
Exploiting autophagy in multiple myeloma


Abstract
Multiple myeloma (MM) is a plasma cell cancer characterized by sustained endoplasmic reticulum (ER) stress and unfolded protein response activation in the setting of high rates of immunoglobulin synthesis. Consequently, MM cells rely heavily on protein quality control pathways for survival as evidenced by the clinical efficacy of proteasome inhibitors (PI). Autophagy is an intracellular self-digestion mechanism that plays a role in the ER protein quality control process. Unsurprisingly then, basal levels of autophagy were recently found to confer a survival and drug-resistance benefit to MM cells. However, excessive induction of autophagy in MM cells leads to autophagic cell death, highlighting the double-edged nature of autophagy modulation in MM. This review provides an overview of the role that autophagy plays in MM pathogenesis, survival, and drug-resistance. We highlight the potential utility of therapeutically targeting autophagy in MM, focusing on preclinical data of autophagic modulators in combination with alkylators, anthracyclines, PI, and immunomodulatory drugs.
Keywords
Multiple myeloma
Multiple myeloma (MM) is a neoplastic disorder characterized by the dysregulated proliferation of a plasma cell clone that typically produces a monoclonal immunoglobulin, ultimately resulting in end-organ damage[1-3]. Clinical suspicion for active MM is often based on the presence of one or more laboratory/imaging abnormalities, termed the CRAB criteria [hypercalcaemia (C), renal impairment (R), anaemia (A), and osteolytic bone lesions (B)], particularly if occurring in a patient with a precursor plasma cell disorder such as monoclonal gammopathy of undetermined significance (MGUS) or smoldering MM (SMM)[4]. A diagnosis of active MM requires the presence of greater than 10% clonal bone marrow (BM) plasma cells in association with either one or more of the CRAB features or a biomarker of malignancy (BM plasmacytosis equal or greater than 60%, ratio of involved vs. uninvolved light chain equal or greater than 100 or the presence of more than 1 focal lesion on magnetic resonance imaging)[5,6]. MM is generally preceded by the asymptomatic precursor conditions MGUS and/or SMM[6]. MGUS is characterized by low levels of monoclonal protein (< 3 g/dL) and less than 10% clonal plasma cells in the BM while SMM is characterized by the presence of > 3 g/dL of monoclonal protein with BM plasmacytosis exceeding 10% but less than 60%[6]. Evidence of end organ damage related to the plasma cell disorder is an exclusion criteria for MGUS/SMM diagnosis. Patients with MGUS and SMM progress to active MM at a rate of 1% and 10% per year, respectively[7].
While single driver mutations have not been identified in MM, marked genomic instability is a hallmark of the disease and contributes to elevated proteotoxic stress[8]. The high frequency of genomic mutations may confer a survival advantage by enabling MM cells to quickly adapt to stresses in the environment. However, this comes at a cost. This deregulation of gene expression results in the accumulation of toxic misfolded proteins that exerts additional stress on MM cells[9-13]. Furthermore, MM are highly secretory cells, characterized by staggering rate of synthesis of clonal immunoglobulins which further contributes to baseline ER stress. Therefore, protein quality control pathways are essential for MM survival[8].
Autophagy
Autophagy is a tightly regulated self-digestion mechanism that promotes the lysosomal degradation of organelles, intracellular pathogens, and misfolded proteins. It is a key cellular mechanism to maintain homeostasis and guarantee energy supply as products of autophagic digestion can be re-utilized in anabolic processes[14-16]. Therefore, nutrient and energy deprivation, ER stress, and hypoxia can all induce autophagy as a means to enable cell survival[17]. In mammalian cells, there are three main types of autophagy, namely macroautophagy, microautophagy, and chaperone-mediated autophagy[15].
Macroautophagy
Macroautophagy is a type of autophagy that delivers cellular contents to the lysosome via the formation of double-membrane structures called autophagosomes which then fuse with lysosomes to form autolysosomes[18,19]. Macroautophagy can be subdivided into non-selective (bulk) and selective autophagy[16]. During non-selective autophagy, bulk cytoplasm is randomly engulfed by a phagophore [Figure 1]. Notably, the mammalian target of rapamycin (mTOR) pathway is a key inhibitor of autophagy[20]. Subsequently, the phagophore matures into an autophagosome and this process is mediated by autophagy-related protein 7 (ATG7), ATG8 (LC3), and ATG12[21,22]. ATG7 functions as an E1-like enzyme by binding and activating ATG12 and ATG8 to facilitate the transfer of ATG12 to ATG5 via the E2 enzyme ATG10[23-27]. The resultant ATG12-ATG5 conjugate forms a large multimeric complex together with ATG16 (ATG12-ATG5-ATG16) which acts as an E3 ligase to facilitate phosphatidylethanolamine (PE) and LC3 conjugation and conversion of LC3-I to LC3-II. LC3-II stably associates with the autophagosome membrane and regulates autophagic membrane expansion, recognition of autophagic cargo, and autolysosome formation[21,22]. Finally, autophagosome and lysosome fusion occurs and the autophagic cargo is degraded by lysosomal hydrolases[21,22]. Selective macroautophagy, on the other hand, specifically targets damaged or redundant organelles such as mitochondria (mitophagy), peroxisomes (pexophagy), ribosomes (ribophagy), aggresomes (aggrephagy), etc.[28]. Specifically, mitophagy is the selective degradation of mitochondria by macroautophagy in a PTEN-induced kinase 1 (PINK1)- and Parkin-dependent fashion[29,30]. Type 1 mitophagy sequesters and removes mitochondria in response to nutrient deprivation, whereas type 2 mitophagy removes damaged mitochondria[31]. Type 3 mitophagy (micromitophagy), on the other hand, eradicates damaged mitochondrial components through the formation of mitochondria-derived vesicles that are subsequently degraded by lysosomes[31].
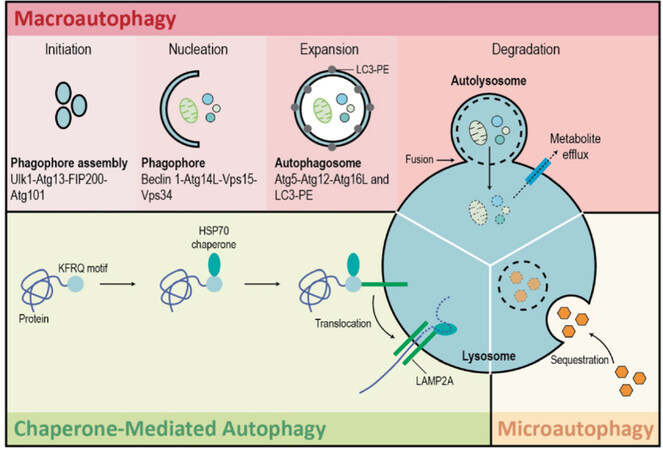
Figure 1. Autophagy. There are three types of autophagy: macroautophagy, microautophagy, and chaperone-mediated autophagy. Macroautophagy is a type of autophagy that delivers cellular contents to the lysosome via the formation of double-membrane structures called autophagosomes which then fuse with lysosomes to form autolysosomes. Macroautophagy takes place in five main steps. Initiation of autophagy occurs in response to metabolic or therapeutic stress and is mediated by ULK1, ATG13, FIP200, and ATG101. During the nucleation step regulated by BECLIN-1, ATG14L, VPS15, and VPS34, the formation of the phagophore occurs. Expansion results in the sequestration of cytosolic contents within the autophagosome and is facilitated by ATG5, ATG12, ATG16L, and LC3-PE. Degradation is the breakdown of autophagosomal contents upon formation of the autolysosome (fusion of autophagosome and lysosome). Microautophagy is a largely non-selective process that facilitates the direct uptake and breakdown of cytosolic cargo by lysosomes. Chaperone-mediated autophagy refers to the chaperone-dependent targeting of specific cytosolic proteins to lysosomes for proteolysis. HSC70 binds to the consensus motif of specific proteins to target them to the lysosome-associated membrane protein type 2A (LAMP-2A) receptor on the lysosomal membrane. Once internalized by the lysosome, these cytosolic proteins are degraded
Microautophagy
In eukaryotic cells, microautophagy is a largely non-selective process that facilitates the direct uptake and breakdown of cytosolic cargo by lysosomes. Specifically, cytosolic material is sequestered by direct invagination of the vacuolar/lysosomal membrane, forming autophagic tubes that pinch off into the lysosomal lumen[32-34].
Chaperone-mediated autophagy
Chaperone-mediated autophagy (CMA) refers to the chaperone-dependent targeting of specific cytosolic proteins to lysosomes for proteolysis[35-37]. This is a mechanistically distinct process that occurs only in mammalian cells. Unlike the other types of autophagy, CMA does not require the formation of vesicles[37]. Instead, HSC70 binds to the consensus motif of specific proteins to target them to the lysosome-associated membrane protein type 2A receptor on the lysosomal membrane[35-37]. Once bound, the targeted proteins start to unfold as they are internalized into the lysosomal lumen and then degraded[35-37].
Immune functions of autophagy
Autophagy as an immune effector
Autophagy plays a key role in eukaryotic cells as a stress response mechanism activated to recycle intracellular organelles in the face of nutrient deprivation[38]. However, advances in our understanding of autophagy reveal also an intricate reciprocal relationship between autophagy and immunity[38]. Specifically, autophagy acts as an immune effector by: (1) facilitating the direct elimination of microbes through xenophagy and LC3-associated phagocytosis[39]; (2) modulating the inflammatory response by amplifying PAMP-TLR signaling whilst inhibiting type I IFN signaling and inflammasome activation[40]; (3) enhancing MHC class II-mediated antigen presentation and cross-presentation[39,40]; and (4) contributing to secretion of pro-inflammatory cytokines such as IL-6, which could be relevant to MM pathogenesis[39].
Autophagy in the maintenance of hematopoiesis
Apart from regulating immune effector functions, autophagy is a key adaptive mechanism critical to the maintenance of hematopoietic hemostasis [Figure 2][41]. Hematopoietic stem cells (HSCs) thread a tight, but dynamic and demand-adapted, balance between quiescence and proliferation throughout the entire life of the organism[41]. Studies have shown that autophagy protects HSCs from exhaustion secondary to metabolic stress[41-43]. Firstly, basal autophagy removes activated mitochondria, regulates the metabolism, and maintains the self-renewal and regenerative capabilities of HSCs[44]. Research shows that conditional deletion of ATG12 in transplanted murine HSCs severely impairs their ability for BM engraftment and self-renewal[45]. Secondly, when subjected to ex vivo cytokine withdrawal or in vivo nutrient deprivation, HSCs robustly upregulate autophagy to circumvent an energy crisis to ensure HSC longevity[43].
