
Chemotherapy-induced immunological breast cancer dormancy: a new function for old drugs?


Abstract
Breast cancer remains the main cause of cancer-related mortality for women world-wide. Main cause of death is the development of therapy-resistant metastases. Relapses occur with a bimodal temporal distribution, with a first peak at 1-2 years after initial therapy and a second peak 2-3 years later. This discontinuous growth kinetics is consistent with the notion that disseminated cancer cells can remain dormant over a prolonged period of time before resuming growth. How cancer cells enter, sustain and exit dormancy, are unanswered questions with relevance to cancer biology, monitoring and therapy. Investigating mechanisms of breast cancer dormancy remains challenging, as in patients the condition is elusive and experimentally there are only a few models that recapitulate the clinical condition. Thus, developing new models to identify clinically relevant mechanisms and candidate therapeutic targets may open new avenues for novel therapies to induce and prolong dormancy. We have observed that cells surviving chemotherapy can enter a state of immunological dormancy. Using this model, we identified IRF-7/Interferon type I/IFNRA as signaling axis essential for this effect. Here we will review concepts and recent developments in cancer metastasis and dormancy with emphasis on breast cancer, and elaborate strategies to exploit them therapeutically.
Keywords
Introduction
With a few exceptions, as in the case of brain or liver cancer, the main cause of cancer-related death is not the primary tumor itself but rather the formation of secondary tumors, so called metastases, in vital organs, in particular lung, liver, bone or brain, leading to organ destruction and failure, resistance to therapy and cachexia[1,2]. Metastasis formation is believed to be a highly inefficient process[3]. The main rate limiting step in the metastatic process appears to be the ability of tumor cells to adapt to the new environment[4,5]. During this adaptation step at the metastatic site, cells have to establish bidirectional paracrine communication with a new tissue different from the primary site, acquire novel survival capacities and escape immune destruction. While cell autonomous processes, such as genetic evolution and epigenetic modifications, altered gene expression and metabolic adaptation, are essential to the metastatic process, the microenvironments of the primary tumor and of the metastatic site are equally critical determinants of metastasis formation[5-8]. Tumor angiogenesis, the remodeling of the extracellular matrix, the activation of local resident cells and the recruitment of inflammatory cells provide essential contributions to the metastatic process, including in breast cancer[9-11]. At diagnosis, only a minority of cancers have already formed clinically overt metastases (i.e., stage IV)[12]. Those that progress to metastatic disease can do with strikingly different kinetic. For example, lung and colorectal cancers mostly relapse within 1-3 years after diagnosis and the 5-year survival rates for these cancers are about 20% and 60%, respectively[13,14]. Conversely, in prostate cancer relapses occur late, with over 90% of the patients still alive 15 years after initial diagnosis[15]. In breast cancer, relapses occur with a peculiar bimodal distribution: a first peak appears generally 1-2 years and a second peak 4-5 years after surgery, followed by a tailed extension up to 15 years[16,17].
Breast cancer subtypes and adjuvant therapies
Breast cancer is the most common cancer diagnosed among women. In spite of improved management over the past 30 years, it remains the leading cause of cancer-related mortality for women world-wide[18]. Therapy and prognosis are largely determined by the biological and molecular characteristics of the primary tumor and its size and spreading at time of diagnosis[19,20]. There are three main clinically relevant biological subtypes: Oestrogen/Progesterone receptor positive breast cancer (ER+/PR+), HER2 amplified (HER2+) breast cancer and Triple Negative Breast Cancer (TNBC; i.e., ER-, PR-, HER2-)[21-23]. Based on gene expression signatures four main molecular subtypes have been reported: Luminal A and B, HER2+, and basal-like, which overlap largely, but not fully with the ER+/PR+; HER2+ and TNBC biological subtypes, respectively[24,25]. Both biological and molecular classifications have prognostic and predictive (therapeutic) relevance[19,20,24,25]: ER+ tumors are treated with adjuvant anti-estrogen therapies (e.g., tamoxifen) while HER2+ tumors are treated with HER2 inhibitors (e.g., trastuzumab), in addition to radiotherapy and chemotherapy, if necessary[19,26]. TNBC, has no molecular target useful for targeted therapy yet and adjuvant radio- and chemotherapies are still the standards of care[22,27]. The rational for administering adjuvant therapy after surgery is to eradicate disseminated tumor cells (DTC) or micro-metastases to decrease the risk of relapse. A large body of evidence from adjuvant studies suggest that ER+ breast cancer benefits less from chemotherapy compared to ER- breast cancer[28]. This is particularly true for the luminal A molecular subtype of ER+ breast cancer, which has a low rate of proliferation. Luminal A tumors have lower rates of pathologic complete response to chemotherapy compared to the highly proliferative ER+ luminal B breast cancer subset, as demonstrated with neo-adjuvant anthracycline/taxane-based chemotherapy[29,30]. For HER2+ breast cancer the introduction of HER2 inhibitors as adjuvant therapy, in combination with taxane-based chemotherapy, has vastly decreased the risk of metastatic progression and greatly improved survival in this cancer subtype[23,31]. TNBC is highly proliferative and respond better to chemotherapy compared to ER+ cancers[22,32]. Today no established targeted therapy exists for TNBC[33]. Interestingly, a fraction of TNBC are rich in tumor infiltrating lymphocytes and this infiltration has been associated with improved disease-free survival and overall survival OS, suggesting that immune cells may contribute to therapy success and implying the possibility of applying check-point inhibitors-based immunotherapies for these patients[10,22].
In spite of an approx 30%-35% decrease in mortality over the past 35 years due to combined systematic early detection and improved adjuvant therapies, there are still about 20%-25% of breast cancer patients, all stages combined, that will eventually succumb to their disease due to formation of therapy resistant metastases. This corresponds to about 95,000 and 40,000 women in Europe[1] (EU 28) and the United States[2], dying every year, respectively[18,19,34]. As of today, there are no effective, curative therapies for metastatic disease. Therapy-resistance and therapy-related toxicity limit therapeutic options[35].
Metastatic dissemination
Cancer metastases is a multistage process. Cancer cells have to first escape from the primary tumor, survive in the circulation as circulating tumor cells (CTC), seed at distant sites as DTC and grow to colonize the new tissue and form secondary tumors[36-38]. Growing evidence indicates that metastases are formed by a subset of tumor cells with “stem cell-like” features[39-41] that also associated with resistance to treatments and dormancy[42-44]. Accordingly, molecules controlling stem cell maintenance and differentiation[10,22,45] have been implicated in metastasis, including Wnts, BMPs, TGFb family members, Notch, CD44 and integrins[46,47]. Cancer stem cells (CSCs), in contrast to normal adult stem cells, seem able to revert the hierarchy so that a differentiated cancer cell can revert and recover the stem-like features, while normal, differentiated somatic cells are not able to do so. Thus, CSCs may be rather defined by function than lineage and may represent a form of adaptation of cancer cells to cellular or microenvironmental stress[48-50]. This plasticity may be one reason why by eliminating CSCs as proposed as a new therapeutic approach to eradicate cancer, may actually not be as effective as anticipated[51-54]. Acquisition of CSCs traits has been associated with Epithelial-to-Mesenchymal Transition (EMT), a condition endowing cancer cells with increased migratory, invasive, metastatic and therapy resistance capacities[53,55,56]. For example, breast cancer cells undergoing EMT acquire a cell surface phenotype (i.e., CD44high/CD24low) associated with CSCs properties[57]. Accordingly, EMT is a reversible process, as cells that disseminated through EMT and lost epithelial features, can revert back to their epithelial phenotype through an opposite process called Mesenchymal-to-Epithelial Transition (MET)[8]. Both CSCs and EMT are features that are heavily influenced by the microenvironment such as the vascular niches or inflammation (See below).
The genetic and epigenetic basis of metastasis is still not fully elucidated[58]. A current paradigm relies on the notion that the accumulation and selection of genetic mutations and epigenetic alterations is the basis of clonal evolution at the primary site and metastatic dissemination is its ultimate expression[59]. This notion is supported by the clinical observation that primary tumor size is a main risk factor for metastasis. This suggests that metastasis formation occurs rather in late disease stage as the end product of an evolutionary processes in the primary tumor (linear model of metastasis)[8,37,60,61]. According to this model many driver mutations found in the primary tumor are present at the metastatic site, and only a few additional mutations accumulate between primary tumor and metastases[62-64], including in breast cancer[65-68]. In the other hand, comparative genomic hybridization analysis in breast and other cancers revealed that DTCs display significantly more genetic aberration than in the primary tumors[69-72]. These observations imply that metastatic cells disseminate early during tumor development and then progress independently from the primary tumor through multiple steps of genetic mutations. Therefore, the parallel progression model of metastasis has been proposed[60]. Importantly, the two models are not mutually exclusive: a first vague of cancer cells may disseminate early during tumor formation, for example at the time of oncogene activation or EMT induction[73-75], followed by the late dissemination of cells that acquired metastatic properties through local evolution[64,76]. Recently, evidence for the parallel progression model in breast cancer was reported by using experimental models. By studying metastasis in a HER2-driven murine model of breast cancer, Harper et al.[77] showed that cancer cells migrate away from early lesions shortly after HER2 activation. In this model over 80% of the metastases were derived from early disseminated cancer cells. Using the MMTV-HER2 breast tumor model, Harper et al.[77] identified a subpopulation of ERBB2+/p-p38low/p-ATFlow/TWIST1high/E-CADlow early cancer cells that are invasive and can spread to distant organs (early disseminated cancer cells - eDCC). By using intra-vital imaging they showed that ErbB2+ eDCC precursors invaded locally, intravasated and lodged in target organs through a WNT-dependent EMT-like dissemination program. Strikingly, although the majority of eDCCs were TWIST1high/E-CADlow and dormant, they eventually formed metastasis. This experimental observation supports the notion that DTC/DCC can remain dormant for prolonged periods before resuming growth to form macroscopic metastases. As current adjuvant treatment in breast cancer seems to have reached a plateau in term of survival benefits, understanding how DTC and micro-metastases adapt to the distant environment, survive and eventually resume growth to form macroscopic metastases may identify new therapeutic opportunities[1,8,78].
In both the linear and parallel tumor progression models, the genomic instability of tumor cells is the basis of the evolutive process. The variability emerging among tumor cells within the same tumor tissue is referred to as intratumoral heterogeneity and is also found within metastases[79]. This heterogeneity may also be responsible to the presence of tumor cells with low and high tumorigenic potentials, the latter being CSCs or cancer initiating cells (CICs)[80]. Moreover, it is believed that heterogeneity also exists inside the CSC population itself. In such a scenario, metastasis progression and resistance to anti-tumor treatment are thought to be due to clonal evolution and selection much alike Darwinian evolution[59]. Intratumoral heterogeneity may also contribute to tumor dormancy or escape from it. Marusyk et al.[81] have used the MDA-MB-468 tumor cell model in vivo to show that a IL-11 expressing tumor population is able to drive non-cell-autonomous tumor growth from dormant tumor cells. This indicates that re-establishment of certain heterogeneity is necessary for tumor growth after seeding or treatment. In line with this observation, Aceto et al.[82] have reported that, CTC clusters have higher ability to seed metastasis compared to single CTC, involving at least in part altered DNA methylation[83]. Furthermore, Kmieciak et al.[84] showed that the heterogeneity of breast cancer cells in the levels of IFN-γ receptor α expression could determine a selective dormancy. Tumor cells expressing high levels of IFN-γ receptor α are eliminated by CD8+ T cells, while tumor cells with low expression levels do not die and remain dormant in the presence of IFN-γ producing CD8+ T cells. Thus, tumor heterogeneity contributes to tumor dormancy by providing cells with different genetic and biological features.
Metastatic breast cancer dormancy
As mentioned before, breast cancer metastasis occurs with a bimodal distribution with two peaks: a first one 1-2 years and a second one at 4-5 years after surgery, followed by a tailed extension up to 15 years[17,85,86]. These observations are inconsistent with a model of continuous growth kinetics and rather suggestive of discontinuous growth, thereby implying a period of dormancy[16,17,85-87]. Clinical observations also revealed that timing of appearance of metastasis has a similar profile for the different breast cancer subtypes, suggesting that after primary tumor removal, DTC from distinct subsets evolve following similar patterns but with different dominances (i.e., TNBC and HER2+ cancers tend to relapse at the early peak, compared to ER+/PR+ cancer which tend to relapse at the second peak or later[85,86,88,89]. These observations also suggest that relapses occurring at peaks may follow inducible and reproducible patterns based on defined mechanisms, for example tumor surgery[90], while relapses occurring in between or in the tailed extension may be due to unpredictable or random events, such as genetic mutations, epigenetic modifications or unrelated inflammatory events[65,67,68,91,92]. Accordingly, these clinical observations were modelled mathematically by considering known basic hazard rates and unknown variables[93,94]. Dormant disseminated cancer cells and micrometastases have been reported in breast cancer patients[95] and in experimental animal models[77,96-98]. Although dormancy is a common phenomenon in breast cancer, the underlying biological mechanisms remain ill characterized. Dormancy might be functionally considered as a transitional, metastable step of cell adaptation to a novel external stress, in particular a “foreign” soil. DTC lacking survival capabilities would rapidly die, while those that have acquired the latter would immediately grow to form macroscopic metastases without latency [Figure 1]. Mechanistically, three forms of cancer dormancy have been described and many genes and molecules involved identified[99-102]: cellular dormancy, microenvironmental (angiogenic) dormancy and immunological dormancy. These three forms of dormancy are not mutually exclusive and it is likely that clinical dormancy is owed by their combination and interrelation.
