
Paxillin serine 178 phosphorylation in control of cell migration and metastasis formation through regulation of EGFR expression in breast cancer
 0
0
Abstract
Aim: Paxillin is a well-known multidomain scaffold protein that is involved in the regulation of cell-matrix adhesion dynamics, a process required for the tumor cell migration and invasion. Phosphorylation of the serine residue 178 requires c-Jun NH2-terminal kinase (JNK) activation, which occurs downstream of epidermal growth factor receptor (EGFR)-mediated signaling and drives cell migration. In this study, we investigated the significance of paxillin Ser178 phosphorylation in breast cancer progression.
Methods: We employed the rat mammary carcinoma MTLn3 cell line with which we established stabile variants of both wild type and mutant GFP-paxillin constructs. With those, we next performed several in vitro assays including cell proliferation, migration and focal adhesion dynamics. Finally, we monitored the metastatic spread of both cell line variants in an othrotopic mouse model for breast cancer.
Results: Here we show that expression of the phospho-defective mutant paxillinS178A in the metastatic mammary adenocarcinoma MTLn3 cell-line significantly decreased EGF-induced cell migration, which was correlated with impaired focal adhesion dynamics. Moreover, paxillinS178A attenuated lung metastasis formation in an orthotopic In vivo mammary gland tumor/metastasis model, demonstrating the importance of JNK-mediated paxillin phosphorylation in breast cancer progression. Expression of paxillinS178A caused a decrease in EGFR expression, while re-expression of EGFR in MTLn3-paxillinS178A cells fully restored EGF-driven cell motility and focal adhesion dynamics. Furthermore, re-expression of EGFR in MTLn3-paxillinS178A rescued spontaneous metastasis from breast to lung.
Conclusion: Overall our data show an important role for JNK-mediated paxillin Ser178 phosphorylation in the regulation of EGFR expression and thereby, in EGF-driven cell migration and metastasis formation.
Keywords
Introduction
Breast cancer represents the most common type of cancer among women. The formation of metastases, which is a determinant of the prognosis of cancer patients, involves distinct cellular processes including cell migration, invasion, intra- and extravasation and proliferation. These processes are regulated by growth factors, cytokines and cellular matrix molecules[1-6]. An important regulator of cell proliferation and migration is the receptor tyrosine kinase epidermal growth factor receptor (EGFR) whose stimulation by epidermal growth factor (EGF) results in the activation of downstream signaling pathways including different mitogen-activated protein kinases cascades: extracellular signal-regulated kinase (ERK), p38 and c-Jun NH2-terminal kinase (JNK)[7-11].
The JNK group of kinases has essential roles in cancer development including regulation of the survival/proliferation balance as well as cell migration[7,9,12,13]. Dual Thr and Tyr phosphorylation of JNK by upstream MAP kinases results in JNK activation and nuclear translocation. In the nucleus, JNKs phosphorylate and activate transcription factors including members of the AP-1 family such as c-Jun. The JNK-AP-1 pathway regulates the expression of genes involved in the cell cycle, survival and apoptosis and extracellular matrix homeostasis[14-22]. Besides phosphorylation of transcription factors, several cytoskeleton-associated adaptor and signaling proteins have recently been identified as direct JNK substrates including β-catenin[23] and paxillin[24,25]. Since both proteins are well known to be involved in cell-matrix and cell-cell contacts, the direct JNK-mediated phosphorylation of these targets may also influence cell migration.
Paxillin is a 68 kD multidomain adaptor protein associated with focal adhesions[26-31]. It is long known to function as a scaffold to integrate multiple signaling pathways involved in matrix adhesion dynamics and cell migration[27,31-36], and recently, it was shown to play a role in the nucleus in relation to mRNA transcription and subsequent translation[37,38]. Few studies have identified paxillin as a JNK substrate[8,24,25]. EGF-driven JNK activation results in JNK-mediated phosphorylation of paxillin at Ser178 and is required for cell migration[9,13,19,25,39]. In addition, expression of a phospho-defective Ser178 to Ala mutant of paxillin inhibited cell migration[9,19,24]. How JNK-mediated phosphorylation of paxillin regulates cell migration is still under investigation. It is still unclear how this phosphorylation event is relevant for breast cancer metastasis formation[40,41].
Here we explored the role and mechanism of paxillin Ser178 phosphorylation in breast tumor progression using the highly metastastic breast tumor cell line MTLn3 as a model. We show that ectopic expression of paxillinS178A significantly decreased EGF-dependent signaling and cell migration. Using an orthotopic mammary gland tumor/metastasis model, we demonstrate that the JNK-mediated phosphorylation of paxillin at Ser178 is essential for efficient metastasis of MTLn3 cells to the lung. Expression of mutant paxillinS178A was found to reduce EGFR expression, whereas re-expression of EGFR rescued the defected tumor cell migration and metastasis formation. Our data indicate that the phosphorylation of Ser178 of paxillin by JNK can regulate cell migration and metastasis formation via modulation of the EGFR-signaling pathway.
Methods
Chemicals and antibodies
Alpha modified minimal essential medium without ribonucleosides and deoxyribonucleosides (α-MEM), fetal bovine serum (FBS), phosphate-buffered saline (PBS), trypsin and geneticin (G418 sulphate) were from Life Technologies. Rat tail collagen type I was from Upstate Biotechnology. LipofectAMINE Plus transfection reagents were from Invitrogen. Primary antibodies were anti-paxillin (BD), anti-tubulin, anti-GFP (Sigma, St. Louis, MO), anti-pT183/pY185-JNK (Promega), anti-Phospho-Thr202/Tyr204 ERK1/2, anti-pSer473-AKT (Cell Signaling), anti-pSer178-paxillin (Abcam, Cambridge, UK), anti-EGFR for Western blot (rabbit polyclonal, Cell Signaling Technology) and FACS/immunostaining (mouse monoclonal, Calbiochem). All secondary antibodies were from Jackson. The Western-Star immunodetection system (Tropix kit) was from Applied Biosystems and ECL Plus reagent was from Amersham. Hoechst 33258 and rhodamine-phalloidin were from Molecular Probes and Aqua Poly/Mount was from Polysciences.
Cell culture
MTLn3 cells were cultured as before[42]. To generate stable cell lines, MTLn3 cells were transfected with GFP-paxillin or GFP-paxillinS178A kindly provided by Huang et al.[24] were transfected using LipofectAMINE plus reagents according to manufacturer’s procedures. Stable transfectants were selected using G418 and individual clones were picked and kept at 100 μg/mL G418. For EGF experiments, 70%-80% confluent cells were starved for 4 h followed by exposure to EGF (10 nmol/L). Human EGFR was expressed in GFP-paxillin S178A using retroviral transfection followed by FACS sorting as described previously[43]. Retroviral transduction of GFP-paxillin cells with a pMSCV-blast-hEGFR retroviral vector, followed by blasticidin selection (12.5 μg/mL) was used to generate GFP-paxillin S178A EGFR cells.
Luciferase reporter assay
MTLn3 cells were transiently transfected with the reporter constructs pGL3-Tata-5xE3AP1[44], pGL3-Tata-5xCol1TRE, pGL3-Tata-5xJun2 kindly provided by Dr. Hans van Dam (LUMC) using Lipofectamine Plus reagent[42]. 1 µg of expression reporter plasmids together with 1 µg of a renilla-luciferase construct (as an internal control for transfection efficiency) were transfected in both clones WT and S178A. After 48 h, cells were serum starved for 4 h then lysed and analyzed for luciferase activity using a luminescence plate reader.
