
Role of autophagy in therapeutic resistance of glioblastoma


Abstract
Patients with glioblastoma (GBM), a malignant brain tumor, exhibit a mean survival of less than 1.5 years. Despite treatment, the disease eventually develops resistance, resulting in disease relapse. Autophagy is a process of degradation and clearance that is activated to maintain cellular homeostasis. Its roles in cancer disease course and the treatment response, however, are controversial. In GBM, accumulating evidence has indicated that autophagy can protect cells, especially those with stemness features, causing the development of cell resistance. In this review, we discuss the impact of the cell reaction to currently active treatments, including temozolomide, radiation, tumor treating fields, bevacizumab (Avastin), etoposide (VP-16), cisplatin (CDDP), and carmustine (BCNU). Most of these induce the up-regulation of autophagy through signaling pathways of DNA damage response, reactive oxygen species, hypoxia, retinoblastoma, AMP-activated protein kinase, AKT/mTOR and MST4 kinase affecting cell fate by altering cell metabolism, cell death, and DNA repair. Treatment-related autophagy may be modulated by combining autophagy inhibitors such as chloroquine or antioxidants to prevent the development of resistance, thus improving cancer treatment.
Keywords
Introduction
The term autophagy is derived from Greek words, “auto” and “phagy”, meaning self-eating[1], representing a process of degradation and clearance that is activated in response to aggregated protein and dysfunctional organelles to maintain cellular homeostasis[2]. The autophagy pathway is initiated by a series of active proteins, such as autophagy-related (ATG) proteins, UVRAG, Beclin 1, PIK3C3, LC3 and p62[3]. Together, they contribute to the formation of double-membrane autophagosomes, and eventually, fusion with lysosomes for degradation of inner substances [Figure 1]. Hence, autophagy has been implicated in several diseases including cancer[4]. In the past, autophagy was believed to be related to apoptosis[5]. Recently, an opposite impact of autophagy, i.e., promoting cell survival, has been widely studied[6]. This “double-edged sword” effect in tumorigenesis varies depending on the cellular reaction to specific stimulation as well as in different cancer types. High levels of autophagy in the tumor environment (with limited nutrient and oxygen conditions) allow cancer cells to survive, resuming proliferation and initiation[7].
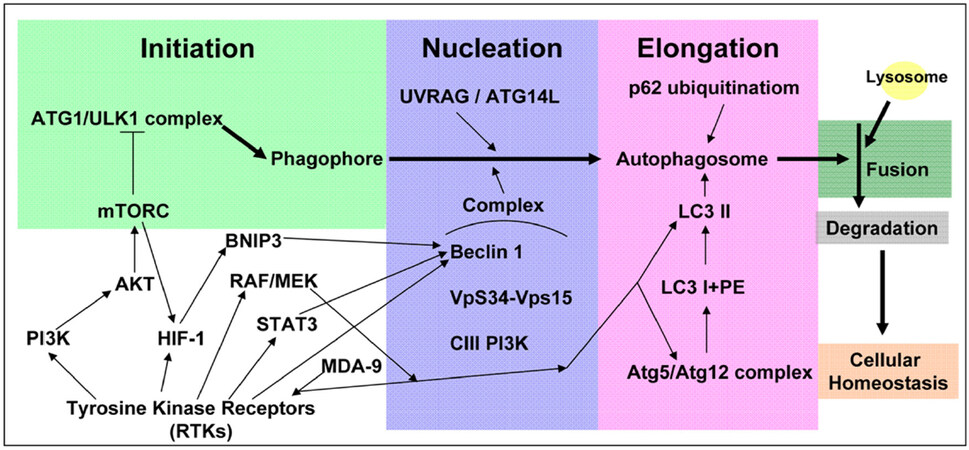
Figure 1. The schematic figure of the principal cell signaling pathways assocated with the autophagic process. The process of autophagy pathway begins from the initiation step to the nucleation step, the elongation step, and to the formation of mature autophagosome which subsequently fuses with lysosome to perform the degradation process. The commonly activated pathways related to aberrant RTKs in GBM are also shown. Their roles to affect the individual steps of the autophagic process are indicated. RTKs: receptor tyrosine kinases; GBM: glioblastoma; HIF-1: hypoxia-inducible factor-1
Glioblastoma (GBM) is a disease composed of extremely varied tumor microenvironments and heterogeneous cancer cells. It is a fatal disease known to have a poor prognosis, and is not considered curable. Despite advanced treatment strategies, a notable improvement was not observed in terms of the outcome. Hence, it is not surprising that the tumor cells in the apex of the hierarchy, or those that are capable of self-renewal and differentiation, display highly-activated autophagy signaling to survive and thrive from the given treatment, such as temozolomide (TMZ)[8]. Moreover, the molecular characters of GBM involved in the growth and survival via intracellular pathways are distinct genetic aberrations in epidermal growth factor receptor (EGFR), PTEN, TP53, IDH1 and so on[9]. Among these frequently reported genes in clinical disease, EGFR is well-known as a driving receptor tyrosine kinase (RTK) in GBM that dictates multiple oncogenic signaling[10]. The signaling amplification of EGFR accounts for approximately 60% of GBM cases. The mutant form EGFRvIII receptor, which is constitutively active that is independent of the ligand binding condition, is also common. In association with EGFR signaling pathway, it was found that levels of MDA-9, a protein related to tumor cell behavior and stemness, were increased in glioma stem-like cells to regulate the protective autophagy[11][Figure 1]. Moreover, it was found that STAT3 levels were related to beclin 1 expression via EGFR amplification[12,13][Figure 1]. Other RTKs in GBM, such as vascular endothelial growth factor receptor (VEGFR)[14], platelet-derived growth factor receptor (PDGFR)[15] and discoidin domain RTK 1 (DDR1)[16], also contributes to modulating the autophagy formation through AKT/mTOR[16,17], RAF/MEK[18] and HIF-1/BCL2 Interacting Protein 3[19-21] signalings [Figure 1]. As thus, therapeutic agents targeting these factors, for example bevacizumab[14] and PDGF neutralizing antibody[15], has been reported to enhance autophagy signaling [Figure 1]. Despite of the implications by disease- or treatment-related autophagic alterations, clinical trials with autophagy inhibitors, such as chloroquine (CQ) or its analogs, showed only limiting benefits[22]. So far, the exact role of autophagy in GBM remains ambiguous, especially regarding treatment resistance[23].