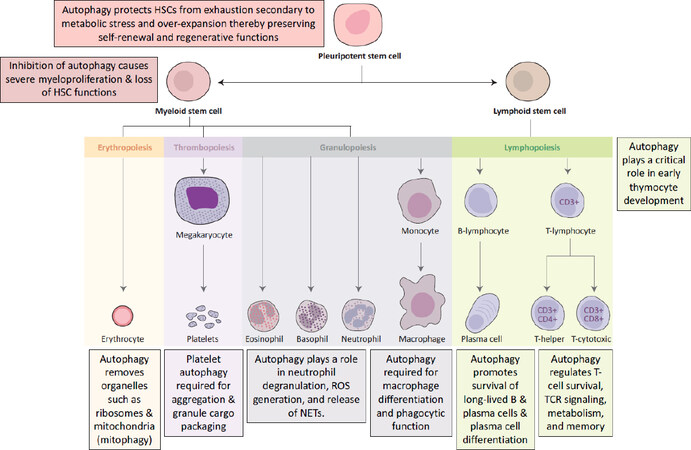
Figure 2. Examples of key functions that autophagy plays in the maintenance and differentiation of hematopoietic stem cells. Image adapted from First Aid for the USMLE Step 1[63]
Deletion of ATG7, a critical component of autophagosome formation, in murine HSCs results in accumulation of dysfunctional mitochondria, upregulation of oxidative stress and DNA damage, and ultimately cell death[46]. Interestingly, aged HSCs were found to maintain a low metabolic state by upregulating autophagy in order to sustain robust long-term self-renewal potential comparable to young HSCs[44]. Downstream of HSCs, autophagy also plays a key role in the development, differentiation, and function of erythrocytes, platelets, granulocytes, macrophages, and T cells as summarized in Figure 2[41].
Since malignant transformation of HSCs or early progenitors results in leukemia, homeostatic mechanisms; such as autophagy, that protect HSCs from metabolic, oxidative, and genotoxic stress; are crucial to prevent hematopoietic malignancies[47]. Indeed, ATG7 deletion in myeloid cells results in dysregulated and invasive myeloproliferation resembling acute myeloid leukemia[42].
Autophagy in plasma cell ontogeny
Autophagy plays a key role in plasma cell (1) differentiation; (2) survival; and (3) protein quality control.
Autophagy in plasma cell differentiation and survival
B lymphocyte to plasma cell differentiation is controlled by a complex genetic reprogramming system leading to downregulation of genes involved in the maintenance of B-cell identity [e.g., paired box protein 5 (PAX5), transcription regulator protein BACH2 (BACH2), B-cell lymphoma protein 6 (BCL6)] and upregulation of genes involved in terminal differentiation of Ig-secretory plasma cells [e.g., B-lymphocyte-induced maturation protein 1 (BLIMP-1), interferon regulatory factor 4 (IRF4), and X-box binding protein 1 (XBP1)][48]. Specifically, BLIMP-1 acts as a molecular switch to repress PAX5 and BCL6, and induces XBP1 to promote antibody production and plasma cell differentiation[49,50]. Interestingly, while BLIMP-1 and IRF4 are essential for plasma cell differentiation, only IRF4 is essential for plasma cell survival[51]. Consistent with this, BLIMP-1 deficient plasma cells remained viable and retained their transcriptional identity but lose the ability to secrete Ig[51].
Beyond epigenetics, autophagy also plays an essential role in plasma cell differentiation and survival [Figure 3]. Studies have found increased expression of autophagic genes in differentiating plasma cells. Conditional deletion of ATG5 in murine B cells results in reduced IgM and IgG responses in the setting of both T-cell dependent and independent immunizations; further suggesting that autophagy is required for B lymphocyte to plasma cell differentiation[52]. Notably, ATG5 was also essential for the homing and/or survival of long-lived plasma cells in the BM[52]. Consistent with this, long-lived plasma cells were found to highly express autophagic genes and display high basal levels of autophagy[53].
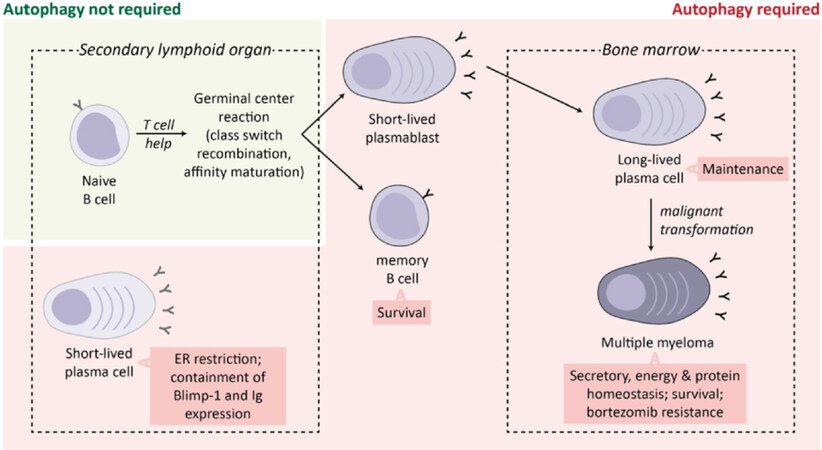
Figure 3. Role of autophagy in plasma cell ontogeny and malignancy. Differentiating plasma cells and MM cells induce autophagy to restrict the ER and downregulate BLIMP-1 expression to decrease immunoglobulin synthesis and deal with excessive proteotoxic stress. Autophagy is also essential for the survival and maintenance of memory B cells and BM long-lived plasma cells. In MM, induction of autophagy has been linked with bortezomib resistance. Image adapted from Milan et al.[60]
Autophagy as a mechanism of protein quality control
Plasma cells are professional antibody secreting cells (ASCs) uniquely optimized towards large-scale immunoglobulin synthesis, folding, assembly, and secretion[54]. Not unique to plasma cells, however, is the fact that the protein synthesis process is intrinsically error prone. In fact, up to 30% of newly-synthesized proteins are defective and need to be degraded[47,54]. Thus, an intricate balance between protein synthesis, folding, and clearance must be maintained to prevent the accumulation of potentially toxic misfolded proteins[47,54]. This is especially crucial for ASCs that cope with increased Ig synthesis by upregulating folding capacity through the induction of unfolded protein response (UPR)-driven ER expansion[11]. However, when Ig synthesis exceeds folding capacity, the integrity of the proteome is preserved through an interconnected network of protein quality control pathways which include the proteasome, autophagy, aggresome, and UPR pathways[47]. These pathways are so important to plasma cells that the amount of newly-synthesized proteins degraded by the proteasome is 15-folds higher in plasma cells compared to resting B-cells[55]. Perhaps not surprisingly then, that MM, a cancer of plasma cells, exhibits the same reliance on the protein quality control as evidenced by the clinical efficacy of proteasome inhibitors (PI). Indeed, sensitivity of MM to PI is determined by a combination of Ig secretory load, protein degradation capacity, and commitment to plasma cell maturation; which, taken together, suggests that being an ASC confers this unique susceptibility to MM[56-59].
While autophagy does not directly dispose of misfolded immunoglobulins, plasma cells deficient in autophagy had greater energy imbalance, enhanced immunoglobulin synthesis, reduced intracellular ATP, and elevated ER stress[52]. Consistent with this, autophagy-deficient plasma cells displayed higher expression of BLIMP-1 and XBP1 with corresponding increase in ER size [Figure 3][52]. These findings suggest that autophagy not only facilitates the acquisition of an Ig-secretory phenotype in the in pre-plasma cells, but also serves to limit the overproduction of Ig and maintain cellular ATP levels to ensure survival in differentiated plasma cells [Figure 3][60]. Autophagy also plays an important role in the removal of misfolded protein aggregates which, due to steric hindrance, are unable to be efficiently degraded by the proteasome[61]. Failure to dispose of misfolded protein aggregates results in proteotoxic stress and induction of terminal ER stress[62].
Autophagy in MM
Autophagy is necessary for the differentiation and maintenance of antigen-specific long-lived plasma cells, the physiologic counterpart to MM cells. As MM cells retain many characteristics of the original plasma cell clone such as Ig secretion and an enlarged cytoplasm and ER, it is reasonable to hypothesize that autophagy might also play a role in MM proteostasis and survival.
Indeed, MM cells exhibit higher levels of basal autophagy compared to other tumors, including lymphoma derived from earlier B-cell progenitors, and autophagy is necessary for the survival of MM cells[64]. Disruption of autophagy through BECLIN-1 knockdown or pharmacologic inhibition (with 3-methyladenine and/or chloroquine) causes MM cell apoptosis[65,66]. Basal autophagy is hypothesized to alleviate proteotoxic stress and promote MM survival by (1) limiting the secretion of immunoglobulins and (2) providing an alternative proteolytic pathway for the clearance of ubiquitinated proteins through a p62-dependent mechanism[60]. Despite this, concomitant inhibition of the proteasome and autophagy in pre-clinical studies have reported inconsistent results, ranging from synergism to antagonism[60]. This may partly be explained by the observation that while basal autophagy is protective, persistent and uncontrolled autophagy results in autophagic cell death[67,68]. Autophagic cell death in MM can be induced by caspase-10 or caspase-8 inhibition[67,68]. Specifically, caspase-10 cleaves and inhibits BCL2 associated transcription factor 1, a BECLIN-1 activator, to temper the autophagic response and avoid cell death[67]. MM cells therefore need to tightly regulate autophagy to maintain viability as dysregulation either way leads to deleterious effects in MM.
Mitophagy has been reported to play conflicting roles in carcinogenesis, survival, and drug-resistance. Some studies report that mitophagy protects untransformed cells from excessive reactive oxidative species damage and genetic instability, and that suppression of mitophagy favors carcinogenesis[69,70]. Conversely, other studies suggest that mitophagy promotes cancer cell survival and drug resistance by protecting cells from apoptosis[71,72]. In MM, while the suppression of mitophagy is associated with bortezomib resistance; doxorubicin, a widely used anti-MM chemotherapy, is a classical inhibitor of mitophagy[73]. This dichotomy highlights the need for a better understanding of the role mitophagy plays in MM.
DNA damage and autophagy
Apart from ongoing proteotoxic stress, other hallmarks of MM are genetic instability and abnormalities (e.g., aneuploidy, translocations), and oxidative stress which lead to replicative stress and constitutive DNA damage[74,75]. Recent studies have shown that DNA damage can activate autophagy through a number of interconnected pathways[76]. Examples of DNA damage repair (DDR) pathway proteins that regulate autophagy are ataxia telangiectasia-mutated (ATM), poly (ADP-ribose) polymerase 1 (PARP1), c-Jun N-terminal kinase (JNK), and p53[76]. Specifically, autophagy can be induced by ATM-mediated phosphorylation of AMP-activated protein kinase (AMPK)[77,78]. AMPK in turn activates both tuberous sclerosis complex 2 (TSC2), to remove the inhibitory effect of MTOR complex 1 (mTORC1) on autophagy, and unc-51 like autophagy activating kinase 1 (ULK1) to promote autophagosome formation[77-79]. ATM also phosphorylates and activates CHE-1 which in turn upregulates the transcription of two mTOR inhibitor genes [regulated in development and DNA damage responses 1 (REDD1) and DEP domain-containing mTOR-interacting protein (DEPTOR)][80]. Nuclear factor-kappa B, a well-known pro-myeloma transcription factor, can be activated by ATM and as a result, upregulate BECLIN-1[81]. Induction of PARP1 promotes autophagy via AMPK activation on the background of ATP depletion and elevated AMP levels[82]. DNA damage-induced JNK phosphorylates BCL-2 resulting in BCL-2 dissociation, relief of BECLIN-1 inhibition, and induction of autophagy[83]. Nuclear p53 activates autophagy through a number of distinct signaling pathways. Firstly, p53 upregulates phosphatase and tensin homolog (PTEN) which leads to PI3K-Akt-mTORC1 inhibition and autophagy induction[84,85]. Secondly, p53 influences AMPK activity (1) directly through transcriptional upregulation, and (2) indirectly by activating Sestrin1 and Sestrin2 which in turn activate AMPK[85,86]. Thirdly, death-associated protein kinase (DAPK) is transcriptionally upregulated by p53 and triggers autophagy by phosphorylating BECLIN-1 to facilitate its dissociation from BCL-2 and BCL-XL[87-89]. DAPK also phosphorylates protein kinase D which activates the VPS34 class III PI3K complex leading to induction of autophagy[87-89]. Lastly, p53 also upregulates damage-regulated autophagy modulator, a lysosomal protein involved in the degradation step of autophagy[90]. However, unlike nuclear p53, cytoplasmic p53 can also activate mTOR to inhibit autophagy[91]. Functionally, autophagy is essential for homologous recombination and nucleotide excision repair and cells deficient in autophagy rely chiefly on the error-prone non-homologous end joining repair pathway, which may explain the genomic instability observed in autophagy-deficient cells[92-98].