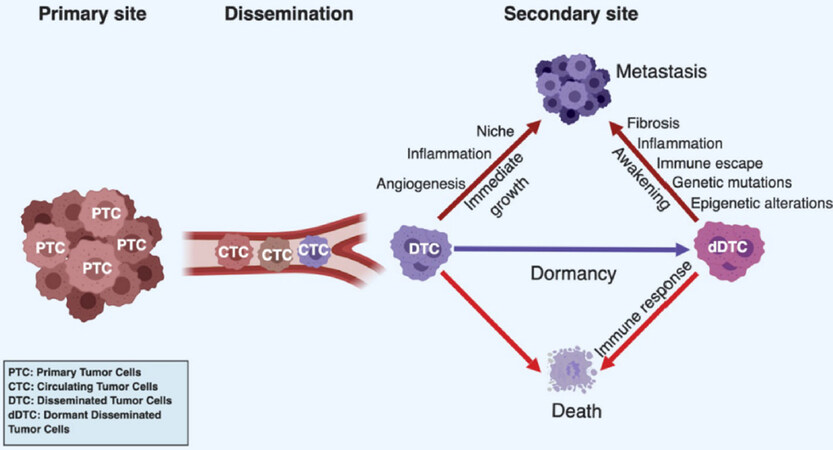
Figure 1. Dormancy in cancer progression. Tumor cells can leave the primary tumor site and enter the systemic circulation as circulating tumor cells (CTCs). Once surviving CTCs have reached a target organ, they seed into a new tissue as disseminated tumor cells (DTCs). Their fate is diverse depending on their cell autonomous capacities and complementary cues provided by the local environment. DTCs can rapidly die if they fail to adapt to the new condition or are killed by the immune system. They can immediately resume proliferation if they have acquired full autonomy for cell survival and proliferation or the local microenvironment provide missing complementary cues. In addition, proliferating cells have to evade the immune system. Alternatively, DTC or small tumor cell clusters, can enter a state of dormancy if cell autonomous or microenvironmental signals are sufficient to maintain survival but do not effectively support growth or the immune system keeps them in check by preventing their expansion. Dormant tumor cells can eventually die by exhaustion, be killed by the immune system or resume proliferation and generate clinically relevant metastases at later time points
Cellular dormancy: survival of non-proliferating solitary cells
Dormant DTC cells have developed mechanisms of survival in a foreign environment, but not yet those allowing unrestricted growth. They enter a state of cell cycle arrest (i.e., G0-G1) and survive as non-proliferating solitary cells or as small cell clusters. Accordingly, solitary dormant cancer cells should be negative for the proliferation marker Ki67 as well as for apoptosis markers such as the terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL). Entry into dormancy and subsequent reactivation seem to be regulated by intrinsic programs and by contextual cues, similar to those involved in the physiological regulation of adult tissue stem cells[103]. Lack of signaling from the matrix seems to play a role in this form of dormancy, as loss of α5β1 integrin expression or function and inhibition of uPAR induce dormancy through the inhibition of the RAS-RAF-ERK signaling pathway, activation of p38/JNK signaling, and induction of p53/Rb-dependent cell-cycle arrest[103-105]. By studying DTC in an experimental model of head and neck squamous cell carcinoma, Sosa et al.[106] have shown that epigenetic upregulation of orphan nuclear receptor NR2F1 (COUP-TF1) plays a critical role in maintaining DTCs in a dormant state. This finding has been further extended by a study in breast cancer patients, in which NR2F1 was tested as a dormancy marker. Breast cancer patients with < 1% NR2F1high DTC in bone marrow aspirate had all systemic relapse within 12 months, while only half of the patients with > 50% NR2F1high remained metastasis-free[107].
Alterations in cell signaling have been found associated with tumor dormancy. High levels of ERK1/2 activity lean toward a higher proliferation, so the ratio of ERK1/2 to p38 MAPK regulates the cell cycle suggesting that the cross talk between mitogenic and stress signals may be relevant to induce cellular dormancy[108]. Impinging on the PI3K signaling cascade was shown to lead to quiescence and the activation of autophagy[109]. Dormant tumor cells express high levels of ARHI, an inhibitor of the PI3K-AKT cascade, and ARHI silencing awakens dormant cells of several tumor types, and promotes their proliferation[110,111]. Consistently, very low or absent AKT signaling in DTC from breast cancer patient has been shown to correlate with the state of dormancy[112]. Strikingly, however, mTOR, a known target of AKT, is found to be activated in quiescent cancer cells. This activation is independent of AKT and is maintained by the small GTPase RHEB with anti-apoptotic activity and itself under control of the stress-regulated transcription factor ATF6α and high p38 kinase activity. Thus, mTOR seems to be a critical node integrating diverse signaling pathways regulating dormancy[111,113,114]. Recently Malladi et al.[115] have shown that human breast and lung carcinoma cells express stem-cell like SOX transcription factors, which induces autocrine expression of DKK1, a WNT inhibitor, resulting in a state of metastatic latency and immune evasion consistent with dormancy.
Interaction with the extracellular matrix is also implicated in controlling dormancy[116], paralleling the role of cell-matrix interaction in physiological CSC niches[117]. For example, the uPAR interaction with a5b1 integrin increases ERK activity and integrin binding to fibronectin fibrils suppresses p38 activity, increases ERK activity and promotes cell proliferation. Accordingly, low uPAR-expressing cells that are growth arrested (dormant) in vivo, have a high p38/ERK activity ratio and fail to assemble fibronectin fibrils and ligate a5b1 integrin[104]. Integrin a5b1 was shown to promote survival of growth-arrested breast cancer cells reminiscent of breast cancer dormancy in bone marrow[118]. Collagen-rich matrix (fibrosis) at the metastatic site is also a critical determinant of DTC transition from dormancy to metastatic growth[116]. For instance, hepatocellular carcinoma cells that colonize rigid matrix resume growth and proliferation through TGF-β1 signaling, while cells colonizing a softer matrix remain dormant[116]. The proliferative switch of dormant DTCs in response to fibrosis in a mouse model of breast cancer was shown to be also mediated by β1 integrin[116]. 3D-in vitro models and further in vivo studies demonstrated the critical role of type I collagen in the proliferative fate of DTCs[119-121]. Another matrix protein, periostin, produced by TGF-β1-stimulated stromal fibroblasts and angiogenic blood vessels[47,122], can drive DTCs escape from dormancy through WNT signaling in breast carcinoma[123]. Interfering with extracellular matrix-integrin interaction, in particular b1 integrins, has been proposed as therapeutic approach to promote dormancy, including in breast cancer[124,125].
Autophagy appears to promote DTCs survival and dormancy by maintaining DTCs viable under conditions of microenvironmental stress[126]. Autophagy also promotes survival of DTC against chemotherapy-induced cell stress[126]. On the other side, in breast cancer the lack of autophagy is associated with early tumor recurrence and escape from dormancy[127]. It is important to note, that cellular dormancy is not just a feature of cancer cells but it also occurs in normal cells. For example, hair follicle (bulge) stem cells, muscle stem cells (satellite cells), hematopoietic and liver stem cells are rather quiescent under homeostatic conditions and can be rapidly activated during tissue regeneration and repair[128-130]. These observations raised the idea that cancer cells may hijack the signaling cascade used physiologically by these cells to enter, maintain and exit dormancy during homeostatic and regenerative conditions[103,131].
Senescence, a special form of cellular dormancy?
The term senescence was originally introduced to describe primary human fibroblasts in culture, that after reaching a maximal number of passages (cell divisions) entered a state of permanent growth arrest[132,133]. Senescent cells remain metabolically and synthetically active but show significant alterations in morphology[132-134]. Following these original observations, scientists discovered that replicative senescence is driven by telomere shortening[135]. Telomerase, an enzyme reconstituting telomers, is expressed in germ line cells, early embryonic cells, but not in normal cells, and is re-expressed in most cancer cells[134]. Besides telomere shortening, other stimuli have been found to induce the senescence[136] including DNA damage[137], chemotherapy[138] and oncogene activation[139]. Despite the nature of the stimulus initiating the senescence cascade, the signals ultimately converge to the p53/p21 and/or p16INK4a/pRB pathways[136]. The tumor suppressor proteins DEC1 (Deleted in Esophageal Cancer) and Decoy Receptor 2 are also used as senescence markers in some cell types[140]. Importantly, when oncogenic HRAS (HRAS12V) was used to transform immortalized embryo fibroblasts into tumorigenic cells[141], unexpectedly, it induced senescence in normal cells associated with the accumulation of p53 and p16INK4a[142]. Thus, senescence may act as a tumor-suppressor mechanism in response to oncogene activation[133]. Generally, overexpression or sustained activation of one of the tumor suppressors p53, p21, p16INK4a, or pRB is sufficient to induce senescence[136,143-145]. Importantly, growth arrest caused by senescence is considered as irreversible as senescent cell could not be stimulated to resume proliferation by exposure to growth factors[136,141,143]. However, the genetic or epigenetic alternations which cause the shift of the senescence maintaining mechanisms, such as the inactivation of tumor suppressor genes p53 and/or p16INK4a, could potentially push the cells to re-enter the cell cycle[136,143].
It is not clear whether senescence is one of the mechanisms that drives tumor dormancy and late relapse, but there are some potential links suggesting so. Senescence-associated secretory phenotype (SASP) defines the spectrum of factors secreted by senescent cells. It consists of a mixture of chemokines, cytokines, growth factors and proteases, many of which are pro-inflammatory[136]. The cytokine GM-CSF (granulocyte-macrophage colony-stimulating factor, also known as CSF2), one of the components from SASP, induces differentiation of myeloid dendritic cells, which present tumor-associated antigens (TAAs), resulting in the activation of the immune system, enhanced immunosurveillance and improved tumor control[146,147]. Furthermore, Braumüller et al.[148] showed that adaptive TH1 cell are capable, via the combined secretion of IFN-γ and TNFR to induce tumor cells senescence. In conclusion, while senesce is a mechanism capable of negatively controlling tumor growth, its relevance in dormancy is not fully demonstrated.
Microenvironmental dormancy: deficient support from the microenvironment
In this form of dormancy, the fate of the DTCs is mainly controlled by the immediate, and possibly distant, host environment[126,149]: cancer cells proliferate but fail to grow as a tumor mass because proliferation is balanced by cell death[99]. This situation has been described first when disseminated cancer cells fail to induce blood vessels[150]. During the early stage of tumor spreading, DTCs associate to preexisting (coopted) blood vessels where oxygen and nutrient levels are highest[151]. Importantly, quiescent mature blood vessels keep DTCs in a state of dormancy through angiocrine communication[122]. With the growing tumor mass, however, the increasing metabolic demand call for the formation of novel blood vessels through an angiogenic switch[152]. Several molecules, including hypoxia-inducible factor 1, vascular endothelial growth factor (VEGF), placental growth factor and angiopoietin-1, are induced upon metabolic stress or hypoxia and initiate endothelial cell sprouting from preexisting vessels[153]. Angiogenic endothelial sprouts secrete periostin and TGF-β1 to create a microenvironment that promotes DTCs exit from dormancy and accelerates proliferation[122]. Thus, the failure of initiating the angiogenic switch will keep the tumor mass small, a condition called angiogenic dormancy[154]. Conversely, a short-term angiogenic burst may awaken dormant tumor cells[155]. For example, in the mouse model of Lewis lung carcinoma, dormant micrometastatic cells can be induced to to grow by the expression of VEGF and the recruitment of bone-marrow-derived endothelial progenitor cells[156].
Importantly, inflammatory cells recruited to the tumor microenvironment are critical inducer of the angiogenic switch and lack of recruitment may therefore contribute to angiogenic dormancy[11,157]. In particular the polarization of tumor associated macrophages toward the M2 phenotype results in the formation of a metastatic niche favoring tumor cell outgrowth[158]. In addition, the angiogenic endothelium triggers a T helper 2-mediated inflammatory response, which can accelerate metastatic outgrowth in tumor models[159]. The hemopoietic stem cell niche also supports quiescence and survival through the CXCL12/CXCR4 pathway and Src pathway[47,160] as well as the TGF-β2-rich bone marrow microenvironment[161]. On the other hand, expression of VCAM1 on DTCs promotes escape from dormancy. This is due to the recruitment of osteoclast progenitors via α4β1 integrin binding to VECAM1 causing the breakdown of the bone matrix and stimulation of DTC to grow and form metastases[162,163]. Likewise, metastatic outgrowth following skeletal traumas was associated with bone remodeling in the reactivation of DTCs via TNFα, IL1β, IL6 and prostaglandin E2 production[163]. Thus, the cellular tumor microenvironment, in particular inflammatory cells, endothelial cells and osteoclasts should be considered as critical regulators of tumor dormancy.
