
Mouse tumor susceptibility genes identify drug combinations for multiple myeloma

Abstract
Long-term genetic studies utilizing backcross and congenic strain analyses coupled with positional cloning strategies and functional studies identified Cdkn2a, Mtor, and Mndal as mouse plasmacytoma susceptibility/resistance genes. Tumor incidence data in congenic strains carrying the resistance alleles of Cdkn2a and Mtor led us to hypothesize that drug combinations affecting these pathways are likely to have an additive, if not synergistic effect in inhibiting tumor cell growth. Traditional and novel systems-level genomic approaches were used to assess combination activity, disease specificity, and clinical potential of a drug combination involving rapamycin/everolimus, an Mtor inhibitor, with entinostat, an histone deacetylase inhibitor. The combination synergistically repressed oncogenic MYC and activated the Cdkn2a tumor suppressor. The identification of MYC as a primary upstream regulator led to the identification of small molecule binders of the G-quadruplex structure that forms in the NHEIII region of the MYC promoter. These studies highlight the importance of identifying drug combinations which simultaneously upregulate tumor suppressors and downregulate oncogenes.
Keywords
Introduction
The majority of human cancers arise in response to exposure to environmental factors and carcinogenic agents that may lead to somatic mutations. Signatures of these mutational processes are often evident in the sequences of cancer genomes[1]. Genetic and epigenetic factors also play an important role in determining which exposed individuals will develop tumors. Most tumor susceptibility models in humans and experimental animals have focused on the inherited abnormality of a single gene such as germline mutations of Rb or p53. These particular single locus lesions are predisposed to tumor formation because they harbor strong “altered function” alleles. However, it is estimated that such strong germline alleles may only account for approximately 2%-14% of human cancers which implies that another paradigm is required to explain the other 86%-98%[2]. The individuals in whom these latter cancers arise must either lack a germline genetic component, or tumor development in these individuals represents an inherited trait that may depend on several genes or epigenetic modifiers, in concert with environmental stressors, thus presenting cancer as a complex genetic trait.
Genome wide association studies of cancer development provide a systematic approach to identifying genes that may influence cancer risk[3]. Genome-wide linkage studies in genetically uniform strains of mice can provide a window into the more complex genetics associated with human cancers and may be used to model certain patient subpopulations. Thus in 1993, we chose to look at the inheritance of mouse plasmacytoma (PCT) susceptibility alleles associated with genetic variants segregating in immunocompetent backcross mice between BALB/c and DBA/2 strains of mice[4].
Experimental system: identification of mouse tumor susceptibility pathways to target
Human multiple myeloma (MM) is a clonal proliferative of neoplastic plasma cells in the bone marrow. Mouse plasma cell tumors model certain aspects of these antibody producing neoplasms. Plasmacytomagenesis in BALB/cAn mice is a complex genetic trait with 40%-60% penetrance in non-specific pathogen free mice[5]. Through our genome-wide mapping studies utilizing genetic crosses with DBA/2 mice (0% tumor incidence), together with the development and use of a series of C.D2 congenic strains, coupled with representational difference analysis and positional cloning, we determined that Cdkn2a (p16), Mtor, and Mndal contribute to PCT susceptibility and resistance [Figures 1 and 2][4-11]. Pctr1-2 are localized in non-contiguous, non-overlapping segments of mouse Chr 4, and Pctm, a modifier of PCT, on Chr 1. The two Pctr loci on Chr 4 are susceptibility loci in BALB mice while in DBA mice, they are resistance loci as evidenced by backcross and congenic strain analyses. The genes identified for Pctr1 and Pctr2 are, Cdkn2a (p16) and Mtor, respectively. The BALB alleles of both p16 and Mtor encode efficiency and hypomorphic alleles whose functional activities are much less active than the respective DBA alleles. In contrast to Pctr1 and Pctr2, the Pctm locus on Chr 1 encodes a resistance allele in BALB and a susceptibility allele in DBA. In fact, the candidate, Mndal, for the Pctm locus is deleted in DBA mice, but is present and functionally active in BALB mice[12].