Exploiting autophagy in MM
Preclinical studies of autophagy modulators in MM
HDAC6 inhibitors
Histone deacetylases (HDACs) catalyze the removal of acetyl groups on lysine residues in target proteins[99]. Furthermore, HDACs also deacetylate non-histone proteins, thereby providing an additional layer of control over protein function, stability, and protein-protein interaction[99]. HDAC6 is a class IIb HDAC that is mainly localized to the cytoplasm, unlike class I and IIa HDACs which shuttle between the cytoplasm and nucleus, suggesting that HDAC6 functions mainly to regulate non-histone proteins[99]. Indeed, HDAC6 possess intrinsic ubiquitin-binding activity and co-localizes with the microtubule network to transport misfolded polyubiquitinated proteins to aggresomes/autophagosomes for subsequent lysosomal degradation[99-101]. HDAC6 therefore promotes ubiquitin-dependent or ubiquitin-independent aggresome formation and while HDAC6 is not required for the initiation of autophagy per se, it is necessary for the targeted delivery of the autophagy machinery to aggresomes[62,100-105]. Importantly, HDAC6 also promotes the formation of an F-actin network essential for autophagosome-lysosome fusion[105]. The development of HDAC6 inhibitors therefore presents an opportunity to exploit autophagy to destabilize protein homeostasis in MM.
Preclinical studies have shown that the HDAC6-selective inhibitors WT161 and Tubacin trigger the accumulation of acetylated tubulin of and inhibits MM cell growth in vitro[106,107]. Additionally, combinatory treatment using WT161 or Tubacin with the PI bortezomib not only induces synergistic cytotoxicity but was also able to overcome bortezomib resistance[106,107]. Mechanistically, the addition of WT161 to bortezomib resulted in further accumulation of polyubiquitinated proteins which led to a further increase in ER stress signaling and the UPR, evidenced by the upregulation of ATF4 and the pro-apoptotic protein CHOP[106]. Interestingly, the ER stress sensor proteins inositol-requiring enzyme 1 (IRE1α) and PRKR-like endoplasmic reticulum kinase (PERK) were found to be downregulated by WT161, suggesting that HDAC6 inhibition also suppresses the UPR, preventing it from functioning as an ER stress mitigator[106].
Another HDAC6-selective inhibitor, ACY-1215, demonstrated synergistic cytotoxicity when used in combination with carfilzomib, an irreversible second generation PI[108]. The addition of ACY-1215 resulted in increased LC3-II consistent with the disruption of autophagic flux secondary to HDAC6 inhibition[108]. Mechanistically, the addition of ACY-1215 to carfilzomib inhibited both aggresome formation and autophagosome-aggresome association[108]. Consistently, a novel HDAC6 inhibitor MPT0G413 was shown to both disrupt bortezomib-induced aggresome formation and exhibit synergism with bortezomib[109]. The combination of MPT0G413 and bortezomib could also overcome cell adhesion-mediated drug resistance in the MM-BMSC co-culture setting[109]. MPT0G413 was further found to decrease MM-BMSC adhesion and downregulate the pro-myeloma cytokines VEGF and IL-6 in the context of the MM BM microenvironment[109].
Autophagy inhibitors + DNA-damaging chemotherapy
DNA damaging agents such as melphalan are highly cytotoxic to MM cells and are a mainstay of treatment in MM, particularly as a conditioning regimen ahead of autologous hematopoietic stem-cell transplantation[110]. Consistent with the aforementioned link between DDR and autophagy, a recent study found that melphalan and doxorubicin induce cytoprotective autophagy as a pro-survival mechanism in MM cells through the upregulation of BECLIN-1-dependent autophagosomes[111]. knockdown of autophagy genes (BECLIN-1 and ATG5) and pharmacologic inhibition of autophagy (via hydroxychloroquine and 3-methyladenine) significantly enhanced the cytotoxicity of melphalan and doxorubicin in vitro and in vivo[111].
High mobility group protein B1 (HMGB1) is a critical regulator of autophagy that is often upregulated in MM[112,113]. Specifically, HMGB1 binds competitively to BECLIN-1 causing BCL-2 displacement and autophagy induction[112]. A study found that HMGB1 overexpression in MM was associated with mTOR inhibition, autophagy induction, and reduced sensitivity to dexamethasone[113]. Conversely, knockdown of HMGB1 increased apoptosis in MM cells exposed to dexamethasone[113].
HMGB1 has also been recognized a marker of induction of immunogenic cell death (ICD). ICD is triggered by exposure to certain cytotoxic agents (e.g., anthracyclines, oxaliplatin, bortezomib)[114,115]. ICD involves the release of soluble mediators along with alterations in the cancer cell surface composition in a way that converts dying cancer cells into therapeutic vaccines that are phagocytosed by antigen presenting cells (APCs) for the cross-priming of CD8+ T-cells to stimulate anti-tumor specific T-cell immune responses[114,115].
HMGB1 is a nuclear nonhistone chromatin-binding protein secreted by dying tumor cells exposed to cytotoxic agents that binds to toll-like receptor 4 on APCs to increase the rate of antigen processing and presentation by APCs to T cells[116]. Interestingly, studies have shown that autophagy in tumor cells regulates HMGB1 secretion and that inhibition of autophagy leads to intracellular sequestration of HMGB1 and the induction of caspase-mediated cell death[117]. Macroautophagy therefore enhances antigen cross-priming after ICD which results in the following conundrum. From a cytotoxicity point of view, it makes sense to combine DNA-damaging agents with autophagy inhibitors. However, from an ICD perspective, the addition of autophagy inhibitors to DNA-damaging agents may be counterproductive to antigen cross-priming.
Autophagy modulators + PI
Bortezomib, a first-in-class PI, was approved for use in MM by the FDA in 2003 and has become a mainstay of therapy at every disease stage ever since. Despite significant clinical efficacy in most naïve patients, most of the patients ultimately become refractory to bortezomib therapy[118-120]. This has prompted the development of second-generation PIs (e.g., carfilzomib and ixazomib) currently in clinical use for relapsed or refractory MM; and next-generation PIs (e.g., marizomib and oprozomib) currently in advanced clinical trials[121]. Based on evidence that susceptibility to PI depends on the ability of MM cells to remove misfolded proteins through protein quality control pathways, novel therapeutic approaches targeting alternative pathways in protein homeostasis are also actively being studied[122].
Research has shown that crosstalk exists between the proteasome, UPR, and autophagy[65,123,124]. Specifically, whenever the proteasome is overwhelmed and/or inhibited, polyubiquitinated and misfolded proteins co-aggregate in the cytosol to form aggresomes which are then degraded through macroautophagy[101,125,126]. Mechanistically, proteasome inhibition induces ER stress and PERK-eIF2α and IRE1-JNK activation, leading to the induction of autophagy[127,128]. Other mechanisms of bortezomib resistance, in the context of autophagy, include the upregulation of PROFILIN-1 which enhances autophagy through BECLIN-1 interaction and increased expression of CIC5, a chloride channel that enhances bortezomib-induced autophagy via AKT-mTOR inhibition[129]. PIs also rapidly induce the expression of SQSMT1/p62 to upregulate p62-dependent autophagy to compensate for proteasome insufficiency[64].
Along the same line, preclinical studies have reported synergistic cytotoxicity with dual blockade of the proteasome and autophagy in MM. MG132, a PI, induced cytoprotective autophagy that can be inhibited, by 3-methyladenine, to enhance apoptotic cell death[128]. Notably, 3-methyladenine resulted in the accumulation of polyubiquitinated proteins and exacerbation of ER stress in cells treated with MG132[128]. Autophagy inhibitors that have demonstrated synergy and enhanced MM cytotoxicity with bortezomib in vitro include Elaiphyllin (macrolide antibiotic) and Metformin[130,131]. Elaiphyllin, in particular, had significant cytotoxic activity even when used alone in mutant p53 MM[130]. Metformin, on the other hand, inhibits GRP78 which is crucial mediator of bortezomib-induced protective autophagy[131].
However, other conflicting studies have also shown that Metformin inhibits MM proliferation by inducing autophagy (and cell cycle arrest) through AMPK activation and dual repression of mTORC1 and mTORC2[132,133]. In line with this, it has been reported that when used in combination with bortezomib, the autophagy inhibitors 3-methyladenine or chloroquine can also have an antagonistic effect[65]. One potential explanation could be that bortezomib induces apoptosis partially through autophagic cell death. Consistent with a role for autophagic cell death in MM cells treated with bortezomib, a novel SCF (Skp2) inhibitor CpdA stabilizes p27 to induce caspase-independent autophagic cell death in MM cells resistant to bortezomib; and also synergized with bortezomib[134]. Another compound, betulinic acid (BetA), activates protein phosphatase 2A (PP2A) to trigger DAPK-dependent autophagic cell death in MM cells with high BCL-2 expression[135].
Autophagy inhibitors have also shown potency in bortezomib and carfilzomib-resistant MM cells[136]. The non-selective HDAC inhibitor SBHA upregulates the BH3-only protein BIM, which in turn sequesters BECLIN-1 to inhibit cytoprotective autophagy, thereby overcoming acquired bortezomib resistance[136]. Chloroquine potentiated carfilzomib cytotoxicity and was able to overcome carfilzomib resistance in vitro[137]. Lastly, pharmacologic inhibition of thioredoxin with PX12 upregulates mitophagy to re-sensitize bortezomib-resistant cells to bortezomib[73]. Combination treatment of MM cells with PX12 and bortezomib led to synergistic toxicity and inhibition of ERK1/2 and mTOR signaling, suggesting the involvement of ERK1/2 and mTOR in mitophagy suppression[73].
PI3K-AKT-mTOR inhibitors
While activating mutations in PI3K and AKT have not been reported in MM, the PI3K-AKT-mTOR pathway is commonly activated in MM through juxtacrine and paracrine signaling within the MM tumor microenvironment[138,139]. Interactions between MM and the stromal and endothelial compartments result in the secretion of IL-6, VEGF, and IGF-1, which in turn activate pro-survival and proliferative pathways such as PI3K-AKT-mTOR, JAK/STAT3, NFkB, and MEK/ERK[139]. In line with this, efforts to inhibit PI3K-AKT-mTOR signaling have led to the study of mTORC1 inhibitors (e.g., rapamycin) in MM. However, results from preclinical studies were disappointing as rapamycin and the other rapalogs demonstrated a cytostatic, but not cytotoxic, response in MM[140]. This lack of potency could be due, in part, to the activation of cytoprotective autophagy. Nonetheless, mTORC1 inhibitors have shown synergistic activity in other cancers when used in combination with clinically approved anti-MM agents such as dexamethasone, lenalidomide, and panobinostat, and pre-clinical agents such as sorafenib (tyrosine kinase inhibitor), 17-AAG (HSP90 inhibitor), NVP-AEW541 (IGF1R inhibitor), and MK2206 (AKT inhibitor)[141-148].