Immunological dormancy: active control by the immune system
The immune system can specifically recognize tumor cells through TAAs and initiate an anti-tumor immune response through a multi-step process called immunoediting[164,165]: initially, the immune response can effectively eliminate cancer cells and no metastases are formed (elimination phase). If a balance between the tumor suppressive immune response and tumor evasion is achieved (equilibrium phase), the tumor mass remains constant resulting in a state of immunological dormancy[166]. Tumor cells may eventually succeed in evading the immune system through a combination of genetic evolution and exhaustion of the immune system, and resuming growth to form macroscopic metastases (escape phase). Evasion involves multiple mechanisms, including the down-regulation of TAAs or MHC molecules, secretion of immunosuppressive cytokines, (e.g., IL-10 or TGF-b), recruitment of regulatory T cells (Treg), myeloid derived suppressor cells (MDSC) or alternatively (M2) - polarized macrophages or by expressing immunosuppressive molecules on the cell surface, such as PDL1 or B7[167-169], in multiple cancer types, including breast cancer[170]. The occurrence and relevance of immunological dormancy in human cancer is difficult to grasp, as the absence of biomarkers of dormancy makes it hard to investigate in patients. Also, there is paucity of clinically relevant in vivo model for experimental studies[100]. Pommier et al.[171] have recently reported that patients and mice with pancreatic ductal adenocarcinoma contained single quiescent DTCs lacking MHC-I expression, which enabled them to escape immunity and establish latent metastases. Four cases of breast cancer transmission to immunosuppressed transplant recipients from a single, clinically healthy donor have been recently described[172]. The latency time to metastasis formation ranged from 16 months to 6 years, and transmissions did not occur in a patient that discontinued immunosuppression. This unintentional situation suggested that donor tissues harbored DTCs, which persisted in a latent state because of immune control, while the immunosuppression of the recipient allowed the DTCs to resume growth[172]. Once cells have escaped immunosurveillance it is unlikely that they can re-enter a second state of immunological dormancy, because the mechanisms that eluded immunosurveillance are generally part of the genetic or epigenetic tumor clonal evolution and therefore irreversible[59,91].
The recent observation that immunosuppressive immune cells promote angiogenesis[157], and that some angiogenic factors, including VEGF, are immunosuppressive, established an important cross-talk between these two systems[173-175]. They implicate that suppression of angiogenesis may stimulate the immune system and that reversal of immunosuppression may inhibit angiogenesis in a bi-directional cross-talk. In support of this notion, several trials have been performed to improve the anti-tumor immune response with anti-angiogenic drugs[176]. Combination of anti-angiogenic agents with immunotherapy is currently being considered as strategy to improve the response rates and duration of immunotherapy[177]. For example, VEGF has been shown to suppress the immune-response by impinging on maturation of dendritic cells[178,179]. Thus, inhibition of VEGF abrogates its immunosuppressive effect and improves the antitumor effect of adoptive cellular immunotherapy[180]. Combining anti-VEGF therapies with check-point inhibitors (e.g., anti-PD-L1) has shown synergy and positive outcomes in phases I to III studies, particularly in patients with high VEGF levels[177]. Using the 4T1 experimental model of TNBC we have demonstrated that inhibition of tumor angiogenesis with the anti-VEGFR-2 antibody DC101, attenuated the inhibitory effect of MDSC on T cell proliferation and decreased the frequency of Tregs in primary tumors and lung metastases[181]. Combined angiopoietin-2 and VEGF inhibition was shown to promote superior vascular regression, tumor necrosis, and enhanced the perivascular recruitment of cytotoxic T lymphocytes, as compared to the single agents in multiple cancer models. Addition of a PD-1 blocker unleashed the cytotoxic activity resulting in improved tumor control[182,183]. These findings support the rationale for combining anti-angiogenic therapy with immune checkpoints inhibitors in cancer therapy, including in breast cancer. In addition to immunosuppressive angiogenic molecules, endothelial cells themselves also play a direct role in modulating cellular dormancy. Quiescent endothelial-derived thrombospondin-1 induces sustained quiescence of DTC in breast cancer, while sprouting vessels wake dormant DTC and promote their outgrowth[122,184,185][Figure 2].
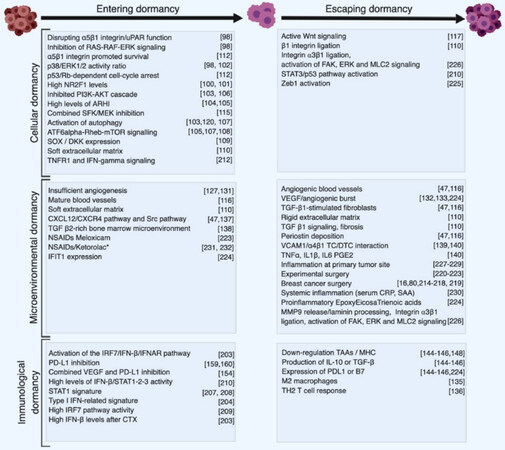
Figure 2. Synthetic summary of cellular and molecular events and interventions associated with dormancy. Cellular and molecular events promoting entering into dormancy or escape from dormancy are listed based on their implication in the three forms of dormancy. Classification is based on the main mechanisms and does not consider potential cross talk between different types of dormancy
Dormant cancer cells are resistant to the cytotoxic effects of chemotherapy
Adjuvant chemotherapy significantly improves breast cancer patient survival, in particular for TNBC, by decreasing the hazard of relapse after surgical removal of the primary tumor[26,32,186-188]. Relapses and metastases however still occur in a fraction of patients and are likely due to tumor cells that had already invaded the surrounding tissue, lymphatic vessels and lymph nodes, or disseminated through the blood stream to the bone marrow and distant sites prior to surgery and resume grow at a later time point[60,77,189]. As it is generally assumed that adjuvant chemotherapy acts by killing tumor cells, a corollary of this assumption is that relapses are due to DTC resuming growth at a later time point after surviving chemotherapy. Dormant, poorly proliferative DTC may not be affected by chemotherapy and may persist upon treatment since chemotherapeutic drugs mainly target highly proliferating cells. This has been shown for instance in acute lymphoblastic leukemia, colorectal, lung, liver and breast cancers[190-196]. In addition, chemotherapy pre-exposed DTC may develop mechanisms of chemoresistance and become less responsive to subsequent chemotherapies, as is often observed at relapse in patients. CSCs have been shown to be chemo-resistant and to be responsible for post-therapy relapses[197]. Chemotherapy causes enrichment of CSCs thereby facilitating recurrences and resistance to further chemotherapies in multiple cancers including glioma, ovarian, liver, colon, breast cancers[197]. Resistance involves multiple mechanisms, such as activation of signaling pathways prominent in stem cells (e.g., WNT, NOTCH, HEDGEHOG)[198-200], but also pathways that are frequently mutated and activated in cancer, in particular the EGFR-HER2/PI3K/PTEN/Akt/mTORC pathway[198,200,201]. CSC are often enriched at sites of chronic hypoxia leading to the activation of the HIF pathway[200,202]. Interestingly, HIF activation leads to the initiation of protective pathways, including WNT and NOTCH, and genes of the ATP-binding cassette (ABC) drug transporter family members, such as MDR1, MRP1 and ABCG2 which are responsible for the efflux of cytotoxic drugs from the cells[203-208]. Additional, HIF-independent mechanisms have been reported[209]. In short, the mechanisms behind the long-term beneficial effects of adjuvant chemotherapy remain in part elusive and cannot be fully explained by the direct cytotoxic activity of chemotherapy as dormant/CSC that are mostly responsible for late relapses that are highly resistant to chemotherapy.
Immune response and chemotherapy in breast cancer
Cumulating evidence indicates that tumor infiltrating lymphocytes (TILs) play an active role in controlling progression and clinical outcome in breast cancer, particularly in highly proliferative TNBC and HER2+ cancers[210-212], and in conjunction with chemotherapy[213,214]. This is particularly relevant to TNBC, as these cancers present the richest presence of TILs, most notably CD8+ T lymphocytes, and tertiary lymphoid structures[211,215,216]. Increased numbers of infiltrating TIL in TNBC, are associated with an improved pathological complete response to chemotherapy[217], decreased rates of recurrences and improved survival[210,216,218]. Evaluation of TILs in breast cancer has been recommended as an immunological biomarker with prognostic and potentially predictive values, mainly in TNBC and HER2-amplified breast cancers[219]. In TNBC, expression of antigen presenting MHC class II pathway molecules is associated with a better outcome, consistent with the hypothesis that a functional antigen presentation pathway may trigger a protective antitumor immune response in response to chemotherapy[220]. Ladoire et al.[221] demonstrated that pathological complete response to neoadjuvant chemotherapy of breast carcinoma resulted in the disappearance of tumor-infiltrating FOXP3+ regulatory T cells and the increase in tumor infiltrating cytotoxic TiA1+ and granzyme B+ T cells, consistent with the induction of an antitumor immune response by chemotherapy. While the association between lymphocytic infiltrates, and improved outcome after chemotherapy appears to be strongest in TNBC and HER2+ breast cancer subtypes, the association with luminal tumors is less clear, and may be limited by the reduced immune infiltration or by the greater tumor heterogeneity of these tumors[222]. Several recent experimental studies have shown that chemotherapy can induce a therapeutic anti-tumor immune response. For example, Ma et al.[223,224], reported that anthracycline-based chemotherapy induces the release of ATP by dying breast cancer cells, which promotes the recruitment and differentiation of CD11c+CD11b+Ly6Chigh antigen presenting cells. Depletion or preventing tumor infiltration by these cells abolished the anti-tumor immune responses elicited by anthracyclines[223,224].
Besides these desired immunological effects, chemotherapy can also induce unwanted, immune-mediated tumor-promoting effects. For example, increased expression of TNFa in the breast tumor microenvironment due to chemotherapy, induces CXCL1/2 expression via NF-κB activation in breast cancer cells and initiates a paracrine loop involving myeloid cell-derived S100A8/9 to enhance cancer cell survival and chemo-resistance. Inhibition of CXCR2 blunt this TNFa-induced response and augments the efficacy of chemotherapy, particularly against breast cancer metastasis[225].
Taken together, there is growing evidence to support the notion that chemotherapy, in particular those based on anthracyclines, can elicit an effective anti-tumor immune response in breast cancer, mainly in the TNBC and HER2+ subtypes.
Chemotherapy-induced immunological breast cancer dormancy
One important question emerging from these studies is whether the cytotoxic activity of chemotherapy and the elicited immune response, dominated by CD8+ T lymphocytes, may be sufficient to effectively kill cancer cells or whether additional mechanisms may be involved. This is particularly relevant to dormant DTC as these cells are naturally less responsive to chemotherapy due to the fact that they are not or low proliferative. We were interested in the possibility of whether a short chemotherapy treatment, induce a long-lasting immune response leading to immunological dormancy. We recently addressed this question experimentally[226]. To this end, we treated the TNBC-like 4T1 cells with high dose Methotrexate and Doxorubicin in vitro and characterized the in vitro and in vivo behavior of the surviving cells (MR20 and DR500 derived from Methotrexate and Doxorubicin treatment, respectively). The hallmark of the surviving cell lines was the dormant phenotype at the primary (MR20 in the mammary gland) or at the metastatic (MR20 and DR500 in the lung) site. MR20 cells grew significantly slower in vitro compared to parental 4T1 cells and this was due to increased cell death, while there was no significant alteration in the cell cycle. When injected orthotopically in the mammary fat pad of immunocompetent BALB/c mice, only about half of the MR20-injected mice developed tumors with a longer latency (between 6 week and 4 months) compared to parental 4T1 cells (between 2 and 4 weeks). These tumor cells grew with nearly the same rate as parental 4T1 tumors and were highly metastatic. DR500 cells formed primary tumors but no lung metastases. Both conditions were consistent with dormancy. In BALB/c mice MR20 and DR500 cells twisted the immune response from CD11b+ Gr1+ MDSC-dominated to CD4/8 T cell, B cell and DC dominated response. When injected in vivo in immunodeficient NSG mice, however, treated cells formed tumors without latency. We then performed a genome wide gene expression analysis of parental 4T1, MR20 and DR500 cells and the dominant trait observed was a type I IFN gene expression signature and the sustained activation of the IRF7/IFN-β/IFNAR pathway. Upregulated IRF7 expression in treated cancer cells was responsible for suppressed mobilization of CD11b+ Gr1+ MDSCs, increased expansion of DCs, T and B lymphocytes and chemo resistance. Silencing IRF7 or blocking IFNAR1 resulted in escape from dormancy in vivo. Elevated levels of IFN-β were present in the blood of mice injected with dormant cells, while MR20 cells escaping dormancy and forming late tumors no longer expressed IFN-β. Interestingly, the dormant D2.0R murine breast cancer cells that are generally considered as a model of cellular dormancy, also induced a T-cell twisted immune response and had a constitutive active IRF7/IFN-β/IFNAR pathway [Figure 3]. To collect evidence that the activation of the type I IFN pathway was associated with a better response to chemotherapy in patients, we monitored IFN-β levels in serum samples of ER- breast cancer patients treated with neoadjuvant chemotherapy (TOP trial NCT00162812). Patients with detectable IFN-β during neoadjuvant therapy had significantly longer distant metastasis free survival (DMFS) compared to patients with undetectable levels[226].