The effect of autophagy on tumors is not universal. On one hand, autophagy protects cells by clearing damaged organelles, thereby promoting cell survival. On the other hand, it achieves damage control by eliciting self-eating, leading to the process of autophagic cell death. The pros and cons of this biologic reaction does not have a clear boundary and may be dynamic. Regarding drug treatment, the appearance of autophagy also had divergent effect. For example, in a study of PI3K/mTOR inhibitor BGT226, application of the drug led to autophagic cell death in head and neck cancer cell lines[24]. In contrast, application of PI3K/AKT/mTOR inhibitors sensitized cells to radiotherapy as well as reduced autophagy formation[25]. Various effects of autophagy in the cancer treatment have been demonstrated, making the reaction complicated in each circumstance. However, in the case of GBM, the appearance of autophagy during specific treatment seems to support the development of resistance[26]. In this article, we will discuss the molecular significance of autophagy in the current treatment of GBM. We will also discuss the formation of the reaction and the potential benefit of inhibiting autophagy along with the treatment.
TMZ
Being the most standard chemotherapy for GBM, it is mandatory to understand how autophagy is induced by TMZ. The first-line drug acts by inducing lethal DNA damage and subsequent reactive oxygen species (ROS) production[27]. The effect is, however, mostly only transient and resistance is almost inevitable, with up to 90% of the patients experiencing early disease recurrence[28]. Development of TMZ resistance in GBM is complicated and less understood, but till date, clear factors leading to resistance are still limited to pre-existing O6-methylguanine-DNA methyltransferase (MGMT)[29]. According to previous studies, the DNA damage response (DDR) can induce autophagy in an yeast system[30] and the enhanced autophagy can increase DNA repair ability to cause chemo-resistance[31]. In GBM, TMZ-induced autophagy in glioma cell lines is dependent on MGMT expression, mismatch repair system and Rad51-mediated homologous recombination[32]. The glioma cell lines resistant to TMZ cotreated with O6-benzylguanine, an O6-alkylguanine–DNA alkyltransferase (a DNA repair enzyme) inhibitor, can be re-susceptive to TMZ via autophagy regulation[33]. The cascades of TMZ-induced autophagy showed that the phosphorylation of H2AFX at Ser139 following TMZ treatment initiates DDR [Figure 2], sequentially, leading to phosphorylation of PRKAA (Thr172), ULK1 (Ser555/575), MAPK14 (The180/Tyr182), RPTOR (Ser792) and suppressive phosphorylation of AKT (Ser473) and mTOR (Ser2448) to induce autophagy and inhibit apoptosis[34]. However, further investigation is needed to examine whether there are other signaling pathways involved between autophagy and DDR.
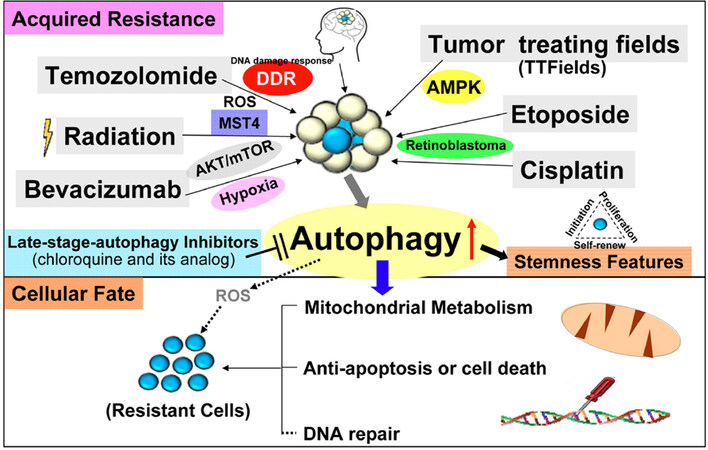
Figure 2. The schematic diagram demonstrates that in primary or recurrent GBM patients, the standard regimens consists of TMZ, radiation, TTFields, bevacizumab, etoposide and cisplatin, which enhance cell stemness, metabolism (energy), anti-apoptosis and DNA repair to acquire therapeutic resistance through the signaling pathways of DDR, ROS, hypoxia, AKT/mTOR, MST4 kinase, AMPK and RB. Nowadays, the inhibitors of late-stage autophagy, such as CQ or its analogs, have been tested in clinical trials of GBM patients. TMZ: temozolomide; GBM: glioblastoma; TTFields: tumor treating fields; ROS: reactive oxygen species; DDR: DNA damage response; AMPK: AMP-activated protein kinase; RB: retinoblastoma; CQ: chloroquine
In addition to MGMT that contributes to naïve TMZ resistance, the mechanism for tumor acquiring resistance is more complicated. In our previous studies, we identified transcriptional binding factor specific protein 1 and its downstream modulator, superoxide dismutase 2 (SOD2), as the critical factors in the tolerance of TMZ-induced ROS in models of resistance[35,36]. In fact, it has also been reported that TMZ can induce autophagy via ROS signaling[37][Figure 2]. The detailed mechanism between autophagy and ROS, however, needs further investigation. SOD2 is known to specifically function in mitochondria to regulate oxidative stress and energy metabolism[38]. Altered metabolic reprogramming by mitochondrial control in cancer may play a role in chemotherapy resistance[39]. For instance, increased autophagy derived from glucose starvation condition causes GBM cell quiescence, survival and chemoresistance[40] and complex I linked oxidative phosphorylation is required to maintain mitochondrial respiration (electron transfer system) through autophagy regulation[41]. Furthermore, presence of glioma stem-like cells may have crucial role in acquiring resistance[42]. An emerging concept in cancer biology suggests these specific subgroups with stemness features are responsible for acquisition of resistance[42]. The stem cells are characterized by self-renewal and multipotent abilities[43]. These allow cells to be resistant to standard therapy, and are associated with the treatment outcome[42]. In glioma stem-like cells, autophagy-associated proteins, i.e., DNA Damage Regulated Autophagy Modulator 1 and p62, can regulate the cell migration and invasion by modulating energy metabolism (ATP) and affect mitochondrial morphology by regulating mitochondrial fusion[44]. Some studies have indicated that autophagy can regulate epithelial-to-mesenchymal transition[45] and the capabilities of migration and invasion in those cells under nutrient deprivation are damaged when autophagy is activated through regulation of ATG proteins or beclin 1[46]. However, it was found that TMZ treatment plus autophagy inhibitor could suppress cell migration and invasion[47], suggesting further investigation was needed to determine the role of TMZ-induced autophagy in this aspect. Moreover, stemness features in the glioma stem-like cells are reduced by autophagy inhibition and apoptosis is enhanced by blockage of autophagy[48]. Recently, it is found that the blockage of autophagy up-regulates the TMZ-sensitivity in these stem-like cells through metabolic dysfunction-related ferroptosis[49] in the stem-like cells, demonstrating the close correlation between autophagy and metabolism. In summary, application of these the explains how the disease is capable of surviving TMZ toxicity regardless of MGMT status.