Proliferation, attachment and wound healing assay
For proliferation assay, cells were plated in complete medium in 6 wells-plates and cultured for 24, 48, 72 or 96 h at which time point the amount of cells was determined by counting. For cell attachment assay, 1 h serum-starved cells were seeded in complete medium on collagen-coated 6 wells-plates for 30, 60, 90 and 120 min, and following a PBS wash, attached cells were trypsinized and counted. For wound healing assays, monolayer cells were scratched using a pipette tip to generate a wound followed by a wash with medium and incubation in α-MEM supplemented with 1% (v/v) FBS for 20 hrs. Wounds were photographed using phase contrast microscopy with a Nikon Coolpix digital camera directly after scratching and after 20 h. Wound closure was determined using Image J software.
Live cell imaging
Random cell migration
Cells were cultured in glass-bottom plates overnight and starved for 4 h followed by imaging for 1-3 h on a Nikon TE 2000-E microscope in a humid climate of 37 °C and 5% CO2 with either DIC (Differential Interference Contrast) or fluorescence microscopy. Subsequently, cells were treated with EGF and time-lapses were captured with 20× objective. Per biological replicate, there were 3 wells treated similarly and 2 positions per well were imaged. About 30-40 cells were followed over time in each field of view, which means that we analyzed the behavior of more than 120 cells per biological replicate. When used, the JNK inhibitor SP600125 (20 μmol/L) was added 30 min prior to stimulation. Cell speed was determined with a homemade macro written in Image-Pro Plus (Media Cybernetics Inc., Silver Spring, MD).
Total internal reflection fluorescence and fluorescence recovery after photobleaching
Total internal reflection fluorescence (TIRF) microscopy was performed with a Nikon TE 2000-E microscope in a climate control chamber. To determine the turnover of GFP-tagged paxillin proteins in individual focal adhesions, fluorescence recovery after photobleaching (FRAP) was performed as follows: photobleaching was applied to a small area covering a single focal adhesion for 1 s with laser intensity of 50 μW. Redistribution of fluorescence was monitored with 100 ms time intervals at 7.5 μW starting directly after the bleach pulse. Approximately 20 focal adhesions (each in distinct cells) were averaged to generate one FRAP curve for a single experiment. All measurements were performed at 37 °C and the experiment was performed on three different days. The relative fluorescence intensity of individual focal adhesion was calculated at each time interval as follows: Irel(t) = (FAt/FA0), where FAt is the intensity of the focal adhesion at time point t after bleaching and FA0 is the average intensity of the focal adhesion before bleaching.
Gel electrophoresis and immunoblotting
Equal protein amounts (25 μg; Bradford protein assay) were separated on 7.5% polyacrylamide gels and transferred to PVDF membranes (Millipore). Membranes were blocked in 5% (w/v) BSA in TBS-T and probed with primary antibody overnight followed by sufficient washes and incubation with secondary antibodies. Alkaline phosphatase-conjugated secondary antibodies for phospho-proteins were detected with the Western-Star immunodetection system. For detection of horseradish peroxidase-conjugated antibodies, ECL Plus reagent was used, followed by visualization on a Typhoon Imager 9400.
Immunofluorescence
Cells were plated on collagen-coated glass coverslips. Cells were briefly washed in PBS, followed by fixation in 3.7% formaldehyde for 10 min at room temperature. After washing, coverslips were blocked in TBP (0.1% (w/v) Triton X-100, 0.5% (w/v) BSA in PBS, pH 7.4). Incubation with primary antibodies diluted in TBP containing 0.05% (w/v) NaN3 was carried out overnight at 4 °C. Coverslips were mounted on glass slides using Aqua Poly/Mount.
RNA isolation and DNA array analysis
Total RNA was isolated from all MTLn3 clones using TRIzol reagent (Invitrogen Corp.). Five microgram of RNA was used for cDNA synthesis. A custom cDNA kit (Invitrogen Corp.) with T7-(dT)24 primer was used for this reaction. Biotinylated cRNA was generated from the cDNA reaction using the BioArray high yield RNA transcript kit (Affymetrix Inc., Santa Clara, CA). cRNA was then fragmented (5X fragmentation buffer: 200 mmol/L Tris acetate, pH 8.1, 500 mmol/L potassium acetate, 150 mmol/L magnesium acetate) at 94 °C for 35 min before chip hybridization. Following the manufacturer's protocol, fragmented cRNA was added to the hybridization mixture. For DNA array, HG-U133A from Affymetrix were hybridized for 16 h in a GeneChip Fluidics Station 400 and scanned with a GeneArray Scanner. The Human Genome U133A set of microarray represents ~14,500 human genes. Affymetrix GeneChip Microarray software was used for basic analysis. Samples were normalized to the average hybridization intensity on each chip. The study was performed for all 6 clones in duplicate. Gene Spring 6.0 (Silicon Genetics, Redwood City, CA) software was used for data analysis. Data mining of the list of genes was done using Enrichr (http://amp.pharm.mssm.edu/Enrichr/), an online gene set enrichment analysis web tool from the Ma'ayan Lab[45,46].
Stable shRNA-mediated gene knockdown
MC7 cells were transduced with lentiviral shRNA constructs coding for a non-targeting control sequences shCtrl (SHC002) and a sequence targeting the coding region of PXN (TRCN0000123138) (Mission/Sigma-Aldrich, Zwijndrecht, The Netherlands). The cells were selected by puromycin (sc-108071, Santa Cruz Biotechnology, Heidelberg, Germany). Knockdown efficiency was verified by Western Blot and immunofluorescent staining.
In vivo tumor growth and metastasis formation
Six-week-old Rag2-/-ɣc-/-mice were obtained from in-house breeding. Animals were housed in individually ventilated cages under sterile conditions containing 3 mice per cage. Sterilised food and water were provided ad libitum. To measure spontaneous metastasis, tumor cells (5 × 105) were injected into the right thoracic mammary fat pads as described previously[10]. After 4 weeks, the lungs were excised as well as the primary tumor which was also weighed. For quantification of all GFP-paxillin positive macro- and micro-metastases, the flat side of the right lung was analysed with the immunofluorescence microscope using a 10× objective lens (NA 0.25). Next the right lung and primary tumor were fixated in 4% paraformaldehyde. Paraffin sections of the lungs (5 μm) were stained with hematoxylin and eosin followed by histological analysis.
Statistical analysis
When not indicated, all experiments were performed in biological triplicates. As all data sets in this study follow a normal distribution, therefore were compared with Student’s t-test (two-tailed, equal variances) using GraphPad Prism 6.0. Results were considered to be significant if P value < 0.05.