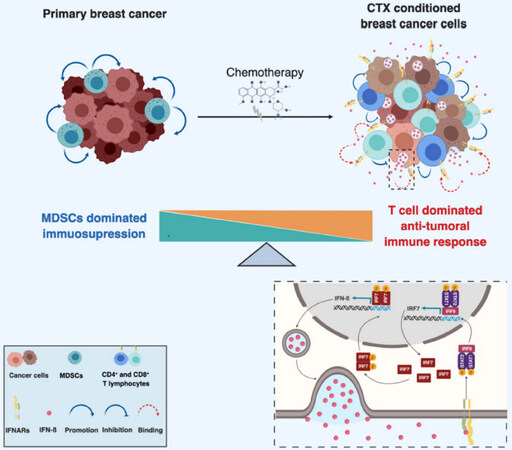
Figure 3. Schematic model of chemotherapy-induced immunological dormancy. Primary breast cancer cells escape immune elimination by inducing the expansion of myeloid derived suppressive cells (MDSC) which also promote tumor growth. Chemotherapy induces a type I IFN response in treated tumor cells, resulting in an autocrine and self-sustained increase of IRF7 expression and activation, which in turn induces expression and secretion of IFN-β. Secreted IFN-β binds to IFNAR and induces signaling in both immune cells and tumor cells. IFNARs signaling in tumor cells activates STAT1/STAT2/IRF9 complex which further induces the expression of IFN-β responsive genes including IRF7 resulting in a sustained autocrine IFN-β expression and secretion. Paracrine activation of IFNARs on immune cells stimulates the expansion of tumor suppressive lymphocytes (e.g., CD4+ and CD8+ T cells) and prevents the mobilization of MDSCs, resulting in the switch of the immune response from immunosuppressive to anti-tumoral
Type I IFN response to chemotherapy mediates immunological dormancy in breast cancer
Taken together our results demonstrate that sustained activation of the IFN-β/IFNAR/IRF7 signaling axis in chemotherapy-treated TNBC-like murine breast cancer cells instigates immunological dormancy. Elevated levels of IFN-β in the serum of TNBC patients during adjuvant therapy correlates with a shorter DMFS. Acute exposure of breast cancer cells to chemotherapy induced IFN-β expression[226]. Thus IFN-β may be considered as a potential therapeutic tool to improve chemotherapy efficacy and clinical outcome as well as a potential predictive biomarker to identify responding vs. non-responding patients. While the implication of type I IFN in dormancy is novel, previous reports have implicated it in acute anti-tumor response to chemotherapy. Sistigu et al.[227] reported that anthracycline-based chemotherapy rapidly induced the production of type I IFN in cancer cells via TLR3 activation resulting in CXCL10 secretion promoting an anti-tumor immune response mimicking those induced by viruses. Moreover, a type I IFN-related signature predicted clinical responses to anthracycline-based chemotherapy in breast cancer patients. Using a panel of ER- breast cancer PDXs before and after chemotherapy, Legrier et al.[228] demonstrated activation of the IFN/STAT1 pathway and expression of IFN-inducible genes, early after chemotherapy treatment. IFN-a deficient DC were shown to accumulate in aggressive breast cancers favoring the expansion of Tregs implicating that IFN-α deficiency may contribute to tumor immune tolerance and poor clinical outcome[229]. Previous studies based on tumor-derived IFN signatures have shown that IFN-regulated genes may correlate with favorable outcomes. A STAT1 signature before therapy was associated with a better response to neoadjuvant chemotherapy[230] and better prognosis in TNBC and HER2+ breast cancers[231]. High IRF7 pathway activity in human breast cancer predicted bone relapse-free survival and, and protected mice against bone metastasis[232]. In the same study, treatment with IFN-α improved bone metastasis-free survival[232]. High level of IFN-β activates STAT1, STAT2 and STAT3 to facilitate cell autonomous cellular dormancy of melanoma repopulating cells[233]. Taken together, our recent data extend the role of type I IFN in immunoediting in cancer[234] to a putative role in inducing immunological dormancy after chemotherapy in TNBC. An important outstanding question raised by our study, concerns the effector mechanism of T cell mediated dormancy. While it is likely that direct CD8+ T cell mediated killing and immune control is involved, particularly since CD4+ Th1 T cells produce IFN-γ which upregulates MHC expression on tumor cells thereby facilitating killing by CD8+ T cells[235], it is plausible that additional mechanisms may be involved. One possibility is suppression of tumor angiogenesis by the secretion of anti-angiogenic factors, such as CXCL9 and CXCL10 by CD4+ T cells[235]. Another putative mechanism is induction of senescence. CD4+ Th1 T cells also produce TNF-α, which in combination with IFN-γ induces an irreversible state of cellular senescence keeping DTC dormant[235,236]. Ongoing projects in our lab are aimed at unraveling the effectors steps.
Breaking dormancy: impact of inflammation
Clinical and experimental observations indicate that dormant tumor cells can resume proliferation and generate macroscopic metastases at various times after primary tumor treatment[17,85]. Escape from dormancy could be initiated by cell autonomous events, such as oncogenic mutation or inactivation of tumor suppressor genes. Alternatively, alterations in the host micro- or macro-environment may promote escape from dormancy. Metastatic growth following surgical removal of the primary tumor itself is often observed in clinical settings with predictable patterns, including in breast cancer[16,86,237-241]. Reconstructive surgery after mastectomy for breast cancer significantly accelerates relapse rates proportionally to the extent of surgery, when compared to primary surgery. However, no worsening in long-term survival was reported, consistent with an effect on breaking dormancy and accelerating progression but not altering the overall outcome[242]. Enhancement of metastatic growth induced by experimental surgery has been long observed in animal models, suggesting that surgical wounding itself may be directly involved in breaking dormancy[243-245]. Recently Krall et al.[246] described an experimental model that links the wound-healing response after surgery to the outgrowth of DTC at distant sites. The link is mediated by the systemic inflammatory response induced after surgery suppressing a tumor-specific T cell response, thereby resulting in tumor growth. Consistent with these findings, perioperative anti-inflammatory treatment significantly reduced tumor outgrowth. Epoxyeicosatrienoic acids, a family of pro-inflammatory molecules, stimulated escape from dormancy in several tumor models independently of the primary tumor and was associated with increased production of VEGF by endothelial cells[247]. Recently, it was reported that lung inflammation promotes escape from tumor latency by inducing ZEB1 expression, a regulator of the EMT[248]. Another recent study reported that neutrophil extracellular traps (NET) produced by neutrophils during repeated acute inflammation awaken dormant cancer cells in a mouse model of breast cancer[249]. The releasing of neutrophil elastase and MMP9 from NET then remodel laminin in the extracellular matrix rendering it accessible to a3β1 integrin. Ligated a3β1 integrin leads to the activation of FAK, ERK and MLC2 signaling resulting in the awakening of the dormant cancer cells[249].
Clinical and experimental evidence suggests that sustained inflammation may also promote relapses[91]. Correlations between inflammation at primary tumor site and risk or recurrences were reported for several cancers including oral[250], endometrial[251] and breast cancers[250,252]. Elevated levels of circulating C-reactive protein and serum amyloid A, two proteins of the inflammatory response, are associated with reduced overall survival in breast cancer, independently of body mass index, age and tumor type and stage, consistent with inflammation being involved in breaking dormancy[253]. Interestingly, perioperative administration of the analgesic nonsteroidal anti-inflammatory drug (NSAID) ketorolac was reported to suppress early breast cancer relapse particularly in TNBC patients[254] and to reduce distant recurrences in patients with increased BMI[255]. Taken together, there is compelling clinical and experimental evidence indicating that inflammation promotes breast (and other) cancer relapses by breaking dormancy.
Outlook and perspectives
Tumor dormancy is widely accepted as one discrete step during multistage tumor progression and bears considerable therapeutic potential[256]. The rapid translation of this innovative concept to the patient is limited by the paucity of therapeutic tools currently available in the clinic. Treatments directly aimed at DTC by critical cell survival and proliferation pathways (e.g., PI3K-AKT or MAPK pathways), stem cell pathways (e.g., WNT, NOTCH) or cell adhesion molecules (e.g., b1 integrin) would be virtually excluded for such an approach given their expected long-term systemic toxicities. We are proposing here selected strategies based on limiting the host (unwanted) inflammatory response and stimulating the (wanted) anti-tumoral-immune response that may be rapidly tested in clinical-translation studies in breast cancer [Figure 4].
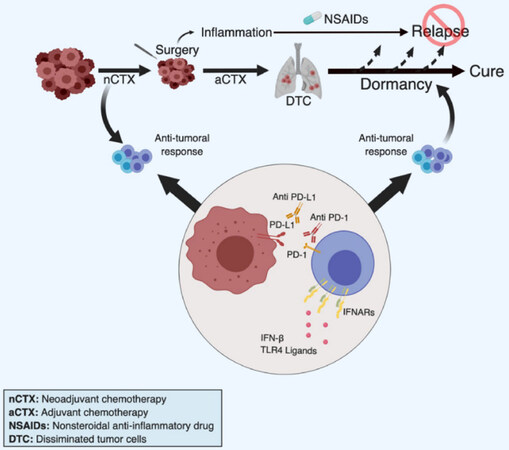
Figure 4. Strategies to improve chemotherapy-induced immunological dormancy. Based on work by others and us, we propose four clinically feasible approaches to induce or maintain breast cancer dormancy, primarily in TNBC. Firstly, we propose neo-adjuvant chemotherapy (nCTX) to promote chemotherapy-induced immune response. Chemotherapy may be pursued as adjuvant therapy (aCTX) if necessary. Secondly, during and following surgery we propose the administration of NSAIDs (in particular Ketorolac) to prevent surgery-associated inflammation that may potentially promote relapses. Thirdly, the cytotoxic immune response could be stimulated by providing type I IFN, or inducers of an interferon response such as TLR4 ligands, in particularly in low IFN-producing patients. Fourthly, addition of checkpoint inhibitors, such as anti-PD1/PDL-1 antibodies, may be applied to maintain the immune response active
NSAIDs
There is growing clinical and experimental evidence that inflammation can trigger cancer relapse, in particular in breast cancer, and that NSAIDs treatment can prolong dormancy and delay or reduce relapses[246,248,249,252,254,255,257]. The overall positive safety profile of aspirin and other NSAIDs would make them realistic candidate drugs for such long-term therapies[258]. Even more interesting, as a short perioperative treatment with ketorolac has been shown to significantly decrease the risk of breast cancer relapses particularly in obese patients[255], a short term NSAID treatment at time of surgery may have long-lasting effects through suppression of surgery-associated inflammation.
Neoadjuvant chemotherapy
A second approach to consider, is to exploit the ability of chemotherapy to elicit an effective immune response in breast cancer, particularly in lymphocyte-infiltrated TNBC or HER2+ tumors[211,212,222,224,226,227,259]. Our and other’s results indicate that chemotherapy induces an effective anti-tumor immune response upon tumor cell treatment[214,226,227]. Clinically this implies that neo-adjuvant/pre-operative chemotherapies may be more effective in inducing a long-lasting, protective immune response compared to classical adjuvant/post-operative therapies, due to the larger targeted tumor mass[224,260]. Indeed, neoadjuvant chemotherapy is already used in highly proliferative breast cancers (i.e., Luminal B, HER2+ and TNBC) with high frequencies of pathological complete responses (pCR). Interestingly, paclitaxel, in combination with trastuzumab, induced a high rate of pCR in HER2+ patients, likely due to the synergy between the immunomodulating properties of these drugs[260]. Ladoire et al.[221] reported that pCR to breast cancer neoadjuvant chemotherapy was associated with the disappearance of tumor-infiltrating FOXP3+ Tregs and recruitment of CD8+ T cells, consistent with the induction of an antitumor immune response by chemotherapy.
Type I IFN
In our experimental model we have shown that Type I IFN response is essential to elicit a state of immunological breast cancer dormancy[226]. Others have shown that exogenous addition of type I IFN boosted an insufficient response to chemotherapy in an experimental model of breast cancer and that a type I IFN-related signature predicted clinical responses to anthracycline-based chemotherapy in breast cancer patients[227]. We demonstrated that patients with high levels of serum IFN-β during neoadjuvant therapy have a better outcome compared to patients with low levels[226]. This suggests that administration of type I IFN during neo-adjuvant therapy may be effective in mounting a long-lasting immune response in particular in those patients with low endogenous type I IFN levels. Type I IFN, in particular IFN-α has been already tested as immunostimulatory anti-cancer agent, especially in melanoma and kidney cancers, albeit with mild results, in part also due to the need of repeated administrations and its intrinsic toxicity[261-263]. Alternatively, less toxic inducers of Type I IFN response, such as TLR-ligands of STING stimulators may be considered[227,264].
Check point inhibitors
A complementary strategy to IFN administration could be the use of check point inhibitors, in particular anti-PD-1/PD-L1 antibodies to relieve tumor-induced immunosuppression[265]. In breast cancer, immunotherapy is being explored, in particular in patients with tumors expressing PD-L1 and infiltrated with lymphocytes[266]. Potential response to PD-1 or PD-L1 inhibitors was demonstrated in metastatic TNBC[267] and HER2+ breast cancers[268]. However, because the number of potential neoantigens available for immune response in most breast cancers, responses are modest compared with other cancers such as lung and melanoma, the use of check-point inhibitors may require combination with other therapies[269]. Therefore, combination with neo-adjuvant chemotherapy (causing the release of tumor antigens), anti-angiogenesis therapies (suppressing inhibitory cues)[182] or Type I IFN (acting immunostimulating)[270] may be more effective and should be explored.
Biomarkers of dormancy
There are currently no specific non-invasive biomarkers to monitor breast cancer dormancy of clinical utility[271]. Such markers would allow personalized follow up and accelerate therapeutic decisions in case of evidence of disease progression. CD44+/CD24- CTC subsets along with combinatorial expression of uPAR and b1 integrin, as well as proliferation and apoptosis markers in CTC of early breast cancer patients, have been explored for potential use as biomarker of dormancy or aggressiveness[272,273]. Genomic analysis (i.e., SNP/CNV) of circulating ctDNA was shown to potentially identify breast cancer patients with dormant/minimal residual disease[274]. Also, serum inflammatory markers might serve as biomarkers of relapse in disease-free patients, as inflammation is associated with escape from dormancy but will likely be unspecific and of limited sensitivity[275]. Serum IFN-β levels were associated with longer DMFS (as a surrogate of dormancy) in our model of chemotherapy-induced dormancy and during neoadjuvant therapy in patients with favorable outcome[226]. Also, we observed a shift in peripheral blood leucocyte populations in our experimental model, from a Gr1+CD11b+ cell dominated response in mice with progressive tumors toward a CD4+/CD8+-, B cell and CD11c+ cell-dominated response in mice with dormant tumors[226]. Thus, Serum IFN-β levels and immunophenotyping should be explored for their potential use as biomarkers of breast cancer dormancy.