Radiation
Radiation can cause similar treatment response to TMZ. Radiation treatment in GBM cells can induce autophagy through the PI3K/AKT/mTOR pathway, ROS[50,51] and MST4 kinase-ATG4B signaling[52][Figure 2]. Inhibition of autophagy makes these cells susceptible to stimulation[53,54]. Moreover, it is reported that GBM cells are susceptible to radiation following inhibition of MDA-9 expression[55]. Radiation has various effect to autophagy. Studies have indicated that the relatively radio-sensitive cells or small radiotherapy fraction (2 Gy) displays enhanced autophagic flux, while more radio-resistant cells or largeer radiotherapy fraction exhibited an inhibition of autophagic flux[56]. Regarding the glioma stem-like cells, it was reported the subsets were enhanced for autophagy after radiation to promote metabolism, anti-apoptosis and stemness[57], while the other reports showed that CD133+ cells or sphere cells from one patient have a lower level of autophagy[58,59]. Notably, it is also found that CD133+ glioma stem-like cells can be resensitized to radiation treatment by using autophagy inhibitors (bafilomycin) or down-regulation of ATG protein levels[60]. Though the exact effect remains unclear, the association between autophagy, radiation, and the stemness features is intriguing.
Whether targeting autophagy will benefit radiotherapy effect is also controversial. On one hand, a study has found that autophagy in malignant glioma cells is a transition status promoting apoptotic cell death following radiation, which can be reversed by suppressing autophagy function[61]. On the other hand, from the aspect of PI3K/AKT/mTOR signaling (it is active in most GBM patients[62]), Cerniglia et al.[63] and Gil del Alcazar et al.[64] have indicated that NVP-BEZ235, a dual PI3K/mTOR inhibitor, can promote GBM cells sensitive to radiation and up-regulate autophagy by directly affecting the function of DNA damage repair (DNA-dependent protein kinase catalytic subunit and ATM kinase) and autophagy related proteins (ATG5 and Beclin1), respectively. The results demonstrated that there are different responses after administrating the inhibitor and radiation leading to cell death. Furthermore, Zhuang et al.[65] also reported that rapamycin can lead to the differentiation of glioma stem-like cells, up-regulating the cell radiosensitivity and reducing tumorgenesis through activation of autophagy. However, the application of the autophagy inducers on mice experiment or clinical seems to be rare and limited in information[62,66]. In addition, most clinical trials nowadays prefer the combination of autophagy inhibitors with radiochemotherapy but not radiotherapy only[67]. Therefore, the directions of future research must focus on further exploration of how radiation induces autophagy (time point, tumor suppress genes, DNA damage repair and so on) and how the affected autophagy changes the cell fate.
Tumor treating fields
OPtune, a tumor treating fields (TTFields) device, is used as an advanced GBM treatment with only minor side effects noted in newly diagnosed or recurrent GBM patients. The device in combination with TMZ increased the median overall survival by almost 5 months compared to the TMZ alone group[68]. At 200 kHZ frequency, TTFields treatment has been reported to cause damaged mitosis and up-regulation of autophagy[69]. It is also found that the enhancement of autophagy is induced by TTFields treatment through AMP-activated protein kinase (AMPK) signaling[70][Figure 2] and down-regulation of the pathway inhibits the protective autophagy to reduce TTFields-induced acquired resistance. By combining with sorafenib, a multi-kinase inhibitor that can induce autophagy in prostate cancer cells through increased levels of LC3[71], the device exhibited enhanced autophagy and more inhibited tumor behavior to promote a better therapeutic strategy[72]. Regarding the mechanism, it was reported that TTFields can suppress DDR in non-small cell lung cancer cell lines[73] and the combination of radiation can inhibit DNA damage repair in glioma cell lines[74]. Furthermore, it is found that TTFields can cause ROS production to promote apoptosis in the cell line study[72]. This may have impact in inducing the clearance of damaged mitochondria (mitophagy)[75]. It is not clear, however, whether TTFields-increased autophagy is associated with inhibition of DNA damage repair. Further explorative experiments are thus needed to elucidate the mechanism and the role of autophagy in the disease-controlling mechanism of the device.
