
Cellular plasticity and metastasis in breast cancer: a pre- and post-malignant problem

Abstract
As a field we have made tremendous strides in treating breast cancer, with a decline in the past 30 years of overall breast cancer mortality. However, this progress is met with little affect once the disease spreads beyond the primary site. With a 5-year survival rate of 22%, 10-year of 13%, for those patients with metastatic breast cancer (mBC), our ability to effectively treat wide spread disease is minimal. A major contributing factor to this ineffectiveness is the complex make-up, or heterogeneity, of the primary site. Within a primary tumor, secreted factors, malignant and pre-malignant epithelial cells, immune cells, stromal fibroblasts and many others all reside alongside each other creating a dynamic environment contributing to metastasis. Furthermore, heterogeneity contributes to our lack of understanding regarding the cells’ remarkable ability to undergo epithelial/non-cancer stem cell (CSC) to mesenchymal/CSC (E-M/CSC) plasticity. The enhanced invasion & motility, tumor-initiating potential, and acquired therapeutic resistance which accompanies E-M/CSC plasticity implicates a significant role in metastasis. While most work trying to understand E-M/CSC plasticity has been done on malignant cells, recent evidence is emerging concerning the ability for pre-malignant cells to undergo E-M/CSC plasticity and contribute to the metastatic process. Here we will discuss the importance of E-M/CSC plasticity within malignant and pre-malignant populations of the tumor. Moreover, we will discuss how one may potentially target these populations, ultimately disrupting the metastatic cascade and increasing patient survival for those with mBC.
Keywords
Introduction: the mortality of metastatic breast cancer
Breast cancer (BC) is the most commonly diagnosed cancer among women, and the second leading cause of cancer related deaths in women[1]. The statistics highlight the importance of metastasis in BC mortality: in patients with distant metastasis, 5-year survival rates are only 22% (13% at 10 years), compared to 90% for patients with local disease[2]. Furthermore, for those patients with metastatic BC (mBC), there are currently no effective treatment options. Understanding how BC cells escape the primary tumor, spread to distant organs, initiate outgrowth at a distant site, and then developing therapies to target those metastatic processes remains a significant clinical challenge. Our understanding of the metastatic cascade has increased in recent years: cells must degrade the extracellular matrix (ECM) surrounding them, extravasate into the circulatory or lymphatic system and circulate throughout the body, intravasate into the new organ tissue, and regain their proliferative capacity[3-5]. Yet, the molecular mechanisms driving each of these processes, all of which are important for a successful metastatic event, remain unresolved.
A complicating factor to our understanding of BC metastasis is the heterogeneous milieu of the primary tumor site, or tumor micro-environment (TME), which is comprised of epithelial, endothelial, immune, and stromal cells. It is important to note that the epithelial populations can be subdivided into two distinct groups, malignant and pre-malignant. The malignant population has completed the transformation process through loss of tumor suppressive mechanisms and a gain of oncogenic signaling, via genetic mutation or sustained growth factor or cytokine signaling. Conversely, pre-malignant refers to a spectrum of points on the path towards transformation. Pre-malignant cells drift further from normalcy as they acquire mutations and engage aberrant signaling. If intact, a tumor suppressive response may be engaged to halt the transformation process, however if lost, the cell may progress to a fully transformed endpoint. The complex cellular composition within the TME results in a network of secreted factors and ECM proteins, which also profoundly influence metastatic potential[6-9]. Numerous TME factors can drive epithelial cells to undergo epithelial-mesenchymal transition (EMT) and acquire cancer stem cell (CSC) properties, which we refer to as epithelial-mesenchymal (E-M)/CSC plasticity[10-14]. Seminal work has defined E-M/CSC plasticity as an important step in metastasis and is often investigated from the perspective of a malignant population. However, malignant cells are not the only populations capable of undergoing E-M/CSC reprogramming. Recent evidence has demonstrated a remarkable ability of pre-malignant epithelial cells to take on a more invasive phenotype able to intravasate and disseminate to secondary sites following signaling cues from the TME[15,16]. Here, we discuss the challenges of targeting various cell populations and the signaling pathways that contribute to the cellular plasticity driving mBC. Importantly, we will explore the impact of pre-malignant cells escaping senescence by undergoing E-M/CSC reprogramming to gain invasive, metastatic, and tumor-initiating properties. Identifying determinants of metastasis, such as E-M/CSC plasticity, and advancing our ability to target the drivers of plasticity will have a significant impact on survival for those with mBC.
Setting the stage: heterogeneity within the primary site
A major challenge in dealing with BC is the heterogeneity that accompanies it. With three distinct clinical subtypes, estrogen and progesterone receptor positive (ER+/PR+), human epidermal growth factor receptor 2 positive (HER2+), and triple negative breast cancer (TNBC; negative for ER, PR, HER2 expression), finding the proper treatment options can be difficult[17-19]. Therapies targeting ER/PR hormone signaling via selective estrogen receptor modulators (SERMs) or aromatase inhibitors (i.e., Tamoxafin and Arimidex) have significantly improved patient survival and have made this subtype more manageable. Likewise, HER2+ tumors can be treated with antibodies or kinase inhibitors targeting HER2 signaling (i.e., Trastuzamab and Lapatinib) and are often met with success provided the disease is caught early; the efficacy of these therapies drastically decreases with late stage, metastatic HER2+ BC[20-23]. Conversely, TNBC currently lacks a targeted therapy tailored to a specific driver oncogene and is most often treated with cytotoxic chemotherapies. Patients with TNBC exhibit an increased risk of metastatic dissemination resulting in higher clinical stage at diagnosis and lower disease-free survival compared to patients with non-TNBC cancers[24]. Much like metastatic HER2+ BC, metastatic TNBC is not effectively treated, highlighting the need for better therapies targeting those cells which progress beyond the primary site and are ultimately responsible for patient mortality and morbidity. Further challenges in treating BC involve the heterogeneous nature of the tumor cells themselves, a phenomenon often referred to as intra-tumoral heterogeneity (ITH)[25]. Evidence suggests that across a panel of human cancers, including breast, increased ITH correlates with decreased overall survival, and therapy resistance[26,27]. Furthermore, high ITH inversely correlates with low tumor infiltrating lymphocytes, which are often associated with increased patient survival[28-36].
The path to ITH is complex and involves a series of genetic and epigenetic events throughout the transformation process which permit normal human mammary epithelial cells (HMEC) to develop into fully malignant cancer cells[37-44]. Progress in RNA and DNA sequencing technologies have helped shape the evolutionary picture of HMEC; losing tumor suppressor function (TP53 mutations or RB loss) and acquiring oncogenic drivers (MYC, HER2, or CCND1 amplification or PIK3CA mutations)[45-49]. Genetic alterations lead to the expansion of a pre-malignant population which progressively acquires additional genetic and epigenetic changes until one or more cells become fully transformed[50,51]. These additional mutations are numerous, and genomic profiling has found a wide variety of changes in copy number, chromatin alterations, chromosomal rearrangements, and point mutations throughout the genome from single cell sequencing of bulk tumor tissue in TNBC[36,52,53]. Not only does this dynamic process of transformation alter the cancer cell itself, but transforming cells have a substantial impact on the surrounding environment. Evidence suggests that the accumulation of mutations within epithelial cells can lead to a dysregulated secretory network, including a number of inflammatory cytokines linked to poor prognosis, therapy failure, and disease recurrence (IL-6, IL-8, TGF-β, CCL2, TNF-α, IL-17 and others)[54-59]. This dysregulated secretory network in turn, changes the cellular composition of the TME, leading to a reciprocal cross-talk between non-cancerous stromal cells and the transforming epithelial cells. Overall, this demonstrates the immense complexity of the tumor, as the heterogeneity described above culminates in a highly diverse TME, with an array of cell types, secreted factors, and structural make up.
Importantly, not all epithelial cells that begin the transformation process reach full malignancy. As a cell senses aberrant activation of signaling pathways/gene induction, it may enact intact tumor suppressive mechanisms, resulting in senescence[60-66]. Senescence is a major growth-inhibiting and tumor-suppressive barrier that must be bypassed in vivo during transformation en-route to tumor development[63,67-73]. Large senescent cell populations can be found at various stages of tumor development, further contributing to tumor heterogeneity. Remarkably, an investigation by Cotarelo et al.[74] was able to identify the presence of senescent cells in approximately 83.7% of the human invasive breast carcinomas examined, suggesting their involvement throughout the transformation process and as tumors evolve and progress. Since the 129 tumors surveyed in this study were from untreated patients, the origin of senescence within these invasive BC is an in vivo physiological response.
Long thought inert, bystanders within the tumor, senescent cells have gained considerable interest for their potential impact on the tumor as a whole. Despite being growth-arrested, senescent cells remain viable, metabolically active, and play an important role in the developing TME[75-77]. A hallmark of senescent cells is the secretion of a wide variety of growth factors, pro-inflammatory cytokines, chemokines, and proteinases, a characteristic termed the senescence-associated secretory phenotype (SASP) [Figure 1][78,79]. Under normal conditions, the SASP-factors act in an autocrine manner to maintain the senescence program and recruit immune cells into the local environment[80-83]. However, paracrine signaling by SASP components can also influence the behavior of adjacent cells, engaging signaling programs that contribute to tumor progression and therapy failure[64,84-89]. A collection of recent studies has demonstrated the ability of senescent cells and SASP components in the TME to drive cellular E-M plasticity and the expansion of a CSC-like cell population[90,91]. In fact, the SASP program can promote stemness within both senescent cells and neighboring cells, both in vitro and in vivo, through secretion of potent inflammatory cytokines associated with disease recurrence, and overall poor prognosis[92,93]. More specifically, less aggressive luminal MCF-7 cells were treated with conditioned medium harvested from senescent populations experiencing SASP. Exposure to conditioned media led to a more CD24LO/CD44HI invasive/stem like population similar to already aggressive MDA-MB-231 cells, which was dependent upon IL6 and IL8, two well defined SASP-factors, secretion[94]. Furthermore, sustained hyper-activation of signal transducer and activator of transcription 3 (STAT3) by SASP components plays a critical role in induction of an invasive and stem-like program[95]. Taken together, the presence of malignant, pre-malignant, and senescent epithelial cells creates a diverse TME suitable to drive E-M/CSC plasticity within the tumor and contribute to metastatic, therapy-resistant, and tumor-initiating phenotypes. Below we will discuss how E-M/CSC plasticity contributes to these deadly phenotypes responsible for patient mortality.
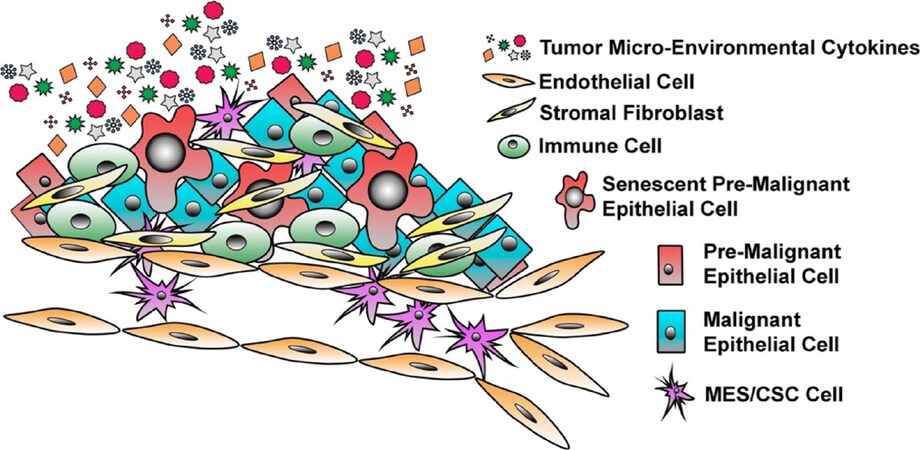
Figure 1. The Heterogeneity of Breast Cancer. Breast cancer is a heterogeneous disease with a highly dynamic tumor micro-environment (TME). Within the primary site, one can observe the presence of epithelial cells that have undergone E-M/CSC plasticity, malignant epithelial cells, pre-malignant cells, senescent epithelial cells, stromal fibroblasts, infiltrating immune cells, and endothelial cells. The presence of all of these diverse cell types results in a distinct and complex milieu of secreted factors within the TME that influence, tumor progression, disease recurrence, and cell plasticity
An identity crisis: mesenchymal vs. epithelial
Each step along the metastatic cascade presents a new environmental context and challenge that a potentially metastatic cell must adapt to in order to thrive. This adaptation involves changes in a cell’s state. Cellular plasticity is defined as the ability of a cell to acquire new biological properties due to intrinsic and extrinsic cues. It is important to recognize that plasticity is most often a highly dynamic and reversible process that can be used to describe multiple cellular changes (i.e., differentiation, metabolism, response to immune cells, motility, and cell fate). Throughout this review, we will refer to cellular plasticity as the ability for cells to undergo E-M/CSC plasticity, that is, cells shifting between epithelial/non-CSC and mesenchymal/CSC states. E-M/CSC plasticity is important in imparting invasive and motile phenotypes as well as cells acquiring tumor-initiating potential and reduced sensitivities to therapy.