Conclusion
Advances in the understanding of the mechanisms of breast cancer dormancy have raised the hope to therapeutically exploit dormancy to prevent relapses and overt metastatic disease. To date many potential therapeutic targets and strategies have been considered and proposed for clinical testing[100,185]. However, many of these approaches would be difficult to apply to patients due to lack of suitable drugs, potential long-term toxicity and over all complexity in the in their clinical translation. Recent reports implicating a T cell based immune response in the therapeutic effects of chemotherapy including dormancy, have opened novel perspectives for a feasible clinical translation. Administration of chemotherapy before tumor removal (i.e., neoadjuvant chemotherapy) may be explored for improved effects on immunological dormancy compared to conventional, post-surgery, adjuvant chemotherapy. Drugs with potential beneficial effects on promoting or prolonging dormancy, such as NSAIDs to suppress inflammation, type I interferons, check-point inhibitors or anti-angiogenic drugs to stimulate the immune response, are available for clinical use and could be tested in combination with chemotherapy. Thus, the observation that chemotherapy can induce a state of immunological dormancy adds a new therapeutic effect to the older class of anti-cancer drugs and opens unanticipated therapeutic opportunities for clinical translation in breast cancer (and possibly other cancers).
Declarations
AcknowledgmentsWe apologize for not being able to cite all published work relevant to this topic due to space limitation and selective focus of the article.
Authors’ contributionsConceiving the paper: Peyvandi S, Lan Q, Lorusso G, Rüegg C
Writing the paper: Peyvandi S, Lan Q, Lorusso G, Rüegg C
Performed the work of immunological dormancy in the laboratory: Peyvandi S, Lan Q, Lorusso G, Rüegg C
Availability of data and materialsNot applicable.
Financial support and sponsorshipThis work was supported by the Swiss National Sciences foundation (31003A_159824/1, 31003A_179248/1); the Swiss Cancer League (KSF3513-08-2014, KSF-4400-02-2018); NCCR Molecular Oncology, NCCR Bio-Inspired materials, the Medic Foundation, the Sassella Stiftung, the 3R foundation, and the European Union under the auspices of the FP7 collaborative project TuMIC (HEALTH-F2-2008-201662).
Conflicts of interestAll authors declare that there are no conflicts of interest.
Ethical approval and consent to participateNot applicable.
Consent for publicationNot applicable.
CopyrightThe Author(s) 2019.
REFERENCES
3. Chaffer CL, Weinberg RA. How does multistep tumorigenesis really proceed? Cancer Discov 2015;5:22-4.
4. Massague J, Obenauf AC. Metastatic colonization by circulating tumour cells. Nature 2016;529:298-306.
5. Obenauf AC, Massague J. Surviving at a distance: organ-specific metastasis. Trends Cancer 2015;1:76-91.
6. Lorusso G, Rüegg C. New insights into the mechanisms of organ-specific breast cancer metastasis. Seminar Cancer Biol 2012;22:226-33.
8. Sleeman JP, Christofori G, Fodde R, Collard JG, Berx G, et al. Concepts of metastasis in flux: the stromal progression model. Semin Cancer Biol 2012;22:174-86.
9. Bussard KM, Mutkus L, Stumpf K, Gomez-Manzano C, Marini FC. Tumor-associated stromal cells as key contributors to the tumor microenvironment. Breast Cancer Res 2016;18:84.
10. Mao Y, Keller ET, Garfield DH, Shen K, Wang J. Stromal cells in tumor microenvironment and breast cancer. Cancer Metastasis Rev 2013;32:303-15.
11. Lorusso G, Ruegg C. The tumor microenvironment and its contribution to tumor evolution toward metastasis. Histochem Cell Biol 2008;130:1091-103.
12. Cserni G, Chmielik E, Cserni B, Tot T. The new TNM-based staging of breast cancer. Virchows Arch 2018;472:697-703.
16. Demicheli R, Biganzoli E, Ardoino I, Boracchi P, Coradini D, et al. Recurrence and mortality dynamics for breast cancer patients undergoing mastectomy according to estrogen receptor status: different mortality but similar recurrence. Cancer Sci 2010;101:826-30.
17. Retsky M, Demicheli R. Multimodal hazard rate for relapse in breast cancer: quality of data and calibration of computer simulation. Cancers (Basel) 2014;6:2343-55.
18. DeSantis C, Ma J, Bryan L, Jemal A. Breast cancer statistics, 2013. CA Cancer J Clin 2014;64:52-62.
20. Yersal O, Barutca S. Biological subtypes of breast cancer: prognostic and therapeutic implications. World J Clin Oncol 2014;5:412-24.
21. Prat A, Pineda E, Adamo B, Galvan P, Fernandez A, et al. Clinical implications of the intrinsic molecular subtypes of breast cancer. Breast 2015;24 Suppl 2:S26-35.
22. Foulkes WD, Smith IE, Reis-Filho JS. Triple-negative breast cancer. N Engl J Med 2010;363:1938-48.
24. Friend S, Royce M. The changing landscape of breast cancer: how biology drives therapy. Medicines (Basel) 2016;3:E2.
25. Guler EN. Gene expression profiling in breast cancer and its effect on therapy selection in early-stage breast cancer. Eur J Breast Health 2017;13:168-74.
26. Colozza M, de Azambuja E, Cardoso F, Bernard C, Piccart MJ. Breast cancer: achievements in adjuvant systemic therapies in the pre-genomic era. Oncologist 2006;11:111-25.
27. Brady-West DC, McGrowder DA. Triple negative breast cancer: therapeutic and prognostic implications. Asian Pac J Cancer Prev 2011;12:2139-43.
28. Goldvaser H, Ribnikar D, Majeed H, Ocana A, Amir E. Absolute benefit from adjuvant chemotherapy in contemporary clinical trials: A systemic review and meta-analysis. Cancer Treat Rev 2018;71:68-75.
29. Coates AS, Winer EP, Goldhirsch A, Gelber RD, Gnant M, et al. Tailoring therapies-improving the management of early breast cancer: St Gallen international expert consensus on the primary therapy of early breast cancer 2015. Ann Oncol 2015;26:1533-46.
30. von Minckwitz G, Loibl S, Maisch A, Untch M. Lessons from the neoadjuvant setting on how best to choose adjuvant therapies. Breast 2011;20 Suppl 3:S142-5.
31. Zardavas D, Fouad TM, Piccart M. Optimal adjuvant treatment for patients with HER2-positive breast cancer in 2015. Breast 2015;24 Suppl 2:S143-8.
32. Wahba HA, El-Hadaad HA. Current approaches in treatment of triple-negative breast cancer. Cancer Biol Med 2015;12:106-16.
33. Hirshfield KM, Ganesan S. Triple-negative breast cancer: molecular subtypes and targeted therapy. Curr Opin Obstet Gynecol 2014;26:34-40.
34. Malvezzi M, Bertuccio P, Levi F, La Vecchia C, Negri E. European cancer mortality predictions for the year 2012. Ann Oncol 2012;23:1044-52.
35. Di Leo A, Curigliano G, Dieras V, Malorni L, Sotiriou C, et al. New approaches for improving outcomes in breast cancer in Europe. Breast 2015;24:321-30.
38. Kang Y, Pantel K. Tumor cell dissemination: emerging biological insights from animal models and cancer patients. Cancer Cell 2013;23:573-81.
39. Ajani JA, Song S, Hochster HS, Steinberg IB. Cancer stem cells: the promise and the potential. Semin Oncol 2015;42 Suppl 1:S3-17.
41. Magee JA, Piskounova E, Morrison SJ. Cancer stem cells: impact, heterogeneity, and uncertainty. Cancer Cell 2012;21:283-96.
42. Lytle NK, Barber AG, Reya T. Stem cell fate in cancer growth, progression and therapy resistance. Nat Rev Cancer 2018;18:669-80.
43. Monteiro J, Fodde R. Cancer stemness and metastasis: therapeutic consequences and perspectives. Eur J Cancer 2010;46:1198-203.
44. Allan AL, Vantyghem SA, Tuck AB, Chambers AF. Tumor dormancy and cancer stem cells: implications for the biology and treatment of breast cancer metastasis. Breast Dis 2006;26:87-98.
46. Valastyan S, Weinberg RA. Tumor metastasis: molecular insights and evolving paradigms. Cell 2011;147:275-92.
47. Lambert AW, Pattabiraman DR, Weinberg RA. Emerging biological principles of metastasis. Cell 2017;168:670-91.
49. Antoniou A, Hebrant A, Dom G, Dumont JE, Maenhaut C. Cancer stem cells, a fuzzy evolving concept: a cell population or a cell property? Cell Cycle 2013;12:3743-8.
50. Huang Z, Wu T, Liu AY, Ouyang G. Differentiation and transdifferentiation potentials of cancer stem cells. Oncotarget 2015;6:39550-63.
51. Bai X, Ni J, Beretov J, Graham P, Li Y. Cancer stem cell in breast cancer therapeutic resistance. Cancer Treat Rev 2018;69:152-63.
52. Smalley M, Piggott L, Clarkson R. Breast cancer stem cells: obstacles to therapy. Cancer Lett 2013;338:57-62.
53. Luo M, Brooks M, Wicha MS. Epithelial-mesenchymal plasticity of breast cancer stem cells: implications for metastasis and therapeutic resistance. Curr Pharm Des 2015;21:1301-10.
54. Margaryan NV, Seftor EA, Seftor REB, Hendrix MJC. Targeting the stem cell properties of adult breast cancer cells: using combinatorial strategies to overcome drug resistance. Curr Mol Biol Rep 2017;3:159-64.
55. Pinto CA, Widodo E, Waltham M, Thompson EW. Breast cancer stem cells and epithelial mesenchymal plasticity - Implications for chemoresistance. Cancer Lett 2013;341:56-62.
56. Hong D, Fritz AJ, Zaidi SK, van Wijnen AJ, Nickerson JA, et al. Epithelial-to-mesenchymal transition and cancer stem cells contribute to breast cancer heterogeneity. J Cell Physiol 2018;233:9136-44.
57. Morel AP, Lievre M, Thomas C, Hinkal G, Ansieau S, et al. Generation of breast cancer stem cells through epithelial-mesenchymal transition. PLoS One 2008;3:e2888.
61. Reeves MQ, Kandyba E, Harris S, Del Rosario R, Balmain A. Multicolour lineage tracing reveals clonal dynamics of squamous carcinoma evolution from initiation to metastasis. Nat Cell Biol 2018;20:699-709.
62. Reiter JG, Makohon-Moore AP, Gerold JM, Heyde A, Attiyeh MA, et al. Minimal functional driver gene heterogeneity among untreated metastases. Science 2018;361:1033-7.
63. Yachida S, Jones S, Bozic I, Antal T, Leary R, et al. Distant metastasis occurs late during the genetic evolution of pancreatic cancer. Nature 2010;467:1114-7.
64. Leung ML, Davis A, Gao R, Casasent A, Wang Y, et al. Single-cell DNA sequencing reveals a late-dissemination model in metastatic colorectal cancer. Genome Res 2017;27:1287-99.
65. Desmedt C, Yates L, Kulka J. Catalog of genetic progression of human cancers: breast cancer. Cancer Metastasis Rev 2016;35:49-62.
66. Brown D, Smeets D, Szekely B, Larsimont D, Szasz AM, et al. Phylogenetic analysis of metastatic progression in breast cancer using somatic mutations and copy number aberrations. Nat Commun 2017;8:14944.
67. Yates LR, Knappskog S, Wedge D, Farmery JHR, Gonzalez S, et al. Genomic evolution of breast cancer metastasis and relapse. Cancer Cell 2017;32:169-84.e7.
68. Kroigard AB, Larsen MJ, Laenkholm AV, Knoop AS, Jensen JD, et al. Clonal expansion and linear genome evolution through breast cancer progression from pre-invasive stages to asynchronous metastasis. Oncotarget 2015;6:5634-49.
69. Weckermann D, Polzer B, Ragg T, Blana A, Schlimok G, et al. Perioperative activation of disseminated tumor cells in bone marrow of patients with prostate cancer. J Clin Oncol 2009;27:1549-56.
70. Stoecklein NH, Hosch SB, Bezler M, Stern F, Hartmann CH, et al. Direct genetic analysis of single disseminated cancer cells for prediction of outcome and therapy selection in esophageal cancer. Cancer Cell 2008;13:441-53.
71. Schmidt-Kittler O, Ragg T, Daskalakis A, Granzow M, Ahr A, et al. From latent disseminated cells to overt metastasis: genetic analysis of systemic breast cancer progression. Proc Natl Acad Sci U S A 2003;100:7737-42.
72. Schardt JA, Meyer M, Hartmann CH, Schubert F, Schmidt-Kittler O, et al. Genomic analysis of single cytokeratin-positive cells from bone marrow reveals early mutational events in breast cancer. Cancer Cell 2005;8:227-39.
73. Biondini M, Duclos G, Meyer-Schaller N, Silberzan P, Camonis J, et al. RalB regulates contractility-driven cancer dissemination upon TGFbeta stimulation via the RhoGEF GEF-H1. Sci Rep 2015;5:11759.
74. Oft M, Akhurst RJ, Balmain A. Metastasis is driven by sequential elevation of H-ras and Smad2 levels. Nat Cell Biol 2002;4:487-94.
75. Ansieau S, Bastid J, Doreau A, Morel AP, Bouchet BP, et al. Induction of EMT by twist proteins as a collateral effect of tumor-promoting inactivation of premature senescence. Cancer Cell 2008;14:79-89.