Bevacizumab
Bevacizumab can directly promote apoptosis and antiangiogenesis by regulating the associated proteins in GBM cells[19,76]. This drug has been reported to cause resistance in the cells through suppressing AKT-mTOR signaling[76] or through inducing hypoxia-inducible factor-1α/AMPK pathway[19] to increase autophagy influx [Figure 2]. It was also reported that bevacizumab could cause a hypoxia microenvironment, which in turn, was related to enhanced autophagy[77,78]. Thus, inhibition of the autophagy process was postulated to resensitize bevacizumab to GBM cells.
Up-regulation of glucose metabolism before autophagy induction was observed in bevacizumab-treated GBM cell lines[79]. Further investigation is needed to link the mechanism between autophagy and the cell metabolism after the treatment. It was also noted that bevacizumab can induce autophagy in glioma stem-like cells by enhancing VEGF-independent angiogenesis (vasculogenic mimicry, an alternative vasculature) through KDR/VEGFR-2 phosphorylation, which the formation is closely related with GBM resistance[80]. This highlights the therapeutic value of autophagy-inhibiting strategies in regard with the anti-angiogenesis therapy.
Etoposide
Etoposide (VP-16) treatment has been reported to induce autophagy, thereby increasing the therapeutic resistance in GBM cells[81,82]. The drug is a topoisomerase II inhibitor that also enhances autophagy by modulating though retinoblastoma protein (RB)[81][Figure 2]. Down-regulation of RB can inhibit autophagy by preventing autophagosomes fusion from lysosome and promoting cell death in GBM cells and glioma stem-like cells[81]. Furthermore, knockdown of RB can enhance apoptotic cell death and up-regulate the immunostaining intensity of γ-H2AX (a DNA double-strand break marker) induced by etoposide[81]. Earlier studies have demonstrated a close link between etoposide-induced autophagy, RB, and DNA damage.
Cisplatin
Cisplatin (CDDP) is a DNA alkylating agent that is widely used for human malignant tumors including GBM[83]. CDDP-associated resistance often develops through autophagy induction in multiple cancer cell types[84-87]. In cervical cancer cells, it is found that blockage of autophagy can increase CDDP-induced cytotoxicity via endoplasmic reticulum stress[88]. One study indicated that autophagy was associated with CDDP resistance in GBM and down-regulation of RB suppressed autophagy induced by CDDP[87][Figure 2]. Moreover, it was found that CDDP could induce long non-coding RNA to suppress CDDP-induced autophagy, which promoted apoptosis in GBM cells[89]. CDDP could also enhance the level of death receptor (DR5), directing glioma stem-like cells to tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) apoptotic pathway[90]. TRAIL, on the other hand, was reported to enhance the expression of autophagy leading to resistance of breast cancer cells, and blockage of autophagy could reduce the resistance[91]. The association between autophagy and TRAIL in GBM has thus been inferred. In addition, the mechanism of DNA repair induced by CDDP could promote resistance of GBM cells via the Jun kinase/stress-activated protein kinase signaling pathway[92]. Further experiments are required to verify whether autophagy is related to the DNA repair induced by CDDP.
Carmustine
Another DNA damaging nitrosoureas agent carmustine (BCNU) is reported to promote ROS-mediated autophagic cell death in solid tumor cells in combination with arsenic trioxide[93]. In terms of the role of ROS in the drug reaction, it was supportively reported that in human glioma cell line U98MG, the overexpression of Nrf 2, an anti-oxidant transcriptional factor, could reduce the carmustine-induced cytotoxicity[94]. How autophagy was utilized in carmustine-related oxidative stress and anti-oxidant reaction remains to be elucidated.
Autophagy inhibitors in clinical trial
Traditionally, CQ is used for malaria treatment. Due to the basic property of CQ, it stays in lysosomes leading to enhanced lysosomal pH and inhibition of autophagy[95,96]. Hence, CQ has been investigated in clinical trials for cancer therapy including GBM. An early clinical trial of CQ in a small population of GBM patients (NCT00224978) showed that the median survival increased in patients receiving CQ in combination with standard regimens[97]. Currently, there are 3 ongoing clinical trials involving CQ in GBM or astrocytoma patients (NCT02378532 (Phase 1), NCT02432417 (Phase 2) and NCT03243461 (Phase 3). In addition to CQ, more candidate autophagy modulators are expected with or without the standard drugs to be investigated in clinical trials.
Conclusion
The current standard treatments of primary or recurrent GBM can cause autophagy induction and the enhanced autophagy through DDR, ROS, hypoxia, AKT/mTOR, AMPK and RB pathways may result in cell resistance, resulting in enhancement of stemness features, increased abilities of metabolism, anti-apoptosis, and DNA repair. Therefore, in order to reduce the resistance caused by treatments, the usage of standard treatment plus autophagy inhibitors and signaling inhibitors may be a potential strategy for GBM therapy.
Declarations
Authors’ contributionsManuscript writing: Chien CH, Hsueh WT, Chuang JY, Chang KY
Supervised the final version: Chang KY
Availability of data and materialsNot applicable.
Financial support and sponsorshipThis work was supported by grants from National Health Research Institutes, Taiwan (CA-107-PP-08) and the Ministry of Science and Technology, Taiwan (MOST 108-2314-B-400-026).
Conflicts of interestAll authors declared that there are no conflicts of interest.
Ethical approval and consent to participateNot applicable.
Consent for publicationNot applicable.
Copyright© The Author(s) 2019.
REFERENCES
1. Eisenberg-Lerner A, Bialik S, Simon HU, Kimchi A. Life and death partners: apoptosis, autophagy and the cross-talk between them. Cell Death Differ 2009;16:966-75.
2. Ryter SW, Cloonan SM, Choi AM. Autophagy: a critical regulator of cellular metabolism and homeostasis. Mol Cells 2013;36:7-16.
3. Parzych KR, Klionsky DJ. An overview of autophagy: morphology, mechanism, and regulation. Antioxid Redox Signal 2014;20:460-73.