Results
EGF-induced cell migration of MTLn3 cells is dependent on JNK activation and associated with paxillin Ser178 phosphorylation
First we determined the role of JNK-paxillin signaling in EGF-induced migration of the highly metastatic MTLn3 rat mammary adenocarcinoma cell line. EGF exposure caused membrane ruffling [Supplementary Figure 1], random cell migration and scattering of MTLn3 cells [Figure 1A and Video 1]. An inhibitor of JNK, SP600125, blocked the migration almost completely, which was associated with increased cell clustering of the MTLn3 cells. This clustered phenotype was associated with increased localization of β-catenin at cell-cell contacts [Figure 1B and Supplementary Figure 2]. Visual inspection of the movies indicates that inhibition of JNK reduces persistent movement [Figure 1C] and quantitative analysis of the cellular tracks reveal that the velocity of both individual and clustered cells was significantly impaired [Figure 1D]. By immunofluorescence, we observed that active phosphorylated JNK co-localizes with paxillin at focal adhesions in MTLn3 cells [Supplementary Figure 3B] as was previously observed in renal epithelial cells[47,48]. EGF treatment induced a transient phosphorylation of paxillin at Ser178 in association with JNK activation, while SP600125 prevented phosphorylation of JNK, c-Jun as well as paxillin at Ser178 indicating the importance of EGF-induced JNK activation for paxillin phosphorylation [Figure 1E and Supplementary Figure 3C]. Together, these data indicate that in MTLn3 cells EGF-induced JNK activation mediates cell migration possibly via the phosphorylation of paxillin at Ser178.
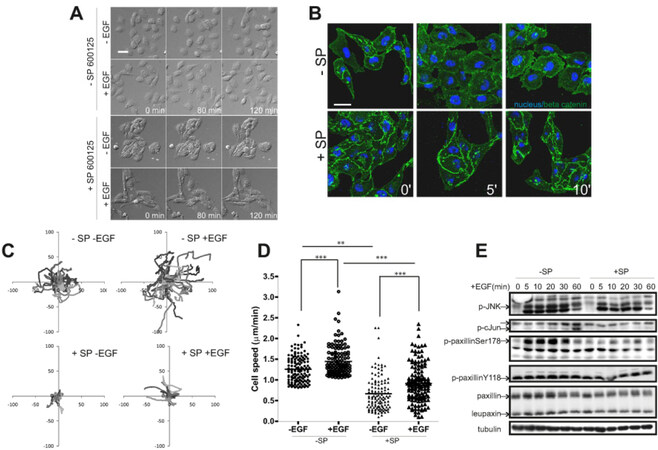
Figure 1. c-Jun NH2-terminal kinase (JNK)-mediated phosphorylation of paxillin Ser178 plays a role in tumor cell migration. MTLn3 cells were either untreated or treated with EGF (10 nmol/L) in the absence or presence of the JNK inhibitor SP600125 (20 μmol/L). A: migration of these cells was observed by live DIC microscopy. Snapshots of the time-lapse made for 2 h are shown, scale bar is 50 μm. See movie M1; B: at 0, 5 and 10 min after treatment cells were fixed and stained for the nucleus (blue) and β-catenin (green). Scale bar is 20 μm; C: overall migration trajectories of individual cells of one representative experiment (only one position from the 6 technical replicates of one biological replicate); D: average cell speeds of about 100 cells per treatment imaged in one biological replicate were plotted. This graph shows the data for one representative biological replicate.*P < 0.05, **P < 0.01, ***P < 0.001; E: the JNK signaling pathway was analyzed by Western Blotting using the indicated antibodies. The arrows indicate the phospho specific bands of the different antibodies. The paxillin antibody detects also a paxillin family member leupaxin encoded by LPXN, which has a much lower molecular weight than paxillin encoded by PXN
PaxillinS178A mutant inhibits MTLn3 cell motility in vitro
To further investigate the role of paxillin Ser178 in cell migration, we generated MTLn3 cell-lines stably expressing either GFP-tagged paxillin-wt or phospho-defective mutant GFP-paxillin in which the serine residue 178 was replaced by alanine (further referred to as paxillinS178A). Three independently obtained paxillin-wt clones and paxillinS178A clones were selected for further experiments. MTLn3 clones stably expressing GFP-wt-paxillin or GFP-paxillinS178A were evaluated by flow cytometry, Western blotting [Figure 1E and [Supplementary Figure 4A] and immunofluorescence [Figure 2A]. Expression levels were approximately equal in all clones. Although GFP-paxillinS178A still localized at focal adhesions, cells were smaller and clustered with enhanced β-catenin-containing cell-cell contacts while cells expressing the GFP-wt-paxillin were stretched with large lamellipodia and almost no cell-cell contacts [Figure 2A and Supplementary Figure 4B]. The paxillinS178A-induced cell clusters [Figure 2B and Supplementary Figure 4C] resembled those formed in SP600125 treated cells [Figure 1B]. Given the prominent role of paxillin in focal adhesion formation and dynamics, a process required for cell spreading, we next examined the effect of paxillinS178A expression on cell attachment and spreading. Significantly less paxillinS178A cells adhered shortly after plating compared to paxillin-wt cells [Figure 2Ca]. Furthermore, while most of paxillin-wt cells had already spread most of the paxillinS178A cells remained rounded and presented a smaller surface area even after three hours of spreading [Figure 2Cb]. We also determined the effect of paxillinS178A on directed cell migration in an artificial wound healing assay [Figure 2D and Supplementary Figure 4D]. While wt-paxillin cells closed the wound by 83%, paxillin-S178A cells had only closed 25% of the wound after 20 h.
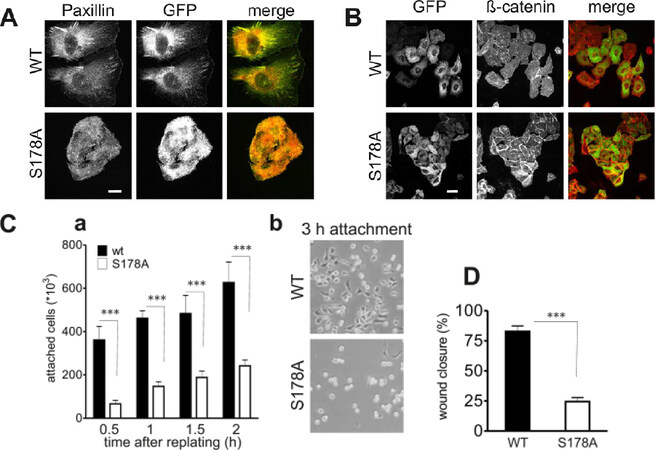
Figure 2. Expression of paxillinS178A decreases cell spreading and directed cell migration. GFP-paxillin-wt and GFP-paxillinS178A MTLn3 cells were generated and three independent clones were used for further research. A: endogenous paxillin (red) colocalized with ectopically expressed GFP-paxillin-wt and GFP-paxillinS178A (green). Scale bar is 10 μm; B: cell clusters were detected using β-catenin (red) and GFP-paxillin (green) staining. Scale bar is 20 μm; C: cells were analyzed for cell adhesion (a). Cells were replated on collagen-coated plastic culture dishes. The number of attached cells was counted at different time points after replating. Columns show the mean of three independent experiments; bars show SE, ***P < 0.001. The spreading after 3 hrs of both wildtype and mutant cells was assessed using phase-contrast pictures (b); D: directed cell migration capacity was assessed using a woundhealing assay. The wound closure was measured at three different location in the wound after 24 h. The assay was repeated three times. Columns show the mean of three independent experiments; bars show SE, *** P < 0.001. All three adhesion related assays were demonstrating a defect in the GFP-paxillinS178A cells
PaxillinS178A affects EGF-induced cell migration and focal adhesion dynamics
In a random cell migration assay paxillin-wt cells rapidly formed lamellipodia and became highly motile while paxillinS178A cells showed decreased cell motility and responded less to EGF stimulation [Figure 3A and Video 2]. Since paxillinS178A most likely acts as a dominant negative construct in these cells, it may compete for the localization of endogenous paxillin at focal adhesions and prevent the phosphorylation of endogenous paxillin at Ser178 by JNK. Indeed, EGF stimulation of paxillinS178A cells induced negligible Ser178 phosphorylation of endogenous paxillin whereas in paxillin-wt cells, both endogenous and GFP-paxillin-wt were phosphorylated at Ser178 after EGF treatment [Supplementary Figure 5]. Importantly, an EGF-induced mobility shift of endogenous paxillin was observed in both WT and S178A cell-lines, indicating that most of the other paxillin modifications were unaffected [Supplementary Figure 5].
