF7 cells developed metastases after survival surgery to remove primary tumors in xenograft models. The Y537S mutant greatly potentiated both tumor growth and metastasis compared to D538G mutant[64].
Mechanisms and therapeutic vulnerabilities of breast cancers harboring ESR1 point mutations
Structural analysis has revealed that the formation of hydrogen bonds between S537 or G538 and D351 located within helix 12 of ESR1 LBD confers an agonist conformation to ESR1 mutant proteins[53]. In wild-type ER, the binding of ligand alters the position of helix 12 into an open pocket, favoring recruitment of transcriptional coactivators such as p160 family members that include SRC-3, and histone acetylases CBP and p300. In contrast, tamoxifen results in disposition of helix 12 that hinders coactivators binding and results in recruitment of corepressors such as N-CoR/SMRT[65]. The substitution of D538 to glycine mimics the active conformation of wild-type ER bound by estrogen[54].
To better understand the consequences of coactivator recruitment to mutant ER proteins, a proteomic profiling approach was used and revealed enhanced recruitment of transcriptional coactivators, histone H3 lysine 4 (H3K4) methyltransferase KMT2D/2C complex, as well as steroid receptor coactivators (SRCs), to ERE-bound ESR1-Y537S and ESR1-D538G mutants compared to ERE-bound wild-type ER[66]. Genetic inhibition of SRC-3 in HeLa cells expressing ESR1-Y537S and ESR1-D538G significantly suppressed activity of an ERE-luciferase reporter. Pharmacological inhibition using a pan-SRC inhibitor, SI-1, also suppressed transcriptional activation in ESR1 mutant expressing HeLa cell lines and blocked cell proliferation in ER+ breast cancer cells stably expressing ESR1-Y537S and ESR1-D538G. Using a PDX naturally harboring the ESR1-Y537S mutation (WHIM20), treatment with an improved pan-SRC inhibitor, SI-2, suppressed growth in vivo. Suppression of WHIM20 tumor growth was even greater when SI-2 was administered in combination with an oral SERD, AZD9496, compared to either single agent alone, suggesting that targeting coactivator recruitment in combination with endocrine therapy could be a promising therapeutic strategy for breast tumors harboring ESR1 LBD mutants such as Y537S and D538G[66]. Another study identified that the transcription factor TFIIH was also recruited by the ESR1-Y537S mutant[62]. Phosphorylation of Ser118 was found to be mediated by TFIIH kinase, cyclin-dependent kinase (CDK) 7 and subsequent ESR1-Y537S driven cell proliferation was suppressed by a CDK7 inhibitor, THZ1[62]. These results suggest that CDK7 may represent another target that is associated with ESR1 mutant proteins for therapeutic intervention.
Targeting non-genomic signaling pathways activated by ESR1 mutants has also been investigated. As discussed above, interactions between ER with RTKs such as EGFR, HER2, and IGF1-R can activate downstream kinases. This results in phosphorylation of multiple transcriptional factors, including ER, and coregulators leading to changes in gene expression in a hormone-independent manner[67]. A recent study demonstrated that IGF1 signaling was the most activated pathway in ESR1 mutant MCF7 cells[61]. IGF1 stimulation lead to increased phosphorylation of both IGF1-Rβ and insulin receptor substrate-1 (pIRS-1). Treatment with an IGF1-Rβ inhibitor (GSK1838705A) monotherapy was able to block Y537S-driven cell motility and combinatorial treatment with tamoxifen abrogated transcriptional activity and cell growth driven by Y537S, Y537N, and D538G mutants[61] . These results suggest that targeting non-genomic signaling pathways activated by ESR1 mutants may be an additional therapeutic strategy to block ESR1 mutant driven breast tumors.
Fulvestrant is used to treat metastatic ER+ breast cancer patients who have developed resistance to AI and tamoxifen. In preclinical models, transcriptional activity and cell proliferation of ESR1 LBD mutant cells are partially sensitive to fulvestrant, requiring higher doses of fulvestrant compared to controls[37,47,63]. Moreover, fulvestrant did not completely block transcriptional activity nor cell proliferation compared to control cells expressing wild-type ESR1. Of note, ESR1 mutants showed differential responses to fulvestrant. Y537S required the highest dose to completely block transcriptional activity and cell proliferation compared to other mutants, D538G, E380Q and S463P[63]. Using an MCF7 xenograft model, ESR1 mutants also showed differential responses to fulvestrant. Tumor growth of E380Q, S463P and D538G expressing tumors were significantly reduced while Y537S tumors showed resistance to treatment[63]. Given the inconvenience and poor bioavailability of intramuscular fulvestrant injections, second-generation SERDs, such AZD9496, that can be orally administrated have been tested and showed anti-proliferative ability in endocrine resistant experimental models cell xenograft models[63,68]. AZD9496 which has improved bioavailability compared to fulvestrant, was able to provide greater suppression of tumor growth in the Y537S MCF7 xenograft model and in a D538G PDX model compared to fulvestrant treatment[63]. A phase I clinical trial with AZD9496 in extensively pretreated advanced ER+ breast cancer patients has recently been completed with promising results, providing disease stabilization to the study cohort[69]. These results suggest that newer generation SERDs with improved bioavailability could be an attractive therapeutic option to treat endocrine-refractory breast tumors driven by ESR1 mutations.