4. Mizushima N, Levine B, Cuervo AM, Klionsky DJ. Autophagy fights disease through cellular self-digestion. Nature 2008;451:1069-75.
7. Mathew R, Karantza-Wadsworth V, White E. Role of autophagy in cancer. Nature reviews. Cancer 2007;7:961-67.
8. Buccarelli M, Marconi M, Pacioni S, De Pascalis I, D'Alessandris QG, et al. Inhibition of autophagy increases susceptibility of glioblastoma stem cells to temozolomide by igniting ferroptosis. Cell Death Dis 2018;9:841.
9. Zinn PO, Singh SK, Kotrotsou A, Abrol S, Thomas G, et al. Distinct Radiomic Phenotypes Define Glioblastoma TP53-PTEN-EGFR Mutational Landscape. Neurosurgery 2017;64:203-10.
10. Hatanpaa KJ, Burma S, Zhao D, Habib AA. Epidermal growth factor receptor in glioma: signal transduction, neuropathology, imaging, and radioresistance. Neoplasia 2010;12:675-84.
11. Talukdar S, Pradhan AK, Bhoopathi P, Shen XN, August LA, et al. MDA-9/Syntenin regulates protective autophagy in anoikis-resistant glioma stem cells. Proc Natl Acad Sci U S A 2018;115:5768-73.
12. Caldera V, Mellai M, Annovazzi L, Valente G, Tessitore L, et al. Stat3 expression and its correlation with proliferation and apoptosis/autophagy in gliomas. J Onco 2008;2008:219241.
13. Tini P, Belmonte G, Toscano M, Miracco C, Palumbo S, et al. Combined epidermal growth factor receptor and Beclin1 autophagic protein expression analysis identifies different clinical presentations, responses to chemo- and radiotherapy, and prognosis in glioblastoma. Biomed Res Int 2015;2015:208076.
14. Muller-Greven G, Carlin CR, Burgett ME, Ahluwalia MS, Lauko A, et al. Macropinocytosis of Bevacizumab by Glioblastoma Cells in the Perivascular Niche Affects their Survival. Clin Cancer Res 2017;23:7059-71.
15. Takeuchi H, Kanzawa T, Kondo Y, Kondo S. Inhibition of platelet-derived growth factor signalling induces autophagy in malignant glioma cells. Br J Cancer 2004;90:1069-75.
16. Vehlow A, Cordes N. DDR1 (discoidin domain receptor tyrosine kinase 1) drives glioblastoma therapy resistance by modulating autophagy. Autophagy 2019;15:1487-88.
17. Fan QW, Weiss WA. Inhibition of PI3K-Akt-mTOR signaling in glioblastoma by mTORC1/2 inhibitors. Methods Mol Biol 2012;821:349-59.
18. Shingu T, Holmes L, Henry V, Wang Q, Latha K, et al. Suppression of RAF/MEK or PI3K synergizes cytotoxicity of receptor tyrosine kinase inhibitors in glioma tumor-initiating cells. J Transl Med 2016;14:46.
19. Hu YL, DeLay M, Jahangiri A, Molinaro AM, Rose SD, et al. Hypoxia-induced autophagy promotes tumor cell survival and adaptation to antiangiogenic treatment in glioblastoma. Cancer Res 2012;72:1773-83.
20. Azad MB, Gibson SB. Role of BNIP3 in proliferation and hypoxia-induced autophagy: implications for personalized cancer therapies. Ann N Y Acad Sci 2010;1210:8-16.
21. Womeldorff M, Gillespie D, Jensen RL. Hypoxia-inducible factor-1 and associated upstream and downstream proteins in the pathophysiology and management of glioblastoma. Neurosurg Focus 2014;37:E8.
22. Verbaanderd C, Maes H, Schaaf MB, Sukhatme VP, Pantziarka P, et al. Repurposing Drugs in Oncology (ReDO)-chloroquine and hydroxychloroquine as anti-cancer agents. Ecancermedicalscience 2017;11:781.
23. Yan Y, Xu Z, Dai S, Qian L, Sun L, et al. Targeting autophagy to sensitive glioma to temozolomide treatment. J Exp Clin Cancer Res 2016;35:23.
24. Chang KY, Tsai SY, Wu CM, Yen CJ, Chuang BF, et al. Novel phosphoinositide 3-kinase/mTOR dual inhibitor, NVP-BGT226, displays potent growth-inhibitory activity against human head and neck cancer cells in vitro and in vivo. Clin Cancer Res 2011;17:7116-26.
25. Chang L, Graham PH, Hao J, Ni J, Bucci J, et al. PI3K/Akt/mTOR pathway inhibitors enhance radiosensitivity in radioresistant prostate cancer cells through inducing apoptosis, reducing autophagy, suppressing NHEJ and HR repair pathways. Cell Death Dis 2014;5:e1437.
26. Golden EB, Cho HY, Jahanian A, Hofman FM, Louie SG, et al. Chloroquine enhances temozolomide cytotoxicity in malignant gliomas by blocking autophagy. Neurosurg Focus 2014;37:E12.
27. Zhang WB, Wang Z, Shu F, Jin YH, Liu HY, et al. Activation of AMP-activated protein kinase by temozolomide contributes to apoptosis in glioblastoma cells via p53 activation and mTORC1 inhibition. J Biol Chem 2010;285:40461-71.
28. Stupp R, Hegi ME, Mason WP, van den Bent MJ, Taphoorn MJ, et al. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol 2009;10:459-66.
29. Wick W, Weller M, van den Bent M, Sanson M, Weiler M, et al. MGMT testing--the challenges for biomarker-based glioma treatment. Nat Rev Neurol 2014;10:372-85.
30. Eapen VV, Waterman DP, Bernard A, Schiffmann N, Sayas E, et al. A pathway of targeted autophagy is induced by DNA damage in budding yeast. Proc Natl Acad Sci U S A 2017;114:E1158-67.