The switch from epithelial to mesenchymal state facilitates dissolution of cell-cell junctions (due to the repression of cell adhesion proteins E-Cadherin, EPCAM, and CD24) and increased ability to remodel the surrounding ECM [due to matrix-metalloprotease proteins (MMP), adamalysins (ADAMs), and differential integrin expression][3,96-101]. The corresponding cytoskeletal and ECM remodeling imparts a migratory and invasive phenotype important for metastasis. Lineage tracing experiments confirm that epithelial-to-mesenchymal transition (EMT) occurs in vivo, and that it precedes metastasis in murine BC models[102-104]. Through intravital imaging, Beerling and colleagues showed that mesenchymal tumor cells have a unique and specific migratory behavior that results in greater circulating tumor cells (CTC), increased tumor cells within the lungs, and metastasis; in contrast, the more abundant epithelial cell population remained non-motile and less metastatic[104]. It should be noted that others, Zheng et al.[105] and Fischer et al.[106] have employed in vivo lineage tracing models and reported that EMT is not required for metastasis. As Beerling and colleagues discuss, many of these reports rely on fixed gene manipulation (for example, gene silencing or protein overexpression) to experimentally test an EMT-underlies-metastasis hypothesis. It is possible that such artificial manipulation is not able to recapitulate physiologic events and, in this way, contributes to discrepancies in findings. Other small, but crucial, details could play a further role in some discrepancies: (1) EMT may be indispensable to metastasis for select cancer subtypes, but dispensable for others; (2) reliance on activation of a single gene reporter (e.g., Fsp1) to capture and "tag" an EMT event restricts the sensitivity of the model system; (3) criteria for how the EMT program is identified, such as the panoply of specific “epithelial” or “mesenchymal” proteins that are induced or suppressed, may also lead to false-negatives if these identifying protein sets are incongruent across cancers and cancer subtypes. Regarding the latter point, Zheng et al.[105] and Fischer et al.[106] reported on Vimentin and E-cadherin status, but each represents just one exemplar for mesenchymal or epithelial cell state and ultimately, these may not be the most relevant. Finally, evidence that metastases occur in the absence of EMT does not preclude a potential for EMT to enhance cancer cell metastasis.
The gain of migratory and invasive properties which accompany E-M plasticity occurs concomitantly with changes in global signaling programs and gene expression. Several key transcription factors (TF) have been identified as master regulators of EMT: SNAI1 (Snail), SNAI2 (Slug), ZEB1, ZEB2, TWIST1, and TWIST2[107-112]. These TF are typically kept silenced in adult cells when plasticity is unnecessary but become aberrantly activated by TME factors to induce EMT[113,114]. EMT-TF expression strongly correlates to regions of the tumor with mesenchymal marker positivity, notably, the invasive front of the tumor where mesenchymal cells act as ‘trailblazers’ that initiate and guide local metastasis[108,115-118].
Tissue-invasive cells that encounter vessels may intravasate into vascular or lymph networks and be disseminated throughout the body as CTC. CTC are not only present in measurable quantities in patients with BC, but their abundance is predictive of overall survival and directly correlates with the likelihood of relapse following treatment[119-123]. Moreover, CTC display a wide range of markers associated with cells that have or are undergoing E-M/CSC reprogramming. CTC often exhibit a decrease in epithelial markers CD24, E-Cadherin (CDH1), EPCAM and an increase in well-known mesenchymal and CSC markers (ZEB1, Snail, CD44, Vimentin)[116,124-127]. Critically, CTC are capable of tumor initiation at a secondary site, and more importantly demonstrate remarkable plasticity by engaging specific molecular programs that dictate the organ-specificity of metastases[128-133]. Overall, mesenchymal cells are simply more well-suited to the task of escaping the primary site and reaching distant organs.
However, escaping the primary site and circulating throughout the lymph or cardiovascular system is not sufficient for metastases to develop. Once a mesenchymal cell has reached a secondary organ, it must embed itself in the new tissue and flourish in order to establish a metastatic outgrowth; not all cells have this capability[3,134]. CSC have been deemed the “roots” of primary and secondary site outgrowth due to their tumor-initiating capacity and their ability to differentiate into multiple lineages, recreating the heterogeneity seen in the primary site[135-137]. Since Al-Hajj et al.[138]’s initial isolation of CSC from BC models, marked by CD44HI/CD24LO cell surface expression profile, the field has made significant insight into the CSC paradigm[138]. Shortly after, Ginestier et al.[139] demonstrated an increase in aldehyde dehydrogenase 1 (ALDH1) activity strongly correlated with both normal and malignant stem/progenitor cells within BC. CSC have been isolated from nearly every human cancer through identification of surface marker expression of nearly 40 different markers which vary from cancer to cancer (CD133, CD44, CXCR4, CD90, etc.)[140]. Further complicating matters, our understanding of the dynamics of breast cancer stem cell (BCSC) populations is poorly understood. For instance, Liu et al.[141] identified ALDH1 activity to inversely correlate with CD44 expression in BC models, suggesting a complex make up of phenotypes contributing to tumor initiation, which further work is required to better understand. Regardless of differences in marker expression, isolated CSC populations all exhibit similar characteristics in terms of their ability to initiate tumor formation in limiting dilution, produce multiple cell lineages, maintain tumor-initiating potential through periods of metastatic latency, and survive cytotoxic therapies due to a wide range of resistance mechanisms[142,143]. Seminal papers from the Weinberg and Puisieux groups provided the first evidence of a link between shifting from epithelial to mesenchymal identity and the acquisition of CSC properties in BC[144,145]. Prior to this, the existence of CSC and the occurrence of EMT in malignant neoplasms had each been garnering significant attention, but an association between the two phenotypes had yet to be demonstrated. Specifically, induction of EMT led to a shift in surface marker profiles from CD24HI/CD44LO to CD24LO/CD44HI, resulting in (1) the ability to generate tumorspheres and tumors (self-renewal potential); and (2) the ability to give rise to differentiated daughter cells[144-148].
One implication of this finding is that breast CSC may not solely arise through malignant transformation of a pre-existing ‘normal’ mammary stem cell. More recent work, including ours, has described the ability for transformed epithelial/non-CSC populations to acquire mesenchymal/CSC properties in response to autocrine or paracrine signaling initiated from secreted factors within the TME. Importantly, the ability to fluidly move between epithelial/non-CSC and mesenchymal/CSC states has been shown to facilitate metastatic outgrowth [Figure 2A], as cells are believed to reactivate a proliferative program and reacquire epithelial cell phenotypes at the site of metastasis[12-14,137,141,149-163].
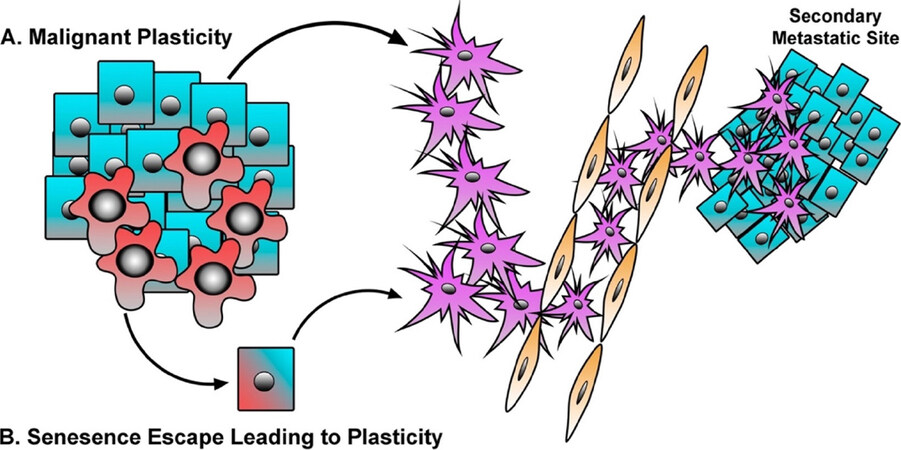
Figure 2. Malignant and Senescent Epithelial Cells Contribute to Metastasis. A: malignant cells respond to TME cytokines or intrinsic genetic alterations in order to drive E-M/CSC plasticity, resulting in greater metastasis, enhanced disease recurrence, and therapeutic resistance; B: pre-malignant cells undergo senescence in response to aberrant oncogene or cytokine signaling. However, in some instances a small population of senescent cells may undergo E-M/CSC plasticity, and thus escape the tumor suppressive barrier of senescence and further contribute to the metastatic phenotype
The quiet neighbor: pre-malignant cells and plasticity
E-M/CSC plasticity is a major contributor to tumor heterogeneity, progression towards metastasis, and therapy failure. Yet, cellular plasticity also plays a crucial role in normal physiology, such as embryonic development, wound healing, and tissue remodeling[164,165]. For instance, differentiated epithelial cells can de-differentiate or trans-differentiate, wherein a differentiated cell may change state into an alternate lineage. These forms of plasticity are seen routinely in skin cells, hepatocytes, colon epithelium, pancreatic acinar cells, and others as they undergo a phenotypic transition in order to repair and sustain the homeostatic nature of the tissue[166-170]. In fact, cellular plasticity of mammary epithelial cells has been suggested to be a driver of the heterogeneity seen in BC[171-173].
Similarly, normal mammary gland organogenesis and homeostasis requires the presence of mammary stem cells (MaSCs) in the mammary gland epithelium that differentiate into both progenitor cells and differentiated cells[174]. Remarkably, many of the markers used to distinguish normal MaSCs overlap with those used to identify breast CSC including mRNA expression profiles[174]. Interestingly E-M plasticity is also essential for normal mammary gland development and maintenance. For example, E-M plasticity is crucial for MaSC migration as well as the expansion and invasion of terminal end buds (TEB) during mammary gland development[174]. Much like the mechanisms that guide stemness in MaSC, the upregulation of multiple EMT-TFs, such as Snail and Twist, was uncovered in the microenvironment of the TEB by genomic transcriptional analysis[175]. E-M plasticity in non-MaSC cells that comprise the normal mammary gland guides cell polarity and the organization of mammary cells into their well-defined glandular structures[176]. Given its importance in tissue remodeling and homeostasis, the normal processes governing cellular plasticity are strictly regulated[177-179].
As discussed above, senescence serves as an important tumor suppressive barrier to prevent transformation and the outgrowth of a dysregulated and uncontrolled population. Interestingly, the induction of senescence in normal cells often results in the simultaneous emergence of mesenchymal/stem-cell markers in conjunction with senescence markers, and loss of proliferative capacity. For example, senescence leads to the induction of CD44 expression, a cell surface marker regularly used to distinguish breast CSC from non-CSC, and often expressed as malignant cells acquire MES/CSC properties and undergo plasticity[138,180-183]. As small subsets of senescent cells dismantle the senescence program, the emergent population of proliferating cells can harbor a mesenchymal/stem-cell phenotype that can persist throughout the remainder of the transformation process, ultimately yielding a more invasive and aggressive cancer cell[184-187]. Li et al.[15] demonstrated that many of the transcriptional changes observed in BC are also initiated in normal HMEC as they escape senescence. In their study, HMEC were shown to have a “pre-transformation” transcriptome and exhibited a partial EMT following their senescence escape[15]. Similarly, normal HMEC that have spontaneously escaped replicative senescence exhibit a greater mesenchymal and CD24LO/CD44HI CSC-phenotype[16,188]. Further studies have suggested cell cycle regulator Cyclin A1 and tumor suppressors p53 and p16 can act as “gatekeepers” to maintain cells in an epithelial state. Dysregulation of these proteins in epithelial cells can result in the initiation of the mesenchymal/stem-cell program, which interestingly corresponds with the escape from senescence[189,190]. Moreover, induction of EMT (via Snail, Twist, and ZEB1 expression) prior to a cell’s engagement of oncogene-induced senescence, prevents senescence altogether and results in the induction of a CSC program and tumor initiation[191-196].