Upstream of mTOR, AKT inhibitors have also been studied in MM. In preclinical studies, perifosine, an AKT inhibitor, was cytotoxic to MM cells as a single-agent and also synergized with bortezomib, dexamethasone, doxorubicin, melphalan, and U0126 (MEK1/2 inhibitor)[149]. Another AKT inhibitor TAS-117 enhanced ER stress and MM apoptosis when added to bortezomib or carfilzomib[150]. These studies once again highlight the duality of autophagy in MM, suggesting that excessive induction of autophagy, rather than protecting cells from excessive ER stress, pushes cells towards autophagic cell death.
The next class of PI3K-AKT-mTOR pathway inhibitors are the dual PI3K-mTOR inhibitors. One such inhibitor, BEZ235, demonstrated good preclinical single-agent activity in MM and also synergized with bortezomib, doxorubicin, and melphalan[151,152]. Another study reported a pro-survival function of autophagy in MM cells treated with PI-103, a competitive dual PI3K and mTOR inhibitor, which inhibits the proteasome, induces UPR, and upregulates autophagy[153]. Importantly, Bafilomycin-A, an autophagy inhibitor, enhanced apoptosis in cells treated with PI-103[153].
Heat-shock protein inhibitors
Heat shock proteins are molecular chaperones that play indispensable roles in protein folding/unfolding, multiprotein complex assembly, and protein sorting[154]. As a function of protein sorting, HSP70 and HSP90 also participate in chaperone-mediated autophagy[155-157]. Heat-shock proteins (HSPs) therefore help alleviate proteotoxic stress to prevent apoptosis in MM[158]. Consistent with this, HSP70 and/or HSP90 inhibition induces UPR and apoptosis in MM[159-161]. In preclinical studies, combination of HSP90 inhibitors KW-2478, Retaspimycin, and 17-AAG with bortezomib demonstrated synergistic cytotoxicity[162-164]. Another HSP90 inhibitor, NVP-HSP990, displayed potent, in vitro anti-myeloma activity and synergism with melphalan, HDAC inhibitors, and PI3K/mTOR inhibitors[165,166]. Other HSP90 inhibitors such as PU-H71, SNX5422, and NVP-AUY922 have also shown promising pre-clinical results in MM[158,167-169]. Apart from HSP90, HSP70 has recently emerged as a promising therapeutic target in MM and a number of HSP70 inhibitors (e.g., PET-16, Ver-155008, MAL3-101) have shown good pre-clinical anti-myeloma activity[160,161,170,171].
Interestingly, treatment of MM cells with HSP90 inhibitors (e.g., 17-AAG, NVP-AUY922), bortezomib, or dexamethasone results in compensatory upregulation of HSP70 which confers a degree of drug-resistance and protects MM cells from apoptosis[172,173]. HSP70 is a molecular chaperone of HSP90. Consequently, inhibition of HSP70 leads to the downregulation of HSP90 while inhibition of HSP90 results in upregulation of HSP70[160,171,174]. It has been shown however, that simultaneous inhibition of both HSP70 and HSP90 leads to greater MM cytotoxicity compared to inhibiting HSP90 alone[160,171,174]. To this end, there has been increasing interest in developing inhibitors against heat shock factor 1 (HSF), the “master regulator” of the heat shock response that controls the expression of both HSP90 and HSP70[158]. In preclinical studies, several novel HSF1 inhibitors (e.g., CCT251236, KRIBB11) were found to induce MM cell death that was associated with the induction of the UPR[175].
Autophagy modulators currently in clinical trials
Several clinical trials have highlighted the potential role of autophagy modulators in the treatment of MM. For the purpose of consistency while discussing these trials, overall response rate (ORR) is defined as a partial response (PR) or better, and clinical benefit rate (CBR) is defined as stable disease (SD) or better.
Hydroxychloroquine/Chloroquine
Hydroxychloroquine (HCQ)/Chloroquine (CQ) have been clinically studied for their potential role in inhibiting autophagy. Although the exact mechanism of HCQ/CQ has not been elucidated, it is thought to function by alkalinizing intracellular compartments which disrupts the autophagic proteolytic process[176]. HCQ/CQ have been extensively studied in myeloma to potentiate the effects of other anti-myeloma drugs, particularly PI.
A phase I clinical trial assessed the efficacy of HCQ in combination with bortezomib in patients with relapsed or refractory MM[177]. Patients were given a 2-week run in with HCQ alone, followed by combination therapy with bortezomib. The combination of HCQ and bortezomib was well tolerated, with no adverse events meeting the criteria of a dose-limiting toxicity. Of the 22 patients assessed at the end of the study, 13.6% had a very good partial response (VGPR), 13.6% had minimal response (MR), and 45.5% had stable disease (SD). Interestingly, all of the patients who had a VGPR were bortezomib-naïve. Twenty-seven percent of patients in the study had progressive disease (PD). Of these, two thirds were bortezomib-refractory. This study also found a therapy-associated increase in autophagic vesicles in BM plasma cells. However, due to the small sample size, the authors were unable to correlate number of autophagic vesicles with clinical response.
A smaller phase II trial looked at CQ in combination with bortezomib and cyclophosphamide in refractory MM[178]. Of the 11 patients enrolled, 8 completed at least 2 cycles and were assessed for clinical response. The most common side effects included fatigue, constipation, myalgia, anorexia, anemia and thrombocytopenia, but was generally well tolerated. 37.5% of patients achieved a PR with a median duration of response of 4 months. One patient (12.5%) had SD, and 50% of patients experienced PD, with an overall clinical benefit rate of 40%. These studies demonstrate that HCQ/CQ are well tolerated in combinations with PI and may help potentiate the effects of existing myeloma therapies.
A recent study looked at dual autophagic inhibition via HCQ and rapamycin with cyclophosphamide and dexamethasone in patients with relapsed or refractory MM based on their preclinical, synergistic anti-tumor activity[179,180]. The quadruple therapy was generally well-tolerated with one case of hematologic dose limiting toxicity (thrombocytopenia) occurring at a HCQ dose of 800 mg, which was then established as the maximum tolerated dose. Of the 18 patients enrolled in the trial, 1 had a VGPR and 3 had a PR, for an ORR of 22%. Seven patients had MR, and 5 had SD, for a CBR of 89%. Median duration of a response was 4.5 months, and median time to best response was 1.9 months. Two patients had immediate progression of disease. Unfortunately, correlative studies were unable to predict the depth of treatment response based on the number of autophagic vesicles in BM-derived MM cells. The promising results of this study warrant further investigation of dual autophagy modulation in MM.
HDAC6 inhibitors
HDAC inhibitors represent an important new group of anti-cancer drugs. There are 18 different isoforms of HDACs in human cells that are subdivided into 4 classes based on subcellular localization and non-cell-based enzymatic activity[181]. Clinical studies with pan-HDAC inhibitors vorinostat and panobinostat outlined a narrow therapeutic window due to frequent and often severe hematologic and non-hematologic (particularly gastrointestinal/GI) toxicities, eliciting clinical interest in isoform-specific HDAC inhibition[182].
Ricolinostat is an oral, selective HDAC6 inhibitor that has been extensively studied in MM. A phase 1b multicenter trial of escalating doses of ricolinostat with lenalidomide and dexamethasone in patients with relapsed or refractory MM showed 55% ORR[182]. In lenalidomide-naïve and lenalidomide-sensitive patients, the ORR was 69%, compared to 25% in lenalidomide-refractory patients. In patients that responded, the median time to response was 7 weeks with a median duration of response of 24 months. Treatment with ricolinostat was overall well tolerated with 2 patients experiencing a dose-limiting toxicity at the highest tested dose which included syncope and myalgia.
Similarly promising results emerged from a phase 1/2 clinical trial assessing the safety and efficacy of ricolinostat with bortezomib and dexamethasone in patients with MM relapsed or refractory to PI, immunomodulatory drugs, or stem cell transplant[183]. In this study, 15 patients were recruited for dose escalation monotherapy with ricolinostat, and 57 patients were given combination therapy. Monotherapy with ricolinostat was well tolerated with no dose-limiting toxicities up to 360 mg Q.D. Patients on combination therapy did not have any dose-limiting toxicities during escalation studies. Most common side effects associated with combination therapy were gastrointestinal toxicities, cytopenia, and fatigue. Of the patients treated with monotherapy, 6 patients (6/15, 40%) had stable disease with a median response duration of response of 11 weeks. Patients on combination therapy had an ORR of 29%, and a clinical response rate of 39%. Interestingly, patients on combination therapy who were previously refractory to bortezomib had an ORR of 14%. The response seen in patients with bortezomib-refractory MM suggests that HDAC6 inhibitors can overcome resistance to PI.
Taken together, these results demonstrate a promising role for HDAC6 inhibitors in the treatment of relapsed and/or refractory MM. There are currently ongoing trials assessing the role of HDAC6 inhibitors in combination with anti-myeloma treatments such as pomalidomide and dexamethasone (NCT01997840, NCT02189343).
HSP90 inhibitors
A phase 2 clinical trial looked at the HSP90 inhibitor tanespimycin in combination with bortezomib in patients with relapsed and/or refractory MM[184]. All patients enrolled in the study experienced at least one side effect, with most common grade 3/4 toxicities being fatigue, thrombocytopenia, neutropenia, and abdominal pain. Four patients had significant liver toxicity with higher doses of tanespimycin, however this was manageable and reversible. The best responses observed to treatment were 1 MR in the 340 mg/m2 group, and 2 PRs in the 175 mg/m2 group. An additional 10 patients across all treatment groups had stable disease. A later phase 3 study evaluating tanespimycin and bortezomib has been completed, however results are not yet available.
KW-278 is a novel, non-anisomycin, non-purine based HSP90 inhibitor that has a more favorable pharmacokinetic and safety profile compared to tanespimycin[185]. A phase 1 trial examined the safety and efficacy of KW-278 in B-cell malignancies, including 22 MM patients[186]. The most common side effects included diarrhea, headache, rhinitis, and fatigue. Ten patients experienced grade 3 and 4 toxicities such as lethargy, syncope, QT prolongation, and neutropenia. Six patients experienced eye disorders (decreased visual acuity, blurry vision, dry eyes) that were deemed related to KW-278. All of the eye disorders were reversible with the exception of dry eyes. Two patients died during the trial and both deaths were determined to be unrelated to the study medication. Of the 21 MM patients evaluated, 20 patients (95%) had stable disease, and 1 patient had progressive disease.
A subsequent phase 1/2 study evaluated KW-278 with bortezomib in patients with relapsed or refractory MM who had failed at least 1 previous treatment[187]. The most common side effects were fatigue and gastrointestinal toxicity, while the most common grade 3-4 side toxicities were cytopenias. There was no sign of significant ophthalmologic side effects in the 95 patients enrolled in this study. There were 28 serious treatment-related adverse events such as lung infections, GI toxicity, anemia, hematuria, syndrome of inappropriate diuretic hormone, pancreatitis, and transient ischemic attack. Efficacy of the treatment was based on a KW-278 dose of 175 mg/m2 with bortezomib at the standard concentration. The ORR was 39%, the CBR was 92%. Median progression-free survival was 6.8 months, and median duration of response was 5.6 months. Patients who were lenalidomide-naïve had an ORR of 45.5 as compared to 25% for patients previously exposed to lenalidomide. Bortezomib-naïve patients had an ORR of 44% vs. 33% in patients previously exposed to bortezomib. These findings suggest a potential therapeutic benefit of KW-278 with bortezomib in the treatment of relapsed or refractory MM.