31. Liu EY, Xu N, O'Prey J, Lao LY, Joshi S, et al. Loss of autophagy causes a synthetic lethal deficiency in DNA repair. Proc Natl Acad Sci U S A 2015;112:773-8.
32. Knizhnik AV, Roos WP, Nikolova T, Quiros S, Tomaszowski KH, et al. Survival and death strategies in glioma cells: autophagy, senescence and apoptosis triggered by a single type of temozolomide-induced DNA damage. PloS one 2013;8:e55665.
33. Kanzawa T, Bedwell J, Kondo Y, Kondo S, Germano IM. Inhibition of DNA repair for sensitizing resistant glioma cells to temozolomide. J Neurosurg 2003;99:1047-52.
34. Filippi-Chiela EC, Bueno e Silva MM, Thome MP, Lenz G. Single-cell analysis challenges the connection between autophagy and senescence induced by DNA damage. Autophagy 2015;11:1099-113.
35. Chang KY, Hsu TI, Hsu CC, Tsai SY, Liu JJ, et al. Specificity protein 1-modulated superoxide dismutase 2 enhances temozolomide resistance in glioblastoma, which is independent of O(6)-methylguanine-DNA methyltransferase. Redox Biol 2017;13:655-64.
36. Chang KY, Huang CT, Hsu TI, Hsu CC, Liu JJ, et al. Stress stimuli induce cancer-stemness gene expression via Sp1 activation leading to therapeutic resistance in glioblastoma. Biochem Biophys Res Commun 2017;493:14-19.
37. Lin CJ, Lee CC, Shih YL, Lin CH, Wang SH, et al. Inhibition of mitochondria- and endoplasmic reticulum stress-mediated autophagy augments temozolomide-induced apoptosis in glioma cells. PLoS One 2012;7:e38706.
38. Quijano C, Trujillo M, Castro L, Trostchansky A. Interplay between oxidant species and energy metabolism. Redox Biol 2016;8:28-42.
39. Guerra F, Arbini AA, Moro L. Mitochondria and cancer chemoresistance. Biochimica et biophysica acta. Biochim Biophys Acta Bioenerg 2017;1858:686-99.
40. Wang L, Shang Z, Zhou Y, Hu X, Chen Y, et al. Autophagy mediates glucose starvation-induced glioblastoma cell quiescence and chemoresistance through coordinating cell metabolism, cell cycle, and survival. Cell Death Dis 2018;9:213.
41. Kriel J, Müller-Nedebock K, Maarman G, Mbizana S, Ojuka E, et al. Coordinated autophagy modulation overcomes glioblastoma chemoresistance through disruption of mitochondrial bioenergetics. Sci Rep 2018;8:10348.
42. Vidal SJ, Rodriguez-Bravo V, Galsky M, Cordon-Cardo C, Domingo-Domenech J. Targeting cancer stem cells to suppress acquired chemotherapy resistance. Oncogene 2014;33:4451-63.
43. Lathia JD, Mack SC, Mulkearns-Hubert EE, Valentim CL, Rich JN. Cancer stem cells in glioblastoma. Genes Dev 2015;29:1203-17.
44. Galavotti S, Bartesaghi S, Faccenda D, Shaked-Rabi M, Sanzone S, et al. The autophagy-associated factors DRAM1 and p62 regulate cell migration and invasion in glioblastoma stem cells. Oncogene 2013;32:699-712.
45. Colella B, Faienza F, Di Bartolomeo S. EMT Regulation by Autophagy: A New Perspective in Glioblastoma Biology. Cancers (Basel) 2019;11:E312.
46. Catalano M, D'Alessandro G, Lepore F, Corazzari M, Caldarola S, et al. Autophagy induction impairs migration and invasion by reversing EMT in glioblastoma cells. Mol Oncol 2015;9:1612-25.
47. Jiang C, Shen F, Du J, Fang X, Li X, et al. Upregulation of CASC2 sensitized glioma to temozolomide cytotoxicity through autophagy inhibition by sponging miR-193a-5p and regulating mTOR expression. Biomed Pharmacother 2018;97:844-50.
48. Filippi-Chiela EC, Villodre ES, Zamin LL, Lenz G. Autophagy interplay with apoptosis and cell cycle regulation in the growth inhibiting effect of resveratrol in glioma cells. PLoS One 2011;6:e20849.
49. Buccarelli M, Marconi M, Pacioni S, De Pascalis I, D'Alessandris QG, et al. Inhibition of autophagy increases susceptibility of glioblastoma stem cells to temozolomide by igniting ferroptosis. Cell Death Dis 2018;9:841.
50. Wang WJ, Long LM, Yang N, Zhang QQ, Ji WJ, et al. NVP-BEZ235, a novel dual PI3K/mTOR inhibitor, enhances the radiosensitivity of human glioma stem cells in vitro. Acta Pharmacol Sin 2013;34:681-90.
51. Singer E, Judkins J, Salomonis N, Matlaf L, Soteropoulos P, et al. Reactive oxygen species-mediated therapeutic response and resistance in glioblastoma. Cell Death Dis 2015;6:e1601.
52. Huang T, Kim CK, Alvarez AA, Pangeni RP, Wan X, et al. MST4 Phosphorylation of ATG4B Regulates Autophagic Activity, Tumorigenicity, and Radioresistance in Glioblastoma. Cancer Cell 2017;2:840-55.e848.
53. Paglin S, Hollister T, Delohery T, Hackett N, McMahill M, et al. A novel response of cancer cells to radiation involves autophagy and formation of acidic vesicles. Cancer Res 2001;61:439-44.
54. Ito H, Daido S, Kanzawa T, Kondo S, Kondo Y. Radiation-induced autophagy is associated with LC3 and its inhibition sensitizes malignant glioma cells. Int J Oncol 2005;26:1401-10.
55. Kegelman TP, Wu B, Das SK, Talukdar S, Beckta JM, et al. Inhibition of radiation-induced glioblastoma invasion by genetic and pharmacological targeting of MDA-9/Syntenin. Proc Natl Acad Sci U S A 2017;114:370-75.