In addition to pre-malignant cells, cytotoxic therapies can drive cancer cells into a therapy-induced senescence (TIS). Again, cells that escape TIS have acquired a senescence-associated stemness[197-199]. Milanovic et al.[200] have shown that cells undergoing TIS express a variety of stem-cell associated markers, and that TIS in B-cell lymphomas exhibit a gene signature which mirrors that of adult tissue stem cells, conferring a highly aggressive phenotype responsible for tumor relapse. The emergence of a stem-like population following the engagement of senescence may be an inherent phenomenon that also occurs during other stress-induced senescence responses (i.e., oncogenes, replication stress, γ-irradiation) among different cell types[200-202]. Altogether, these findings suggest that, the mesenchymal/stem-cell program engaged during senescence may have significant negative consequences if those cells can overcome the signals maintaining senescence, resulting in cells with greater ability metastasize and survive therapy [Figure 2B]. Thus, we suggest that therapies that target senescent cells would limit the reservoir of aggressive cells harboring a senescence-associated stemness responsible for therapy failure and relapse. In the following section we will discuss the potential of targeting E-M/CSC plasticity within malignant, pre-malignant, and senescent populations.
Halting metastasis: targeting pre- and post-malignant cell plasticity
Liquid biopsies from patients with early-stage BC receiving neoadjuvant therapy can be used to identify subjects at high risk of recurrence by quantifying the number of CTC. Furthermore, expression of mesenchymal markers in the CTC correlates with poor prognosis and response to therapy[138,203-206]. With the advent of single cell analysis techniques, our understanding of the evolution and diversity of tumor cells that are responsible for invasion, metastasis, and therapy failure is expanding. For example, single-cell qPCR has identified mesenchymal/CSC gene expression patterns in early-stage breast cancer micro-metastases[137]. In contrast, later-stage metastases (from the same PDX tissue) are more heterogeneous, more proliferative, express differentiation markers, and display greater similarity to the primary tumors. The findings are consistent with the idea that mesenchymal/CSC initiate metastatic outgrowth at a secondary site, followed later by increased proliferation and differentiation. More recent single cell RNA-sequencing (scRNA-seq) studies have confirmed that EMT in primary tumors proceeds through distinct, hybrid states, ranging from completely epithelial to completely mesenchymal[207]. These E-M hybrids, which harbor the greatest level of plasticity, are more efficient at intravasating, extravasating to the lungs, and forming metastases[208]. Underlying this biology, E-M hybrids have distinct chromatin landscapes and transcriptional profiles. In situ analysis identified increasing vascularization and immune cell infiltration (particularly macrophages) nearest the E-M hybrids and fully mesenchymal cells[208]. A separate scRNA-seq study determined that, in response to chemotherapy, emerging chemo-resistant cells undergo transcriptional changes consistent with EMT. In most patients, this chemo-resistant transcriptional program was not evident before treatment but acquired via transcriptional reprogramming following treatment[209]. These studies and others make a strong case that epithelial tumor cells can be induced into a drug-tolerant, E-M hybrid cell state by chemotherapy[141,209-214].
Identifying and targeting the pathways responsible for this chemo-resistant reprogramming would help improve the efficacy of chemotherapy. In a recent example, SRC kinase inhibition prevented the de novo generation of chemo-resistant cells[209]. Importantly, this chemo-sensitization was temporally dependent, and only effective if SRC inhibition occurred after chemotherapy, when the signaling responsible for generating the chemo-resistance phenotype had become activated. More recently, Cazet et al.[215] identified cross-talk between TNBC models and cancer-associated fibroblasts (CAF), which promoted stemness and drug resistance in the cancer cells via paracrine Hedgehog (Hh) signaling. A key developmental pathway, Hh signaling requires receptor mediated binding of Hh to Patched (PTCH), resulting in Smoothened (SMO)-mediated nuclear translocation of Gli1. Gli1 then acts as a transcription factor, mediating Hh pathway transcriptional changes[216]. Hh signaling is often reactivated in a subset of BC, and specifically in the context of TNBC, a paracrine manner[217]. After targeting paracrine Hh signaling in vivo, via two clinically available smoothened inhibitors (SMOi) Vismodegib and Sonidegib, Cazet et al.[215] observed a suppression of cancer cell plasticity and increased sensitivity to docetaxel. Most importantly, in a phase I clinical trial, 3 of 12 patients with metastatic TNBC observed clinical benefit from combinatorial therapy of SMOi and docetaxel (one with complete response), similar to treatment paradigms we suggest above. SMOi have been beneficial for basal cell carcinomas and medulloblastomas where tumors rely on cell-autonomous hedgehog signaling, however the work here suggests the ability to target the TME in order to dampen cancer cell plasticity, and achieve a greater therapeutic response[216,218,219]. Below we will address additional potential therapeutic avenues one may use to dismantle E-M/CSC plasticity in order to prevent metastatic dissemination, secondary site outgrowth, or re-sensitize cancer cells to standard of care therapies.
Targeting malignant populations
STAT3
STAT3 is persistently activated in cancer cells, as it is a downstream effector of several receptor tyrosine kinases (RTKs) commonly activated by growth factors and cytokines[220-222]. We, as well as others have demonstrated that, persistent STAT3 activation in cancer cells induces mesenchymal and CSC properties, inhibits apoptosis, and maintains a more un-differentiated phenotype[12,14]. Therefore, STAT3 is a promising therapeutic target. A number of small-molecule inhibitors of STAT3 (KI16; BP-5-087; WP1066) are currently in development and combination therapies, with BCR-ABL1 or BRAF inhibitors, have shown positive results in the treatment of several cancer types[223-225]. In the context of TNBC, a recent phase Ib/II study combining the cancer stemness inhibitor Napubacasin (BB608), which prevents STAT3 activation, with weekly administrations of paclitaxel showed improvement in metastatic patients whose cancer had progressed while on a taxane-based regimen (NCT01325441).
PI3K, Akt and mTOR
The PI3K/Akt/mTOR pathway is involved in several cell processes, including proliferation, metabolism and motility, therefore it is not surprising that its dysregulation corresponds to uncontrolled proliferation and propagation in a wide spectrum of cancers. The role of the PI3K/Akt/mTOR pathway in maintaining cell plasticity in cancer has been documented in several publications[226-228]. In the context of breast cancer, PIK3CA mutations have been observed in each of the different subtypes, but mostly in hormone receptor-positive tumors where it’s associated with disease progression and resistance to endocrine therapy. Each PIK3CA mutation results in an abnormal activation of the alpha subunit of PI3K, that with the beta subunit is the most common in breast tissue[229]. PIK3CA mutations appear to hold prognostic and predictive value in hormone receptor-positive, HER2-negative advanced or metastatic breast cancer. Several studies show how targeting tumors carrying a PIK3CA mutation with PIK3CA inhibitors increased the PFS of patients[230]. In January 2019, results from the phase III SANDPIPER clinical trial (NCT02340221) were posted. This international, multicenter, randomized, double-blinded, placebo-controlled study was designed to test the efficacy of a combo of the PIK3CA SMI taselisib and the synthetic estrogen receptor antagonist fulvestran versus placebo and fulvestran in the treatment of ER-positive, HER-2-negative locally advanced or metastatic breast cancer harboring a PIK3CA mutation in patients with disease recurrence after or during treatment with aromatase inhibitor (AI). SANDIPIPER is the first placebo-controlled trial testing the efficacy of a mutant-specific PI3K inhibitor[231]. Taselisib is specifically directed against the alpha isoform of PIK3CA, however it can inhibit the gamma and delta isoforms as well, thus causing an increase in toxicity mostly involving the gastro-intestinal tract. Another phase III clinical trial, BELLE-2 (NCT01610284) is analyzing the effects of the pan-PI3K inhibitor buparlisib in combination with fulvestran compared to fulvestran and placebo combo. Unfortunately, also buparlisib showed important side effects, particularly hyperglycemia[229]. Several other PI3K inhibitors are currently under investigation in clinical trials, including apelisib (NCT02437318 - SOLAR-1), which showed encouraging clinical benefits in the majority of patients enrolled[232]. In the future, research efforts should be more focused at inhibiting exclusively the alpha subunit of PIK3CA, thus reducing the risk of toxicity and side effects. Interestingly, GDC-0077 from Genentech appears to be extremely more specific towards the alpha subunit of PIK3A over other subunits, thus representing a potentially less toxic alternative to other inhibitors and is currently investigated in a phase I clinical trial alone or in combination with other agents, such as palbociclib, letrozole and fulvestran (NCT03006172).
While the role of mTOR signaling in promoting a CSC phenotype is still controversial, its activation in BC appears to be essential for colony formation in vitro and tumorigenicity[233]. Furthermore, mTOR signaling increases aldehyde dehydrogenase 1 (ALDH1) activity[234,235]. IGF-1R activation signaling through PI3K/Akt/mTOR represents a promising target in BC as an abnormal activation of PI3K can also lead to an increased activation of STAT3 through enhanced expression of the chemokine (C-X-C motif) receptor 4 (CXCR4)[236,237]. IGF-1R is activated in the 50% of breast cancer patients. Several phase III clinical trials targeting IGF-1R with small molecule inhibitors have so far failed due to side effects such as hyperglycemia and metabolic syndrome caused by the homology of IGF-1R and insulin receptor (IR) and IGF elevation in response to disruption of glucose homeostasis. On the other hand, monoclonal antibodies specifically targeting IGF-1R or its ligand have shown a higher specificity by sparing the insulin metabolism from any inhibitory effect in preclinical models[238]. Early phase trials have been setup to determine the efficacy of an antibody-based inhibition of IGF-1R signaling by targeting either the receptor or the ligand in combination with aromatase inhibitors or mTOR inhibitors but with small success[239]. A more complex therapeutic protocol combining inhibitors of PI3K or other downstream effectors of IGF-1R would probably be more beneficial.
Several drugs targeting the PI3K/mTOR pathway have shown a selective effect on CSCs, inhibiting their growth and sensitizing them to traditional chemotherapy. Since 2007, when the Food and Drug Administration (FDA) approved the mTOR inhibitor temsirolimus for the treatment of advanced renal carcinoma, new generations of compounds have been developed. Everolimus, is a dual PI3K/mTOR inhibitor, which blocks all PI3K class I isoforms as well as mTORC1 and 2, thus preventing the development of CSCs when combined with letrozole[240,241]. Interestingly, mTOR is also inhibited by metformin (1,1-dimethylbiguanide hydrochloride), usually prescribed for the treatment of type 2 diabetes. Metformin preferentially kills CSCs over non-CSCs and reduces tumorsphere formation and CSC marker expression (CD133, CD44 and ALDH1)[242]. Similar effects have been achieved with the antibiotic salinomycin, which selectively kills breast CSC[243]. The plant-derived chemotherapeutic molecule rottlerin induces apoptosis in breast CSC by inhibiting PI3K/Akt/mTOR pathway[244]. More recently, the PI3K/mTOR dual inhibitor VS5584 has shown promising results by delaying tumor recurrence through selective killing of CSC after chemotherapy[245].
Notch
Notch signaling is known to be involved in different cellular processes, including differentiation and proliferation. Perturbation in these processes can be caused by mutations in Notch or one of its effectors. Mutations of the Notch pathway are the hallmark of many subtypes of cancer, including BC, where cells overexpressing Notch have increased CSC markers (SOX2 and OCT4) and phenotypes (tumorsphere formation)[246-248]. Moreover, Notch promotes EMT and metastasis in TNBC cells and Notch inhibition can prevent EMT both in vitro and in vivo[249]. For example, 3,6-dihydroxiflavone (3,6-DHF) causes a significant reduction of CSCs in vitro and blocks lung metastasis by specifically down regulating Notch, Hes-1 and c-Myc[250]. 3,6 DHF shows potent chemo-preventive properties against breast carcinogenesis both in vitro and in vivo, although a mechanism has not been identified yet, besides an epigenetic increase in the synthesis of miR-34a, a potent down-regulator of Notch and thus of EMT in breast cancer[251]. Furthermore, 3,6-DHF down-regulates the expression of Notch’s target genes Hes1, c-Myc and the EMT mediators SNAIL, Twist and Slug by compromising the formation of the transcriptional complex NICD-CSL-MAML[252,253]. More recently, it has been shown that Notch3 inhibition by siRNA silencing increases TNBC sensitivity to gefitinib in EGFR-Tyrosine kinase inhibitors (TKI)-resistant cells by blocking the nuclear translocation of activated EGFR[254].