Treatment of late-stage ER+ breast cancer patients with CDK4/6 inhibitors in combination with endocrine therapy has been tremendously successful. CDK4/6 inhibitors have also been tested in PDX breast cancer models harboring ESR1 point mutations. Wardell et al.[70] reported the suppressive effects of a CDK4/6 inhibitor, palbociclib, on endocrine-refractory PDX tumors as long as the downstream target retinoblastoma (Rb) protein was expressed. Used as monotherapy or in combination with a hybrid SERM/SERD, bazedoxifene, palbociclib suppressed tumor growth of a WHIM20 PDX tumor harboring an ESR1-Y537S mutant. In contrast, palbociclib was ineffective in inhibiting the growth of WHIM43, a PDX naturally bearing ESR1-D538G mutant due to the lack of Rb protein expression, suggesting that Rb is a determinant of CDK4/6 treatment response. CDK4/6 inhibitors also showed favorable therapeutic effects in treatment-resistant ER+ patients harboring ESR1 point mutations[59].
Currently, screening of ESR1 point mutations have not been used as biomarkers to predict response to therapy in the clinic. Wild-type ER, human epidermal growth factor receptor 2 (HER2), and progesterone receptor (PR), are histopathological markers that guide therapeutic selection. In clinical management of metastatic ER+ breast cancer, SERDs, such as fulvestrant is used for patients with resistance to AIs and tamoxifen without regard for ESR1 mutation status. An analysis of BOLERO-2, a phase III clinical trial that enrolled ER+ breast cancer patients with locally advanced or metastatic disease whom progressed on AI, evaluated the prevalence of the two most frequent ESR1 point mutations, Y537S and D538G and their effects on patient outcomes in ER+ metastatic patients[56]. Having either one or two of these mutations was associated with decreased overall survival. In the PALOMA-3 clinical trial which enrolled ER+ breast cancer patients with advanced, endocrine refractory disease, palbociclib combined with fulvestrant led to longer PFS than fulvestrant alone[59,71]. 69% of patients from the PALOMA-3 were analyzed for ESR1 mutation status, which showed that 25% of these cases harbored ESR1 mutations consisting mainly of Y537S, Y537N, D538G, and E380Q mutations[59]. However, palbociclib was found to provide equal benefit regardless of ESR1 mutation status. Although these studies indicate that the presence of ESR1 mutations may predict poor outcomes, they also highlight the need for more analyses of studies investigating the predictive value of ESR1 mutation status and response to therapy once the disease has become endocrine therapy resistant.
The development of sequencing technologies and the various models to recapitulate ESR1 mutant bearing tumors allow insightful studies into the landscape and targeted therapies of activating point mutations in the ESR1 LBD. Further studies are needed to address the use of ESR1 mutations as predictive biomarkers to stratify patient subsets and predict ESR1 mutation specific therapeutic vulnerabilities.
ESR1 structural rearrangements and ESR1 fusions
In contrast to well-studied ESR1 point mutations, structural rearrangements involving ESR1 are understudied. A variety of ESR1 gene fusion transcripts have been identified in luminal breast tumors[72,73]. Analysis of RNA-seq data from 990 primary TCGA breast samples revealed that 21 of these tumors (2.1%), all of the luminal B subtype, contained recurrent fusion transcripts involving the first two non-coding exons of ESR1 fused to various C-termini sequences from the coiled-coil domain containing 170 gene, CCDC170 (ESR1-e2>CCDC170)[73]. These fusion transcripts do not provide sufficient coding sequences to generate chimeric ER fusion proteins but instead generate truncated forms of CCDC170 proteins (ΔCCDC170). Exogenous expression of ΔCCDC170 in ER+ breast cancer cells led to enhanced growth and reduced sensitivity to tamoxifen[73] suggesting a role for ESR1-e2>CCDC170 in endocrine therapy resistance. Another independent study that examined early stage and non-metastatic ER+ breast samples also identified two ESR1-e2>CCDC170 fusion transcripts as well as ESR1-e2>C6orf211 and another fusion containing the first 6 exons of ESR1 fused to AKAP12 (ESR1-e6>AKAP12)[72]. These ESR1 fusions were identified in 4 out of 62 surgical samples (6.5%) that were resistant to letrozole aromatase inhibitor treatment 10-21 days post treatment as defined by Ki67 labeling[74], suggesting a higher frequency for these ESR1 fusions gene events in endocrine-refractory tumors compared to primary, untreated samples. However, detailed functional characterization and evidence demonstrating a causal role for ESR1 fusions in endocrine therapy resistance has been lacking and the incidence of ESR1 fusions from late-stage ER+ breast cancer still remains unclear. Furthermore, therapeutic strategies to treat ESR1 translocated tumors remains poorly understood.
Using a PDX model to better understand endocrine therapy resistance, we previously reported a somatic gain-of-function event in the form of a chromosomal translocation identified in a patient presenting with aggressive endocrine therapy resistant, metastatic ER+ disease. This translocation produced an in-frame fusion gene consisting of exons 1-6 of ESR1 (ESR1-e6) and the C-terminus of the Hippo pathway coactivator gene, YAP1 (ESR1-e6>YAP1), thereby generating a stable ESR1 fusion protein that was a highly active constitutive transcription factor[47][Figure 1D]. Our group more recently discovered another in-frame ESR1 fusion gene involving the protocadherin 11 X-linked gene, PCDH11X (ESR1-e6>PCDH11X) provided by inter-chromosomal translocation that also produced stable ESR1 fusion protein identified in a patient with endocrine-refractory, metastatic ER+ breast cancer[75]. In both ESR1-e6>YAP1 and ESR1-e6>PCDH11X fusions, the LBD of ESR1 is replaced with in-frame sequences from another gene, and therefore the drug binding domain that endocrine therapies recognize is absent. These two fusions promoted endocrine therapy resistant cell proliferation and constitutively activated ER target genes. Interestingly, both fusions also upregulated an epithelial-to-mesenchymal transition (EMT)-like transcriptional signature, induced cell motility, and increased lung metastatic frequency[75]. These results suggest that ESR1 fusions are able to drive not only endocrine therapy resistance, but also drive metastasis, linking these two lethal processes together.