PI3K-AKT-mTOR inhibitors
The PI3K-AKT-mTOR inhibitor everolimus was tested as a single agent in a phase 1 trial in patients with relapsed and/or refractory MM[188]. Most side effects were mild to moderate with gastrointestinal upset, elevated muscle and liver enzymes, and cytopenias being the most common. One patient developed an atypical pneumonia that was thought to be drug-related. Of the 15 patients assessed, 10 (67%) experienced clinical benefit with one patient achieving a partial remission after 4 cycles.
A phase I trial assessed the safety and efficacy of everolimus in combination with lenalidomide in patients with relapsed or refractory MM[189]. Most common side effects were fatigue, cytopenia, diarrhea and neuropathy. One patient discontinued treatment due to non-infectious pneumonitis that was related to everolimus, and another discontinued due to grade 3 myalgia. Of the 23 patients that completed 2 cycles of treatment, the CBR was 74% with 1 CR and 4 PR. In patients who were lenalidomide-naïve, the ORR was 90%, compared to 37.5% for patients who had previously received lenalidomide. Median progression-free survival was 5.5 months, and median overall survival was 29.5 months. Promising results from these studies demonstrate the potential for mTOR inhibitors to be used the treatment of relapsed or refractory MM. A clinical trial looking at the role of mTOR inhibitors in combination with pomalidomide and dexamethasone is currently enrolling (NCT03657420).
Repurposing of FDA-approved drugs for use in MM
Metformin, the oral biguanide drug used in the treatment of diabetes, has been shown to exert anti-MM effects both in vitro and in vivo. The exact mechanism of metformin’s anti-tumour activity has not been elucidated, but it is hypothesized to induce autophagy through the inhibition of STAT3 and BCL-2[132]. The anti-retroviral drug nelfinavir has also shown anti-MM effects in vivo by triggering the UPR, and has been studied extensively clinical trials for relapsed MM[190,191]. A new phase 1 clinical trial is assessing the safety and efficacy of metformin, nelfinavir and bortezomib in with relapsed or refractory MM (NCT03829020). This is only one example of the potential utility of drug repurposing strategies in cancer, rationally designed based on theoretical synergistic mechanisms of activity.
Conclusion
Autophagy plays a crucial pro-survival role in MM. Increased protein synthesis and proteotoxic stress are hallmarks of cancer, and MM is the prototypic cancer with impaired protein homeostasis based on its staggering rate of immunoglobulin synthesis and baseline level of proteotoxicity. Importantly, autophagy protects MM cells from excessive ER stress by limiting the secretion of immunoglobulins and providing an alternative proteolytic pathway for the clearance of ubiquitinated proteins. Consistent with a therapeutic role in targeting protein homeostasis, PI are highly effective anti-MM agents that are FDA approved for the treatment of MM in all its stages. However, clinical resistance to PI is inevitable, leading to research interest in targeting alternative pathways contributing to protein quality control such as autophagy, alone or in combination with PI. As with other mechanisms of protein homeostasis, such as the UPR; while a basal level of autophagy is cytoprotective and inhibiting autophagy to blunt non- proteasomal proteolysis has also been shown to have therapeutic benefit, sustained autophagy as in the setting of persistent proteasome inhibition, may result in autophagic cell death. This highlights the “Janus-faced” role of autophagy in MM. Autophagy therefore represents an opportunity to therapeutically exploit MM’s unique “Achilles’ heel”: its dependence on protein quality control. With a better understanding of the molecular sequalae of autophagy inhibition/induction in MM, we are eagerly awaiting the development or repurposing of drugs to target autophagy in an effort to overcome PI resistance and improve outcome for our patients with MM.
Declarations
Authors’ contributionsAll authors contributed to the writing of this review article.
Availability of data and materialsNot applicable.
Financial support and sponsorshipNone.
Conflicts of interestAll authors declared that there are no conflicts of interest.
Ethical approval and consent to participateNot applicable.
Consent for publicationNot applicable.
Copyright© The Author(s) 2019.
REFERENCES
2. Bianchi G, Munshi NC. Pathogenesis beyond the cancer clone(s) in multiple myeloma. Blood 2015;125:3049-58.
3. Bianchi G, Anderson KC. Understanding biology to tackle the disease: multiple myeloma from bench to bedside, and back. CA Cancer J Clin 2014;64:422-44.
5. Rajkumar SV, Dimopoulos MA, Palumbo A, Blade J, Merlini G, et al. International Myeloma Working Group updated criteria for the diagnosis of multiple myeloma. Lancet Oncol 2014;15:e538-48.
6. Multiple myeloma: 2018 update on diagnosis, risk-stratification, and management. Am J Hematol 2018;93:981-1114.
7. Dhodapkar MV. MGUS to myeloma: a mysterious gammopathy of underexplored significance. Blood 2016;128:2599-606.
8. Guang MHZ, Bianchi G. Targeting protein synthesis and degradation in multiple myeloma: a look at What’s on the Horizon. Am J Hematol Oncol 2017;13:11.
9. Pollack JR, Sorlie T, Perou CM, Rees CA, Jeffrey SS, et al. Microarray analysis reveals a major direct role of DNA copy number alteration in the transcriptional program of human breast tumors. Proc Natl Acad Sci U S A 2002;99:12963-8.
10. Torres EM, Sokolsky T, Tucker CM, Chan LY, Boselli M, et al. Effects of aneuploidy on cellular physiology and cell division in haploid yeast. Science 2007;317:916-24.
11. Tsafrir D, Bacolod M, Selvanayagam Z, Tsafrir I, Shia J, et al. Relationship of gene expression and chromosomal abnormalities in colorectal cancer. Cancer Res 2006;66:2129-37.
12. Denoyelle C, Abou-Rjaily G, Bezrookove V, Verhaegen M, Johnson TM, et al. Anti-oncogenic role of the endoplasmic reticulum differentially activated by mutations in the MAPK pathway. Nat Cell Biol 2006;8:1053-63.
13. Papp I, Markkanen M, von Bonsdorff M. Clinical environment as a learning environment: student nurses’ perceptions concerning clinical learning experiences. Nurse Educ Today 2003;23:262-8.
14. Desantis V, Saltarella I, Lamanuzzi A, Mariggio MA, Racanelli V, et al. Autophagy: a new mechanism of prosurvival and drug resistance in multiple myeloma. Transl Oncol 2018;11:1350-7.
15. Jacob JA, Salmani JMM, Jiang Z, Feng L, Song J, et al. Autophagy: an overview and its roles in cancer and obesity. Clin Chim Acta 2017;468:85-9.
16. Glick D, Barth S, Macleod KF. Autophagy: cellular and molecular mechanisms. J Pathol 2010;221:3-12.
17. Yun Z, Zhichao J, Hao Y, Ou J, Ran Y, et al. Targeting autophagy in multiple myeloma. Leuk Res 2017;59:97-104.
18. Xie Z, Klionsky DJ. Autophagosome formation: core machinery and adaptations. Nat Cell Biol 2007;9:1102-9.
19. Mehrpour M, Esclatine A, Beau I, Codogno P. Autophagy in health and disease. 1. Regulation and significance of autophagy: an overview. Am J Physiol Cell Physiol 2010;298:C776-85.
20. Jung CH, Ro SH, Cao J, Otto NM, Kim DH. mTOR regulation of autophagy. FEBS Lett 2010;584:1287-95.
21. Nakatogawa H, Suzuki K, Kamada Y, Ohsumi Y. Dynamics and diversity in autophagy mechanisms: lessons from yeast. Nat Rev Mol Cell Biol 2009;10:458.
22. Itakura E, Mizushima N. Characterization of autophagosome formation site by a hierarchical analysis of mammalian Atg proteins. Autophagy 2010;6:764-76.
23. Yang S, Wang X, Contino G, Liesa M, Sahin E, et al. Pancreatic cancers require autophagy for tumor growth. Genes Dev 2011;25:717-29.
24. Lee JA, Gao FB. Inhibition of autophagy induction delays neuronal cell loss caused by dysfunctional ESCRT-III in frontotemporal dementia. J Neurosci 2009;29:8506-11.
25. Razi M, Chan EY, Tooze SA. Early endosomes and endosomal coatomer are required for autophagy. J Cell Biol 2009;185:305-21.
26. Park KJ, Lee SH, Kim TI, Lee HW, Lee CH, et al. A human scFv antibody against TRAIL receptor 2 induces autophagic cell death in both TRAIL-sensitive and TRAIL-resistant cancer cells. Cancer Res 2007;67:7327-34.
27. Chen Y, Sawada O, Kohno H, Le YZ, Subauste C, et al. Autophagy protects the retina from light-induced degeneration. J Biol Chem 2013;288:7506-18.
31. Lemasters JJ. Variants of mitochondrial autophagy: types 1 and 2 mitophagy and micromitophagy (Type 3). Redox Biol 2014;2:749-54.
33. Li WW, Li J, Bao JK. Microautophagy: lesser-known self-eating. Cell Mol Life Sci 2012;69:1125-36.
34. Sahu R, Kaushik S, Clement CC, Cannizzo ES, Scharf B, et al. Microautophagy of cytosolic proteins by late endosomes. Dev Cell 2011;20:131-9.
35. Todde V, Veenhuis M, van der Klei IJ. Autophagy: principles and significance in health and disease. Biochim Biophys Acta 2009;1792:3-13.
36. Kaushik S, Cuervo AM. The coming of age of chaperone-mediated autophagy. Nat Rev Mol Cell Biol 2018;19:365-81.
37. Cuervo AM, Wong E. Chaperone-mediated autophagy: roles in disease and aging. Cell Res 2014;24:92-104.
38. Levine B, Mizushima N, Virgin HW. Autophagy in immunity and inflammation. Nature 2011;469:323-35.
39. Deretic V, Saitoh T, Akira S. Autophagy in infection, inflammation and immunity. Nat Rev Immunol 2013;13:722-37.
40. Wu TT, Li WM, Yao YM. Interactions between autophagy and inhibitory cytokines. Int J Biol Sci 2016;12:884-97.
41. Djavaheri-Mergny M, Giuriato S, Tschan MP, Humbert M. Therapeutic modulation of autophagy in leukaemia and lymphoma. Cells 2019;8:E103.
42. Mortensen M, Watson AS, Simon AK. Lack of autophagy in the hematopoietic system leads to loss of hematopoietic stem cell function and dysregulated myeloid proliferation. Autophagy 2011;7:1069-70.
43. Warr MR, Binnewies M, Flach J, Reynaud D, Garg T, et al. FOXO3A directs a protective autophagy program in haematopoietic stem cells. Nature 2013;494:323-7.
44. Ho TT, Warr MR, Adelman ER, Lansinger OM, Flach J, et al. Autophagy maintains the metabolism and function of young and old stem cells. Nature 2017;543:205.
45. Ianniciello A, Rattigan KM, Helgason GV. The Ins and Outs of autophagy and metabolism in hematopoietic and leukemic stem cells: food for thought. Front Cell Dev Biol 2018;6:120.
46. Mortensen M, Soilleux EJ, Djordjevic G, Tripp R, Lutteropp M, et al. The autophagy protein Atg7 is essential for hematopoietic stem cell maintenance. J Exp Med 2011;208:455-67.
47. Ho Zhi Guang M, Kavanagh EL, Dunne LP, Dowling P, Zhang L, et al. Targeting proteotoxic stress in cancer: a review of the role that protein quality control pathways play in oncogenesis. Cancers 2019;11:66.