Wnt and β-catenin
The Wnt pathway is involved in the maintenance of breast CSCs by promoting self-renewal and plasticity through PAF (proliferating cell nuclear antigen-associated factor)[255]. The Wnt ligand, Frizzled, is upregulated in high-grade tumors, including more aggressive forms of BC, and can cause EMT and metastasis through non-canonical STAT3 activation[256]. Inhibitors targeting the Wnt pathway have been developed and are showing promise in BC models. In particular, the Wnt/beta-catenin inhibitor CWP232228, which blocks beta-catenin binding to T-cell factor (TCF) in the nucleus, prevents the proliferation of breast CSC, selectively depleting CD133-positive and ALDH1-high cells both in vitro and in vivo[257,258].
Targeting senescent populations
As described in the previous section, cells escaping senescence exhibit increased invasive and tumor-initiating properties. On top of that, senescent cells secrete a variety of cytokines and growth factors as part of the SASP. The production of these factors into the TME has been shown to drive E-M/CSC plasticity in neighboring cells, as well as the senescent cells themselves, expanding the population that can facilitate metastasis and drug resistance. Moreover, the chronic presence of senescent cells can impair local tissue function, create a highly inflammatory environment, and in some instances exacerbate the side effects of chemotherapies[259,260]. Considering conventional treatments such as chemo- and radio-therapy often induce senescence in both cancer and normal cells, it seems pertinent to target senescent cells and clear them from the local tissue. The concept for targeting senescent cells was brought to light by Lee et al.[261] by exploiting the high lysosomal β-galactosidase activity in senescent cells, cytotoxic drugs encapsulated in galacto-oligosaccharides particles (galNP beads) can target chemotherapy-induced senescent cells in mice[262]. Pre-clinical results showed a significant regression of tumor xenografts after treatment with galNP beads loaded with doxorubicin in combination with palbociclib[263]. Moreover, senescent-cell accumulation in mice can be reduced by treatment with potential senolytic agent, Navitoclax (ABT-263), a small molecule inhibitor of the anti-apoptotic proteins BCL-2 and BCLxL[264,265]. However, targeting senescent cells is a relatively new concept, and further insights into the signaling mechanisms which senescent cells rely on is needed.
Conclusion
To date our ability to target BC metastases have been largely unsuccessful. With patient survival falling to 22% for those that reach distant and wide-spread disease, the ability to target cells at various stages of the metastatic cascade is greatly needed. Here, we have focused on an epithelial cancer cell’s ability to out-maneuver cytotoxic agents by changing cell state; E-M/CSC plasticity. This induced reprogramming often reduces sensitivity to therapy by a number of mechanisms, creating an immense problem with effectively removing the disease. We propose to remove the molecular “escape hatch” which provides cells that have undergone E-M/CSC reprogramming a sustained advantage in survival and resurgence. An approach that combines readily available small molecule inhibitors of plasticity-inducing pathways in conjunction with commonly used front-line therapies should increase therapeutic sensitivities. Often, pathway-selective small molecule inhibitors that make it to the clinic are used with the objective of inducing growth inhibition or cell death. However, many of these inhibitors may present a wide range of side effects by acting on non-tumor cells or show little efficacy as a single agent[266-268]. Instead, a low dosage may prevent toxicity or off-target effects while reducing a tumor cells ability to undergo reprograming to a more MES/CSC-like state. In doing so, these combination therapies may overcome the MES/CSC-like programs which promote therapy failure and metastatic disease progression, ultimately rendering populations sensitive to currently used chemotherapies and increasing overall patient survival.
Cell plasticity is garnering significant interest in the field of cancer biology, as we attempt to better understand the metastatic process, and those drivers behind it. However, we do not yet fully understand what allows an epithelial cell to undergo MES/CSC reprograming or what pathways maintain this newly attained phenotype. While most of the work understanding plasticity has been done on malignant populations, recent studies have hinted at the ability of pre-malignant populations to undergo MES/stem-cell reprograming, albeit often halted by intact tumor suppressive mechanisms, leading to senescence. Here, we discussed the ability of pre-malignant epithelial cells to undergo a MES/stem-cell reprogramming that is comparable to that observed with malignant cells. However, much remains to be discovered about the importance of these pre-malignant populations. We still do not yet know which cells within a pre-malignant population have escaped the senescence “barrier”, which in turn generate more aggressive sub-populations capable of driving metastasis. Identifying, or molecularly defining, which cells possess the capability to escape senescence may allow us to predict the aggressiveness of a patient’s metastatic burden and open potential avenues for targeting cells with the potential to escape senescence. Ultimately, targeting the signaling responsible for plasticity, both in pre-malignant and fully transformed cells, will enhance the efficacy of chemotherapies and suppress a key driving force responsible for patient death.
Declarations
Authors’ contributionsFigure concept/design and writing of the manuscript: Smigiel JM
Writing of the manuscript: Taylor SE, Bryson BL, Tamagno I, Polak K, Jackson MW
Availability of data and materialsNot applicable.
Financial support and sponsorshipThis work is supported by F30 (NIH NCIF30 CA224979) to Taylor SE.
Conflicts of interestAll authors declared that there are no conflicts of interest.
Ethical approval and consent to participateNot applicable.
Consent for publicationNot applicable.
Copyright© The Author(s) 2019.
REFERENCES
1. Jemal A, Bray F, Center MM, Ferlay J, Ward E, et al. Global cancer statistics. CA Cancer J Clin 2011;61:69-90.
2. Eng LG, Dawood S, Sopik V, Haaland B, Tan PS, et al. Ten-year survival in women with primary stage IV breast cancer. Breast Cancer Res Treat 2016;160:145-52.
3. Lambert AW, Pattabiraman DR, Weinberg RA. Emerging biological principles of metastasis. Cell 2017;168:670-91.
4. Kienast Y, von Baumgarten L, Fuhrmann M, Klinkert WE, Goldbrunner R, et al. Real-time imaging reveals the single steps of brain metastasis formation. Nat Med 2010;16:116-22.
6. Nakamura T, Fidler IJ, Coombes KR. Gene expression profile of metastatic human pancreatic cancer cells depends on the organ microenvironment. Cancer Res 2007;67:139-48.
7. Quail DF, Joyce JA. Microenvironmental regulation of tumor progression and metastasis. Nat Med 2013;19:1423-37.
8. Salvatore V, Teti G, Focaroli S, Mazzotti MC, Mazzotti A, et al. The tumor microenvironment promotes cancer progression and cell migration. Oncotarget 2017;8:9608-16.
10. Jung HY, Fattet L, Yang J. Molecular pathways: linking tumor microenvironment to epithelial-mesenchymal transition in metastasis. Clin Cancer Res 2015;21:962-8.
11. Qiao Y, Shiue CN, Zhu J, Zhuang T, Jonsson P, et al. AP-1-mediated chromatin looping regulates ZEB2 transcription: new insights into TNFalpha-induced epithelial-mesenchymal transition in triple-negative breast cancer. Oncotarget 2015;6:7804-14.
12. Smigiel JM, Parameswaran N, Jackson MW. Potent EMT and CSC phenotypes are induced by oncostatin-m in pancreatic cancer. Mol Cancer Res 2017;15:478-88.
13. Junk DJ, Cipriano R, Bryson BL, Gilmore HL, Jackson MW. Tumor microenvironmental signaling elicits epithelial-mesenchymal plasticity through cooperation with transforming genetic events. Neoplasia 2013;15:1100-9.
14. Junk DJ, Bryson BL, Smigiel JM, Parameswaran N, Bartel CA, et al. Oncostatin M promotes cancer cell plasticity through cooperative STAT3-SMAD3 signaling. Oncogene 2017;36:4001-13.
15. Li Y, Pan J, Li JL, Lee JH, Tunkey C, et al. Transcriptional changes associated with breast cancer occur as normal human mammary epithelial cells overcome senescence barriers and become immortalized. Mol Cancer 2007;6:7.
16. Romanov SR, Kozakiewicz BK, Holst CR, Stampfer MR, Haupt LM, et al. Normal human mammary epithelial cells spontaneously escape senescence and acquire genomic changes. Nature 2001;409:633-7.
17. Prat A, Adamo B, Cheang MC, Anders CK, Carey LA, et al. Molecular characterization of basal-like and non-basal-like triple-negative breast cancer. Oncologist 2013;18:123-33.
18. Perou CM, Sorlie T, Eisen MB, van de Rijn M, Jeffrey SS, et al. Molecular portraits of human breast tumours. Nature 2000;406:747-52.
19. Nielsen TO, Parker JS, Leung S, Voduc D, Ebbert M, et al. A comparison of PAM50 intrinsic subtyping with immunohistochemistry and clinical prognostic factors in tamoxifen-treated estrogen receptor-positive breast cancer. Clin Cancer Res 2010;16:5222-32.
20. Slamon DJ, Leyland-Jones B, Shak S, Fuchs H, Paton V, et al. Use of chemotherapy plus a monoclonal antibody against HER2 for metastatic breast cancer that overexpresses HER2. N Engl J Med 2001;344:783-92.
21. Geyer CE, Forster J, Lindquist D, Chan S, Romieu CG, et al. Lapatinib plus capecitabine for HER2-positive advanced breast cancer. N Engl J Med 2006;355:2733-43.
22. Baselga J, Cortes J, Kim SB, Im SA, Hegg R, et al. Pertuzumab plus trastuzumab plus docetaxel for metastatic breast cancer. N Engl J Med 2012;366:109-19.
23. Diéras V, Miles D, Verma S, Pegram M, Welslau M, et al. Trastuzumab emtansine versus capecitabine plus lapatinib in patients with previously treated HER2-positive advanced breast cancer (EMILIA): a descriptive analysis of final overall survival results from a randomised, open-label, phase 3 trial. Lancet Oncol 2017;18:732-42.
24. Qiu J, Xue X, Hu C, Xu H, Kou D, et al. Comparison of clinicopathological features and prognosis in triple-negative and non-triple negative breast cancer. J Cancer 2016;7:167-73.
25. Alizadeh AA, Aranda V, Bardelli A, Blanpain C, Bock C, et al. Toward understanding and exploiting tumor heterogeneity. Nat Med 2015;21:846-53.
26. Junttila MR, de Sauvage FJ. Influence of tumour micro-environment heterogeneity on therapeutic response. Nature 2013;501:346-54.
27. Morris LG, Riaz N, Desrichard A, Senbabaoglu Y, Hakimi AA, et al. Pan-cancer analysis of intratumor heterogeneity as a prognostic determinant of survival. Oncotarget 2016;7:10051-63.
28. Erez N, Truitt M, Olson P, Arron ST, Hanahan D. Cancer-associated fibroblasts are activated in incipient neoplasia to orchestrate tumor-promoting inflammation in an NF-kappaB-dependent manner. Cancer Cell 2010;17:135-47.
29. Gajewski TF, Schreiber H, Fu YX. Innate and adaptive immune cells in the tumor microenvironment. Nat Immunol 2013;14:1014-22.
30. Bianchini G, Balko JM, Mayer IA, Sanders ME, Gianni L. Triple-negative breast cancer: challenges and opportunities of a heterogeneous disease. Nat Rev Clin Oncol 2016;13:674-90.
31. Sikov WM, Barry WT, Hoadley KA, Pitcher BN, Singh B, et al. Abstract S4-05: Impact of intrinsic subtype by PAM50 and other gene signatures on pathologic complete response (pCR) rates in triple-negative breast cancer (TNBC) after neoadjuvant chemotherapy (NACT) +/- carboplatin (Cb) or bevacizumab (Bev): CALGB 40603/150709 (Allianc. Cancer Research 2015;75:S4-05-S4-05.
32. Sabatier R, Finetti P, Mamessier E, Adelaide J, Chaffanet M, et al. Prognostic and predictive value of PDL1 expression in breast cancer. Oncotarget 2015;6:5449-64.