Importantly, ESR1 fusion-driven growth could be suppressed by CDK4/6 inhibition. This suggests that targeting downstream kinases of ER could be a potential therapeutic strategy to treat ESR1 translocated tumors and further suggests that ESR1 fusion status may be a potential biomarker to stratify patients to CDK4/6 inhibitor therapy. To further explore therapeutic strategies to target ESR1 fusions, a collaborative study was performed to examine interacting proteins with ESR1 fusion transcriptional complexes[66]. Results from that study showed enhanced recruitment of 26S proteasomal subunits to ESR1-e6>YAP1 driving transcriptional activation and cell proliferation. Subsequent pharmacological inhibition with a broad-spectrum proteasome inhibitor, MG132, blocked ESR1-e6>YAP1-mediated activation of an ERE-luciferase reporter. Furthermore, bortezomib, a specific 26S proteasome inhibitor in phase II clinical trial used to treat endocrine-refractory, metastatic ER+ breast cancer in combination with fulvestrant[76] suppressed growth driven by ESR1-e6>YAP1. Taken together, these results suggest that downstream ER kinases such as CDK4/6 as well as transcriptional coregulators such as the 26S proteasome are attractive therapeutic targets to treat ESR1 fusion positive, metastatic breast tumors.
Additional in-frame ESR1 translocations with diverse partner genes have now been identified in late-stage, endocrine-refractory, ER+ metastatic cases. These include ESR1-e6>DAB2, ESR1-e6>GYG1, and ESR1-e6>SOX9[77]. Like the ESR1-e6>YAP1 and ESR1-e6>PCDH11X fusions, the ESR1-e6>DAB2 and ESR1-e6>GYG1 fusions produce stable ESR1 fusion proteins and all three were able to drive hormone-independent activation of a ERE-luciferase reporter[77]. Remarkably, these ESR1 fusions all follow a pattern preserving the first six exons of ESR1, containing the N-terminal DNA binding domain fused in-frame to C-terminal partner genes, thus excluding the LBD in ESR1[Figure 1D]. Therefore, these additional ESR1 fusion proteins likely drive pan-endocrine therapy resistance like our previously discovered ESR1-e6>YAP1 and ESR1-e6>PCDH11X fusions[75]. The functional and therapeutic significance of these additional ESR1 fusions are the focus of ongoing investigation by our group and others.
In contrast to transcriptionally active ESR1 fusions, we also identified an in-frame ESR1-e6 fusion, ESR1-e6>NOP2 in a treatment naïve primary breast tumor that was transcriptionally inactive despite producing stable ESR1 fusion protein[75]. ESR1-e6>NOP2 did not promote endocrine therapy resistant growth and was found to bind relatively few sites in a genome-wide DNA binding assay, potentially explaining the weak functional activity measured by our experimental systems. In addition, out-of-frame ESR1 fusions identified in primary tumors preserving diverse exons of ESR1 gene, ESR1-e3, ESR1-e4, ESR1-e5, and ESR1-e6 did not facilitate estrogen-independent proliferation[75]. More studies are required to fully understand the contribution of transcriptionally inactive in-frame and out-of-frame ESR1 fusions in breast cancer.
ESR1 fusion structural studies revealed that driver ESR1 fusions from metastatic patients follow the same fusion pattern containing the first 6 exons of ESR1 (ESR1-e6) fused to C-termini of diverse gene partners suggesting this pattern is strongly connected to endocrine therapy resistant, metastatic ER+ breast tumors. The observation of a highly consistent and recurrent ESR1 breakpoint, together with the promiscuity of ESR1 for a variety of fusion partners is certainly interesting. In prostate cancer, recurrent fusions involving promoter regions of an androgen regulated gene, transmembrane protease serine 2 gene (TMPRSS2) fused to coding sequences of erythroblastosis virus E26 gene (ETS) family members have been identified in more than 50% of prostate cancer cases[78]. Androgen receptor (AR) signaling has been shown to bring the androgen regulated gene TMPRSS2 and the ERG gene in close proximity in prostate cancer cell line models[79]. Androgen signaling also generates DNA damage in the form of double strand breaks (DSBs) at sites of TMPRSS2-ERG genomic breakpoints. These DSBs have been shown to be mediated by the class II topoisomerase beta, TOP2B, which is recruited to AR, inducing DSBs[80]. TMPRSS2-ERG gene fusions can then arise from dysfunction of mechanisms to repair DSBs, such as homologous recombination (HR) pathway and the error-prone non-homologous end-joining (NHEJ) pathway. AR-mediated DSBs in prostate cancer may provide clues to the recurrent ESR1 breakpoints for ESR1 fusions seen in breast cancer. Recruitment of TOP2B to ER and subsequent DSBs have been shown to occur at regulatory regions of ER target genes as a consequence of ER-mediated transcriptional activation[81]. Since regulatory regions of ESR1 itself has also been shown to be bound by ER[82], transcription-induced DSBs by ER, coupled with dysregulation of DSB repair mechanisms may contribute to the highly recurrent ESR1 breakpoints. Although none of the fusion partners from endocrine-refractory, metastatic disease observed in our studies are known ER targets, additional studies are needed to better understand the diversity of preferred ESR1 partner genes.