48. Nutt SL, Taubenheim N, Hasbold J, Corcoran LM, Hodgkin PD. The genetic network controlling plasma cell differentiation. Semin Immunol 2011;23:341-9.
49. Reimold AM, Iwakoshi NN, Manis J, Vallabhajosyula P, Szomolanyi-Tsuda E, et al. Plasma cell differentiation requires the transcription factor XBP-1. Nature 2001;412:300-7.
50. Shaffer AL, Shapiro-Shelef M, Iwakoshi NN, Lee AH, Qian SB, et al. XBP1, downstream of Blimp-1, expands the secretory apparatus and other organelles, and increases protein synthesis in plasma cell differentiation. Immunity 2004;21:81-93.
51. Tellier J, Shi W, Minnich M, Liao Y, Crawford S, et al. Blimp-1 controls plasma cell function through the regulation of immunoglobulin secretion and the unfolded protein response. Nat Immunol 2016;17:323-30.
52. Pengo N, Scolari M, Oliva L, Milan E, Mainoldi F, et al. Plasma cells require autophagy for sustainable immunoglobulin production. Nat Immunol 2013;14:298-305.
53. Halliley JL, Tipton CM, Liesveld J, Rosenberg AF, Darce J, et al. Long-lived plasma cells are contained within the CD19(-)CD38(hi)CD138(+) subset in human bone marrow. Immunity 2015;43:132-45.
54. Pengo N, Cenci S. Chapter 6 - autophagy in plasma cells. Autophagy: cancer, other pathologies, inflammation, immunity, infection, and aging Academic Press; 2017. pp. 175-86.
55. Cenci S, Oliva L, Cerruti F, Milan E, Bianchi G, et al. Pivotal advance: protein synthesis modulates responsiveness of differentiating and malignant plasma cells to proteasome inhibitors. J Leukoc Biol 2012;92:921-31.
56. Leung-Hagesteijn C, Erdmann N, Cheung G, Keats JJ, Stewart AK, et al. Xbp1s-negative tumor B cells and pre-plasmablasts mediate therapeutic proteasome inhibitor resistance in multiple myeloma. Cancer Cell 2013;24:289-304.
57. Meister S, Schubert U, Neubert K, Herrmann K, Burger R, et al. Extensive immunoglobulin production sensitizes myeloma cells for proteasome inhibition. Cancer Res 2007;67:1783-92.
59. Ling SC, Lau EK, Al-Shabeeb A, Nikolic A, Catalano A, et al. Response of myeloma to the proteasome inhibitor bortezomib is correlated with the unfolded protein response regulator XBP-1. Haematologica 2012;97:64-72.
60. Milan E, Fabbri M, Cenci S. Autophagy in plasma cell ontogeny and malignancy. J Clin Immunol 2016;36 Suppl 1:18-24.
61. Snyder H, Mensah K, Theisler C, Lee J, Matouschek A, et al. Aggregated and monomeric alpha-synuclein bind to the S6’ proteasomal protein and inhibit proteasomal function. J Biol Chem 2003;278:11753-9.
62. Yao TP. The role of ubiquitin in autophagy-dependent protein aggregate processing. Genes Cancer 2010;1:779-86.
63. Le T, Bhushan V, Sochat M. . ISBN: 978-1-26-014367-6
64. Milan E, Perini T, Resnati M, Orfanelli U, Oliva L, et al. A plastic SQSTM1/p62-dependent autophagic reserve maintains proteostasis and determines proteasome inhibitor susceptibility in multiple myeloma cells. Autophagy 2015;11:1161-78.
65. Hoang B, Benavides A, Shi Y, Frost P, Lichtenstein A. Effect of autophagy on multiple myeloma cell viability. Mol Cancer Ther 2009;8:1974-84.
66. Caro LH, Plomp PJ, Wolvetang EJ, Kerkhof C, Meijer AJ. 3-Methyladenine, an inhibitor of autophagy, has multiple effects on metabolism. Eur J Biochem 1988;175:325-9.
67. Lamy L, Ngo VN, Emre NC, Shaffer AL 3rd, Yang Y, et al. Control of autophagic cell death by caspase-10 in multiple myeloma. Cancer Cell 2013;23:435-49.
68. Yu L, Alva A, Su H, Dutt P, Freundt E, et al. Regulation of an ATG7-beclin 1 program of autophagic cell death by caspase-8. Science 2004;304:1500-2.
70. Kryston TB, Georgiev AB, Pissis P, Georgakilas AG. Role of oxidative stress and DNA damage in human carcinogenesis. Mutat Res 2011;711:193-201.
71. Wu W, Tian W, Hu Z, Chen G, Huang L, et al. ULK1 translocates to mitochondria and phosphorylates FUNDC1 to regulate mitophagy. EMBO Rep 2014;15:566-75.
72. Maes H, Rubio N, Garg AD, Agostinis P. Autophagy: shaping the tumor microenvironment and therapeutic response. Trends Mol Med 2013;19:428-46.
73. Zheng Z, Fan S, Zheng J, Huang W, Gasparetto C, et al. Inhibition of thioredoxin activates mitophagy and overcomes adaptive bortezomib resistance in multiple myeloma. J Hematol Oncol 2018;11:29.
74. Gourzones-Dmitriev C, Kassambara A, Sahota S, Reme T, Moreaux J, et al. DNA repair pathways in human multiple myeloma: role in oncogenesis and potential targets for treatment. Cell Cycle 2013;12:2760-73.
75. Cottini F, Hideshima T, Suzuki R, Tai YT, Bianchini G, et al. Synthetic lethal approaches exploiting DNA damage in aggressive myeloma. Cancer Discov 2015;5:972-87.
76. Eliopoulos AG, Havaki S, Gorgoulis VG. DNA damage response and autophagy: a meaningful partnership. Front Genet 2016;7:204.
77. Alexander DE, Gong E, Martin LD, Burnham DA, Falk AR. Model tests of gliding with different hindwing configurations in the four-winged dromaeosaurid microraptor gui. Proc Natl Acad Sci U S A 2010;107:2972-6.
78. Alexander A, Kim J, Walker CL. ATM engages the TSC2/mTORC1 signaling node to regulate autophagy. Autophagy 2010;6:672-3.
79. Kim J, Kundu M, Viollet B, Guan KL. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol 2011;13:132-41.
80. Desantis A, Bruno T, Catena V, De Nicola F, Goeman F, et al. Che-1-induced inhibition of mTOR pathway enables stress-induced autophagy. EMBO J 2015;34:1214-30.
81. Copetti T, Bertoli C, Dalla E, Demarchi F, Schneider C. p65/RelA modulates BECN1 transcription and autophagy. Mol Cell Biol 2009;29:2594-608.
82. Rodriguez-Vargas JM, Ruiz-Magana MJ, Ruiz-Ruiz C, Majuelos-Melguizo J, Peralta-Leal A, et al. ROS-induced DNA damage and PARP-1 are required for optimal induction of starvation-induced autophagy. Cell Res 2012;22:1181-98.
83. Wei Y, Sinha S, Levine B. Dual role of JNK1-mediated phosphorylation of Bcl-2 in autophagy and apoptosis regulation. Autophagy 2008;4:949-51.
84. Stambolic V, MacPherson D, Sas D, Lin Y, Snow B, et al. Regulation of PTEN transcription by p53. Mol Cell 2001;8:317-25.
85. Feng Z, Hu W, de Stanchina E, Teresky AK, Jin S, et al. The regulation of AMPK beta1, TSC2, and PTEN expression by p53: stress, cell and tissue specificity, and the role of these gene products in modulating the IGF-1-AKT-mTOR pathways. Cancer Res 2007;67:3043-53.
86. Buschmann T, Potapova O, Bar-Shira A, Ivanov VN, Fuchs SY, et al. Jun NH2-terminal kinase phosphorylation of p53 on Thr-81 is important for p53 stabilization and transcriptional activities in response to stress. Mol Cell Biol 2001;21:2743-54.
87. Zalckvar E, Berissi H, Eisenstein M, Kimchi A. Phosphorylation of Beclin 1 by DAP-kinase promotes autophagy by weakening its interactions with Bcl-2 and Bcl-XL. Autophagy 2009;5:720-2.
88. Zalckvar E, Berissi H, Mizrachy L, Idelchuk Y, Koren I, et al. DAP-kinase-mediated phosphorylation on the BH3 domain of beclin 1 promotes dissociation of beclin 1 from Bcl-XL and induction of autophagy. EMBO Rep 2009;10:285-92.
89. Eisenberg-Lerner A, Kimchi A. PKD is a kinase of Vps34 that mediates ROS-induced autophagy downstream of DAPk. Cell Death Differ 2012;19:788-97.
90. Crighton D, Wilkinson S, O’Prey J, Syed N, Smith P, et al. DRAM, a p53-induced modulator of autophagy, is critical for apoptosis. Cell 2006;126:121-34.
91. Kim JS, Ro SH, Kim M, Park HW, Semple IA, et al. Sestrin2 inhibits mTORC1 through modulation of GATOR complexes. Sci Rep 2015;5:9502.
92. Park BC, Kim TH, Sim KI, Kang B, Kim JW, et al. Terahertz single conductance quantum and topological phase transitions in topological insulator Bi(2)Se(3) ultrathin films. Nat Commun 2015;6:6552.
93. Liu IB, Gharbi MA, Ngo VL, Kamien RD, Yang S, et al. Elastocapillary interactions on nematic films. Proc Natl Acad Sci U S A 2015;112:6336-40.
94. Hewitt G, Carroll B, Sarallah R, Correia-Melo C, Ogrodnik M, et al. SQSTM1/p62 mediates crosstalk between autophagy and the UPS in DNA repair. Autophagy 2016;12:1917-30.
95. Qiang L, Zhao B, Shah P, Sample A, Yang S, et al. Autophagy positively regulates DNA damage recognition by nucleotide excision repair. Autophagy 2016;12:357-68.
96. Wang Z, Miao G, Xue X, Guo X, Yuan C, et al. The Vici syndrome protein EPG5 is a Rab7 effector that determines the fusion specificity of autophagosomes with late endosomes/lysosomes. Mol Cell 2016;63:781-95.
97. Mathew R, Kongara S, Beaudoin B, Karp CM, Bray K, et al. Autophagy suppresses tumor progression by limiting chromosomal instability. Genes Dev 2007;21:1367-81.
98. Karantza-Wadsworth V, Patel S, Kravchuk O, Chen G, Mathew R, et al. Autophagy mitigates metabolic stress and genome damage in mammary tumorigenesis. Genes Dev 2007;21:1621-35.
99. Harada T, Hideshima T, Anderson KC. Histone deacetylase inhibitors in multiple myeloma: from bench to bedside. Int J Hematol 2016;104:300-9.
100. Kaliszczak M, van Hechanova E, Li Y, Alsadah H, Parzych K, et al. The HDAC6 inhibitor C1A modulates autophagy substrates in diverse cancer cells and induces cell death. Br J Cancer 2018;119:1278-87.
101. Iwata A, Riley BE, Johnston JA, Kopito RR. HDAC6 and microtubules are required for autophagic degradation of aggregated huntingtin. J Biol Chem 2005;280:40282-92.
102. Kawaguchi Y, Kovacs JJ, McLaurin A, Vance JM, Ito A, et al. The deacetylase HDAC6 regulates aggresome formation and cell viability in response to misfolded protein stress. Cell 2003;115:727-38.