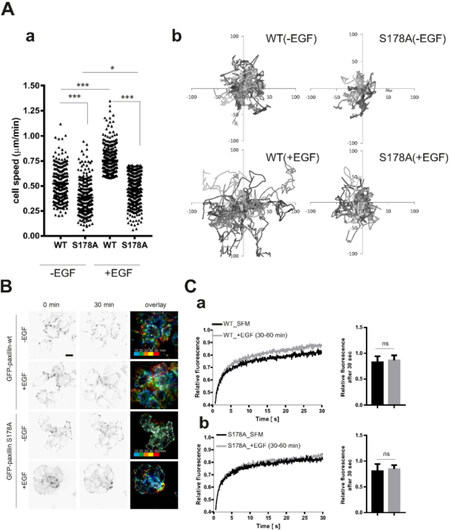
Figure 3. EGF-driven cell migration is inhibited in paxillinS178A cells because of impaired focal adhesion turnover. MTLn3 cells were either untreated or treated with EGF (10 nmol/L). A: migration was observed for 10 h by epi-fluorescence microscopy PaxillinS178A reduced cell speed average (about 100 cells per condition were imaged in one biological replicate). This graph shows the data for one representative biological replicate.*P < 0.05, **P < 0.01, ***P < 0.001 (a) and directional cell movement (b). See also Supplementary movie M2; B: matrix adhesions dynamics in MTLn3 cells was visualized with TIRF microscopy. See alsoSuplementary movie M3. Overlay of different timeframes were generated in red, green and blue. Focal adhesions in white, as observed for PaxillinSer178, represent unchanged (less dynamic) focal adhesions. Scale bar is 20 μm; C: protein dynamics was measured with the spot bleaching technique and showed similar dynamics for both GFP-paxillinS178A (b) and GFP-paxillin-wt (a). Approximately 20 focal adhesions (each in distinct cells) were averaged to generate one FRAP curve for a single experiment.The mean relative fluorescence of both GFP-paxillin-wt and GFP-paxillinS178A both in SFM and upon EGF stimulation shows no significant difference after 30 s of recovery
To understand the mechanism of the inhibitory effect of paxillinS178A on cell migration, we determined the dynamics of focal adhesions in WT and S178A cells using TIRF microscopy. MTLn3 cells expressing paxillin-wt showed a high focal adhesion turnover which was enhanced upon EGF stimulation. In contrast, paxillinS178A cells showed a much slower rate of FA disassembly either in the presence or absence of EGF [Figure 3B and Video 3]. The decreased focal adhesion dynamics could not be explained by a changed in mobility of GFP-paxillinS178A as determined by FRAP experiments. Indeed, both under serum-free conditions and upon EGF stimulation, the rates and percentages of fluorescence recovery of GFP-paxillin-wt and GFP-paxillinS178A were similar [Figure 3C].
GFP-paxillinS178A expression impairs metastasis formation of MTLn3 cells in an orthotopic breast tumor model
We next determined whether paxillin Ser178 was important for spontaneous lung metastasis formation. The MTLn3 cell line has been established as a suitable cell model to study metastasis formation from mammary gland tumors to the lung[10]. We injected GFP-paxillin-wt (clone #2) and GFP-paxillinS178A (clone #2) cells into the mammary fat pads of immunodeficient Rag2-/-γ-/-mice. After three weeks mice were sacrificed for the analysis of the primary mammary gland tumors as well as lung metastases. All primary tumors remained GFP-positive, indicating expression of wt or paxillinS178A GFP-paxillin continuously during the experiment. The edges of the GFP-paxillin-wt tumors were more invasive-like compared to those of GFP-paxillinS178A tumors [Figure 4A]. Yet, the weight of the primary tumor was not significantly altered by GFP-paxillinS178A [Figure 4B]. GFP-paxillinS178A MTLn3 cells formed significantly less spontaneous lung metastases than GFP-paxillin-wt MTLn3 cells [Figure 4C and D], and these metastases also had a less invasive phenotype [Figure 4C]. Our data show for the first time an important role for phosphorylation of paxillin on Ser178 in breast cancer progression.

Figure 4. Expression of paxillinS178A impairs lung metastasis formation in vivo. One representative clone of either GFP-paxillin-wt or GFP-paxillinS178A MTLn3 cell-lines were used for in vivo experiments. A: Primary tumors were imaged using two-photon intravital imaging. Scale bar is 100 μm; B: no significant difference in tumor weight was measured between the wt mice (n = 10) and S178A mice (n = 12); C: two-photon intravital microscopy, H&E staining of lung slices; D: quantification of the number of GFP-positive lung metastases demonstrated that paxilinS178A reduced lung metastasis formation. Scale bar is 100 μm
Ectopic paxillinS178A expression results in EGFR down regulation at mRNA and protein levels
Next we sought to determine the possible mechanism by which paxillinS178A affects tumor cell migration and metastasis formation. Using Affimetrix microarrays, we analyzed the differentially expressed genes (DEG) between GFP-paxillinWT and GFP-paxillinS178A MTLn3 clones. Using a very low p-value, a comparison between WT and mutant clones delineated 134 genes that were differentially expressed of which 84 were down-regulated and 50 up-regulated [Supplementary Table 1]. In Figure 5Aa, we plotted the fold changes of the 10 most down- and 10 most up-regulated genes. Strikingly, EGFR was significantly down-regulated in the mutant clones. A pathway analysis of the complete set of genes using Gene Ontology revealed that pathways involved in intermediate filament assembly, cell proliferation and regulation of β-catenin transport were significantly altered when compared to the wt clones [Figure 5Ab]. Furthermore, a protein-protein interaction analysis of those same DEGs highlighted the EGFR network as most significantly altered [Figure 5B]. Paxillin localizes at focal adhesions and EGFR signaling is regulated and trans-activated at focal adhesions by both integrins[49,50] and FAK[51,52]. Therefore we reasoned that paxillinS178A may disturb the EGFR signaling pathway. In agreement with our microarray analysis, all three paxillinS178A clones had much lower levels of EGFR protein than wt clones. As a consequence, downstream EGFR signaling towards Akt and ERK were also reduced in S178A clones [Figure 5C]. Since EGF also caused JNK activation in control MTLn3 cells [Figure 1], we also determined the activation of JNK signaling pathway in these cells. Indeed, paxillinS178A expression also inhibited EGF-induced JNK activation [Figure 5C], which was associated with a reduction in c-Jun transcriptional activity as determined by luciferase reporter assays [Figure 5D]. These data indicate that paxillinS178A affects the expression of EGFR possibly through the regulation of c-Jun-mediated EGFR transcription[53,54], thereby disturbing downstream signaling pathways that are essential in the cell migration process.