33. Callari M, Cappelletti V, D'Aiuto F, Musella V, Lembo A, et al. Subtype-Specific Metagene-Based Prediction of Outcome after Neoadjuvant and Adjuvant Treatment in Breast Cancer. Clin Cancer Res 2016;22:337-45.
34. Gu-Trantien C, Loi S, Garaud S, Equeter C, Libin M, et al. CD4(+) follicular helper T cell infiltration predicts breast cancer survival. J Clin Invest 2013;123:2873-92.
35. Denkert C, von Minckwitz G, Brase JC, Sinn BV, Gade S, et al. Tumor-infiltrating lymphocytes and response to neoadjuvant chemotherapy with or without carboplatin in human epidermal growth factor receptor 2-positive and triple-negative primary breast cancers. J Clin Oncol 2015;33:983-91.
36. Chung W, Eum HH, Lee HO, Lee KM, Lee HB, et al. Single-cell RNA-seq enables comprehensive tumour and immune cell profiling in primary breast cancer. Nat Commun 2017;8:15081.
37. Yaswen P, Stampfer MR. Molecular changes accompanying senescence and immortalization of cultured human mammary epithelial cells. The International Journal of Biochemistry & Cell Biology 2002;34:1382-94.
38. Novak P, Jensen TJ, Garbe JC, Stampfer MR, Futscher BW. Stepwise DNA methylation changes are linked to escape from defined proliferation barriers and mammary epithelial cell immortalization. Cancer Res 2009;69:5251-8.
39. Garbe JC, Vrba L, Sputova K, Fuchs L, Novak P, et al. Immortalization of normal human mammary epithelial cells in two steps by direct targeting of senescence barriers does not require gross genomic alterations. Cell Cycle 2014;13:3423-35.
40. Saab R. Senescence and pre-malignancy: how do tumors progress? Semin Cancer Biol 2011;21:385-91.
43. Schmitt CA. Senescence, apoptosis and therapy--cutting the lifelines of cancer. Nat Rev Cancer 2003;3:286-95.
44. Ito Y, Hoare M, Narita M. Spatial and temporal control of senescence. Trends Cell Biol 2017;27:820-32.
46. Shah SP, Roth A, Goya R, Oloumi A, Ha G, et al. The clonal and mutational evolution spectrum of primary triple-negative breast cancers. Nature 2012;486:395-9.
47. Rajendran BK, Deng CX. Characterization of potential driver mutations involved in human breast cancer by computational approaches. Oncotarget 2017;8:50252-72.
48. Yates LR, Knappskog S, Wedge D, Farmery JHR, Gonzalez S, et al. Genomic Evolution of Breast Cancer Metastasis and Relapse. Cancer Cell 2017;32:169-84 e7.
49. Nik-Zainal S, Davies H, Staaf J, Ramakrishna M, Glodzik D, et al. Landscape of somatic mutations in 560 breast cancer whole-genome sequences. Nature 2016;534:47-54.
50. Reya T, Morrison SJ, Clarke MF, Weissman IL. Stem cells, cancer, and cancer stem cells. Nature 2001;414:105-11.
51. Stephens PJ, Tarpey PS, Davies H, Van Loo P, Greenman C, et al. The landscape of cancer genes and mutational processes in breast cancer. Nature 2012;486:400-4.
52. Ellsworth DL, Blackburn HL, Shriver CD, Rabizadeh S, Soon-Shiong P, et al. Single-cell sequencing and tumorigenesis: improved understanding of tumor evolution and metastasis. Clin Transl Med 2017;6:15.
53. Karaayvaz M, Cristea S, Gillespie SM, Patel AP, Mylvaganam R, et al. Unravelling subclonal heterogeneity and aggressive disease states in TNBC through single-cell RNA-seq. Nat Commun 2018;9:3588.
54. Polyak K, Haviv I, Campbell IG. Co-evolution of tumor cells and their microenvironment. Trends Genet 2009;25:30-8.
55. Allinen M, Beroukhim R, Cai L, Brennan C, Lahti-Domenici J, et al. Molecular characterization of the tumor microenvironment in breast cancer. Cancer Cell 2004;6:17-32.
56. Chen W, Qin Y, Liu S. Cytokines, breast cancer stem cells (BCSCs) and chemoresistance. Clin Transl Med 2018;7:27.
57. Tan C, Hu W, He Y, Zhang Y, Zhang G, et al. Cytokine-mediated therapeutic resistance in breast cancer. Cytokine 2018;108:151-9.
58. Esquivel-Velazquez M, Ostoa-Saloma P, Palacios-Arreola MI, Nava-Castro KE, Castro JI, et al. The role of cytokines in breast cancer development and progression. J Interferon Cytokine Res 2015;35:1-16.
59. Al-Hassan AA. Prognostic value of proinflammatory cytokines in breast cancer. J Biomol Res Ther 2013;1:104.
60. Larsson LG. Oncogene- and tumor suppressor gene-mediated suppression of cellular senescence. Semin Cancer Biol 2011;21:367-76.
61. Serrano M, Lin AW, McCurrach ME, Beach D, Lowe SW. Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell 1997;88:593-602.
62. Feldser DM, Greider CW. Short telomeres limit tumor progression in vivo by inducing senescence. Cancer Cell 2007;11:461-9.
63. Campisi J. Cellular senescence as a tumor-suppressor mechanism. Trends Cell Biol 2001;11:S27-31.
66. Ishikawa F. Cellular senescence, an unpopular yet trustworthy tumor suppressor mechanism. Cancer Science 2003;94:944-7.
67. Westphalen CB, Renz BW, Reichert M, Rustgi AK, Wang TC. Cellular plasticity and heterogeneity in pancreatic regeneration and malignancy. Cancer Cell Microenvironment 2016;3:e1472.
68. Salama R, Sadaie M, Hoare M, Narita M. Cellular senescence and its effector programs. Genes Development 2014;28:99-114.
70. Prieur A, Peeper DS. Cellular senescence in vivo: a barrier to tumorigenesis. Curr Opin Cell Biol 2008;20:150-5.
71. Gruber HE, Hoelscher GL, Ingram JA, Zinchenko N, Hanley EN Jr. Senescent vs. non-senescent cells in the human annulus in vivo: cell harvest with laser capture microdissection and gene expression studies with microarray analysis. BMC Biotechnol 2010;10:5.
72. Zarling JM, Shoyab M, Marquardt H, Hanson MB, Lioubin MN, et al. Oncostatin M: a growth regulator produced by differentiated histiocytic lymphoma cells. Proc Natl Acad Sci U S A 1986;83:9739-43.
73. Efimova EV, Mauceri HJ, Golden DW, Labay E, Bindokas VP, et al. Poly(ADP-ribose) polymerase inhibitor induces accelerated senescence in irradiated breast cancer cells and tumors. Cancer Res 2010;70:6277-82.
74. Cotarelo CL, Schad A, Kirkpatrick CJ, Sleeman JP, Springer E, et al. Detection of cellular senescence within human invasive breast carcinomas distinguishes different breast tumor subtypes. Oncotarget 2016;7:74846-59.
75. Acosta JC, Banito A, Wuestefeld T, Georgilis A, Janich P, et al. A complex secretory program orchestrated by the inflammasome controls paracrine senescence. Nat Cell Biol 2013;15:978-90.
76. Iannello A, Thompson TW, Ardolino M, Lowe SW, Raulet DH. p53-dependent chemokine production by senescent tumor cells supports NKG2D-dependent tumor elimination by natural killer cells. J Exp Med 2013;210:2057-69.
77. Demaria M, Ohtani N, Youssef SA, Rodier F, Toussaint W, et al. An essential role for senescent cells in optimal wound healing through secretion of PDGF-AA. Dev Cell 2014;31:722-33.
78. Tchkonia T, Zhu Y, van Deursen J, Campisi J, Kirkland JL. Cellular senescence and the senescent secretory phenotype: therapeutic opportunities. J Clin Invest 2013;123:966-72.
79. Watanabe S, Kawamoto S, Ohtani N, Hara E. Impact of senescence-associated secretory phenotype and its potential as a therapeutic target for senescence-associated diseases. Cancer Sci 2017;108:563-9.
80. Coppe JP, Patil CK, Rodier F, Sun Y, Munoz DP, et al. Senescence-associated secretory phenotypes reveal cell-nonautonomous functions of oncogenic RAS and the p53 tumor suppressor. PLoS Biol 2008;6:2853-68.
81. Kuilman T, Peeper DS. Senescence-messaging secretome: SMS-ing cellular stress. Nat Rev Cancer 2009;9:81-94.
82. Hernandez-Segura A, Nehme J, Demaria M. Hallmarks of Cellular Senescence. Trends Cell Biol 2018;28:436-53.
83. Sagiv A, Krizhanovsky V. Immunosurveillance of senescent cells: the bright side of the senescence program. Biogerontology 2013;14:617-28.
84. Alspach E, Fu Y, Stewart SA. Senescence and the pro-tumorigenic stroma. Crit Rev Oncog 2013;18:549-58.
85. Ruhland MK, Loza AJ, Capietto AH, Luo X, Knolhoff BL, et al. Stromal senescence establishes an immunosuppressive microenvironment that drives tumorigenesis. Nat Commun 2016;7:11762.
86. Laberge RM, Awad P, Campisi J, Desprez PY. Epithelial-mesenchymal transition induced by senescent fibroblasts. Cancer Microenviron 2012;5:39-44.
87. Coppe JP, Desprez PY, Krtolica A, Campisi J. The senescence-associated secretory phenotype: the dark side of tumor suppression. Annu Rev Pathol 2010;5:99-118.
88. Pribluda A, Elyada E, Wiener Z, Hamza H, Goldstein RE, et al. A senescence-inflammatory switch from cancer-inhibitory to cancer-promoting mechanism. Cancer Cell 2013;24:242-56.
89. Hoare M, Narita M. Transmitting senescence to the cell neighbourhood. Nat Cell Biol 2013;15:887-9.
90. Cahu J, Bustany S, Sola B. Senescence-associated secretory phenotype favors the emergence of cancer stem-like cells. Cell Death Dis 2012;3:e446.
91. Ritschka B, Storer M, Mas A, Heinzmann F, Ortells MC, et al. The senescence-associated secretory phenotype induces cellular plasticity and tissue regeneration. Genes Dev 2017;31:172-83.
92. Castro-Vega LJ, Jouravleva K, Ortiz-Montero P, Liu WY, Galeano JL, et al. The senescent microenvironment promotes the emergence of heterogeneous cancer stem-like cells. Carcinogenesis 2015;36:1180-92.
93. Mosteiro L, Pantoja C, Alcazar N, Marion RM, Chondronasiou D, et al. Tissue damage and senescence provide critical signals for cellular reprogramming in vivo. Science 2016;354.
94. Ortiz-Montero P, Londono-Vallejo A, Vernot JP. Senescence-associated IL-6 and IL-8 cytokines induce a self- and cross-reinforced senescence/inflammatory milieu strengthening tumorigenic capabilities in the MCF-7 breast cancer cell line. Cell Commun Signal 2017;15:17.
95. Canino C, Mori F, Cambria A, Diamantini A, Germoni S, et al. SASP mediates chemoresistance and tumor-initiating-activity of mesothelioma cells. Oncogene 2012;31:3148-63.
96. Thiery JP, Acloque H, Huang RY, Nieto MA. Epithelial-mesenchymal transitions in development and disease. Cell 2009;139:871-90.
97. Perl AK, Wilgenbus P, Dahl U, Semb H, Christofori G. A causal role for E-cadherin in the transition from adenoma to carcinoma. Nature 1998;392:190-3.
98. Onder TT, Gupta PB, Mani SA, Yang J, Lander ES, et al. Loss of E-cadherin promotes metastasis via multiple downstream transcriptional pathways. Cancer Res 2008;68:3645-54.
99. Petrova YI, Schecterson L, Gumbiner BM. Roles for E-cadherin cell surface regulation in cancer. Mol Biol Cell 2016;27:3233-44.
100. Bruner HC, Derksen PWB. Loss of e-cadherin-dependent cell-cell adhesion and the development and progression of cancer. Cold Spring Harb Perspect Biol 2018;10.
101. Moreno-Bueno G, Portillo F, Cano A. Transcriptional regulation of cell polarity in EMT and cancer. Oncogene 2008;27:6958-69.
102. Trimboli AJ, Fukino K, de Bruin A, Wei G, Shen L, et al. Direct evidence for epithelial-mesenchymal transitions in breast cancer. Cancer Res 2008;68:937-45.