103. Ouyang H, Ali YO, Ravichandran M, Dong A, Qiu W, et al. Protein aggregates are recruited to aggresome by histone deacetylase 6 via unanchored ubiquitin C termini. J Biol Chem 2012;287:2317-27.
104. Banreti A, Sass M, Graba Y. The emerging role of acetylation in the regulation of autophagy. Autophagy 2013;9:819-29.
105. Lee JY, Koga H, Kawaguchi Y, Tang W, Wong E, et al. HDAC6 controls autophagosome maturation essential for ubiquitin-selective quality-control autophagy. EMBO J 2010;29:969-80.
106. Hideshima T, Qi J, Paranal RM, Tang W, Greenberg E, et al. Discovery of selective small-molecule HDAC6 inhibitor for overcoming proteasome inhibitor resistance in multiple myeloma. Proc Natl Acad Sci U S A 2016;113:13162-7.
107. Hideshima T, Bradner JE, Wong J, Chauhan D, Richardson P, et al. Small-molecule inhibition of proteasome and aggresome function induces synergistic antitumor activity in multiple myeloma. Proc Natl Acad Sci U S A 2005;102:8567-72.
108. Santo L, Cirstea DD, Eda H, Nemani N, Arastu-Kapur S, et al. Inhibition of autophagy by ACY-1215, a selective HDAC6 inhibitor accelerates carfilzomib-induced cell death in multiple myeloma. Blood 2013;122:4431.
109. Huang FI, Wu YW, Sung TY, Liou JP, Lin MH, et al. MPT0G413, a novel HDAC6-selective inhibitor, and Bortezomib synergistically exert anti-tumor activity in multiple myeloma cells. Front Oncol 2019;9:249.
110. Bayraktar UD, Bashir Q, Qazilbash M, Champlin RE, Ciurea SO. Fifty years of melphalan use in hematopoietic stem cell transplantation. Biol Blood Marrow Transplant 2013;19:344-56.
111. Pan Y, Gao Y, Chen L, Gao G, Dong H, et al. Targeting autophagy augments in vitro and in vivo antimyeloma activity of DNA-damaging chemotherapy. Clin Cancer Res 2011;17:3248-58.
112. Tang D, Kang R, Livesey KM, Cheh CW, Farkas A, et al. Endogenous HMGB1 regulates autophagy. J Cell Biol 2010;190:881-92.
113. Guo X, He D, Zhang E, Chen J, Chen Q, et al. HMGB1 knockdown increases MM cell vulnerability by regulating autophagy and DNA damage repair. J Exp Clin Cancer Res 2018;37:205.
114. Liu P, Zhao L, Pol J, Levesque S, Petrazzuolo A, et al. Crizotinib-induced immunogenic cell death in non-small cell lung cancer. Nature Communications 2019;10:1486.
115. Kroemer G, Galluzzi L, Kepp O, Zitvogel L. Immunogenic cell death in cancer therapy. Annu Rev Immunol 2013;31:51-72.
116. Yamazaki T, Hannani D, Poirier-Colame V, Ladoire S, Locher C, et al. Defective immunogenic cell death of HMGB1-deficient tumors: compensatory therapy with TLR4 agonists. Cell Death Differ 2013;21:69.
117. Joubert PE, Albert ML. Antigen cross-priming of cell-associated proteins is enhanced by macroautophagy within the antigen donor cell. Front Immunol 2012;3:61.
118. Niewerth D, Jansen G, Assaraf YG, Zweegman S, Kaspers GJ, et al. Molecular basis of resistance to proteasome inhibitors in hematological malignancies. Drug Resist Updat 2015;18:18-35.
119. Orlowski RZ, Kuhn DJ. Proteasome inhibitors in cancer therapy: lessons from the first decade. Clin Cancer Res 2008;14:1649-57.
120. McConkey DJ, Zhu K. Mechanisms of proteasome inhibitor action and resistance in cancer. Drug Resist Updat 2008;11:164-79.
121. Yong K, Gonzalez-McQuire S, Szabo Z, Schoen P, Hajek R. The start of a new wave: developments in proteasome inhibition in multiple myeloma. Eur J Haematol 2018; doi: 10.1111/ejh.13071.
122. Bianchi G, Oliva L, Cascio P, Pengo N, Fontana F, et al. The proteasome load versus capacity balance determines apoptotic sensitivity of multiple myeloma cells to proteasome inhibition. Blood 2009;113:3040-9.
123. Kawaguchi T, Miyazawa K, Moriya S, Ohtomo T, Che XF, et al. Combined treatment with bortezomib plus bafilomycin A1 enhances the cytocidal effect and induces endoplasmic reticulum stress in U266 myeloma cells: crosstalk among proteasome, autophagy-lysosome and ER stress. Int J Oncol 2011;38:643-54.
124. Qiao L, Zhang J. Inhibition of lysosomal functions reduces proteasomal activity. Neurosci Lett 2009;456:15-9.
125. Pandey UB, Nie Z, Batlevi Y, McCray BA, Ritson GP, et al. HDAC6 rescues neurodegeneration and provides an essential link between autophagy and the UPS. Nature 2007;447:859-63.
126. Wu WK, Cho CH, Lee CW, Wu YC, Yu L, et al. Macroautophagy and ERK phosphorylation counteract the antiproliferative effect of proteasome inhibitor in gastric cancer cells. Autophagy 2010;6:228-38.
127. Kouroku Y, Fujita E, Tanida I, Ueno T, Isoai A, et al. ER stress (PERK/eIF2alpha phosphorylation) mediates the polyglutamine-induced LC3 conversion, an essential step for autophagy formation. Cell Death Differ 2007;14:230-9.
128. Ding WX, Ni HM, Gao W, Yoshimori T, Stolz DB, et al. Linking of autophagy to ubiquitin-proteasome system is important for the regulation of endoplasmic reticulum stress and cell viability. Am J Pathol 2007;171:513-24.
129. Lu Y, Wang Y, Xu H, Shi C, Jin F, et al. Profilin 1 induces drug resistance through Beclin1 complex-mediated autophagy in multiple myeloma. Cancer Sci 2018;109:2706-16.
130. Wang G, Zhou P, Chen X, Zhao L, Tan J, et al. The novel autophagy inhibitor elaiophylin exerts antitumor activity against multiple myeloma with mutant TP53 in part through endoplasmic reticulum stress-induced apoptosis. Cancer Biol Ther 2017;18:584-95.
131. Jagannathan S, Abdel-Malek MA, Malek E, Vad N, Latif T, et al. Pharmacologic screens reveal metformin that suppresses GRP78-dependent autophagy to enhance the anti-myeloma effect of bortezomib. Leukemia 2015;29:2184-91.
132. Wang Y, Xu W, Yan Z, Zhao W, Mi J, et al. Metformin induces autophagy and G0/G1 phase cell cycle arrest in myeloma by targeting the AMPK/mTORC1 and mTORC2 pathways. J Exp Clin Cancer Res 2018;37:63.
133. Granato M, Gilardini Montani MS, Romeo MA, Santarelli R, Gonnella R, et al. Metformin triggers apoptosis in PEL cells and alters bortezomib-induced unfolded protein response increasing its cytotoxicity and inhibiting KSHV lytic cycle activation. Cell Signal 2017;40:239-47.
134. Chen Q, Xie W, Kuhn DJ, Voorhees PM, Lopez-Girona A, et al. Targeting the p27 E3 ligase SCF(Skp2) results in p27- and Skp2-mediated cell-cycle arrest and activation of autophagy. Blood 2008;111:4690-9.
135. Zhou H, Luo W, Zeng C, Zhang Y, Wang L, et al. PP2A mediates apoptosis or autophagic cell death in multiple myeloma cell lines. Oncotarget 2017;8:80770-89.
136. Chen S, Zhang Y, Zhou L, Leng Y, Lin H, et al. A Bim-targeting strategy overcomes adaptive bortezomib resistance in myeloma through a novel link between autophagy and apoptosis. Blood 2014;124:2687-97.
137. Baranowska K, Misund K, Starheim KK, Holien T, Johansson I, et al. Hydroxychloroquine potentiates carfilzomib toxicity towards myeloma cells. Oncotarget 2016;7:70845-56.
138. Ismail SI, Mahmoud IS, Msallam MM, Sughayer MA. Hotspot mutations of PIK3CA and AKT1 genes are absent in multiple myeloma. Leuk Res 2010;34:824-6.
139. Hideshima T, Mitsiades C, Tonon G, Richardson PG, Anderson KC. Understanding multiple myeloma pathogenesis in the bone marrow to identify new therapeutic targets. Nat Rev Cancer 2007;7:585.
140. Ramakrishnan V, Kumar S. PI3K/AKT/mTOR pathway in multiple myeloma: from basic biology to clinical promise. Leuk Lymphoma 2018;59:2524-34.
141. Raje N, Kumar S, Hideshima T, Ishitsuka K, Chauhan D, et al. Combination of the mTOR inhibitor rapamycin and CC-5013 has synergistic activity in multiple myeloma. Blood 2004;104:4188-93.
142. Yan H, Frost P, Shi Y, Hoang B, Sharma S, et al. Mechanism by which mammalian target of rapamycin inhibitors sensitize multiple myeloma cells to dexamethasone-induced apoptosis. Cancer Res 2006;66:2305-13.
143. Ramakrishnan V, Kimlinger T, Timm M, Haug J, Rajkumar SV, et al. Multiple mechanisms contribute to the synergistic anti-myeloma activity of the pan-histone deacetylase inhibitor LBH589 and the rapalog RAD001. Leuk Res 2014;38:1358-66.
144. Simmons JK, Patel J, Michalowski A, Zhang S, Wei BR, et al. TORC1 and class I HDAC inhibitors synergize to suppress mature B cell neoplasms. Mol Oncol 2014;8:261-72.
145. Ramakrishnan V, Timm M, Haug JL, Kimlinger TK, Wellik LE, et al. Sorafenib, a dual Raf kinase/vascular endothelial growth factor receptor inhibitor has significant anti-myeloma activity and synergizes with common anti-myeloma drugs. Oncogene 2010;29:1190-202.
146. Francis LK, Alsayed Y, Leleu X, Jia X, Singha UK, et al. Combination mammalian target of rapamycin inhibitor rapamycin and HSP90 inhibitor 17-allylamino-17-demethoxygeldanamycin has synergistic activity in multiple myeloma. Clin Cancer Res 2006;12:6826-35.
147. Baumann P, Hagemeier H, Mandl-Weber S, Franke D, Schmidmaier R. Myeloma cell growth inhibition is augmented by synchronous inhibition of the insulin-like growth factor-1 receptor by NVP-AEW541 and inhibition of mammalian target of rapamycin by Rad001. Anticancer Drugs 2009;20:259-66.
148. Ramakrishnan V, Kimlinger T, Haug J, Painuly U, Wellik L, et al. Anti-myeloma activity of Akt inhibition is linked to the activation status of PI3K/Akt and MEK/ERK pathway. PLoS One 2012;7:e50005.
149. Hideshima T, Catley L, Yasui H, Ishitsuka K, Raje N, et al. Perifosine, an oral bioactive novel alkylphospholipid, inhibits Akt and induces in vitro and in vivo cytotoxicity in human multiple myeloma cells. Blood 2006;107:4053-62.
150. Mimura N, Hideshima T, Shimomura T, Suzuki R, Ohguchi H, et al. Selective and potent Akt inhibition triggers anti-myeloma activities and enhances fatal endoplasmic reticulum stress induced by proteasome inhibition. Cancer Res 2014;74:4458-69.