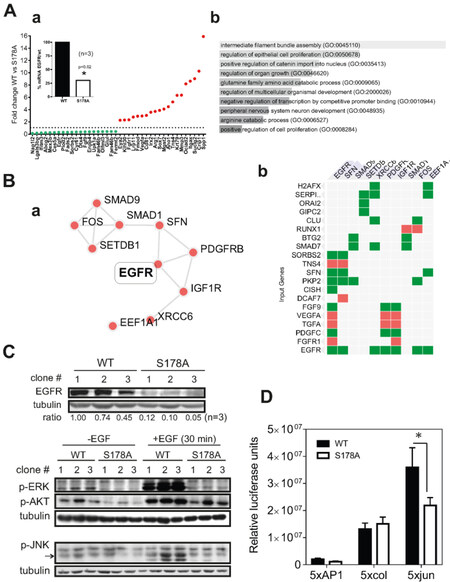
Figure 5. EGFR and EGFR-signaling is downregulated in paxillinS178A mutant cells. A: bar graph displaying the 10 most down- and up-regulating genes in mutant cells when compared with wt cells (a); and Enrichr analysis using the GO Biological process function (processes are sorted by combined score) (b); B: Enrichr analysis using the PPI Hub proteins function (a), EGFR being the first PPI with the highest score (b); C: Western blot analysis confirms a decreased EGFR expression in all S178A clones. Ratios of EGFR levels relative to wt1 clone are indicated below the blot. EGFR downstream signaling towards Akt, ERK and JNK is affected in S178A clones; D: activity of AP-1 member c-Jun was reduced in paxillinS178A cells as determined by TATA-luciferase reporter assay
Ectopic expression of human wt-EGFR in paxillinS178A cells restores EGF-driven cell motility and lung metastasis formation
To determine whether paxillinS178A reduced tumor cell migration and metastases formation via EGFR downregulation, we re-expressed EGFR in the mutant cells [Supplementary Figure 6A]. The EGFR re-expression induced a more spread phenotype in paxillinS178A cells [Figure 6A and Supplementary Figure 6B]. The EGF-driven cell migration was rescued and the protein turnover of paxillinS178A at focal adhesions was slightly faster only upon EGF stimulation [Figure 6B and Supplementary Figure 6C]. This was associated with a sustained activation of both JNK and ERK after EGF exposure [Supplementary Figure 6D]. Next we determined whether EGFR re-expression also restored the capacity of MTLn3 paxillinS178A cells to metastasize to the lungs. For this purpose, we injected GFP-paxillinS178A cells and EGFR-GFP-paxillinS178A cells into the mammary fat pads of immunodeficient Rag2-/-γ-/-mice, although we were aware that EGFR expression would decrease during the course of the experiment. The tumor growth of paxillinS178A and EGFR-paxillinS178A cells was similar in the both groups [Figure 6Cb] and all tumor cells in the primary tumors were GFP-positive [Figure 6Ca]. Mice injected with GFP-paxillinS178A cells that expressed EGFR showed an approximately threefold increase in the lung tumor burden in [Figure 6D]. These data indicate that the reduced metastasis formation of GFP-paxillinS178A cells is directly related to the expression levels of EGFR.
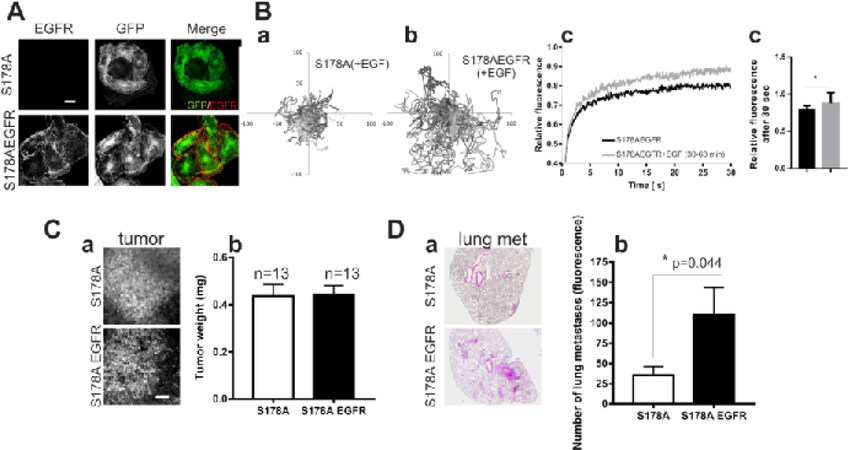
Figure 6. Re-expression of wt-EGFR fully restores EGF-driven cell motility and lung metastasis formation. A: immunostaining with a specific mouse monoclonal antibody against human EGFR confirmed the ectopic expression of EGFR in S178A cells; paxillinS178A (green) and human EGFR (red). Scale bar is 10 μm; B: trajectories of the movements of S178A and S178AEGFR cells are plotted in (a) and (b) respectively (X and Y are in μm). Protein dynamics was quantified with spot bleaching technique and the fluorescence recovery after 30 sec was slightly faster upon EGF stimulation, * P < 0.05. (c); C: two-photon imaging of GFP-positive lung metastases, scale bar is 100 μm. (a). There was no significant difference in tumor weight of S178A (n = 13) and S178AEGFR (n = 13) groups (b); D: H&E staining of lung slices of S178A and S178AEGFR groups (a). Number of GFP-positive metastases counted in the fresh lungs at time of sacrifice is shown (b)
Discussion
Our data indicate that EGF-induced JNK activation is essential for MTLn3 cell migration. Phosphorylation of paxillin on Ser178 in MTLn3 cells was induced upon EGF stimulation in a manner sensitive to the JNK inhibitor, and expression of paxillinS178A inhibited cell motility. Such a role for paxillin Ser178 in cell migration is in agreement with observations made in different tumor cell lines[7,9,13,24,25,40,55-60]. Here we further demonstrate that the JNK-paxillin axis regulates both the dynamics of focal adhesions through modulation of paxillin protein dynamics at focal adhesions as well as the stability of β-catenin-based adherence junction formation. We propose that Ser178 phosphorylation may induce a conformational change, thereby affecting paxillin interactions with other focal adhesion components. Alternatively, the decreased EGFR expression in paxillinS178A cells affects downstream signaling pathways that indirectly modifies focal adhesion dynamics as well as adherence junction stability. Indeed, re-expression of EGFR in paxillinS178A cells reversed the epithelial-like phenotype and rendered paxillinS178A again less immobile at focal adhesions, suggesting that this is not an intrinsic characteristic of paxillinS178A, but is rather due to altered signaling in cells mediated by downstream EGFR signaling pathways. This needs further investigation.