103. Zhao Z, Zhu X, Cui K, Mancuso J, Federley R, et al. In Vivo Visualization and Characterization of Epithelial-Mesenchymal Transition in Breast Tumors. Cancer Res 2016;76:2094-104.
104. Beerling E, Seinstra D, de Wit E, Kester L, van der Velden D, et al. Plasticity between Epithelial and Mesenchymal States Unlinks EMT from Metastasis-Enhancing Stem Cell Capacity. Cell Rep 2016;14:2281-8.
105. Zheng XF, Carstens JL, Kim J, Scheible M, Kaye J, et al. Epithelial-to-mesenchymal transition is dispensable for metastasis but induces chemoresistance in pancreatic cancer. Nature 2015;527:525.
106. Fischer KR, Durrans A, Lee S, Sheng J, Li F, et al. Epithelial-to-mesenchymal transition is not required for lung metastasis but contributes to chemoresistance. Nature 2015;527:472-6.
107. Cano A, Perez-Moreno MA, Rodrigo I, Locascio A, Blanco MJ, et al. The transcription factor snail controls epithelial-mesenchymal transitions by repressing E-cadherin expression. Nat Cell Biol 2000;2:76-83.
108. Comijn J, Berx G, Vermassen P, Verschueren K, van Grunsven L, et al. The two-handed E box binding zinc finger protein SIP1 downregulates E-cadherin and induces invasion. Mol Cell 2001;7:1267-78.
109. Bolos V. The transcription factor Slug represses E-cadherin expression and induces epithelial to mesenchymal transitions: a comparison with Snail and E47 repressors. Journal of Cell Science 2002;116:499-511.
110. Yang J, Mani SA, Donaher JL, Ramaswamy S, Itzykson RA, et al. Twist, a master regulator of morphogenesis, plays an essential role in tumor metastasis. Cell 2004;117:927-39.
111. Peinado H, Olmeda D, Cano A. Snail, Zeb and bHLH factors in tumour progression: an alliance against the epithelial phenotype? Nat Rev Cancer 2007;7:415-28.
112. Ye X, Tam WL, Shibue T, Kaygusuz Y, Reinhardt F, et al. Distinct EMT programs control normal mammary stem cells and tumour-initiating cells. Nature 2015;525:256-60.
113. Ansieau S, Morel AP, Hinkal G, Bastid J, Puisieux A. TWISTing an embryonic transcription factor into an oncoprotein. Oncogene 2010;29:3173-84.
114. De Craene B, Berx G. Regulatory networks defining EMT during cancer initiation and progression. Nat Rev Cancer 2013;13:97-110.
115. Blanco MJ, Moreno-Bueno G, Sarrio D, Locascio A, Cano A, et al. Correlation of Snail expression with histological grade and lymph node status in breast carcinomas. Oncogene 2002;21:3241-6.
116. Bonnomet A, Syne L, Brysse A, Feyereisen E, Thompson EW, et al. A dynamic in vivo model of epithelial-to-mesenchymal transitions in circulating tumor cells and metastases of breast cancer. Oncogene 2012;31:3741-53.
117. Cheung KJ, Gabrielson E, Werb Z, Ewald AJ. Collective invasion in breast cancer requires a conserved basal epithelial program. Cell 2013;155:1639-51.
118. Westcott JM, Prechtl AM, Maine EA, Dang TT, Esparza MA, et al. An epigenetically distinct breast cancer cell subpopulation promotes collective invasion. J Clin Invest 2015;125:1927-43.
119. Cristofanilli M, Budd GT, Ellis MJ, Stopeck A, Matera J, et al. Circulating tumor cells, disease progression, and survival in metastatic breast cancer. N Engl J Med 2004;351:781-91.
120. Cristofanilli M, Hayes DF, Budd GT, Ellis MJ, Stopeck A, et al. Circulating tumor cells: a novel prognostic factor for newly diagnosed metastatic breast cancer. J Clin Oncol 2005;23:1420-30.
121. Budd GT, Cristofanilli M, Ellis MJ, Stopeck A, Borden E, et al. Circulating tumor cells versus imaging--predicting overall survival in metastatic breast cancer. Clin Cancer Res 2006;12:6403-9.
122. Paoletti C, Muñiz MC, Thomas DG, Griffith KA, Kidwell KM, et al. Development of circulating tumor cell-endocrine therapy index in patients with hormone receptor–positive breast cancer. Clinical Cancer Res 2015;21:2487-98.
123. Liu MC, Shields PG, Warren RD, Cohen P, Wilkinson M, et al. Circulating tumor cells: a useful predictor of treatment efficacy in metastatic breast cancer. J Clin Oncol 2009;27:5153-9.
124. Theodoropoulos PA, Polioudaki H, Agelaki S, Kallergi G, Saridaki Z, et al. Circulating tumor cells with a putative stem cell phenotype in peripheral blood of patients with breast cancer. Cancer Lett 2010;288:99-106.
125. Kallergi G, Papadaki MA, Politaki E, Mavroudis D, Georgoulias V, et al. Epithelial to mesenchymal transition markers expressed in circulating tumour cells of early and metastatic breast cancer patients. Breast Cancer Res 2011;13:R59.
126. Yu M, Bardia A, Wittner BS, Stott SL, Smas ME, et al. Circulating breast tumor cells exhibit dynamic changes in epithelial and mesenchymal composition. Science 2013;339:580-4.
127. Papadaki MA, Kallergi G, Zafeiriou Z, Manouras L, Theodoropoulos PA, et al. Co-expression of putative stemness and epithelial-to-mesenchymal transition markers on single circulating tumour cells from patients with early and metastatic breast cancer. BMC Cancer 2014;14:651.
128. Baccelli I, Schneeweiss A, Riethdorf S, Stenzinger A, Schillert A, et al. Identification of a population of blood circulating tumor cells from breast cancer patients that initiates metastasis in a xenograft assay. Nat Biotechnol 2013;31:539-44.
129. Bos PD, Zhang XH, Nadal C, Shu W, Gomis RR, et al. Genes that mediate breast cancer metastasis to the brain. Nature 2009;459:1005-9.
130. Kang Y, Siegel PM, Shu W, Drobnjak M, Kakonen SM, et al. A multigenic program mediating breast cancer metastasis to bone. Cancer Cell 2003;3:537-49.
131. Padua D, Zhang XH, Wang Q, Nadal C, Gerald WL, et al. TGFbeta primes breast tumors for lung metastasis seeding through angiopoietin-like 4. Cell 2008;133:66-77.
132. Minn AJ, Kang Y, Serganova I, Gupta GP, Giri DD, et al. Distinct organ-specific metastatic potential of individual breast cancer cells and primary tumors. J Clin Invest 2005;115:44-55.
133. Minn AJ, Gupta GP, Siegel PM, Bos PD, Shu W, et al. Genes that mediate breast cancer metastasis to lung. Nature 2005;436:518-24.
134. Shibue T, Weinberg RA. Metastatic colonization: settlement, adaptation and propagation of tumor cells in a foreign tissue environment. Semin Cancer Biol 2011;21:99-106.
135. Malanchi I, Santamaria-Martinez A, Susanto E, Peng H, Lehr HA, et al. Interactions between cancer stem cells and their niche govern metastatic colonization. Nature 2011;481:85-9.
136. Liu H, Patel MR, Prescher JA, Patsialou A, Qian D, et al. Cancer stem cells from human breast tumors are involved in spontaneous metastases in orthotopic mouse models. Proc Natl Acad Sci U S A 2010;107:18115-20.
137. Lawson DA, Bhakta NR, Kessenbrock K, Prummel KD, Yu Y, et al. Single-cell analysis reveals a stem-cell program in human metastatic breast cancer cells. Nature 2015;526:131-5.
138. Al-Hajj M, Wicha MS, Benito-Hernandez A, Morrison SJ, Clarke MF. Prospective identification of tumorigenic breast cancer cells. Proc Natl Acad Sci U S A 2003;100:3983-8.
139. Ginestier C, Hur MH, Charafe-Jauffret E, Monville F, Dutcher J, et al. ALDH1 is a marker of normal and malignant human mammary stem cells and a predictor of poor clinical outcome. Cell Stem Cell 2007;1:555-67.
140. Kim W-T, Ryu CJ. Cancer stem cell surface markers on normal stem cells. BMB Reports 2017;50:285-98.
141. Liu S, Cong Y, Wang D, Sun Y, Deng L, et al. Breast cancer stem cells transition between epithelial and mesenchymal states reflective of their normal counterparts. Stem Cell Reports 2014;2:78-91.
142. Malladi S, Macalinao DG, Jin X, He L, Basnet H, et al. Metastatic Latency and Immune Evasion through Autocrine Inhibition of WNT. Cell 2016;165:45-60.
143. Shibue T, Weinberg RA. EMT, CSCs, and drug resistance: the mechanistic link and clinical implications. Nat Rev Clin Oncol 2017;14:611-29.
144. Mani SA, Guo W, Liao MJ, Eaton EN, Ayyanan A, et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell 2008;133:704-15.
145. Morel AP, Lievre M, Thomas C, Hinkal G, Ansieau S, et al. Generation of breast cancer stem cells through epithelial-mesenchymal transition. PLoS One 2008;3:e2888.
146. Morel AP, Hinkal GW, Thomas C, Fauvet F, Courtois-Cox S, et al. EMT inducers catalyze malignant transformation of mammary epithelial cells and drive tumorigenesis towards claudin-low tumors in transgenic mice. PLoS Genet 2012;8:e1002723.
147. Chaffer CL, Marjanovic ND, Lee T, Bell G, Kleer CG, et al. Poised chromatin at the ZEB1 promoter enables breast cancer cell plasticity and enhances tumorigenicity. Cell 2013;154:61-74.
148. Marjanovic ND, Weinberg RA, Chaffer CL. Cell plasticity and heterogeneity in cancer. Clin Chem 2013;59:168-79.
149. Chaffer CL, Brueckmann I, Scheel C, Kaestli AJ, Wiggins PA, et al. Normal and neoplastic nonstem cells can spontaneously convert to a stem-like state. Proc Natl Acad Sci U S A 2011;108:7950-5.
150. Liu S, Ginestier C, Ou SJ, Clouthier SG, Patel SH, et al. Breast cancer stem cells are regulated by mesenchymal stem cells through cytokine networks. Cancer Res 2011;71:614-24.
151. Sullivan NJ, Sasser AK, Axel AE, Vesuna F, Raman V, et al. Interleukin-6 induces an epithelial-mesenchymal transition phenotype in human breast cancer cells. Oncogene 2009;28:2940-7.
152. Vlaicu P, Mertins P, Mayr T, Widschwendter P, Ataseven B, et al. Monocytes/macrophages support mammary tumor invasivity by co-secreting lineage-specific EGFR ligands and a STAT3 activator. BMC Cancer 2013;13:197.
153. Ghajar CM, Peinado H, Mori H, Matei IR, Evason KJ, et al. The perivascular niche regulates breast tumour dormancy. Nat Cell Biol 2013;15:807-17.
154. Zhang J, Tian XJ, Zhang H, Teng Y, Li R, et al. TGF-beta-induced epithelial-to-mesenchymal transition proceeds through stepwise activation of multiple feedback loops. Sci Signal 2014;7:ra91.
155. West NR, Murray JI, Watson PH. Oncostatin-M promotes phenotypic changes associated with mesenchymal and stem cell-like differentiation in breast cancer. Oncogene 2014;33:1485-94.
156. Lu H, Clauser KR, Tam WL, Frose J, Ye X, et al. A breast cancer stem cell niche supported by juxtacrine signalling from monocytes and macrophages. Nat Cell Biol 2014;16:1105-17.
157. Yu Y, Xiao CH, Tan LD, Wang QS, Li XQ, et al. Cancer-associated fibroblasts induce epithelial-mesenchymal transition of breast cancer cells through paracrine TGF-beta signalling. Br J Cancer 2014;110:724-32.
158. Plaks V, Kong N, Werb Z. The cancer stem cell niche: how essential is the niche in regulating stemness of tumor cells? Cell Stem Cell 2015;16:225-38.
159. Grosse-Wilde A, Fouquier d'Herouel A, McIntosh E, Ertaylan G, Skupin A, et al. Stemness of the hybrid epithelial/mesenchymal state in breast cancer and its association with poor survival. PLoS One 2015;10:e0126522.