151. McMillin DW, Ooi M, Delmore J, Negri J, Hayden P, et al. Antimyeloma activity of the orally bioavailable dual phosphatidylinositol 3-kinase/mammalian target of rapamycin inhibitor NVP-BEZ235. Cancer Res 2009;69:5835-42.
152. Baumann P, Mandl-Weber S, Oduncu F, Schmidmaier R. The novel orally bioavailable inhibitor of phosphoinositol-3-kinase and mammalian target of rapamycin, NVP-BEZ235, inhibits growth and proliferation in multiple myeloma. Exp Cell Res 2009;315:485-97.
153. Aronson LI, Davenport EL, Mirabella F, Morgan GJ, Davies FE. Understanding the interplay between the proteasome pathway and autophagy in response to dual PI3K/mTOR inhibition in myeloma cells is essential for their effective clinical application. Leukemia 2013;27:2397.
154. Li Z, Srivastava P. Heat-shock proteins. Curr Protoc Immunol 2004; . Appendix 1:Appendix 1T
155. Agarraberes FA, Terlecky SR, Dice JF. An intralysosomal hsp70 is required for a selective pathway of lysosomal protein degradation. J Cell Biol 1997;137:825-34.
156. Chiang HL, Terlecky SR, Plant CP, Dice JF. A role for a 70-kilodalton heat shock protein in lysosomal degradation of intracellular proteins. Science 1989;246:382-5.
157. Wang B, Chen Z, Yu F, Chen Q, Tian Y, et al. Hsp90 regulates autophagy and plays a role in cancer therapy. Tumour Biol 2016;37:1-6.
158. Zhang L, Fok JH, Davies FE. Heat shock proteins in multiple myeloma. Oncotarget 2014;5:1132-48.
159. Marcu MG, Doyle M, Bertolotti A, Ron D, Hendershot L, et al. Heat shock protein 90 modulates the unfolded protein response by stabilizing IRE1alpha. Mol Cell Biol 2002;22:8506-13.
160. Chatterjee M, Andrulis M, Stuhmer T, Muller E, Hofmann C, et al. The PI3K/Akt signaling pathway regulates the expression of Hsp70, which critically contributes to Hsp90-chaperone function and tumor cell survival in multiple myeloma. Haematologica 2013;98:1132-41.
161. Braunstein MJ, Scott SS, Scott CM, Behrman S, Walter P, et al. Antimyeloma effects of the heat shock protein 70 molecular chaperone inhibitor MAL3-101. J Oncol 2011;2011:232037.
162. Ishii T, Seike T, Nakashima T, Juliger S, Maharaj L, et al. Anti-tumor activity against multiple myeloma by combination of KW-2478, an Hsp90 inhibitor, with bortezomib. Blood Cancer J 2012;2:e68.
163. Sydor JR, Normant E, Pien CS, Porter JR, Ge J, et al. Development of 17-allylamino-17-demethoxygeldanamycin hydroquinone hydrochloride (IPI-504), an anti-cancer agent directed against Hsp90. Proc Natl Acad Sci U S A 2006;103:17408-13.
164. Mimnaugh EG, Xu W, Vos M, Yuan X, Isaacs JS, et al. Simultaneous inhibition of hsp 90 and the proteasome promotes protein ubiquitination, causes endoplasmic reticulum-derived cytosolic vacuolization, and enhances antitumor activity. Mol Cancer Ther 2004;3:551-66.
165. Lamottke B, Kaiser M, Mieth M, Heider U, Gao Z, et al. The novel, orally bioavailable HSP90 inhibitor NVP-HSP990 induces cell cycle arrest and apoptosis in multiple myeloma cells and acts synergistically with melphalan by increased cleavage of caspases. Eur J Haematol 2012;88:406-15.
166. Stuhmer T, Iskandarov K, Gao Z, Bumm T, Grella E, et al. Preclinical activity of the novel orally bioavailable HSP90 inhibitor NVP-HSP990 against multiple myeloma cells. Anticancer Res 2012;32:453-62.
167. Beebe K, Mollapour M, Scroggins B, Prodromou C, Xu W, et al. Posttranslational modification and conformational state of heat shock protein 90 differentially affect binding of chemically diverse small molecule inhibitors. Oncotarget 2013;4:1065-74.
168. Usmani SZ, Bona RD, Chiosis G, Li Z. The anti-myeloma activity of a novel purine scaffold HSP90 inhibitor PU-H71 is via inhibition of both HSP90A and HSP90B1. J Hematol Oncol 2010;3:40.
169. Stuhmer T, Zollinger A, Siegmund D, Chatterjee M, Grella E, et al. Signalling profile and antitumour activity of the novel Hsp90 inhibitor NVP-AUY922 in multiple myeloma. Leukemia 2008;22:1604-12.
170. Bailey CK, Budina-Kolomets A, Murphy ME, Nefedova Y. Efficacy of the HSP70 inhibitor PET-16 in multiple myeloma. Cancer Biol Ther 2015;16:1422-6.
171. Zhang L, Fok JJ, Mirabella F, Aronson LI, Fryer RA, et al. Hsp70 inhibition induces myeloma cell death via the intracellular accumulation of immunoglobulin and the generation of proteotoxic stress. Cancer Lett 2013;339:49-59.
172. Davenport EL, Zeisig A, Aronson LI, Moore HE, Hockley S, et al. Targeting heat shock protein 72 enhances Hsp90 inhibitor-induced apoptosis in myeloma. Leukemia 2010;24:1804-7.
173. Yasui H, Hideshima T, Ikeda H, Jin J, Ocio EM, et al. BIRB 796 enhances cytotoxicity triggered by bortezomib, heat shock protein (Hsp) 90 inhibitor, and dexamethasone via inhibition of p38 mitogen-activated protein kinase/Hsp27 pathway in multiple myeloma cell lines and inhibits paracrine tumour growth. Br J Haematol 2007;136:414-23.
174. Powers MV, Clarke PA, Workman P. Dual targeting of HSC70 and HSP72 inhibits HSP90 function and induces tumor-specific apoptosis. Cancer Cell 2008;14:250-62.
175. Fok JHL, Hedayat S, Zhang L, Aronson LI, Mirabella F, et al. HSF1 is essential for myeloma cell survival and a promising therapeutic target. Clin Cancer Res 2018;24:2395-407.
176. Onorati AV, Dyczynski M, Ojha R, Amaravadi RK. Targeting autophagy in cancer. Cancer 2018;124:3307-18.
177. Vogl DT, Stadtmauer EA, Tan KS, Heitjan DF, Davis LE, et al. Combined autophagy and proteasome inhibition: a phase 1 trial of hydroxychloroquine and bortezomib in patients with relapsed/refractory myeloma. Autophagy 2014;10:1380-90.
178. Montanari F, Lu M, Marcus S, Saran A, Malankar A, et al. A phase II trial of chloroquine in combination with bortezomib and cyclophosphamide in patients with relapsed and refractory multiple myeloma. Blood 2014;124:5775.
179. Scott EC, Maziarz RT, Spurgeon SE, Medvedova E, Gajewski J, et al. Double autophagy stimulation using chemotherapy and mTOR inhibition combined with hydroxychloroquine for autophagy modulation in patients with relapsed or refractory multiple myeloma. Haematologica 2017;102:e261-5.
180. Xie X, White EP, Mehnert JM. Coordinate autophagy and mTOR pathway inhibition enhances cell death in melanoma. PLoS One 2013;8:e55096.
181. Harada T, Hideshima T, Anderson KC. Histone deacetylase inhibitors in multiple myeloma: from bench to bedside. Int J Hematol 2016;104:300-9.
182. Yee AJ, Bensinger WI, Supko JG, Voorhees PM, Berdeja JG, et al. Ricolinostat plus lenalidomide, and dexamethasone in relapsed or refractory multiple myeloma: a multicentre phase 1b trial. Lancet Oncol 2016;17:1569-78.
183. Vogl DT, Raje N, Jagannath S, Richardson P, Hari P, et al. Ricolinostat, the first selective histone deacetylase 6 inhibitor, in combination with Bortezomib and Dexamethasone for relapsed or refractory multiple myeloma. Clin Cancer Res 2017;23:3307-15.
184. Richardson PG, Badros AZ, Jagannath S, Tarantolo S, Wolf JL, et al. Tanespimycin with Bortezomib: activity in relapsed/refractory patients with multiple myeloma. Br J Haematol 2010;150:428-37.
185. Nakashima T, Ishii T, Tagaya H, Seike T, Nakagawa H, et al. New molecular and biological mechanism of antitumor activities of KW-2478, a novel nonansamycin heat shock protein 90 inhibitor, in multiple myeloma cells. Clin Cancer Res 2010;16:2792-802.
186. Yong K, Cavet J, Johnson P, Morgan G, Williams C, et al. Phase I study of KW-2478, a novel Hsp90 inhibitor, in patients with B-cell malignancies. Br J Cancer 2016;114:7-13.
187. Cavenagh J, Oakervee H, Baetiong-Caguioa P, Davies F, Gharibo M, et al. A phase I/II study of KW-2478, an Hsp90 inhibitor, in combination with bortezomib in patients with relapsed/refractory multiple myeloma. Br J Cancer 2017;117:1295-302.
188. Günther A, Baumann P, Burger R, Kellner C, Klapper W, et al. Activity of everolimus (RAD001) in relapsed and/or refractory multiple myeloma: a phase I study. Haematologica 2015;100:541-7.
189. Yee AJ, Hari P, Marcheselli R, Mahindra AK, Cirstea DD, et al. Outcomes in patients with relapsed or refractory multiple myeloma in a phase I study of everolimus in combination with lenalidomide. Br J Haematol 2014;166:401-9.
190. Hitz F, Pabst T, Hess D, Kraus M, Besse L, et al. Nelfinavir and Lenalidomide/Dexamethasone in patients with lenalidomide-refractory multiple myeloma. A Phase I/II Trial - SAKK 39/10. Blood 2017;130:1884.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Ho M, Patel A, Hanley C, Murphy A, McSweeney T, Zhang L, McCann A, O’Gorman P, Bianchi G. Exploiting autophagy in multiple myeloma. J Cancer Metastasis Treat 2019;5:70. http://dx.doi.org/10.20517/2394-4722.2019.25
AMA Style
Ho M, Patel A, Hanley C, Murphy A, McSweeney T, Zhang L, McCann A, O’Gorman P, Bianchi G. Exploiting autophagy in multiple myeloma. Journal of Cancer Metastasis and Treatment. 2019; 5: 70. http://dx.doi.org/10.20517/2394-4722.2019.25
Chicago/Turabian Style
Ho, Matthew, Ashish Patel, Cathal Hanley, Adam Murphy, Tara McSweeney, Li Zhang, Amanda McCann, Peter O’Gorman, Giada Bianchi. 2019. "Exploiting autophagy in multiple myeloma" Journal of Cancer Metastasis and Treatment. 5: 70. http://dx.doi.org/10.20517/2394-4722.2019.25
ACS Style
Ho, M.; Patel A.; Hanley C.; Murphy A.; McSweeney T.; Zhang L.; McCann A.; O’Gorman P.; Bianchi G. Exploiting autophagy in multiple myeloma. J. Cancer. Metastasis. Treat. 2019, 5, 70. http://dx.doi.org/10.20517/2394-4722.2019.25
About This Article
Special Issue
Copyright

Data & Comments
Data


 Cite This Article 32 clicks
Cite This Article 32 clicks

 Like This Article 21
likes
Like This Article 21
likes






Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.