So far no in vivo data on the specific role of paxillin Ser178 in metastasis formation have been presented. Here, we demonstrate that the Ser178 of paxillin is essential for spontaneous metastasis formation in an orthotopic breast tumor/metastasis model. Our in vivo data on the specific role of paxillin Ser178 in metastasis formation is in line with previously published data using a knock-down of MLK3 in MDA-MB-231. Indeed in the later study, they found that Ser178 phosphorylation of paxillin was associated with the metastatic phenotype[40,59]. Importantly in our study, we demonstrate that specifically the Ser178 of paxillin is essential for spontaneous metastasis formation in an orthotopic breast tumor/metastasis model. Our findings are indicative for a role for the JNK-paxillin pathway in the regulation of the expression of EGFR, thereby severely affecting the capacity of tumor cells to migrate and metastasize. Indeed, reduced levels of EGFR were observed in three independent paxillinS178A MTLn3 clones, resulting in decreased EGF-induced activation of signaling pathways downstream of EGFR. Given the fact that EGFR is often highly expressed in advanced breast cancer, and that EGFR antagonists inhibit spontaneous metastasis formation of MTLn3 cells, we anticipated that the reduced EGFR expression was an essential component of the inhibited cell migration and metastasis formation. Indeed, re-expression of the wt-EGFR in the paxillinS178A cells did rescue the migratory phenotype of MTLn3 paxillinS178A cells as well as their metastatic capacity under in vivo conditions. This regulation of the EGFR by paxillin phosphorylation has not been described before. Curiously, a recent study demonstrated that knockdown of MCLK in mammary MCF10A cells induces increase in cell migration through enhanced phosphorylation of paxillin at Ser178 which was linked to an increase in JNK activity and very interestingly to a significant up-regulation of EGFR at protein level[60]. This is striking and yet would suggest that our findings might not be specific to our cell model. In our study, paxillinS178A MTLn3 cells showed reduced c-Jun transcriptional activity. In MCF7 cells stable overexpression of c-Jun induces an increase in EGFR expression suggesting that AP-1 transcription factors can regulate EGFR transcription levels[53]; such a regulation is also observed in keratinocytes[54]. Therefore, we propose a role of the JNK-c-Jun signaling pathway in the regulation of EGFR expression at transcriptional level. Our own data in MCF7 cells that have increased ectopic expression of EGFR show that depletion of paxillin results in significant downregulation of EGFR expression as well as downstream signaling such as AKT and ERK [Supplementary Figure 7]. Thus, an alternative explanation may be that JNK-mediated phosphorylation of paxillin Ser178 is required for efficient EGFR endocytosis and recycling, an essential component in its activation[61]. Hence this will affect proper JNK activation and AP-1 transcription factor activation thereby providing a positive feedback for EGFR expression[62]. Alternatively, paxillin may affect the expression of EGFR by its known role in the regulation of gene expression through its interaction with ERK[63,64], poly-A-binding protein[65], Abl[66,67] and steroid receptors, or through its own ability to undergo nucleocytoplasmic shuttling[37,38,68-71].
Our findings indicate that the JNK-paxillin axis modulates the scattered phenotype of MTLn3 cells. Both inhibition of JNK as well as ectopic expression of paxillinS178A reversed the scattered phenotype towards a more epithelial-like morphology with the formation of E-cadherin/β-catenin cell-cell junctions. This morphological switch was observed under serum-starved and EGF-treated conditions. The (in)direct tyrosine phosphorylation of β-catenin by EGFR and other receptor tyrosine kinases such as c-Met is known to destabilize its binding to E-cadherin. Because of the low EGFR expression in the paxillin mutant cells, we anticipate that this results in de-phosphorylation of β-catenin leading to its localization to the cell-cell contacts. These data suggest that paxillinS178A does not affect the intrinsic molecular components and machinery required for the scattered phenotype and support the notion that the effect of paxillinS178A is rather related to the defects in the EGFR signaling pathways. Consequently, the defects in in vivo metastasis formation of MTLn3 paxillinS178A cells are most likely largely due to defects EGF signaling. This fits with our observations that ectopic EGFR expression itself is sufficient to again allow metastasis formation of paxillinS178A cells.
In summary, in MTLn3, engagement of EGF receptors by EGF triggers rapid activation of JNK, leading to the phosphorylation of paxillin on Ser 178 which facilitates adhesion turnover thus promoting rapid migration. In the MTLn3 cell lines that ectopically express GFP-paxillin-S178A mutants, we found that the EGF receptor is downregulated at both the protein and mRNA levels. Paxillin S178A mutant protein might associate with JNK preventing its activation and consequently affecting AP1 activity (c-Jun phosphorylation) and consequently EGFR expression. In our model [Figure 7], still some links need to be further determined such as how exactly paxillin regulates via JNK EGFR expression. Given the essential role of Ser178 phosphorylation in the migration and metastasis formation, monitoring this paxillin phosphorylation in tumor samples from patients may be indicative of the activation of this pro-metastatic pathway and possibly predictive for the disease prognosis. Moreover, with the further development of specific JNK inhibitors[72,73] it is anticipated that novel targeted therapies that antagonize the migratory/invasive behavior of tumor cells may be used in the clinic in the future. Further work in this area is required.
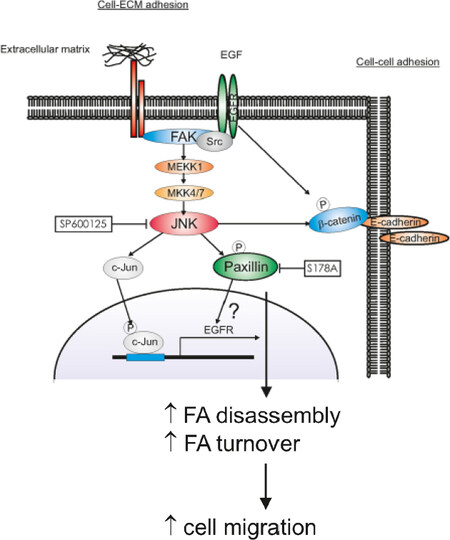
Figure 7. Diagram depicts the hypothesized signaling pathways for JNK-paxillin dependent cell migration. Engagement of EGF receptors by EGF triggers rapid activation of JNK, leading to the phosphorylation of paxillin on Ser 178 which might facilitate adhesion turnover thus promoting rapid migration. Upon EGF stimulation, JNK phophorylates also c-Jun which regulates EGFR transcription and β-catenin which delocalizes from the cell-cell contact breaking down the adherens junctions. Paxillin S178A mutant protein might associate with JNK preventing its activation and consequently affecting AP1 activity and EGFR expression. Inactive JNK as well as down-regulation of EGFR result in de-phosphorylation of β-catenin and formation of stabile cell-cell contact in MTLn3 cells
Declarations
AcknowledgmentsWe would like to thank H. de Bont for assistance with live cell imaging experiments and ImagePro Plus analysis. We thank H. van Dam for luciferase reporter constructs.
Authors’ contributionsCarried out the experiments: Verkoeijen S, Ma YF, van Roosmalen W, Lalai R, van Miltenburg MHAM, Le Dévédec SE
Wrote the manuscript with support from van de Water B: Verkoeijen S, Ma YF, Le Dévédec SE
Conceived the original idea and helped supervise the project: van de Water B
Supervised and finalized the project: Le Dévédec SE
Availability of data and materialsAll data are made available through supplemental data. Materials are available on request.
Financial support and sponsorshipThis work was supported by the EU FP7 Metafight project (HEALTH-F2-2007-201862), Dutch Cancer Society (KWF-UL2007-3860) and NWO grant (911-02-022).
Conflicts of interestAll authors declared that there are no conflicts of interest.
Ethical approval and consent to participateNot applicable.
Consent for publicationNot applicable.
Copyright© The Author(s) 2019.
REFERENCES
3. Fidler IJ, Kripke ML. The challenge of targeting metastasis. Cancer Metastasis Rev 2015;34:635-41.
4. Hamidi H, Ivaska J. Every step of the way: integrins in cancer progression and metastasis. Nat Rev Cancer 2018;18:533-48.