ESR1 fusions that contain the first six exons of ESR1 fused in-frame to partner genes are almost exclusively observed in endocrine therapy resistant, metastatic ER+ breast cancer, with the exception of ESR1-e6>NOP2, as described above, likely suggesting a role in driving disease pathogenesis. However, very few functionally significant ESR1 fusions have been studied to date and therefore ESR1 fusion events remains an understudied form of somatic mutation in breast cancer. The incidence of ESR1 fusions is also still not well understood, especially in the metastatic setting, but the studies discussed here collectively suggest ESR1 fusions to be present in at least 1% of metastatic breast cancer cases[77], with the actual frequency likely to be higher as more studies on ESR1 fusions emerge. Additional studies on ESR1 fusions will further support the causal role ESR1 fusions and have significant diagnostic and clinical implications since pathogenic ESR1 fusions could be used as biomarkers to stratify patients for individualized healthcare in ER+ breast cancer. Therapeutic vulnerabilities from ESR1 translocated tumors could be an alternative to chemotherapy in patients with rapidly progressing, endocrine therapy resistant disease.
Conclusion
Endocrine therapy resistance and metastasis in ER+ breast cancer patients remain significant clinical problems. This review has focused on studies describing a spectrum of ESR1 alterations including amplification, point-mutations, and structural rearrangements in endocrine-refractory, metastatic ER+ breast cancer cases. Results from these studies have provided insights into the underlying mechanisms that contribute to endocrine therapy resistance and metastasis.
Amplification of the ESR1 locus results in overexpression of oncogenic ER protein in the breast and potentially reducing sensitivity of ESR1 amplified breast tumors to endocrine therapies and therefore likely leads to disease progression and metastasis. Point mutations in the LBD of ESR1, the most common of which are Y537S and D538G, confer an agonist confirmation to such ESR1 mutant proteins resulting in constitutively active mutant ER transcription factors that lead to activation of ER target genes in a hormone-independent manner while also promoting activation of metastasis-associated genes[64]. The finding that ER LBD mutant proteins are constitutively active in an estrogen-independent manner suggest that therapeutic strategies which work by blocking estrogen production, such as ovarian ablation and treatment with AIs, are likely to be ineffective in breast tumors harboring ESR1 point mutations. Indeed, a significant proportion of ESR1 LBD point mutations were identified in metastatic tumors that were extensively treated with AIs, suggesting that such mutations may be enriched in breast tumors upon AI treatment[53]. ESR1-Y537S and ESR1-D538G are partially sensitive to fulvestrant[37,47,63], and newer oral SERDs that have better bioavailability compared to fulvestrant, such as AZD9496, have shown promising results in treating tumor growth driven by ESR1 LBD point mutants in experimental models[66]. Although fulvestrant is used exclusively in the metastatic setting for ER+ disease, treating primary breast tumors upfront with fulvestrant or more potent SERDs like AZD9496 may reduce the incidence of disease driven by ESR1 LBD point mutations.
Despite the potential effectiveness of fulvestrant in targeting ER proteins with point mutations in the LBD, it is completely ineffective against ER fusion proteins generated from in-frame ESR1 fusion transcripts arising from ESR1 translocations[75]. These ESR1 fusions transcripts, ESR1-e6>YAP1 and ESR1-e6>PCDH11X, were identified in patients with metastatic ER+ breast tumors that were pan-endocrine therapy resistant[75]. Both fusions retain the first 6 exons of ESR1 fused in-frame to C-terminal sequences of the partner gene but lack exons encoding the LBD, rendering these fusions insensitive to all endocrine therapies that target the LBD, including fulvestrant and most likely AZD9496. These ESR1 fusions were found to generate hyperactive ESR1 fusion proteins that not only drive endocrine therapy resistant growth, but also play a role in the metastatic process, reprogramming the ER cistrome to drive EMT and metastasis to lung[75]. Despite the lack of an ESR1 LBD, blocking signaling downstream of ESR1 fusions with a CDK4/6 inhibitor, palbociclib, suppressed ESR1 fusion-driven growth at primary and metastatic sites in experimental models[75]. Similar to ESR1 point mutations, ESR1 fusion formation is likely a mechanism of acquired endocrine therapy resistance. To date, ESR1 fusion transcripts that produce stable ESR1 fusion proteins have only been detected in metastatic breast tumors resistant to multiple lines of endocrine therapies. This suggests that ESR1 fusions may be enriched in tumors from the selective pressure of endocrine treatment. Since the ESR1-e6>YAP1 and ESR1-e6>PCDHnX fusions were identified from a small cohort of late-stage ER+ patients, more RNA-seq data from primary and late-stage, treatment-refractory tumors are clearly required, particularly with longer sequencing reads, which increase fusion gene detection sensitivity to better understand the incidence of ESR1 fusions in both primary and metastatic breast cancer.
The underlying mechanism of how ESR1 fusions arise remains unclear. However, as mentioned earlier, DSBs mediated by recruitment of TOP2B to ER transcriptional complexes may contribute to formation of ESR1 fusion genes, and therefore TOP2B could potentially be an attractive therapeutic target to prevent the formation of ESR1 fusion events. More studies are required to test this hypothesis. Daunorubicin, an FDA-approved chemotherapeutic drug indicated for treating leukemia, targets TOP2B, however, this agent is very toxic. Developing less toxic agents that target TOP2B may represent a therapeutic strategy to prevent ESR1 translocation events and deserves further study in the context of ER+ breast cancer.
Therapeutic targeting these aberrant forms of ER have shown promise in pre-clinical experimental models with more studies required to translate such findings to the clinic. Collectively, these studies deepen our understanding of how ESR1 alterations trigger breast cancer to become lethal metastatic disease and will guide development of therapeutic strategies to treat a subset of patients with tumors that contains these ESR1 alterations.