76. Naxerova K, Jain RK. Using tumour phylogenetics to identify the roots of metastasis in humans. Nat Rev Clin Oncol 2015;12:258-72.
77. Harper KL, Sosa MS, Entenberg D, Hosseini H, Cheung JF, et al. Mechanism of early dissemination and metastasis in Her2+ mammary cancer. Nature 2016;540:588-92.
79. Dagogo-Jack I, Shaw AT. Tumour heterogeneity and resistance to cancer therapies. Nat Rev Clin Oncol 2018;15:81-94.
80. Meacham CE, Morrison SJ. Tumour heterogeneity and cancer cell plasticity. Nature 2013;501:328-37.
81. Marusyk A, Tabassum DP, Altrock PM, Almendro V, Michor F, et al. Non-cell-autonomous driving of tumour growth supports sub-clonal heterogeneity. Nature 2014;514:54-8.
82. Aceto N, Bardia A, Miyamoto DT, Donaldson MC, Wittner BS, et al. Circulating tumor cell clusters are oligoclonal precursors of breast cancer metastasis. Cell 2014;158:1110-22.
83. Gkountela S, Castro-Giner F, Szczerba BM, Vetter M, Landin J, et al. Circulating Tumor Cell Clustering Shapes DNA Methylation to Enable Metastasis Seeding. Cell 2019;176:98-112.e14.
84. Kmieciak M, Payne KK, Wang XY, Manjili MH. IFN-gamma Ralpha is a key determinant of CD8+ T cell-mediated tumor elimination or tumor escape and relapse in FVB mouse. PLoS One 2013;8:e82544.
85. Demicheli R. Tumour dormancy: findings and hypotheses from clinical research on breast cancer. Semin Cancer Biol 2001;11:297-306.
86. Demicheli R, Retsky MW, Hrushesky WJ, Baum M. Tumor dormancy and surgery-driven interruption of dormancy in breast cancer: learning from failures. Nat Clin Pract Oncol 2007;4:699-710.
88. Demicheli R, Biganzoli E, Boracchi P, Greco M, Retsky MW. Recurrence dynamics does not depend on the recurrence site. Breast Cancer Res 2008;10:R83.
89. Dillekas H, Demicheli R, Ardoino I, Jensen SAH, Biganzoli E, et al. The recurrence pattern following delayed breast reconstruction after mastectomy for breast cancer suggests a systemic effect of surgery on occult dormant micrometastases. Breast Cancer Res Treat 2016;158:169-78.
90. Hanin L, Korosteleva O. Does extirpation of the primary breast tumor give boost to growth of metastases? Evidence revealed by mathematical modeling. Math Biosci 2010;223:133-41.
91. Manjili MH. Tumor dormancy and relapse: from a natural byproduct of evolution to a disease state. Cancer Res 2017;77:2564-9.
92. Gelao L, Criscitiello C, Fumagalli L, Locatelli M, Manunta S, et al. Tumour dormancy and clinical implications in breast cancer. Ecancermedicalscience 2013;7:320.
93. Rancoita PM, Valberg M, Demicheli R, Biganzoli E, Di Serio C. Tumor dormancy and frailty models: a novel approach. Biometrics 2017;73:260-70.
94. Klein CA, Holzel D. Systemic cancer progression and tumor dormancy: mathematical models meet single cell genomics. Cell Cycle 2006;5:1788-98.
95. Klauber-DeMore N, Van Zee KJ, Linkov I, Borgen PI, Gerald WL. Biological behavior of human breast cancer micrometastases. Clin Cancer Res 2001;7:2434-9.
96. Naumov GN, MacDonald IC, Weinmeister PM, Kerkvliet N, Nadkarni KV, et al. Persistence of solitary mammary carcinoma cells in a secondary site: a possible contributor to dormancy. Cancer Res 2002;62:2162-8.
97. Goodison S, Kawai K, Hihara J, Jiang P, Yang M, et al. Prolonged dormancy and site-specific growth potential of cancer cells spontaneously disseminated from nonmetastatic breast tumors as revealed by labeling with green fluorescent protein. Clin Cancer Res 2003;9:3808-14.
98. Makita M, Sakai T, Ogiya A, Kitagawa D, Morizono H, et al. Optimal surveillance for postoperative metastasis in breast cancer patients. Breast Cancer 2016;23:286-94.
99. Aguirre-Ghiso JA. Models, mechanisms and clinical evidence for cancer dormancy. Nat Rev Cancer 2007;7:834-46.
100. Hensel JA, Flaig TW, Theodorescu D. Clinical opportunities and challenges in targeting tumour dormancy. Nat Rev Clin Oncol 2013;10:41-51.
101. Dittmer J. Mechanisms governing metastatic dormancy in breast cancer. Semin Cancer Biol 2017;44:72-82.
102. Paez D, Labonte MJ, Bohanes P, Zhang W, Benhanim L, et al. Cancer dormancy: a model of early dissemination and late cancer recurrence. Clin Cancer Res 2012;18:645-53.
104. Aguirre-Ghiso JA, Liu D, Mignatti A, Kovalski K, Ossowski L. Urokinase receptor and fibronectin regulate the ERK(MAPK) to p38(MAPK) activity ratios that determine carcinoma cell proliferation or dormancy in vivo. Mol Biol Cell 2001;12:863-79.
106. Sosa MS, Parikh F, Maia AG, Estrada Y, Bosch A, et al. NR2F1 controls tumour cell dormancy via SOX9- and RARbeta-driven quiescence programmes. Nat Commun 2015;6:6170.
107. Borgen E, Rypdal MC, Sosa MS, Renolen A, Schlichting E, et al. NR2F1 stratifies dormant disseminated tumor cells in breast cancer patients. Breast Cancer Res 2018;20:120.
108. Aguirre-Ghiso JA, Estrada Y, Liu D, Ossowski L. ERK(MAPK) activity as a determinant of tumor growth and dormancy; regulation by p38(SAPK). Cancer Res 2003;63:1684-95.
109. Jo H, Jia Y, Subramanian KK, Hattori H, Luo HR. Cancer cell-derived clusterin modulates the phosphatidylinositol 3'-kinase-Akt pathway through attenuation of insulin-like growth factor 1 during serum deprivation. Mol Cell Biol 2008;28:4285-99.
110. Vera-Ramirez L, Vodnala SK, Nini R, Hunter KW, Green JE. Autophagy promotes the survival of dormant breast cancer cells and metastatic tumour recurrence. Nat Commun 2018;9:1944.
111. Lu Z, Luo RZ, Lu Y, Zhang X, Yu Q, et al. The tumor suppressor gene ARHI regulates autophagy and tumor dormancy in human ovarian cancer cells. J Clin Invest 2008;118:3917-29.
112. Balz LM, Bartkowiak K, Andreas A, Pantel K, Niggemann B, et al. The interplay of HER2/HER3/PI3K and EGFR/HER2/PLC-gamma1 signalling in breast cancer cell migration and dissemination. J Pathol 2012;227:234-44.
113. Amaravadi RK. Autophagy-induced tumor dormancy in ovarian cancer. J Clin Invest 2008;118:3837-40.
114. Schewe DM, Aguirre-Ghiso JA. ATF6alpha-Rheb-mTOR signaling promotes survival of dormant tumor cells in vivo. Proc Natl Acad Sci U S A 2008;105:10519-24.
115. Malladi S, Macalinao DG, Jin X, He L, Basnet H, et al. Metastatic latency and immune evasion through autocrine inhibition of WNT. Cell 2016;165:45-60.
116. Barkan D, Green JE, Chambers AF. Extracellular matrix: a gatekeeper in the transition from dormancy to metastatic growth. Eur J Cancer 2010;46:1181-8.
117. Walker ND, Patel J, Munoz JL, Hu M, Guiro K, et al. The bone marrow niche in support of breast cancer dormancy. Cancer Lett 2016;380:263-71.
118. Korah R, Boots M, Wieder R. Integrin alpha5beta1 promotes survival of growth-arrested breast cancer cells: an in vitro paradigm for breast cancer dormancy in bone marrow. Cancer Res 2004;64:4514-22.
119. Wheeler SE, Clark AM, Taylor DP, Young CL, Pillai VC, et al. Spontaneous dormancy of metastatic breast cancer cells in an all human liver microphysiologic system. Br J Cancer 2014;111:2342-50.
120. Clark AM, Wheeler SE, Young CL, Stockdale L, Shepard Neiman J, et al. A liver microphysiological system of tumor cell dormancy and inflammatory responsiveness is affected by scaffold properties. Lab Chip 2016;17:156-68.
121. El Touny LH, Vieira A, Mendoza A, Khanna C, Hoenerhoff MJ, et al. Combined SFK/MEK inhibition prevents metastatic outgrowth of dormant tumor cells. J Clin Invest 2014;124:156-68.
122. Ghajar CM, Peinado H, Mori H, Matei IR, Evason KJ, et al. The perivascular niche regulates breast tumour dormancy. Nat Cell Biol 2013;15:807-17.
123. Sriram R, Lo V, Pryce B, Antonova L, Mears AJ, et al. Loss of periostin/OSF-2 in ErbB2/Neu-driven tumors results in androgen receptor-positive molecular apocrine-like tumors with reduced Notch1 activity. Breast Cancer Res 2015;17:7.
125. Barkan D, Chambers AF. beta1-integrin: a potential therapeutic target in the battle against cancer recurrence. Clin Cancer Res 2011;17:7219-23.
126. Sosa MS, Bragado P, Debnath J, Aguirre-Ghiso JA. Regulation of tumor cell dormancy by tissue microenvironments and autophagy. Adv Exp Med Biol 2013;734:73-89.
127. Aqbi HF, Tyutyunyk-Massey L, Keim RC, Butler SE, Thekkudan T, et al. Autophagy-deficient breast cancer shows early tumor recurrence and escape from dormancy. Oncotarget 2018;9:22113-22.
128. Tierney MT, Sacco A. Inducing and evaluating skeletal muscle injury by notexin and barium chloride. Methods Mol Biol 2016;1460:53-60.
129. Agudo J, Park ES, Rose SA, Alibo E, Sweeney R, et al. Quiescent tissue stem cells evade immune surveillance. Immunity 2018;48:271-85.e5.
130. Trumpp A, Essers M, Wilson A. Awakening dormant haematopoietic stem cells. Nat Rev Immunol 2010;10:201-9.
134. Campisi J. Cellular senescence as a tumor-suppressor mechanism. Trends Cell Biol 2001;11:S27-31.
137. Di Leonardo A, Linke SP, Clarkin K, Wahl GM. DNA damage triggers a prolonged p53-dependent G1 arrest and long-term induction of Cip1 in normal human fibroblasts. Genes Dev 1994;8:2540-51.
138. Schmitt CA, Fridman JS, Yang M, Lee S, Baranov E, et al. A senescence program controlled by p53 and p16INK4a contributes to the outcome of cancer therapy. Cell 2002;109:335-46.
139. Braig M, Lee S, Loddenkemper C, Rudolph C, Peters AH, et al. Oncogene-induced senescence as an initial barrier in lymphoma development. Nature 2005;436:660-5.
140. Collado M, Gil J, Efeyan A, Guerra C, Schuhmacher AJ, et al. Tumour biology: senescence in premalignant tumours. Nature 2005;436:642.
141. Land H, Parada LF, Weinberg RA. Tumorigenic conversion of primary embryo fibroblasts requires at least two cooperating oncogenes. Nature 1983;304:596-602.
142. Serrano M, Lin AW, McCurrach ME, Beach D, Lowe SW. Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell 1997;88:593-602.
143. Beausejour CM, Krtolica A, Galimi F, Narita M, Lowe SW, et al. Reversal of human cellular senescence: roles of the p53 and p16 pathways. EMBO J 2003;22:4212-22.
144. Mu XC, Staiano-Coico L, Higgins PJ. Increased transcription and modified growth state-dependent expression of the plasminogen activator inhibitor type-1 gene characterize the senescent phenotype in human diploid fibroblasts. J Cell Physiol 1998;174:90-8.
145. McConnell BB, Starborg M, Brookes S, Peters G. Inhibitors of cyclin-dependent kinases induce features of replicative senescence in early passage human diploid fibroblasts. Curr Biol 1998;8:351-4.
146. Zou W. Immunosuppressive networks in the tumour environment and their therapeutic relevance. Nat Rev Cancer 2005;5:263-74.
147. Metcalf D. The molecular biology and functions of the granulocyte-macrophage colony-stimulating factors. Blood 1986;67:257-67.
148. Braumüller H, Wieder T, Brenner E, Assmann S, Hahn M, et al. T-helper-1-cell cytokines drive cancer into senescence. Nature 2013;494:361-5.
149. Bragado P, Sosa MS, Keely P, Condeelis J, Aguirre-Ghiso JA. Microenvironments dictating tumor cell dormancy. Recent Results Cancer Res 2012;195:25-39.
150. Naumov GN, Folkman J, Straume O. Tumor dormancy due to failure of angiogenesis: role of the microenvironment. Clin Exp Metastasis 2009;26:51-60.
151. Kienast Y, von Baumgarten L, Fuhrmann M, Klinkert WE, Goldbrunner R, et al. Real-time imaging reveals the single steps of brain metastasis formation. Nat Med 2010;16:116-22.
153. Carmeliet P, Jain RK. Molecular mechanisms and clinical applications of angiogenesis. Nature 2011;473:298-307.
154. Indraccolo S. Insights into the regulation of tumor dormancy by angiogenesis in experimental tumors. Adv Exp Med Biol 2013;734:37-52.
155. Indraccolo S, Favaro E, Amadori A. Dormant tumors awaken by a short-term angiogenic burst: the spike hypothesis. Cell Cycle 2006;5:1751-5.