5. Nguyen DX, Bos PD, Massague J. Metastasis: from dissemination to organ-specific colonization. Nat Rev Cancer 2009;9:274-84.
6. Sethi N, Kang Y. Unravelling the complexity of metastasis - molecular understanding and targeted therapies. Nat Rev Cancer 2011;11:735-48.
7. Huang C, Jacobson K, Schaller MD. A role for JNK-paxillin signaling in cell migration. Cell Cycle 2004;3:4-6.
9. Huang Z, Yan DP, Ge BX. JNK regulates cell migration through promotion of tyrosine phosphorylation of paxillin. Cell Signal 2008;20:2002-12.
10. Le Devedec SE, van Roosmalen W, Maria N, Grimbergen M, Pont C, et al. An improved model to study tumor cell autonomous metastasis programs using MTLn3 cells and the Rag2(-/-) gammac (-/-) mouse. Clin Exp Metastasis 2009;26:673-84.
11. Omran OM, Al SM. Cytoskeletal focal adhesion proteins fascin-1 and paxillin are predictors of malignant progression and poor prognosis in human breast cancer. J Environ Pathol Toxicol Oncol 2015;34:201-12.
12. Wagner EF, Nebreda AR. Signal integration by JNK and p38 MAPK pathways in cancer development. Nat Rev Cancer 2009;9:537-49.
13. Chen J, Gallo KA. MLK3 regulates paxillin phosphorylation in chemokine-mediated breast cancer cell migration and invasion to drive metastasis. Cancer Res 2012;72:4130-40.
14. Shin EY, Kim SY, Kim EG. c-Jun N-terminal kinase is involved in motility of endothelial cell. Exp Mol Med 2001;33:276-83.
15. Xia Y, Makris C, Su B, Li E, Yang J, et al. MEK kinase 1 is critically required for c-Jun N-terminal kinase activation by proinflammatory stimuli and growth factor-induced cell migration. Proc Natl Acad Sci U S A 2000;97:5243-8.
16. Kavurma MM, Khachigian LM. ERK, JNK, and p38 MAP kinases differentially regulate proliferation and migration of phenotypically distinct smooth muscle cell subtypes. J Cell Biochem 2003;89:289-300.
17. Kawauchi T, Chihama K, Nabeshima Y, Hoshino M. The in vivo roles of STEF/Tiam1, Rac1 and JNK in cortical neuronal migration. EMBO J 2003;22:4190-201.
18. Zhang L, Wang W, Hayashi Y, Jester JV, Birk DE, et al. A role for MEK kinase 1 in TGF-beta/activin-induced epithelium movement and embryonic eyelid closure. EMBO J 2003;22:4443-54.
19. Berginski ME, Vitriol EA, Hahn KM, Gomez SM. High-resolution quantification of focal adhesion spatiotemporal dynamics in living cells. PLoS One 2011;6:e22025.
20. Guo L, Guo Y, Xiao S, Shi X. Protein kinase p-JNK is correlated with the activation of AP-1 and its associated Jun family proteins in hepatocellular carcinoma. Life Sci 2005;77:1869-78.
21. Yin Y, Wang S, Sun Y, Matt Y, Colburn NH, et al. JNK/AP-1 pathway is involved in tumor necrosis factor-alpha induced expression of vascular endothelial growth factor in MCF7 cells. Biomed Pharmacother 2009;63:429-35.
22. Kappelmann M, Bosserhoff A, Kuphal S. AP-1/c-Jun transcription factors: regulation and function in malignant melanoma. Eur J Cell Biol 2014;93:76-81.
23. Lee MH, Koria P, Qu J, Andreadis ST. JNK phosphorylates beta-catenin and regulates adherens junctions. FASEB J 2009;23:3874-83.
24. Huang C, Rajfur Z, Borchers C, Schaller MD, Jacobson K. JNK phosphorylates paxillin and regulates cell migration. Nature 2003;424:219-23.
25. Miyamoto Y, Torii T, Yamamori N, Eguchi T, Nagao M, et al. Paxillin is the target of c-Jun N-terminal kinase in Schwann cells and regulates migration. Cell Signal 2012;24:2061-9.
28. Mitra SK, Schlaepfer DD. Integrin-regulated FAK-Src signaling in normal and cancer cells. Curr Opin Cell Biol 2006;18:516-23.
29. Zaidel-Bar R, Itzkovitz S, Ma'ayan A, Iyengar R, Geiger B. Functional atlas of the integrin adhesome. Nat Cell Biol 2007;9:858-67.
30. Huveneers S, Danen EH. Adhesion signal. J Cell Sci 2009;122:1059-69.
36. Lopez-Colome AM, Lee-Rivera I, Benavides-Hidalgo R, Lopez E. Paxillin: a crossroad in pathological cell migration. J Hematol Oncol 2017;10:50.
37. Hammes SR, Miedlich SU, Sen A. Paxillin and steroid signaling: from frog to human. Methods Mol Biol 2014;1204:95-108.
39. Cai J, Du S, Wang H, Xin B, Wang J, et al. Tenascin-C induces migration and invasion through JNK/c-Jun signalling in pancreatic cancer. Oncotarget 2017;8:74406-22.
40. Chen J, Miller EM, Gallo KA. MLK3 is critical for breast cancer cell migration and promotes a malignant phenotype in mammary epithelial cells. Oncogene 2010;29:4399-411.
41. Rattanasinchai C, Llewellyn BJ, Conrad SE, Gallo KA. MLK3 regulates FRA-1 and MMPs to drive invasion and transendothelial migration in triple-negative breast cancer cells. Oncogenesis 2017;6:e345.
42. Huigsloot M, Tijdens IB, Mulder GJ, van de WB. Differential regulation of doxorubicin-induced mitochondrial dysfunction and apoptosis by Bcl-2 in mammary adenocarcinoma (MTLn3) cells. J Biol Chem 2002;277:35869-79.
43. Xue C, Wyckoff J, Liang F, Sidani M, Violini S, et al. Epidermal growth factor receptor overexpression results in increased tumor cell motility in vivo coordinately with enhanced intravasation and metastasis. Cancer Res 2006;66:192-7.
44. Vasanwala FH, Kusam S, Toney LM, Dent AL. Repression of AP-1 function: a mechanism for the regulation of Blimp-1 expression and B lymphocyte differentiation by the B cell lymphoma-6 protooncogene. J Immunol 2002;169:1922-9.
45. Chen EY, Tan CM, Kou Y, Duan Q, Wang Z, et al. Enrichr: interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinformatics 2013;14:128.
46. Kuleshov MV, Jones MR, Rouillard AD, Fernandez NF, Duan Q, et al. Enrichr: a comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res 2016;44:W90-7.
47. Rosse C, Formstecher E, Boeckeler K, Zhao Y, Kremerskothen J, et al. An aPKC-exocyst complex controls paxillin phosphorylation and migration through localised JNK1 activation. PLoS Biol 2009;7:e1000235.
48. de Graauw M, Tijdens I, Cramer R, Corless S, Timms JF, et al. Heat shock protein 27 is the major differentially phosphorylated protein involved in renal epithelial cellular stress response and controls focal adhesion organization and apoptosis. J Biol Chem 2005;280:29885-98.
49. Yamanaka I, Koizumi M, Baba T, Yamashita S, Suzuki T, et al. Epidermal growth factor increased the expression of alpha2beta1-integrin and modulated integrin-mediated signaling in human cervical adenocarcinoma cells. Exp Cell Res 2003;286:165-74.