Declarations
Authors' contributionsMade substantial contributions to conception: Lei JT, Gou X, Seker S
Provided initial drafts of the work: Lei JT, Gou X, Seker S
Revising the work critically for important intellectual content: Lei JT, Ellis MJ
Final approval of the version: Lei JT, Ellis MJ
Availability of data and materialsNot applicable.
Financial support and sponsorshipThis work was supported by a Susan G. Komen Promise Grant (PG12220321) to Ellis MJ; a Cancer Prevention Institute of Texas (CPRIT) Recruitment of Established Investigators Award (RR140033) to Ellis MJ; a Breast Cancer Research Foundation Grant (BCRF ELFF-16-003) to Ellis MJ; and by a National Institutes of Health Training Grant (T32-GM088129) to Lei JT.
Conflicts of interestEllis MJ received consulting fees from Abbvie, Sermonix, Pfizer, AstraZeneca, Celgene, NanoString, Puma, and Novartis, and is an equity stockholder, consultant, and Board Director member of BioClassifier, and inventor on a patent for the Breast PAM50 assay.
Ethical approval and consent to participateNot applicable.
Consent for publicationNot applicable.
Copyright© The Author(s) 2019.
REFERENCES
1. Harvey JM, Clark GM, Osborne CK, Allred DC. Estrogen receptor status by immunohistochemistry is superior to the ligand-binding assay for predicting response to adjuvant endocrine therapy in breast cancer. J Clin Oncol 1999;17:1474-81.
2. Joel PB, Traish AM, Lannigan DA. Estradiol and phorbol ester cause phosphorylation of serine 118 in the human estrogen receptor. Mol Endocrinol 1995;9:1041-52.
3. Le Goff P, Montano MM, Schodin DJ, Katzenellenbogen BS. Phosphorylation of the human estrogen receptor. Identification of hormone-regulated sites and examination of their influence on transcriptional activity. J Biol Chem 1994;269:4458-66.
4. Kato S, Endoh H, Masuhiro Y, Kitamoto T, Uchiyama S, et al. Activation of the estrogen receptor through phosphorylation by mitogen-activated protein kinase. Science 1995;270:1491-4.
5. Zhou W SJ. Links between oestrogen receptor activation and proteolysis: relevance to hormone-regulated cancer therapy. Nat Rev Cancer 2014;14:26-38.
6. Zwart W, de Leeuw R, Rondaij M, Neefjes J, Mancini MA, et al. The hinge region of the human estrogen receptor determines functional synergy between AF-1 and AF-2 in the quantitative response to estradiol and tamoxifen. J Cell Sci 2010;123:1253-61.
7. Davies C, Godwin J, Gray R, Clarke M, et al; Early Breast Cancer Trialists’ Collaborative G (EBCTCG). Relevance of breast cancer hormone receptors and other factors to the efficacy of adjuvant tamoxifen: patient-level meta-analysis of randomised trials. Lancet 2011;378:771-84.
8. Ma CX, Reinert T, Chmielewska I, Ellis MJ. Mechanisms of aromatase inhibitor resistance. Nat Rev Cancer 2015;15:261-75.
9. Osborne CK, Bardou V, Hopp TA, Chamness GC, Hilsenbeck SG, et al. Role of the estrogen receptor coactivator AIB1 (SRC-3) and HER-2/neu in tamoxifen resistance in breast cancer. J Natl Cancer Inst 2003;95:353-61.
10. Arpino G, Green SJ, Allred DC, Lew D, Martino S, et al. HER-2 amplification, HER-1 expression, and tamoxifen response in estrogen receptor-positive metastatic breast cancer: a southwest oncology group study. Clin Cancer Res 2004;10:5670-6.
11. Fan P, Wang J, Santen RJ, Yue W. Long-term treatment with tamoxifen facilitates translocation of estrogen receptor alpha out of the nucleus and enhances its interaction with EGFR in MCF-7 breast cancer cells. Cancer Res 2007;67:1352-60.
12. Song RX, Barnes CJ, Zhang Z, Bao Y, Kumar R, et al. The role of Shc and insulin-like growth factor 1 receptor in mediating the translocation of estrogen receptor alpha to the plasma membrane. Proc Natl Acad Sci U S A 2004;101:2076-81.
13. Osborne CK, Neven P, Dirix LY, Mackey JR, Robert J, et al. Gefitinib or placebo in combination with tamoxifen in patients with hormone receptor-positive metastatic breast cancer: a randomized phase II study. Clin Cancer Res 2011;17:1147-59.
14. Ellis MJ, Perou CM. The genomic landscape of breast cancer as a therapeutic roadmap. Cancer Discov 2013;3:27-34.
15. Sanchez CG, Ma CX, Crowder RJ, Guintoli T, Phommaly C, et al. Preclinical modeling of combined phosphatidylinositol-3-kinase inhibition with endocrine therapy for estrogen receptor-positive breast cancer. Breast Cancer Res 2011;13:R21.
16. Baselga J, Campone M, Piccart M, Burris HA 3rd, Rugo HS, et al. Everolimus in postmenopausal hormone-receptor-positive advanced breast cancer. N Engl J Med 2012;366:520-9.
17. Di Leo A, Johnston S, Lee KS, Ciruelos E, Lonning PE, et al. Buparlisib plus fulvestrant in postmenopausal women with hormone-receptor-positive, HER2-negative, advanced breast cancer progressing on or after mTOR inhibition (BELLE-3): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol 2018;19:87-100.