156. Gao D, Nolan DJ, Mellick AS, Bambino K, McDonnell K, et al. Endothelial progenitor cells control the angiogenic switch in mouse lung metastasis. Science 2008;319:195-8.
157. Laurent J, Touvrey C, Botta F, Kuonen F, Ruegg C. Emerging paradigms and questions on pro-angiogenic bone marrow-derived myelomonocytic cells. Int J Dev Biol 2011;55:527-34.
158. Qian BZ, Pollard JW. Macrophage diversity enhances tumor progression and metastasis. Cell 2010;141:39-51.
159. DeNardo DG, Barreto JB, Andreu P, Vasquez L, Tawfik D, et al. CD4(+) T cells regulate pulmonary metastasis of mammary carcinomas by enhancing protumor properties of macrophages. Cancer Cell 2009;16:91-102.
160. Zhang XH, Wang Q, Gerald W, Hudis CA, Norton L, et al. Latent bone metastasis in breast cancer tied to Src-dependent survival signals. Cancer Cell 2009;16:67-78.
161. Bragado P, Estrada Y, Parikh F, Krause S, Capobianco C, et al. TGF-beta2 dictates disseminated tumour cell fate in target organs through TGF-beta-RIII and p38alpha/beta signalling. Nat Cell Biol 2013;15:1351-61.
162. Lu X, Mu E, Wei Y, Riethdorf S, Yang Q, et al. VCAM-1 promotes osteolytic expansion of indolent bone micrometastasis of breast cancer by engaging alpha4beta1-positive osteoclast progenitors. Cancer Cell 2011;20:701-14.
163. Sosnoski DM, Norgard RJ, Grove CD, Foster SJ, Mastro AM. Dormancy and growth of metastatic breast cancer cells in a bone-like microenvironment. Clin Exp Metastasis 2015;32:335-44.
164. Dunn GP, Bruce AT, Ikeda H, Old LJ, Schreiber RD. Cancer immunoediting: from immunosurveillance to tumor escape. Nat Immunol 2002;3:991-8.
165. Schreiber RD, Old LJ, Smyth MJ. Cancer immunoediting: integrating immunity's roles in cancer suppression and promotion. Science 2011;331:1565-70.
166. Teng MW, Swann JB, Koebel CM, Schreiber RD, Smyth MJ. Immune-mediated dormancy: an equilibrium with cancer. J Leukoc Biol 2008;84:988-93.
167. Tuccitto A, Shahaj E, Vergani E, Ferro S, Huber V, et al. Immunosuppressive circuits in tumor microenvironment and their influence on cancer treatment efficacy. Virchows Arch 2019;474:407-20.
168. Najafi M, Goradel NH, Farhood B, Salehi E, Solhjoo S, et al. Tumor microenvironment: Interactions and therapy. J Cell Physiol 2019;234:5700-21.
169. Groth C, Hu X, Weber R, Fleming V, Altevogt P, et al. Immunosuppression mediated by myeloid-derived suppressor cells (MDSCs) during tumour progression. Br J Cancer 2019;120:16-25.
170. Bates JP, Derakhshandeh R, Jones L, Webb TJ. Mechanisms of immune evasion in breast cancer. BMC Cancer 2018;18:556.
171. Pommier A, Anaparthy N, Memos N, Kelley ZL, Gouronnec A, et al. Unresolved endoplasmic reticulum stress engenders immune-resistant, latent pancreatic cancer metastases. Science 2018;360:eaao4908.
172. Matser YAH, Terpstra ML, Nadalin S, Nossent GD, de Boer J, et al. Transmission of breast cancer by a single multiorgan donor to 4 transplant recipients. Am J Transplant 2018;18:1810-4.
173. Motz GT, Coukos G. The parallel lives of angiogenesis and immunosuppression: cancer and other tales. Nat Rev Immunol 2011;11:702-11.
174. Griffioen AW, Vyth-Dreese FA. Angiostasis as a way to improve immunotherapy. Thromb Haemost 2009;101:1025-31.
175. De Sanctis F, Ugel S, Facciponte J, Facciabene A. The dark side of tumor-associated endothelial cells. Semin Immunol 2018;35:35-47.
176. Khan KA, Kerbel RS. Improving immunotherapy outcomes with anti-angiogenic treatments and vice versa. Nat Rev Clin Oncol 2018;15:310-24.
177. Chen DS, Hurwitz H. Combinations of bevacizumab with cancer immunotherapy. Cancer J 2018;24:193-204.
178. Gabrilovich DI, Chen HL, Girgis KR, Cunningham HT, Meny GM, et al. Production of vascular endothelial growth factor by human tumors inhibits the functional maturation of dendritic cells. Nat Med 1996;2:1096-103.
179. Bai WK, Zhang W, Hu B. Vascular endothelial growth factor suppresses dendritic cells function of human prostate cancer. Onco Targets Ther 2018;11:1267-74.
180. Shi Y, Yu P, Zeng D, Qian F, Lei X, et al. Suppression of vascular endothelial growth factor abrogates the immunosuppressive capability of murine gastric cancer cells and elicits antitumor immunity. FEBS J 2014;281:3882-93.
181. Secondini C, Coquoz O, Spagnuolo L, Spinetti T, Peyvandi S, et al. Arginase inhibition suppresses lung metastasis in the 4T1 breast cancer model independently of the immunomodulatory and anti-metastatic effects of VEGFR-2 blockade. Oncoimmunology 2017;6:e1316437.
182. Schmittnaegel M, Rigamonti N, Kadioglu E, Cassara A, Wyser Rmili C, et al. Dual angiopoietin-2 and VEGFA inhibition elicits antitumor immunity that is enhanced by PD-1 checkpoint blockade. Sci Transl Med 2017;9:eaak9670.
183. Schmittnaegel M, De Palma M. Reprogramming tumor blood vessels for enhancing immunotherapy. Trends Cancer 2017;3:809-12.
184. Goddard ET, Bozic I, Riddell SR, Ghajar CM. Dormant tumour cells, their niches and the influence of immunity. Nat Cell Biol 2018;20:1240-9.
185. Ghajar CM. Metastasis prevention by targeting the dormant niche. Nat Rev Cancer 2015;15:238-47.
186. Bedard PL, Di Leo A, Piccart-Gebhart MJ. Taxanes: optimizing adjuvant chemotherapy for early-stage breast cancer. Nat Rev Clin Oncol 2010;7:22-36.
189. Hosseini H, Obradovic MM, Hoffmann M, Harper KL, Sosa MS, et al. Early dissemination seeds metastasis in breast cancer. Nature 2016; doi: 10.1038/nature20785.
190. Touil Y, Igoudjil W, Corvaisier M, Dessein AF, Vandomme J, et al. Colon cancer cells escape 5FU chemotherapy-induced cell death by entering stemness and quiescence associated with the c-Yes/YAP axis. Clin Cancer Res 2014;20:837-46.
191. Ebinger S, Ozdemir EZ, Ziegenhain C, Tiedt S, Castro Alves C, et al. Characterization of rare, dormant, and therapy-resistant cells in acute lymphoblastic leukemia. Cancer Cell 2016;30:849-62.
192. Pal D, Heidenreich O, Vormoor J. Dormancy stems the tide of chemotherapy. Cancer Cell 2016;30:825-6.
193. Wu FH, Mu L, Li XL, Hu YB, Liu H, et al. Characterization and functional analysis of a slow-cycling subpopulation in colorectal cancer enriched by cell cycle inducer combined chemotherapy. Oncotarget 2017;8:78466-79.
194. Keeratichamroen S, Lirdprapamongkol K, Svasti J. Mechanism of ECM-induced dormancy and chemoresistance in A549 human lung carcinoma cells. Oncol Rep 2018;39:1765-74.
195. Nakamura T, Shinriki S, Jono H, Guo J, Ueda M, et al. Intrinsic TGF-beta2-triggered SDF-1-CXCR4 signaling axis is crucial for drug resistance and a slow-cycling state in bone marrow-disseminated tumor cells. Oncotarget 2015;6:1008-19.
196. Quayle LA, Ottewell PD, Holen I. Chemotherapy resistance and stemness in mitotically quiescent human breast cancer cells identified by fluorescent dye retention. Clin Exp Metastasis 2018;35:831-46.
197. Steg AD, Bevis KS, Katre AA, Ziebarth A, Dobbin ZC, et al. Stem cell pathways contribute to clinical chemoresistance in ovarian cancer. Clin Cancer Res 2012;18:869-81.
198. McCubrey JA, Abrams SL, Fitzgerald TL, Cocco L, Martelli AM, et al. Roles of signaling pathways in drug resistance, cancer initiating cells and cancer progression and metastasis. Adv Biol Regul 2015;57:75-101.
199. Takebe N, Miele L, Harris PJ, Jeong W, Bando H, et al. Targeting Notch, Hedgehog, and Wnt pathways in cancer stem cells: clinical update. Nat Rev Clin Oncol 2015;12:445-64.
200. Schoning JP, Monteiro M, Gu W. Drug resistance and cancer stem cells: the shared but distinct roles of hypoxia-inducible factors HIF1alpha and HIF2alpha. Clin Exp Pharmacol Physiol 2017;44:153-61.
201. Pan ST, Li ZL, He ZX, Qiu JX, Zhou SF. Molecular mechanisms for tumour resistance to chemotherapy. Clin Exp Pharmacol Physiol 2016;43:723-37.
202. Kolenda J, Jensen SS, Aaberg-Jessen C, Christensen K, Andersen C, et al. Effects of hypoxia on expression of a panel of stem cell and chemoresistance markers in glioblastoma-derived spheroids. J Neurooncol 2011;103:43-58.
203. He M, Wu H, Jiang Q, Liu Y, Han L, et al. Hypoxia-inducible factor-2alpha directly promotes BCRP expression and mediates the resistance of ovarian cancer stem cells to adriamycin. Mol Oncol 2019;13:403-21.
204. Maugeri-Sacca M, Vigneri P, De Maria R. Cancer stem cells and chemosensitivity. Clin Cancer Res 2011;17:4942-7.
205. Crowder SW, Balikov DA, Hwang YS, Sung HJ. Cancer Stem Cells under Hypoxia as a Chemoresistance Factor in Breast and Brain. Curr Pathobiol Rep 2014;2:33-40.
206. Yan Y, Liu F, Han L, Zhao L, Chen J, et al. HIF-2alpha promotes conversion to a stem cell phenotype and induces chemoresistance in breast cancer cells by activating Wnt and Notch pathways. J Exp Clin Cancer Res 2018;37:256.
207. Uribe D, Torres A, Rocha JD, Niechi I, Oyarzun C, et al. Multidrug resistance in glioblastoma stem-like cells: role of the hypoxic microenvironment and adenosine signaling. Mol Aspects Med 2017;55:140-51.
208. Qin J, Liu Y, Lu Y, Liu M, Li M, et al. Hypoxia-inducible factor 1 alpha promotes cancer stem cells-like properties in human ovarian cancer cells by upregulating SIRT1 expression. Sci Rep 2017;7:10592.
209. Doktorova H, Hrabeta J, Khalil MA, Eckschlager T. Hypoxia-induced chemoresistance in cancer cells: The role of not only HIF-1. Biomed Pap Med Fac Univ Palacky Olomouc Czech Repub 2015;159:166-77.
210. Stanton SE, Disis ML. Clinical significance of tumor-infiltrating lymphocytes in breast cancer. J Immunother Cancer 2016;4:59.
211. Dushyanthen S, Beavis PA, Savas P, Teo ZL, Zhou C, et al. Relevance of tumor-infiltrating lymphocytes in breast cancer. BMC Med 2015;13:202.
212. de Melo Gagliato D, Cortes J, Curigliano G, Loi S, Denkert C, et al. Tumor-infiltrating lymphocytes in Breast Cancer and implications for clinical practice. Biochim Biophys Acta Rev Cancer 2017;1868:527-37.
213. Kroemer G, Senovilla L, Galluzzi L, André F, Zitvogel L. Natural and therapy-induced immunosurveillance in breast cancer. Nat Med 2015;21:1128-38.
214. Galluzzi L, Buque A, Kepp O, Zitvogel L, Kroemer G. Immunological Effects of Conventional Chemotherapy and Targeted Anticancer Agents. Cancer Cell 2015;28:690-714.
215. Loi S, Michiels S, Salgado R, Sirtaine N, Jose V, et al. Tumor infiltrating lymphocytes are prognostic in triple negative breast cancer and predictive for trastuzumab benefit in early breast cancer: results from the FinHER trial. Ann Oncol 2014;25:1544-50.
216. Lee HJ, Park IA, Song IH, Shin SJ, Kim JY, et al. Tertiary lymphoid structures: prognostic significance and relationship with tumour-infiltrating lymphocytes in triple-negative breast cancer. J Clin Pathol 2016;69:422-30.
217. Wang K, Xu J, Zhang T, Xue D. Tumor-infiltrating lymphocytes in breast cancer predict the response to chemotherapy and survival outcome: a meta-analysis. Oncotarget 2016;7:44288-98.
218. Adams S, Gray RJ, Demaria S, Goldstein L, Perez EA, et al. Prognostic value of tumor-infiltrating lymphocytes in triple-negative breast cancers from two phase III randomized adjuvant breast cancer trials: ECOG 2197 and ECOG 1199. J Clin Oncol 2014;32:2959-66.
219. Salgado R, Denkert C, Demaria S, Sirtaine N, Klauschen F, et al. The evaluation of tumor-infiltrating lymphocytes (TILs) in breast cancer: recommendations by an International TILs Working Group 2014. Ann Oncol 2015;26:259-71.