50. Ricono JM, Huang M, Barnes LA, Lau SK, Weis SM, et al. Specific cross-talk between epidermal growth factor receptor and integrin alphavbeta5 promotes carcinoma cell invasion and metastasis. Cancer Res 2009;69:1383-91.
51. Lu Z, Jiang G, Blume-Jensen P, Hunter T. Epidermal growth factor-induced tumor cell invasion and metastasis initiated by dephosphorylation and downregulation of focal adhesion kinase. Mol Cell Biol 2001;21:4016-31.
52. Thelemann A, Petti F, Griffin G, Iwata K, Hunt T, et al. Phosphotyrosine signaling networks in epidermal growth factor receptor overexpressing squamous carcinoma cells. Mol Cell Proteomics 2005;4:356-76.
53. Johnson AC, Murphy BA, Matelis CM, Rubin stein Y, Piebenga EC, et al. Activator protein-1 mediates induced but not basal epidermal growth factor receptor gene expression. Mol Med 2000;6:17-27.
54. Zenz R, Scheuch H, Martin P, Frank C, Eferl R, et al. c-Jun regulates eyelid closure and skin tumor development through EGFR signaling. Dev Cell 2003;4:879-89.
55. Kimura K, Teranishi S, Yamauchi J, Nishida T. Role of JNK-dependent serine phosphorylation of paxillin in migration of corneal epithelial cells during wound closure. Invest Ophthalmol Vis Sci 2008;49:125-32.
56. Ching YP, Leong VY, Lee MF, Xu HT, Jin DY, et al. P21-activated protein kinase is overexpressed in hepatocellular carcinoma and enhances cancer metastasis involving c-Jun NH2-terminal kinase activation and paxillin phosphorylation. Cancer Res 2007;67:3601-8.
57. Smadja-Lamere N, Boulanger MC, Champagne C, Branton PE, Lavoie JN. JNK-mediated phosphorylation of paxillin in adhesion assembly and tension-induced cell death by the adenovirus death factor E4orf4. J Biol Chem 2008;283:34352-64.
58. Yamauchi J, Miyamoto Y, Sanbe A, Tanoue A. JNK phosphorylation of paxillin, acting through the Rac1 and Cdc42 signaling cascade, mediates neurite extension in N1E-115 cells. Exp Cell Res 2006;312:2954-61.
59. Rattanasinchai C, Llewellyn BJ, Conrad SE, Gallo KA. MLK3 regulates FRA-1 and MMPs to drive invasion and transendothelial migration in triple-negative breast cancer cells. Oncogenesis 2017;6:e345.
60. Kim DY, Helfman DM. Loss of MLCK leads to disruption of cell-cell adhesion and invasive behavior of breast epithelial cells via increased expression of EGFR and ERK/JNK signaling. Oncogene 2016;35:4495-508.
61. Madshus IH, Stang E. Internalization and intracellular sorting of the EGF receptor: a model for understanding the mechanisms of receptor trafficking. J Cell Sci 2009;122:3433-9.
62. Hess J, Angel P, Schorpp-Kistner M. AP-1 subunits: quarrel and harmony among siblings. J Cell Sci 2004;117:5965-73.
63. Ishibe S, Joly D, Zhu X, Cantley LG. Phosphorylation-dependent paxillin-ERK association mediates hepatocyte growth factor-stimulated epithelial morphogenesis. Mol Cell 2003;12:1275-85.
64. Ishibe S, Joly D, Liu ZX, Cantley LG. Paxillin serves as an ERK-regulated scaffold for coordinating FAK and Rac activation in epithelial morphogenesis. Mol Cell 2004;16:257-67.
65. Woods AJ, Roberts MS, Choudhary J, Barry ST, Mazaki Y, et al. Paxillin associates with poly(A)-binding protein 1 at the dense endoplasmic reticulum and the leading edge of migrating cells. J Biol Chem 2002;277:6428-37.
66. Lewis JM, Baskaran R, Taagepera S, Schwartz MA, Wang JY. Integrin regulation of c-Abl tyrosine kinase activity and cytoplasmic-nuclear transport. Proc Natl Acad Sci U S A 1996;93:15174-9.
67. Lewis JM, Schwartz MA. Integrins regulate the association and phosphorylation of paxillin by c-Abl. J Biol Chem 1998;273:14225-30.
68. Hervy M, Hoffman L, Beckerle MC. From the membrane to the nucleus and back again: bifunctional focal adhesion proteins. Curr Opin Cell Biol 2006;18:524-32.
69. Sathe AR, Shivashankar GV, Sheetz MP. Nuclear transport of paxillin depends on focal adhesion dynamics and FAT domains. J Cell Sci 2016;129:1981-8.
70. Sen A, De Castro I, DeFranco DB, Deng FM, Melamed J, et al. Paxillin mediates extranuclear and intranuclear signaling in prostate cancer proliferation. J Clin Invest 2012;122:2469-81.
71. Wang Y, Gilmore TD. Zyxin and paxillin proteins: focal adhesion plaque LIM domain proteins go nuclear. Biochim Biophys Acta 2003;1593:115-20.
72. Bubici C, Papa S. JNK signalling in cancer: in need of new, smarter therapeutic targets. Br J Pharmacol 2014;171:24-37.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Verkoeijen S, Ma YF, van Roosmalen W, Lalai R, van Miltenburg MHAM, de Graauw M, van de Water B, Le Dévédec SE. Paxillin serine 178 phosphorylation in control of cell migration and metastasis formation through regulation of EGFR expression in breast cancer. J Cancer Metastasis Treat 2019;5:51. http://dx.doi.org/10.20517/2394-4722.2019.06
AMA Style
Verkoeijen S, Ma YF, van Roosmalen W, Lalai R, van Miltenburg MHAM, de Graauw M, van de Water B, Le Dévédec SE. Paxillin serine 178 phosphorylation in control of cell migration and metastasis formation through regulation of EGFR expression in breast cancer. Journal of Cancer Metastasis and Treatment. 2019; 5: 51. http://dx.doi.org/10.20517/2394-4722.2019.06
Chicago/Turabian Style
Verkoeijen, Saertje, Ya-Feng Ma, Wies van Roosmalen, Reshma Lalai, Martine H. A. M. van Miltenburg, Marjo de Graauw, Bob van de Water, Sylvia E. Le Dévédec. 2019. "Paxillin serine 178 phosphorylation in control of cell migration and metastasis formation through regulation of EGFR expression in breast cancer" Journal of Cancer Metastasis and Treatment. 5: 51. http://dx.doi.org/10.20517/2394-4722.2019.06
ACS Style
Verkoeijen, S.; Ma Y.F.; van Roosmalen W.; Lalai R.; van Miltenburg MHAM.; de Graauw M.; van de Water B.; Le Dévédec SE. Paxillin serine 178 phosphorylation in control of cell migration and metastasis formation through regulation of EGFR expression in breast cancer. J. Cancer. Metastasis. Treat. 2019, 5, 51. http://dx.doi.org/10.20517/2394-4722.2019.06
About This Article
Special Issue
Copyright

Data & Comments
Data
0

 Cite This Article 16 clicks
Cite This Article 16 clicks

 Like This Article 14
likes
Like This Article 14
likes






Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.