18. André F, Ciruelos EM, Rubovszky G, Campone M, Loibl S, et al. Alpelisib (ALP) + fulvestrant (FUL) for advanced breast cancer (ABC): results of the phase 3 SOLAR-1 trial. Munich, Germany: ESMO Congress; 2018.
19. Cancer Genome Atlas N. Comprehensive molecular portraits of human breast tumours. Nature 2012;490:61-70.
20. Thangavel C, Dean JL, Ertel A, Knudsen KE, Aldaz CM, et al. Therapeutically activating RB: reestablishing cell cycle control in endocrine therapy-resistant breast cancer. Endocr Relat Cancer 2011;18:333-45.
21. Ma CX, Gao F, Luo J, Northfelt DW, Goetz M, et al. NeoPalAna: neoadjuvant palbociclib, a cyclin-dependent kinase 4/6 inhibitor, and anastrozole for clinical stage 2 or 3 estrogen receptor-positive breast cancer. Clin Cancer Res 2017;23:4055-65.
22. Basudan A, Priedigkeit N, Hartmaier RJ, Sokol ES, Bahreini A, et al. Frequent ESR1 and CDK pathway copy-number alterations in metastatic breast cancer. Mol Cancer Res 2019;17:457-68.
23. Zhang QX, Borg A, Wolf DM, Oesterreich S, Fuqua SA. An estrogen receptor mutant with strong hormone-independent activity from a metastatic breast cancer. Cancer Res 1997;57:1244-9.
24. Pejerrey SM, Dustin D, Kim JA, Gu G, Rechoum Y, et al. The impact of ESR1 mutations on the treatment of metastatic breast cancer. Horm Cancer 2018;9:215-28.
25. Niu J, Andres G, Kramer K, Kundranda MN, Alvarez RH, et al. Incidence and clinical significance of ESR1 mutations in heavily pretreated metastatic breast cancer patients. Onco Targets Ther 2015;8:3323-8.
26. Borg A, Tandon AK, Sigurdsson H, Clark GM, Ferno M, et al. HER-2/neu amplification predicts poor survival in node-positive breast cancer. Cancer Res 1990;50:4332-7.
27. Turner N, Pearson A, Sharpe R, Lambros M, Geyer F, et al. FGFR1 amplification drives endocrine therapy resistance and is a therapeutic target in breast cancer. Cancer Res 2010;70:2085-94.
28. Nembrot M, Quintana B, Mordoh J. Estrogen receptor gene amplification is found in some estrogen receptor-positive human breast tumors. Biochem Biophys Res Commun 1990;166:601-7.
29. Holst F, Stahl PR, Ruiz C, Hellwinkel O, Jehan Z, et al. Estrogen receptor alpha (ESR1) gene amplification is frequent in breast cancer. Nat Genet 2007;39:655-60.
30. Brown LA, Hoog J, Chin SF, Tao Y, Zayed AA, et al. ESR1 gene amplification in breast cancer: a common phenomenon? Nat Genet 2008;40:806-7.
31. Horlings HM, Bergamaschi A, Nordgard SH, Kim YH, Han W, et al. ESR1 gene amplification in breast cancer: a common phenomenon? Nat Genet 2008;40:807-8.
32. Reis-Filho JS, Drury S, Lambros MB, Marchio C, Johnson N, et al. ESR1 gene amplification in breast cancer: a common phenomenon? Nat Genet 2008;40:809-10.
33. Vincent-Salomon A, Raynal V, Lucchesi C, Gruel N, Delattre O. ESR1 gene amplification in breast cancer: a common phenomenon? Nat Genet 2008;40:809.
34. Tsiambas E, Georgiannos SN, Salemis N, Alexopoulou D, Lambropoulou S, et al. Significance of estrogen receptor 1 (ESR-1) gene imbalances in colon and hepatocellular carcinomas based on tissue microarrays analysis. Med Oncol 2011;28:934-40.
35. Tomita S, Zhang Z, Nakano M, Ibusuki M, Kawazoe T, et al. Estrogen receptor alpha gene ESR1 amplification may predict endocrine therapy responsiveness in breast cancer patients. Cancer Sci 2009;100:1012-7.
36. Moelans CB, de Weger RA, Monsuur HN, Vijzelaar R, van Diest PJ. Molecular profiling of invasive breast cancer by multiplex ligation-dependent probe amplification-based copy number analysis of tumor suppressor and oncogenes. Mod Pathol 2010;23:1029-39.
37. Jeselsohn R, Yelensky R, Buchwalter G, Frampton G, Meric-Bernstam F, et al. Emergence of constitutively active estrogen receptor-alpha mutations in pretreated advanced estrogen receptor-positive breast cancer. Clin Cancer Res 2014;20:1757-67.
38. Schiff R, Veeraraghavan J, Fu X. Abstract SY01-01: Endocrine resistance in metastatic breast cancer: Mechanisms and new therapeutic strategies. Chicago, IL: AACR Annual Meeting; 2018.
39. Basudan A, Priedigkeit N, Hartmaier RJ, Sokol ES, Bahreini A, et al. Frequent ESR1 and CDK pathway copy-number alterations in metastatic breast cancer. Mol Cancer Res 2019;17:457-68.
40. Markiewicz A, Welnicka-Jaskiewicz M, Skokowski J, Jaskiewicz J, Szade J, et al. Prognostic significance of ESR1 amplification and ESR1 PvuII, CYP2C19*2, UGT2B15*2 polymorphisms in breast cancer patients. PLoS One 2013;8:e72219.
41. Nielsen KV, Bent Ejlertsen, Sven Müller, Susanne Møller, Birgitte B, et al. Amplification of ESR1 may predict resistance to adjuvant tamoxifen in postmenopausal patients with hormone receptor positive breast cancer. Breast Cancer Res Treat 2011;127:345-55.