56. Koukourakis MI, Mitrakas AG, Giatromanolaki A. Therapeutic interactions of autophagy with radiation and temozolomide in glioblastoma: evidence and issues to resolve. Brit J Cancer 2016;114:485-96.
57. Lomonaco SL, Finniss S, Xiang C, Decarvalho A, Umansky F, et al. The induction of autophagy by gamma-radiation contributes to the radioresistance of glioma stem cells. Int J Cancer 2009;125:717-22.
58. Fu J, Liu ZG, Liu XM, Chen FR, Shi HL, et al. Glioblastoma stem cells resistant to temozolomide-induced autophagy. Chin Med J (Engl) 2009;122:1255-9.
59. Ciechomska IA, Przanowski P, Jackl J, Wojtas B, Kaminska B. BIX01294, an inhibitor of histone methyltransferase, induces autophagy-dependent differentiation of glioma stem-like cells. Sci Rep 2016;6:38723.
60. Palumbo S, Comincini S. Autophagy and ionizing radiation in tumors: the "survive or not survive" dilemma. J Cell Physiol 2013;228:1-8.
61. Jo GH, Bogler O, Chwae YJ, Yoo H, Lee SH, et al. Radiation-induced autophagy contributes to cell death and induces apoptosis partly in malignant glioma cells. Cancer Res Treat 2015;47:221-41.
62. Mendiburu-Elicabe M, Gil-Ranedo J, Izquierdo M. Efficacy of rapamycin against glioblastoma cancer stem cells. Clin Transl Oncol 2014;16:495-502.
63. Cerniglia GJ, Karar J, Tyagi S, Christofidou-Solomidou M, Rengan R, et al. Inhibition of autophagy as a strategy to augment radiosensitization by the dual phosphatidylinositol 3-kinase/mammalian target of rapamycin inhibitor NVP-BEZ235. Mol Pharmacol 2012;82:1230-40.
64. Gil del Alcazar CR, Hardebeck MC, Mukherjee B, Tomimatsu N, Gao X, et al. Inhibition of DNA double-strand break repair by the dual PI3K/mTOR inhibitor NVP-BEZ235 as a strategy for radiosensitization of glioblastoma. Clinical cancer research 2014;20:1235-48.
65. Zhuang W, Li B, Long L, Chen L, Huang Q, et al. Induction of autophagy promotes differentiation of glioma-initiating cells and their radiosensitivity. Int J Cancer 2011;129:2720-31.
66. Laks DR, Oses-Prieto JA, Alvarado AG, Nakashima J, Chand S, et al. A molecular cascade modulates MAP1B and confers resistance to mTOR inhibition in human glioblastoma. Neuro Oncol 2018;20:764-75.
67. Rosenfeld MR, Ye X, Supko JG, Desideri S, Grossman SA, et al. A phase I/II trial of hydroxychloroquine in conjunction with radiation therapy and concurrent and adjuvant temozolomide in patients with newly diagnosed glioblastoma multiforme. Autophagy 2014;10:1359-68.
68. Stupp R, Taillibert S, Kanner AA, Kesari S, Steinberg DM, et al. Maintenance Therapy With Tumor-Treating Fields Plus Temozolomide vs Temozolomide Alone for Glioblastoma: A Randomized Clinical Trial. JAMA 2015;314:2535-43.
69. Kessler AF, Frömbling GE, Gross F, Hahn M, Dzokou W, et al. Effects of tumor treating fields (TTFields) on glioblastoma cells are augmented by mitotic checkpoint inhibition. Cell Death Discov 2018;4:12.
70. Shteingauz A, Porat Y, Voloshin T, Schneiderman RS, Munster M, et al. AMPK-dependent autophagy upregulation serves as a survival mechanism in response to Tumor Treating Fields (TTFields). Cell Death Dis 2018;9:1074.
71. Ullén A, Farnebo M, Thyrell L, Mahmoudi S, Kharaziha P, et al. Sorafenib induces apoptosis and autophagy in prostate cancer cells in vitro. Int J Oncol 2010;37:15-20.
72. Jo Y, Kim EH, Sai S, Kim JS, Cho JM, et al. Functional Biological Activity of Sorafenib as a Tumor-Treating Field Sensitizer for Glioblastoma Therapy. Int J Mol Sci 2018;19:E3684.
73. Karanam NK, Srinivasan K, Ding L, Sishc B, Saha D, et al. Tumor-treating fields elicit a conditional vulnerability to ionizing radiation via the downregulation of BRCA1 signaling and reduced DNA double-strand break repair capacity in non-small cell lung cancer cell lines. Cell Death Dis 2017;8:e2711.
74. Giladi M, Munster M, Schneiderman RS, Voloshin T, Porat Y, et al. Tumor treating fields (TTFields) delay DNA damage repair following radiation treatment of glioma cells. Radiat Oncol 2017;12:206.
75. Narasimha K, Liang-hao D, Brock S, Debabrata S, Michael S. CSIG-01, Tumor Treatment Fields downregulate specific transcription factors leading to reduced DNA repair capacity, increased replication stress, the inhibition of mitophagy and enhanced cell death. Neuro Oncol 2017;19:vi49-50.
76. Huang H, Song J, Liu Z, Pan L, Xu G. Autophagy activation promotes bevacizumab resistance in glioblastoma by suppressing Akt/mTOR signaling pathway. Oncol Lett 2018;15:1487-94.
77. Keunen O, Johansson M, Oudin A, Sanzey M, Rahim SA, et al. Anti-VEGF treatment reduces blood supply and increases tumor cell invasion in glioblastoma. Proc Natl Acad Sci U S A 2011;108:3749-54.
78. Abdul Rahim SA, Dirkse A, Oudin A, Schuster A, Bohler J, et al. Regulation of hypoxia-induced autophagy in glioblastoma involves ATG9A. Br J Cancer 2017;117:813-25.