220. Forero A, Li Y, Chen D, Grizzle WE, Updike KL, et al. Expression of the MHC class II pathway in triple-negative breast cancer tumor cells is associated with a good prognosis and infiltrating lymphocytes. Cancer Immunol Res 2016;4:390-9.
221. Ladoire S, Arnould L, Apetoh L, Coudert B, Martin F, et al. Pathologic complete response to neoadjuvant chemotherapy of breast carcinoma is associated with the disappearance of tumor-infiltrating foxp3+ regulatory T cells. Clin Cancer Res 2008;14:2413-20.
222. Luen SJ, Savas P, Fox SB, Salgado R, Loi S. Tumour-infiltrating lymphocytes and the emerging role of immunotherapy in breast cancer. Pathology 2017;49:141-55.
223. Ma Y, Adjemian S, Yang H, Catani JP, Hannani D, et al. ATP-dependent recruitment, survival and differentiation of dendritic cell precursors in the tumor bed after anticancer chemotherapy. Oncoimmunology 2013;2:e24568.
224. Ma Y, Adjemian S, Mattarollo SR, Yamazaki T, Aymeric L, et al. Anticancer chemotherapy-induced intratumoral recruitment and differentiation of antigen-presenting cells. Immunity 2013;38:729-41.
225. Acharyya S, Oskarsson T, Vanharanta S, Malladi S, Kim J, et al. A CXCL1 paracrine network links cancer chemoresistance and metastasis. Cell 2012;150:165-78.
226. Lan Q, Peyvandi S, Duffey N, Huang YT, Barras D, et al. Type I interferon/IRF7 axis instigates chemotherapy-induced immunological dormancy in breast cancer. Oncogene 2018; doi: 10.1038/s41388-018-0624-2.
227. Sistigu A, Yamazaki T, Vacchelli E, Chaba K, Enot DP, et al. Cancer cell-autonomous contribution of type I interferon signaling to the efficacy of chemotherapy. Nat Med 2014;20:1301-9.
228. Legrier ME, Bieche I, Gaston J, Beurdeley A, Yvonnet V, et al. Activation of IFN/STAT1 signalling predicts response to chemotherapy in oestrogen receptor-negative breast cancer. Br J Cancer 2016;114:177-87.
229. Sisirak V, Faget J, Gobert M, Goutagny N, Vey N, et al. Impaired IFN-alpha production by plasmacytoid dendritic cells favors regulatory T-cell expansion that may contribute to breast cancer progression. Cancer Res 2012;72:5188-97.
230. Ignatiadis M, Singhal SK, Desmedt C, Haibe-Kains B, Criscitiello C, et al. Gene modules and response to neoadjuvant chemotherapy in breast cancer subtypes: a pooled analysis. J Clin Oncol 2012;30:1996-2004.
231. Desmedt C, Haibe-Kains B, Wirapati P, Buyse M, Larsimont D, et al. Biological processes associated with breast cancer clinical outcome depend on the molecular subtypes. Clin Cancer Res 2008;14:5158-65.
232. Bidwell BN, Slaney CY, Withana NP, Forster S, Cao Y, et al. Silencing of Irf7 pathways in breast cancer cells promotes bone metastasis through immune escape. Nat Med 2012;18:1224-31.
233. Liu Y, Lv J, Liu J, Liang X, Jin X, et al. STAT3/p53 pathway activation disrupts IFN-beta-induced dormancy in tumor-repopulating cells. J Clin Invest 2018;128:1057-73.
234. Dunn GP, Koebel CM, Schreiber RD. Interferons, immunity and cancer immunoediting. Nat Rev Immunol 2006;6:836-48.
235. Muller-Hermelink N, Braumuller H, Pichler B, Wieder T, Mailhammer R, et al. TNFR1 signaling and IFN-gamma signaling determine whether T cells induce tumor dormancy or promote multistage carcinogenesis. Cancer Cell 2008;13:507-18.
236. Dormond O, Lejeune FJ, Ruegg C. Modulation of cdk2, cyclin D1, p16INK4a, p21WAF and p27Kip1 expression in endothelial cells by TNF/IFN gamma. Anticancer Res 2002;22:3159-63.
237. Retsky MW, Demicheli R, Hrushesky WJ, Baum M, Gukas ID. Dormancy and surgery-driven escape from dormancy help explain some clinical features of breast cancer. APMIS 2008;116:730-41.
238. Retsky M, Demicheli R, Hrushesky W, Baum M, Gukas I. Surgery triggers outgrowth of latent distant disease in breast cancer: an inconvenient truth? Cancers (Basel) 2010;2:305-37.
239. Kelsey CR, Fornili M, Ambrogi F, Higgins K, Boyd JA, et al. Metastasis dynamics for non-small-cell lung cancer: effect of patient and tumor-related factors. Clin Lung Cancer 2013;14:425-32.
240. Demicheli R, Fornili M, Ambrogi F, Higgins K, Boyd JA, et al. Recurrence dynamics for non-small-cell lung cancer: effect of surgery on the development of metastases. J Thorac Oncol 2012;7:723-30.
241. Hanin L, Bunimovich-Mendrazitsky S. Reconstruction of the natural history of metastatic cancer and assessment of the effects of surgery: Gompertzian growth of the primary tumor. Math Biosci 2014;247:47-58.
242. Dillekas H, Demicheli R, Ardoino I, Jensen SA, Biganzoli E, et al. The recurrence pattern following delayed breast reconstruction after mastectomy for breast cancer suggests a systemic effect of surgery on occult dormant micrometastases. Breast Cancer Res Treat 2016;158:169-78.
243. Van Dierendonck JH, Keijzer R, Cornelisse CJ, Van de Velde CJ. Surgically induced cytokinetic responses in experimental rat mammary tumor models. Cancer 1991;68:759-67.
244. Abramovitch R, Marikovsky M, Meir G, Neeman M. Stimulation of tumour angiogenesis by proximal wounds: spatial and temporal analysis by MRI. Br J Cancer 1998;77:440-7.
245. Gunduz N, Fisher B, Saffer EA. Effect of surgical removal on the growth and kinetics of residual tumor. Cancer Res 1979;39:3861-5.
246. Krall JA, Reinhardt F, Mercury OA, Pattabiraman DR, Brooks MW, et al. The systemic response to surgery triggers the outgrowth of distant immune-controlled tumors in mouse models of dormancy. Sci Transl Med 2018;10.
247. Danish HH, Goyal S, Taunk NK, Wu H, Moran MS, et al. Interferon-induced protein with tetratricopeptide repeats 1 (IFIT1) as a prognostic marker for local control in T1-2 N0 breast cancer treated with breast-conserving surgery and radiation therapy (BCS + RT). Breast J 2013;19:231-9.
248. De Cock JM, Shibue T, Dongre A, Keckesova Z, Reinhardt F, et al. Inflammation triggers Zeb1-dependent escape from tumor latency. Cancer Res 2016;76:6778-84.
249. Albrengues J, Shields MA, Ng D, Park CG, Ambrico A, et al. Neutrophil extracellular traps produced during inflammation awaken dormant cancer cells in mice. Science 2018;361.
250. Okubo M, Kioi M, Nakashima H, Sugiura K, Mitsudo K, et al. M2-polarized macrophages contribute to neovasculogenesis, leading to relapse of oral cancer following radiation. Sci Rep 2016;6:27548.
251. Machida H, De Zoysa MY, Takiuchi T, Hom MS, Tierney KE, et al. Significance of monocyte counts at recurrence on survival outcome of women with endometrial cancer. Int J Gynecol Cancer 2017;27:302-10.
252. Bowers LW, Maximo IX, Brenner AJ, Beeram M, Hursting SD, et al. NSAID use reduces breast cancer recurrence in overweight and obese women: role of prostaglandin-aromatase interactions. Cancer Res 2014;74:4446-57.
253. Pierce BL, Ballard-Barbash R, Bernstein L, Baumgartner RN, Neuhouser ML, et al. Elevated biomarkers of inflammation are associated with reduced survival among breast cancer patients. J Clin Oncol 2009;27:3437-44.
254. Retsky M, Rogers R, Demicheli R, Hrushesky WJ, Gukas I, et al. NSAID analgesic ketorolac used perioperatively may suppress early breast cancer relapse: particular relevance to triple negative subgroup. Breast Cancer Res Treat 2012;134:881-8.
255. Desmedt C, Demicheli R, Fornili M, Bachir I, Duca M, et al. Potential Benefit of Intra-operative Administration of Ketorolac on Breast Cancer Recurrence According to the Patient's Body Mass Index. J Natl Cancer Inst 2018;110:1115-22.
257. Li J, Jiang E, Wang X, Shangguan AJ, Zhang L, et al. Dormant cells: the original cause of tumor recurrence and metastasis. Cell Biochem Biophys 2015;72:317-20.
258. Cuzick J, Otto F, Baron JA, Brown PH, Burn J, et al. Aspirin and non-steroidal anti-inflammatory drugs for cancer prevention: an international consensus statement. Lancet Oncol 2009;10:501-7.
259. Stagg J, Andre F, Loi S. Immunomodulation via chemotherapy and targeted therapy: a new paradigm in breast cancer therapy? Breast Care (Basel) 2012;7:267-72.
260. Miolo G, Muraro E, Martorelli D, Lombardi D, Scalone S, et al. Anthracycline-free neoadjuvant therapy induces pathological complete responses by exploiting immune proficiency in HER2+ breast cancer patients. BMC Cancer 2014;14:954.
261. Wang BX, Rahbar R, Fish EN. Interferon: current status and future prospects in cancer therapy. J Interferon Cytokine Res 2011;31:545-52.
262. Brockwell NK, Parker BS. Tumor inherent interferons: impact on immune reactivity and immunotherapy. Cytokine 2018; doi: 10.1016/j.cyto.2018.04.006.
263. Ramos MC, Mardegan MC, Tirone NR, Michelin MA, Murta EF. The clinical use of type 1 interferon in gynecology. Eur J Gynaecol Oncol 2010;31:145-50.
264. Corrales L, Gajewski TF. Molecular pathways: targeting the stimulator of interferon genes (STING) in the immunotherapy of cancer. Clin Cancer Res 2015;21:4774-9.
265. Alsaab HO, Sau S, Alzhrani R, Tatiparti K, Bhise K, et al. PD-1 and PD-L1 checkpoint signaling inhibition for cancer immunotherapy: mechanism, combinations, and clinical outcome. Front Pharmacol 2017;8:561.
266. Vikas P, Borcherding N, Zhang W. The clinical promise of immunotherapy in triple-negative breast cancer. Cancer Manag Res 2018;10:6823-33.
267. Schutz F, Stefanovic S, Mayer L, von Au A, Domschke C, et al. PD-1/PD-L1 pathway in breast cancer. Oncol Res Treat 2017;40:294-7.
268. Bianchini G, Pusztai L, Pienkowski T, Im YH, Bianchi GV, et al. Immune modulation of pathologic complete response after neoadjuvant HER2-directed therapies in the NeoSphere trial. Ann Oncol 2015;26:2429-36.
269. Sanchez K, Page D, McArthur HL. Immunotherapy in breast cancer: an overview of modern checkpoint blockade strategies and vaccines. Curr Probl Cancer 2016;40:151-62.
270. Brockwell NK, Owen KL, Zanker D, Spurling A, Rautela J, et al. Neoadjuvant interferons: critical for effective PD-1-based immunotherapy in TNBC. Cancer Immunol Res 2017;5:871-84.
271. Alix-Panabieres C, Pantel K. Clinical applications of circulating tumor cells and circulating tumor DNA as liquid biopsy. Cancer Discov 2016;6:479-91.
272. Spiliotaki M, Mavroudis D, Kapranou K, Markomanolaki H, Kallergi G, et al. Evaluation of proliferation and apoptosis markers in circulating tumor cells of women with early breast cancer who are candidates for tumor dormancy. Breast Cancer Res 2014;16:485.
273. Vishnoi M, Peddibhotla S, Yin W, A TS, George GC, et al. The isolation and characterization of CTC subsets related to breast cancer dormancy. Sci Rep 2015;5:17533.
274. Shaw JA, Page K, Blighe K, Hava N, Guttery D, et al. Genomic analysis of circulating cell-free DNA infers breast cancer dormancy. Genome Res 2012;22:220-31.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Peyvandi S, Lan Q, Lorusso G, Rüegg C. Chemotherapy-induced immunological breast cancer dormancy: a new function for old drugs?. J Cancer Metastasis Treat 2019;5:44. http://dx.doi.org/10.20517/2394-4722.2019.16
AMA Style
Peyvandi S, Lan Q, Lorusso G, Rüegg C. Chemotherapy-induced immunological breast cancer dormancy: a new function for old drugs?. Journal of Cancer Metastasis and Treatment. 2019; 5: 44. http://dx.doi.org/10.20517/2394-4722.2019.16
Chicago/Turabian Style
Peyvandi, Sanam, Qiang Lan, Girieca Lorusso, Curzio Rüegg. 2019. "Chemotherapy-induced immunological breast cancer dormancy: a new function for old drugs?" Journal of Cancer Metastasis and Treatment. 5: 44. http://dx.doi.org/10.20517/2394-4722.2019.16
ACS Style
Peyvandi, S.; Lan Q.; Lorusso G.; Rüegg C. Chemotherapy-induced immunological breast cancer dormancy: a new function for old drugs?. J. Cancer. Metastasis. Treat. 2019, 5, 44. http://dx.doi.org/10.20517/2394-4722.2019.16
About This Article
Copyright

Data & Comments
Data


 Cite This Article 38 clicks
Cite This Article 38 clicks

 Like This Article 22
likes
Like This Article 22
likes






Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.