42. Holst F, Singer CF. ESR1-amplification-associated estrogen receptor alpha activity in breast cancer. Trends Endocrinol Metab 2016;27:751-2.
43. Khan SA, Rogers MA, Khurana KK, Meguid MM, Numann PJ. Estrogen receptor expression in benign breast epithelium and breast cancer risk. J Natl Cancer Inst 1998;90:37-42.
44. Shoker BS, Jarvis C, Sibson DR, Walker C, Sloane JP. Oestrogen receptor expression in the normal and pre-cancerous breast. J Pathol 1999;188:237-44.
45. Burkhardt L, Grob TJ, Hermann I, Burandt E, Choschzick M, et al. Gene amplification in ductal carcinoma in situ of the breast. Breast Cancer Res Treat 2010;123:757-65.
46. Quenel-Tueux N, Debled M, Rudewicz J, MacGrogan G, Pulido M, et al. Clinical and genomic analysis of a randomised phase II study evaluating anastrozole and fulvestrant in postmenopausal patients treated for large operable or locally advanced hormone-receptor-positive breast cancer. Br J Cancer 2015;113:585-94.
47. Li S, Shen D, Shao J, Crowder R, Liu W, et al. Endocrine-therapy-resistant ESR1 variants revealed by genomic characterization of breast-cancer-derived xenografts. Cell Rep 2013;4:1116-30.
48. Aguilar H, Sole X, Bonifaci N, Serra-Musach J, Islam A, et al. Biological reprogramming in acquired resistance to endocrine therapy of breast cancer. Oncogene 2010;29:6071-83.
49. Haddow A, Watkinson JM, Paterson E, Koller PC. Influence of synthetic oestrogens on advanced malignant disease. Br Med J 1944;2:393-8.
50. Kota K, Adam Brufsky, Steffi Oesterreich, Adrian Lee. Estradiol as a targeted, late-line therapy in metastatic breast cancer with estrogen receptor amplification. Cureus 2017;9:e1434.
51. Magnani L, Frige G, Gadaleta RM, Corleone G, Fabris S, et al. Acquired CYP19A1 amplification is an early specific mechanism of aromatase inhibitor resistance in ERalpha metastatic breast cancer. Nat Genet 2017;49:444-50.
52. Weis KE, Ekena K, Thomas JA, Lazennec G, Katzenellenbogen BS. Constitutively active human estrogen receptors containing amino acid substitutions for tyrosine 537 in the receptor protein. Mol Endocrinol 1996;10:1388-98.
53. Toy W, Shen Y, Won H, Green B, Sakr RA, et al. ESR1 ligand-binding domain mutations in hormone-resistant breast cancer. Nat Genet 2013;45:1439-45.
54. Merenbakh-Lamin K, Ben-Baruch N, Yeheskel A, Dvir A, Soussan-Gutman L, et al. D538G mutation in estrogen receptor-alpha: a novel mechanism for acquired endocrine resistance in breast cancer. Cancer Res 2013;73:6856-64.
55. Robinson DR, Wu YM, Vats P, Su F, Lonigro RJ, et al. Activating ESR1 mutations in hormone-resistant metastatic breast cancer. Nat Genet 2013;45:1446-51.
56. Chandarlapaty S, Chen D, He W, Sung P, Samoila A, et al. Prevalence of ESR1 mutations in cell-free DNA and outcomes in metastatic breast cancer: a secondary analysis of the BOLERO-2 clinical trial. JAMA Oncol 2016;2:1310-5.
57. Spoerke JM, Gendreau S, Walter K, Qiu J, Wilson TR, et al. Heterogeneity and clinical significance of ESR1 mutations in ER-positive metastatic breast cancer patients receiving fulvestrant. Nat Commun 2016;7:11579.
58. Schiavon G, Hrebien S, Garcia-Murillas I, Cutts RJ, Pearson A, et al. Analysis of ESR1 mutation in circulating tumor DNA demonstrates evolution during therapy for metastatic breast cancer. Sci Transl Med 2015;7:313ra182.
59. Fribbens C, O’Leary B, Kilburn L, Hrebien S, Garcia-Murillas I, et al. Plasma ESR1 mutations and the treatment of estrogen receptor-positive advanced breast cancer. J Clin Oncol 2016;34:2961-8.
60. Wang P, Bahreini A, Gyanchandani R, Lucas PC, Hartmaier RJ, et al. Sensitive detection of mono-and polyclonal ESR1 mutations in primary tumors, metastatic lesions, and cell-free DNA of breast cancer patients. Clin Cancer Res 2016;22:1130-7.
61. Gelsomino L, Gu G, Rechoum Y, Beyer AR, Pejerrey SM, et al. Erratum to: ESR1 mutations affect anti-proliferative responses to tamoxifen through enhanced cross-talk with IGF signaling. Breast Cancer Res Treat 2017;163:639-40.
62. Harrod A, Fulton J, Nguyen VTM, Periyasamy M, Ramos-Garcia L, et al. Genomic modelling of the ESR1 Y537S mutation for evaluating function and new therapeutic approaches for metastatic breast cancer. Oncogene 2017;36:2286-96.
63. Toy W, Weir H, Razavi P, Lawson M, Goeppert AU, et al. Activating ESR1 mutations differentially affect the efficacy of ER antagonists. Cancer Discov 2017;7:277-87.
64. Jeselsohn R, Bergholz JS, Pun M, Cornwell M, Liu W, et al. Allele-specific chromatin recruitment and therapeutic vulnerabilities of ESR1 activating mutations. Cancer Cell 2018;33:173-86.e5.