160. Shibue T, Weinberg RA. Integrin beta1-focal adhesion kinase signaling directs the proliferation of metastatic cancer cells disseminated in the lungs. Proc Natl Acad Sci U S A 2009;106:10290-5.
161. Dykxhoorn DM, Wu Y, Xie H, Yu F, Lal A, et al. miR-200 enhances mouse breast cancer cell colonization to form distant metastases. PLoS One 2009;4:e7181.
162. Ocana OH, Corcoles R, Fabra A, Moreno-Bueno G, Acloque H, et al. Metastatic colonization requires the repression of the epithelial-mesenchymal transition inducer Prrx1. Cancer Cell 2012;22:709-24.
163. Del Pozo Martin Y, Park D, Ramachandran A, Ombrato L, Calvo F, et al. Mesenchymal Cancer Cell-Stroma Crosstalk Promotes Niche Activation, Epithelial Reversion, and Metastatic Colonization. Cell Rep 2015;13:2456-69.
164. Ge Y, Fuchs E. Stretching the limits: from homeostasis to stem cell plasticity in wound healing and cancer. Nat Rev Genet 2018;19:311-25.
165. Thiery JP, Chopin D. .
166. Varga J, Greten FR. Cell plasticity in epithelial homeostasis and tumorigenesis. Nat Cell Biol 2017;19:1133-41.
168. Shook D, Keller R. Mechanisms, mechanics and function of epithelial-mesenchymal transitions in early development. Mechanisms of Development 2003;120:1351-83.
170. Thiery JP, Sleeman JP. Complex networks orchestrate epithelial-mesenchymal transitions. Nat Rev Mol Cell Biol 2006;7:131-42.
171. Rodilla V, Fre S. Cellular plasticity of mammary epithelial cells underlies heterogeneity of breast cancer. Biomedicines 2018;6.
172. Van Keymeulen A, Lee MY, Ousset M, Brohee S, Rorive S, et al. Reactivation of multipotency by oncogenic PIK3CA induces breast tumour heterogeneity. Nature 2015;525:119-23.
173. Koren S, Reavie L, Couto JP, De Silva D, Stadler MB, et al. PIK3CA(H1047R) induces multipotency and multi-lineage mammary tumours. Nature 2015;525:114-8.
174. Celia-Terrassa T. Mammary stem cells and breast cancer stem cells: molecular connections and clinical implications. Biomedicines 2018;6.
175. Gjorevski N, Nelson CM. Integrated morphodynamic signalling of the mammary gland. Nature Reviews Molecular Cell Biology 2011;12:581-93.
176. Godde NJ, Galea RC, Elsum IA, Humbert PO. Cell polarity in motion: redefining mammary tissue organization through EMT and cell polarity transitions. J Mammary Gland Biol Neoplasia 2010;15:149-68.
177. Merrell AJ, Stanger BZ. Adult cell plasticity in vivo: de-differentiation and transdifferentiation are back in style. Nat Rev Mol Cell Biol 2016;17:413-25.
178. Ye X, Weinberg RA. Epithelial–Mesenchymal Plasticity: A Central Regulator of Cancer Progression. Trends in Cell Biology 2015;25:675-86.
179. Visvader JE. Keeping abreast of the mammary epithelial hierarchy and breast tumorigenesis. Genes Dev 2009;23:2563-77.
180. Mun GI, Boo YC. Identification of CD44 as a senescence-induced cell adhesion gene responsible for the enhanced monocyte recruitment to senescent endothelial cells. Am J Physiol Heart Circ Physiol 2010;298:H2102-11.
181. Honeth G, Bendahl PO, Ringner M, Saal LH, Gruvberger-Saal SK, et al. The CD44+/CD24- phenotype is enriched in basal-like breast tumors. Breast Cancer Res 2008;10:R53.
182. Williams K, Motiani K, Giridhar PV, Kasper S. CD44 integrates signaling in normal stem cell, cancer stem cell and (pre)metastatic niches. Exp Biol Med (Maywood) 2013;238:324-38.
183. Morath I, Hartmann TN, Orian-Rousseau V. CD44: More than a mere stem cell marker. Int J Biochem Cell Biol 2016;81:166-73.
184. Gewirtz DA, Alotaibi M, Yakovlev VA, Povirk LF. Tumor Cell Recovery from Senescence Induced by Radiation with PARP Inhibition. Radiat Res 2016;186:327-32.
185. Alotaibi M, Sharma K, Saleh T, Povirk LF, Hendrickson EA, et al. Radiosensitization by PARP inhibition in DNA repair proficient and deficient tumor cells: proliferative recovery in senescent cells. Radiat Res 2016;185:229-45.
186. Ali M, Kamjoo M, Thomas HD, Kyle S, Pavlovska I, et al. The clinically active PARP inhibitor AG014699 ameliorates cardiotoxicity but does not enhance the efficacy of doxorubicin, despite improving tumor perfusion and radiation response in mice. Mol Cancer Ther 2011;10:2320-9.
187. Shelton JW, Waxweiler TV, Landry J, Gao H, Xu Y, et al. In vitro and in vivo enhancement of chemoradiation using the oral PARP inhibitor ABT-888 in colorectal cancer cells. Int J Radiat Oncol Biol Phys 2013;86:469-76.
188. Tlsty TD, Crawford YG, Holst CR, Fordyce CA, Zhang J, et al. Genetic and epigenetic changes in mammary epithelial cells may mimic early events in carcinogenesis. J Mammary Gland Biol Neoplasia 2004;9:263-74.
189. Jiang Z, Jones R, Liu JC, Deng T, Robinson T, et al. RB1 and p53 at the crossroad of EMT and triple-negative breast cancer. Cell Cycle 2011;10:1563-70.
190. Bendris N, Cheung CT, Leong HS, Lewis JD, Chambers AF, et al. Cyclin A2, a novel regulator of EMT. Cell Mol Life Sci 2014;71:4881-94.
191. Emadi Baygi M, Soheili ZS, Schmitz I, Sameie S, Schulz WA. Snail regulates cell survival and inhibits cellular senescence in human metastatic prostate cancer cell lines. Cell Biol Toxicol 2010;26:553-67.
192. Wang T, Li Y, Wang W, Tuerhanjiang A, Wu Z, et al. Twist2, the key Twist isoform related to prognosis, promotes invasion of cervical cancer by inducing epithelial-mesenchymal transition and blocking senescence. Hum Pathol 2014;45:1839-46.
193. Smit MA, Peeper DS. Deregulating EMT and senescence: double impact by a single twist. Cancer Cell 2008;14:5-7.
194. Ansieau S, Courtois-Cox S, Morel AP, Puisieux A. Failsafe program escape and EMT: a deleterious partnership. Semin Cancer Biol 2011;21:392-6.
195. de Carne Trecesson S, Guillemin Y, Belanger A, Bernard AC, Preisser L, et al. Escape from p21-mediated oncogene-induced senescence leads to cell dedifferentiation and dependence on anti-apoptotic Bcl-xL and MCL1 proteins. J Biol Chem 2011;286:12825-38.
196. Weinberg RA. Twisted epithelial-mesenchymal transition blocks senescence. Nat Cell Biol 2008;10:1021-3.
197. Salmina K, Jankevics E, Huna A, Perminov D, Radovica I, et al. Up-regulation of the embryonic self-renewal network through reversible polyploidy in irradiated p53-mutant tumour cells. Exp Cell Res 2010;316:2099-112.
198. Chitikova ZV, Gordeev SA, Bykova TV, Zubova SG, Pospelov VA, et al. Sustained activation of DNA damage response in irradiated apoptosis-resistant cells induces reversible senescence associated with mTOR downregulation and expression of stem cell markers. Cell Cycle 2014;13:1424-39.
199. Le Duff M, Gouju J, Jonchere B, Guillon J, Toutain B, et al. Regulation of senescence escape by the cdk4-EZH2-AP2M1 pathway in response to chemotherapy. Cell Death Dis 2018;9:199.
200. Milanovic M, Fan DNY, Belenki D, Dabritz JHM, Zhao Z, et al. Senescence-associated reprogramming promotes cancer stemness. Nature 2018;553:96-100.
201. Barreto-Andrade JC, Efimova EV, Mauceri HJ, Beckett MA, Sutton HG, et al. Response of human prostate cancer cells and tumors to combining PARP inhibition with ionizing radiation. Mol Cancer Ther 2011;10:1185-93.
202. Wu PC, Wang Q, Grobman L, Chu E, Wu DY. Accelerated cellular senescence in solid tumor therapy. Exp Oncol 2012;34:298-305.
203. Mathiesen RR, Borgen E, Renolen A, Lokkevik E, Nesland JM, et al. Persistence of disseminated tumor cells after neoadjuvant treatment for locally advanced breast cancer predicts poor survival. Breast Cancer Res 2012;14:R117.
204. Husemann Y, Geigl JB, Schubert F, Musiani P, Meyer M, et al. Systemic spread is an early step in breast cancer. Cancer Cell 2008;13:58-68.
205. Heitzer E, Perakis S, Geigl JB, Speicher MR. The potential of liquid biopsies for the early detection of cancer. NPJ Precis Oncol 2017;1:36.
206. Pece S, Tosoni D, Confalonieri S, Mazzarol G, Vecchi M, et al. Biological and molecular heterogeneity of breast cancers correlates with their cancer stem cell content. Cell 2010;140:62-73.
207. Doherty MR, Smigiel JM, Junk DJ, Jackson MW. Cancer Stem Cell Plasticity Drives Therapeutic Resistance. Cancers (Basel) 2016;8.
208. Pastushenko I, Brisebarre A, Sifrim A, Fioramonti M, Revenco T, et al. Identification of the tumour transition states occurring during EMT. Nature 2018;556:463-8.
209. Goldman A, Majumder B, Dhawan A, Ravi S, Goldman D, et al. Temporally sequenced anticancer drugs overcome adaptive resistance by targeting a vulnerable chemotherapy-induced phenotypic transition. Nat Commun 2015;6:6139.
210. Biddle A, Liang X, Gammon L, Fazil B, Harper LJ, et al. Cancer stem cells in squamous cell carcinoma switch between two distinct phenotypes that are preferentially migratory or proliferative. Cancer Res 2011;71:5317-26.
211. Li QQ, Xu JD, Wang WJ, Cao XX, Chen Q, et al. Twist1-mediated adriamycin-induced epithelial-mesenchymal transition relates to multidrug resistance and invasive potential in breast cancer cells. Clin Cancer Res 2009;15:2657-65.
212. Sun L, Yao Y, Liu B, Lin Z, Lin L, et al. MiR-200b and miR-15b regulate chemotherapy-induced epithelial-mesenchymal transition in human tongue cancer cells by targeting BMI1. Oncogene 2012;31:432-45.
213. Sharma SV, Lee DY, Li B, Quinlan MP, Takahashi F, et al. A chromatin-mediated reversible drug-tolerant state in cancer cell subpopulations. Cell 2010;141:69-80.
214. Pisco AO, Huang S. Non-genetic cancer cell plasticity and therapy-induced stemness in tumour relapse: 'What does not kill me strengthens me'. Br J Cancer 2015;112:1725-32.
215. Cazet AS, Hui MN, Elsworth BL, Wu SZ, Roden D, et al. Targeting stromal remodeling and cancer stem cell plasticity overcomes chemoresistance in triple negative breast cancer. Nat Commun 2018;9:2897.
216. Hui M, Cazet A, Nair R, Watkins DN, O'Toole SA, et al. The Hedgehog signalling pathway in breast development, carcinogenesis and cancer therapy. Breast Cancer Res 2013;15:203.
217. O'Toole SA, Machalek DA, Shearer RF, Millar EK, Nair R, et al. Hedgehog overexpression is associated with stromal interactions and predicts for poor outcome in breast cancer. Cancer Res 2011;71:4002-14.
218. Amakye D, Jagani Z, Dorsch M. Unraveling the therapeutic potential of the Hedgehog pathway in cancer. Nat Med 2013;19:1410-22.
219. Chuang PT, Kawcak T, McMahon AP. Feedback control of mammalian Hedgehog signaling by the Hedgehog-binding protein, Hip1, modulates Fgf signaling during branching morphogenesis of the lung. Genes Dev 2003;17:342-7.