79. Miranda-Gonçalves V, Cardoso-Carneiro D, Valbom I, Cury FP, Silva VA, et al. Metabolic alterations underlying Bevacizumab therapy in glioblastoma cells. Oncotarget 2017;8:103657-70.
80. Wu HB, Yang S, Weng HY, Chen Q, Zhao XL, et al. Autophagy-induced KDR/VEGFR-2 activation promotes the formation of vasculogenic mimicry by glioma stem cells. Autophagy 2017;13:1528-42.
81. Biasoli D, Kahn SA, Cornélio TA, Furtado M, Campanati L, et al. Retinoblastoma protein regulates the crosstalk between autophagy and apoptosis, and favors glioblastoma resistance to etoposide. Cell Death Dis 2013;4:e767.
82. Wang Z, Liang P, He X, Wu B, Liu Q, et al. Etoposide loaded layered double hydroxide nanoparticles reversing chemoresistance and eradicating human glioma stem cells in vitro and in vivo. Nanoscale 2018;10:13106-21.
83. Sheleg SV, Korotkevich EA, Zhavrid EA, Muravskaya GV, Smeyanovich AF, et al. Local chemotherapy with cisplatin-depot for glioblastoma multiforme. J Neurooncol 2002;60:53-9.
84. Ren JH, He WS, Nong L, Zhu QY, Hu K, et al. Acquired cisplatin resistance in human lung adenocarcinoma cells is associated with enhanced autophagy. Cancer Biother Radiopharm 2010;25:75-80.
85. Bao LJ, Jaramillo MC, Zhang ZB, Zheng YX, Yao M, et al. Nrf2 induces cisplatin resistance through activation of autophagy in ovarian carcinoma. Int J Clin Exp Pathol 2014;7:1502-13.
86. Yu L, Gu C, Zhong D, Shi L, Kong Y, et al. Induction of autophagy counteracts the anticancer effect of cisplatin in human esophageal cancer cells with acquired drug resistance. Cancer Lett 2014;355:34-45.
87. Liu X, Sun K, Wang H, Dai Y. Knockdown of retinoblastoma protein may sensitize glioma cells to cisplatin through inhibition of autophagy. Neurosci Lett 2016;620:137-42.
88. Xu Y, Yu H, Qin H, Kang J, Yu C, et al. Inhibition of autophagy enhances cisplatin cytotoxicity through endoplasmic reticulum stress in human cervical cancer cells. Cancer Lett 2012;314:232-43.
89. Ma B, Gao Z, Lou J, Zhang H, Yuan Z, et al. Long noncoding RNA MEG3 contributes to cisplatininduced apoptosis via inhibition of autophagy in human glioma cells. Mol Med Rep 2017;16:2946-52.
90. Ding L, Yuan C, Wei F, Wang G, Zhang J, et al. Cisplatin restores TRAIL apoptotic pathway in glioblastoma-derived stem cells through up-regulation of DR5 and down-regulation of c-FLIP. Cancer Invest 2011;29:511-20.
91. Lv S, Wang X, Zhang N, Sun M, Qi W, et al. Autophagy facilitates the development of resistance to the tumor necrosis factor superfamily member TRAIL in breast cancer. Int J Oncol 2015;46:1286-94.
92. Potapova O, Haghighi A, Bost F, Liu C, Birrer MJ, et al. The Jun kinase/stress-activated protein kinase pathway functions to regulate DNA repair and inhibition of the pathway sensitizes tumor cells to cisplatin. J Biol Chem 1997;272:14041-4.
93. Kuo CC, Liu TW, Chen LT, Shiah HS, Wu CM, et al. Combination of arsenic trioxide and BCNU synergistically triggers redox-mediated autophagic cell death in human solid tumors. Free Radic Biol Med 2011;51:2195-209.
94. Sukumari-Ramesh S, Prasad N, Alleyne CH, Vender JR, Dhandapani KM. Overexpression of Nrf2 attenuates Carmustine-induced cytotoxicity in U87MG human glioma cells. BMC Cancer 2015;15:118.
95. Yayon A, Cabantchik ZI, Ginsburg H. Susceptibility of human malaria parasites to chloroquine is pH dependent. Proc Natl Acad Sci U S A 1985;82:2784-8.
96. Geng Y, Kohli L, Klocke BJ, Roth KA. Chloroquine-induced autophagic vacuole accumulation and cell death in glioma cells is p53 independent. Neuro Oncol 2010;12:473-81.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Chien CH, Hsueh WT, Chuang JY, Chang KY. Role of autophagy in therapeutic resistance of glioblastoma. J Cancer Metastasis Treat 2019;5:66. http://dx.doi.org/10.20517/2394-4722.2019.016
AMA Style
Chien CH, Hsueh WT, Chuang JY, Chang KY. Role of autophagy in therapeutic resistance of glioblastoma. Journal of Cancer Metastasis and Treatment. 2019; 5: 66. http://dx.doi.org/10.20517/2394-4722.2019.016
Chicago/Turabian Style
Chien, Chia-Hung, Wei-Ting Hsueh, Jian-Ying Chuang, Kwang-Yu Chang. 2019. "Role of autophagy in therapeutic resistance of glioblastoma" Journal of Cancer Metastasis and Treatment. 5: 66. http://dx.doi.org/10.20517/2394-4722.2019.016
ACS Style
Chien, C.H.; Hsueh W.T.; Chuang J.Y.; Chang K.Y. Role of autophagy in therapeutic resistance of glioblastoma. J. Cancer. Metastasis. Treat. 2019, 5, 66. http://dx.doi.org/10.20517/2394-4722.2019.016
About This Article
Special Issue
Copyright

Data & Comments
Data


 Cite This Article 32 clicks
Cite This Article 32 clicks

 Like This Article 11
likes
Like This Article 11
likes






Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.