65. Shang Y, Hu X, DiRenzo J, Lazar MA, Brown M. Cofactor dynamics and sufficiency in estrogen receptor-regulated transcription. Cell 2000;103:843-52.
66. Gates LA, Gu G, Chen Y, Rohira AD, Lei JT, et al. Proteomic profiling identifies key coactivators utilized by mutant ERalpha proteins as potential new therapeutic targets. Oncogene 2018;37:4581-98.
67. Osborne CK, Schiff R. Mechanisms of endocrine resistance in breast cancer. Annu Rev Med 2011;62:233-47.
68. Nagasawa J, Govek S, Kahraman M, Lai A, Bonnefous C, et al. Identification of an orally bioavailable chromene-based selective estrogen receptor degrader (SERD) that demonstrates robust activity in a model of tamoxifen-resistant breast cancer. J Med Chem 2018;61:7917-28.
69. Hamilton EP, Patel MR, Armstrong AC, Baird RD, Jhaveri K, et al. A first-in-human study of the new oral selective estrogen receptor degrader AZD9496 for ER(+)/HER2(-) advanced breast cancer. Clin Cancer Res 2018;24:3510-8.
70. Wardell SE, Ellis MJ, Alley HM, Eisele K, VanArsdale T, et al. Efficacy of SERD/SERM Hybrid-CDK4/6 inhibitor combinations in models of endocrine therapy-resistant breast cancer. Clin Cancer Res 2015;21:5121-30.
71. Turner NC, Ro J, Verma S, Huang Bartlett C, Cristofanilli M, et al. Palbociclib in hormone-receptor-positive advanced breast cancer. N Engl J Med 2015;373:209-19.
72. Giltnane JM, Hutchinson KE, Stricker TP, Formisano L, Young CD, et al. Genomic profiling of ER(+) breast cancers after short-term estrogen suppression reveals alterations associated with endocrine resistance. Sci Transl Med 2017;9.
73. Veeraraghavan J, Tan Y, Cao XX, Kim JA, Wang X, et al. Recurrent ESR1-CCDC170 rearrangements in an aggressive subset of oestrogen receptor-positive breast cancers. Nat Commun 2014;5:4577.
74. Dowsett M, Smith IE, Ebbs SR, Dixon JM, Skene A, et al. Prognostic value of Ki67 expression after short-term presurgical endocrine therapy for primary breast cancer. J Natl Cancer Inst 2007;99:167-70.
75. Lei JT, Shao J, Zhang J, Iglesia M, Chan DW, et al. Functional annotation of ESR1 gene fusions in estrogen receptor-positive breast cancer. Cell Rep 2018;24:1434-44e7.
76. Adelson K, Ramaswamy B, Sparano JA, Christos PJ, Wright JJ, et al. Randomized phase II trial of fulvestrant alone or in combination with bortezomib in hormone receptor-positive metastatic breast cancer resistant to aromatase inhibitors: a New York cancer consortium trial. NPJ Breast Cancer 2016;2:16037.
77. Hartmaier RJ, Trabucco SE, Priedigkeit N, Chung JH, Parachoniak CA, et al. Recurrent hyperactive ESR1 fusion proteins in endocrine therapy-resistant breast cancer. Ann Oncol 2018;29:872-80.
78. Tomlins SA, Rhodes DR, Perner S, Dhanasekaran SM, Mehra R, et al. Recurrent fusion of TMPRSS2 and ETS transcription factor genes in prostate cancer. Science 2005;310:644-8.
79. Mani RS, Tomlins SA, Callahan K, Ghosh A, Nyati MK, et al. Induced chromosomal proximity and gene fusions in prostate cancer. Science 2009;326:1230.
80. Haffner MC, Aryee MJ, Toubaji A, Esopi DM, Albadine R, et al. Androgen-induced TOP2B-mediated double-strand breaks and prostate cancer gene rearrangements. Nat Genet 2010;42:668-75.
81. Ju BG, Lunyak VV, Perissi V, Garcia-Bassets I, Rose DW, et al. A topoisomerase IIbeta-mediated dsDNA break required for regulated transcription. Science 2006;312:1798-802.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Lei JT, Gou X, Seker S, Ellis MJ. ESR1 alterations and metastasis in estrogen receptor positive breast cancer. J Cancer Metastasis Treat 2019;5:38. http://dx.doi.org/10.20517/2394-4722.2019.12
AMA Style
Lei JT, Gou X, Seker S, Ellis MJ. ESR1 alterations and metastasis in estrogen receptor positive breast cancer. Journal of Cancer Metastasis and Treatment. 2019; 5: 38. http://dx.doi.org/10.20517/2394-4722.2019.12
Chicago/Turabian Style
Lei, Jonathan T., Xuxu Gou, Sinem Seker, Matthew J. Ellis. 2019. "ESR1 alterations and metastasis in estrogen receptor positive breast cancer" Journal of Cancer Metastasis and Treatment. 5: 38. http://dx.doi.org/10.20517/2394-4722.2019.12
ACS Style
Lei, JT.; Gou X.; Seker S.; Ellis MJ. ESR1 alterations and metastasis in estrogen receptor positive breast cancer. J. Cancer. Metastasis. Treat. 2019, 5, 38. http://dx.doi.org/10.20517/2394-4722.2019.12
About This Article
Special Issue
Copyright

Data & Comments
Data


 Cite This Article 108 clicks
Cite This Article 108 clicks

 Like This Article 25
likes
Like This Article 25
likes






Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.