220. Yu H, Jove R. The STATs of cancer--new molecular targets come of age. Nat Rev Cancer 2004;4:97-105.
221. Turkson J. STAT proteins as novel targets for cancer drug discovery. Expert Opin Ther Targets 2004;8:409-22.
222. Buettner R, Mora LB, Jove R. Activated STAT signaling in human tumors provides novel molecular targets for therapeutic intervention. Clin Cancer Res 2002;8:945-54.
223. Kolosenko I, Yu Y, Busker S, Dyczynski M, Liu J, et al. Identification of novel small molecules that inhibit STAT3-dependent transcription and function. PLoS One 2017;12:e0178844.
224. Eiring AM, Page BDG, Kraft IL, Mason CC, Vellore NA, et al. Combined STAT3 and BCR-ABL1 inhibition induces synthetic lethality in therapy-resistant chronic myeloid leukemia. Leukemia 2015;29:586-97.
225. Liu F, Cao J, Wu J, Sullivan K, Shen J, et al. Stat3-targeted therapies overcome the acquired resistance to vemurafenib in melanomas. J Invest Dermatol 2013;133:2041-9.
226. Sunayama J, Matsuda K, Sato A, Tachibana K, Suzuki K, et al. Crosstalk between the PI3K/mTOR and MEK/ERK pathways involved in the maintenance of self-renewal and tumorigenicity of glioblastoma stem-like cells. Stem Cells 2010;28:1930-9.
227. Dubrovska A, Kim S, Salamone RJ, Walker JR, Maira SM, et al. The role of PTEN/Akt/PI3K signaling in the maintenance and viability of prostate cancer stem-like cell populations. Proc Natl Acad Sci U S A 2009;106:268-73.
228. Eser S, Reiff N, Messer M, Seidler B, Gottschalk K, et al. Selective requirement of PI3K/PDK1 signaling for Kras oncogene-driven pancreatic cell plasticity and cancer. Cancer Cell 2013;23:406-20.
229. Baselga J, Im S-A, Iwata H, Cortés J, De Laurentiis M, et al. Buparlisib plus fulvestrant versus placebo plus fulvestrant in postmenopausal, hormone receptor-positive, HER2-negative, advanced breast cancer (BELLE-2): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol 2017;18:904-16.
230. Anderson EJ, Mollon L, Dean JL, Aguilar A, Davis LE, et al. A systematic literature review of the clinical prognosis of HR+/HER2-advanced or metastatic breast cancer with and without PIK3CA mutation. J Clin Oncol 2018;36.
231. Baselga J, Dent SF, Cortés J, Im Y-H, Diéras V, et al. Phase III study of taselisib (GDC-0032) + fulvestrant (FULV) v FULV in patients (pts) with estrogen receptor (ER)-positive, PIK3CA-mutant (MUT), locally advanced or metastatic breast cancer (MBC): primary analysis from SANDPIPER. J Clinl Oncol 2018;36:LBA1006.
232. Sharma P, Abramson VG, O'Dea A, Lewis S, Scott JN, et al. Abstract P6-11-08: Safety and efficacy results from phase I study of BYL 719 plus nab-paclitaxel in HER 2 negative metastatic breast cancer. Cancer Res 2017;77:P6-11-08.
233. Zhou J, Wulfkuhle J, Zhang H, Gu P, Yang Y, et al. Activation of the PTEN/mTOR/STAT3 pathway in breast cancer stem-like cells is required for viability and maintenance. Proc Natl Acad Sci U S A 2007;104:16158-63.
234. Douville J, Beaulieu R, Balicki D. ALDH1 as a functional marker of cancer stem and progenitor cells. Stem Cells Dev 2009;18:17-25.
235. Huang EH, Hynes MJ, Zhang T, Ginestier C, Dontu G, et al. Aldehyde dehydrogenase 1 is a marker for normal and malignant human colonic stem cells (SC) and tracks SC overpopulation during colon tumorigenesis. Cancer Res 2009;69:3382-9.
236. Chang WW, Lin RJ, Yu J, Chang WY, Fu CH, et al. The expression and significance of insulin-like growth factor-1 receptor and its pathway on breast cancer stem/progenitors. Breast Cancer Res 2013;15:R39.
237. Jung MJ, Rho JK, Kim YM, Jung JE, Jin YB, et al. Upregulation of CXCR4 is functionally crucial for maintenance of stemness in drug-resistant non-small cell lung cancer cells. Oncogene 2013;32:209-21.
238. Browne BC, Eustace AJ, Kennedy S, O'Brien NA, Pedersen K, et al. Evaluation of IGF1R and phosphorylated IGF1R as targets in HER2-positive breast cancer cell lines and tumours. Breast Cancer Res Treat 2012;136:717-27.
240. Liu Y, Zhang X, Liu J, Hou G, Zhang S, et al. Everolimus in combination with letrozole inhibit human breast cancer MCF-7/Aro stem cells via PI3K/mTOR pathway: an experimental study. Tumour Biol 2014;35:1275-86.
241. Zhu Y, Zhang X, Liu Y, Zhang S, Liu J, et al. Antitumor effect of the mTOR inhibitor everolimus in combination with trastuzumab on human breast cancer stem cells in vitro and in vivo. Tumour Biol 2012;33:1349-62.
242. Iliopoulos D, Hirsch HA, Struhl K. Metformin decreases the dose of chemotherapy for prolonging tumor remission in mouse xenografts involving multiple cancer cell types. Cancer Res 2011;71:3196-201.
243. Gupta PB, Onder TT, Jiang G, Tao K, Kuperwasser C, et al. Identification of selective inhibitors of cancer stem cells by high-throughput screening. Cell 2009;138:645-59.
244. Kumar D, Shankar S, Srivastava RK. Rottlerin-induced autophagy leads to the apoptosis in breast cancer stem cells: molecular mechanisms. Mol Cancer 2013;12:171.
245. Kolev VN, Wright QG, Vidal CM, Ring JE, Shapiro IM, et al. PI3K/mTOR dual inhibitor VS-5584 preferentially targets cancer stem cells. Cancer Res 2015;75:446-55.
246. Reedijk M, Odorcic S, Chang L, Zhang H, Miller N, et al. High-level coexpression of JAG1 and NOTCH1 is observed in human breast cancer and is associated with poor overall survival. Cancer Res 2005;65:8530-7.
247. D'Angelo RC, Ouzounova M, Davis A, Choi D, Tchuenkam SM, et al. Notch reporter activity in breast cancer cell lines identifies a subset of cells with stem cell activity. Mol Cancer Ther 2015;14:779-87.
248. Parr C, Watkins G, Jiang WG. The possible correlation of Notch-1 and Notch-2 with clinical outcome and tumour clinicopathological parameters in human breast cancer. Int J Mol Med 2004;14:779-86.
249. Zhang J, Shao X, Sun H, Liu K, Ding Z, et al. NUMB negatively regulates the epithelial-mesenchymal transition of triple-negative breast cancer by antagonizing Notch signaling. Oncotarget 2016;7.
250. Chen J, Chang H, Peng X, Gu Y, Yi L, et al. 3,6-dihydroxyflavone suppresses the epithelial-mesenchymal transition in breast cancer cells by inhibiting the Notch signaling pathway. Sci Rep 2016;6:28858.
251. Mao L. NOTCH mutations: multiple faces in human malignancies. Cancer Prev Res (Phila) 2015;8:259-61.
252. Zhang X, Zhao X, Shao S, Zuo X, Ning Q, et al. Notch1 induces epithelial-mesenchymal transition and the cancer stem cell phenotype in breast cancer cells and STAT3 plays a key role. Int J Oncol 2015;46:1141-8.
253. Hui C, Yujie F, Lijia Y, Long Y, Hongxia X, et al. MicroRNA-34a and microRNA-21 play roles in the chemopreventive effects of 3,6-dihydroxyflavone on 1-methyl-1-nitrosourea-induced breast carcinogenesis. Breast Cancer Res 2012;14:R80.
254. Diluvio G, Del Gaudio F, Giuli MV, Franciosa G, Giuliani E, et al. NOTCH3 inactivation increases triple negative breast cancer sensitivity to gefitinib by promoting EGFR tyrosine dephosphorylation and its intracellular arrest. Oncogenesis 2018;7:42.
255. Wang X, Jung YS, Jun S, Lee S, Wang W, et al. PAF-Wnt signaling-induced cell plasticity is required for maintenance of breast cancer cell stemness. Nat Commun 2016;7:10633.
256. Gujral TS, Chan M, Peshkin L, Sorger PK, Kirschner MW, et al. A noncanonical Frizzled2 pathway regulates epithelial-mesenchymal transition and metastasis. Cell 2014;159:844-56.
257. Jang GB, Hong IS, Kim RJ, Lee SY, Park SJ, et al. Wnt/beta-Catenin Small-Molecule Inhibitor CWP232228 Preferentially Inhibits the Growth of Breast Cancer Stem-like Cells. Cancer Res 2015;75:1691-702.
258. Kim JY, Lee HY, Park KK, Choi YK, Nam JS, et al. CWP232228 targets liver cancer stem cells through Wnt/beta-catenin signaling: a novel therapeutic approach for liver cancer treatment. Oncotarget 2016;7:20395-409.
259. Demaria M, O'Leary MN, Chang J, Shao L, Liu S, et al. Cellular senescence promotes adverse effects of chemotherapy and cancer relapse. Cancer Discov 2017;7:165-76.
260. Baar MP, Brandt RMC, Putavet DA, Klein JDD, Derks KWJ, et al. Targeted apoptosis of senescent cells restores tissue homeostasis in response to chemotoxicity and aging. Cell 2017;169:132-47.e16.
261. Lee M, Lee JS. Exploiting tumor cell senescence in anticancer therapy. Bmb Reports 2014;47:51-9.
262. Nardella C, Clohessy JG, Alimonti A, Pandolfi PP. Pro-senescence therapy for cancer treatment. Nat Rev Cancer 2011;11:503-11.
263. Muñoz-Espín D, Rovira M, Galiana I, Giménez C, Lozano-Torres B, et al. A versatile drug delivery system targeting senescent cells. EMBO Mol Med 2018;10:e9355.
264. Zhu Y, Tchkonia T, Fuhrmann-Stroissnigg H, Dai HM, Ling YY, et al. Identification of a novel senolytic agent, navitoclax, targeting the Bcl-2 family of anti-apoptotic factors. Aging Cell 2016;15:428-35.
265. Chang J, Wang Y, Shao L, Laberge RM, Demaria M, et al. Clearance of senescent cells by ABT263 rejuvenates aged hematopoietic stem cells in mice. Nat Med 2016;22:78-83.
266. Karaman MW, Herrgard S, Treiber DK, Gallant P, Atteridge CE, et al. A quantitative analysis of kinase inhibitor selectivity. Nat Biotechnol 2008;26:127-32.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Smigiel JM, Taylor SE, Bryson BL, Tamagno I, Polak K, Jackson MW. Cellular plasticity and metastasis in breast cancer: a pre- and post-malignant problem. J Cancer Metastasis Treat 2019;5:47. http://dx.doi.org/10.20517/2394-4722.2019.26
AMA Style
Smigiel JM, Taylor SE, Bryson BL, Tamagno I, Polak K, Jackson MW. Cellular plasticity and metastasis in breast cancer: a pre- and post-malignant problem. Journal of Cancer Metastasis and Treatment. 2019; 5: 47. http://dx.doi.org/10.20517/2394-4722.2019.26
Chicago/Turabian Style
Smigiel, Jacob M., Sarah E. Taylor, Benjamin L. Bryson, Ilaria Tamagno, Kelsey Polak, Mark W. Jackson. 2019. "Cellular plasticity and metastasis in breast cancer: a pre- and post-malignant problem" Journal of Cancer Metastasis and Treatment. 5: 47. http://dx.doi.org/10.20517/2394-4722.2019.26
ACS Style
Smigiel, JM.; Taylor SE.; Bryson BL.; Tamagno I.; Polak K.; Jackson MW. Cellular plasticity and metastasis in breast cancer: a pre- and post-malignant problem. J. Cancer. Metastasis. Treat. 2019, 5, 47. http://dx.doi.org/10.20517/2394-4722.2019.26
About This Article
Special Issue
Copyright

Data & Comments
Data


 Cite This Article 16 clicks
Cite This Article 16 clicks

 Like This Article 11
likes
Like This Article 11
likes






Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.