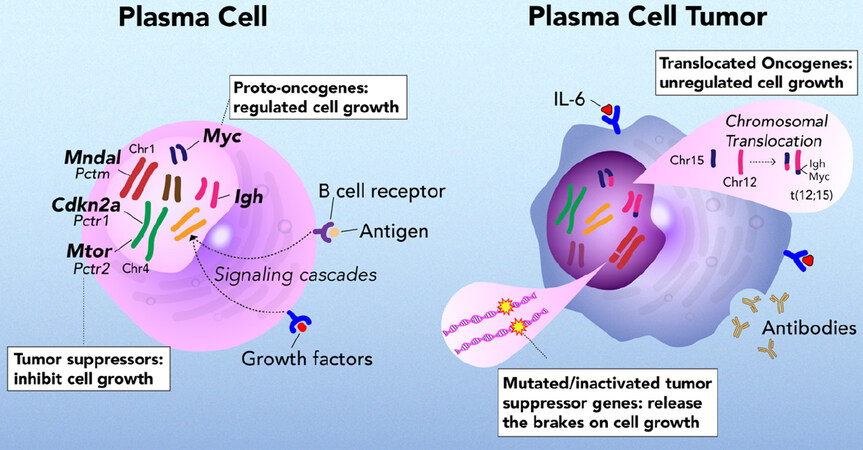
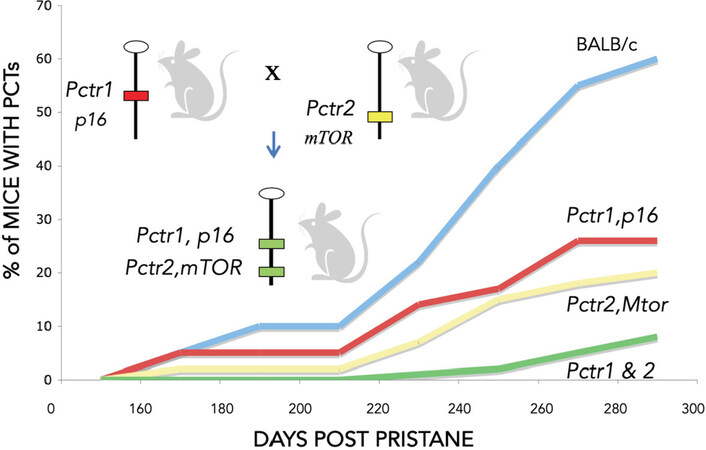
Figure 2. BALB/c congenic strains of mice carrying two (p16 and Mtor) DBA/2 plasmacytoma resistance alleles are more resistant to tumor formation than congenics carrying only one of the Pctr alleles
Compound allelic variation in both coding and promoter sequences, found in Cdkn2a [p16 exon 2: G232A in ANK repeat domain, RREB cis regulatory element (CRE)] and Mtor (exon 11: R628C in HEAT repeat domain; MZF1 CRE)[6,11,16,17], contribute to the complex genetics associated with PCT susceptibility in BALB/c mice[4,9]. Hypomorphic activity of the promoter and coding regions of the BALB alleles of both p16 and Mtor is associated with tumorigenesis after exposure to pristane, suggesting that both Cdkn2a (p16) and Mtor can act as tumor suppressors in PCT development in response to stress and in an allele-dependent manner[11,15-17].
BALB/c mice are susceptible because they harbor several tumor susceptibility loci that act in concert to produce the susceptible phenotype [Figure 2]. We hypothesize the combination of these relatively subtle allelic defects tip the balance toward both uncontrolled cell growth and a lack of appropriately timed cell death and removal from the cell cycle.
BALB/c congenic strains of mice carrying two tumor resistance alleles (Pctr1 and Pctr2) are more resistant than mice carrying only one of the resistance alleles [Figure 2]. This led us to hypothesize that drug combinations targeting these pathways are likely to have a cooperative effect in inhibiting tumor cell growth. The p16/Rb and Mtor/PI3K pathways are frequently dysregulated in both mouse plasma cell tumors and in human multiple myeloma[18-20].
Synergistic drug combination phenocopies resistance alleles
The activity of combining Mtor and histone deacetylase (HDAC) inhibitors, rapamycin and entinostat respectively, chosen to target the mouse tumor susceptibility pathways (p16/Rb and Mtor/PI3K) was found to be synergistic in limiting the growth of a number of B lineage tumor cell lines, including mouse plasma cell tumors, and the human B cell neoplasms, mantle cell lymphoma, and multiple myeloma[20]. We found that combining rapamycin and entinostat elicited responses distinct from a simple combination or the additive effects of the two drugs[19]. As such, we developed a rational, unbiased approach to uncover mechanisms of drug synergy for this combination.
Systems approach
We evaluated the synergistic activity of combining Mtor and HDAC inhibitors at the organismal, cellular, and molecular levels with a cross-disciplinary “systems pharmacology” approach [Figure 3][19]. While the future impact of these specific Mtor/HDAC findings is intrinsically linked to the outcome of clinical investigations, there is broader potential for further application and development of our approach. The integration of patient datasets in the identification of a core synergistic response signature offers particular opportunities for the development of companion diagnostics to aid in the clinical development of these combinations. Gene expression-based signatures of cooperative drug responses may prove beneficial for pre-treatment stratification of patients most likely to benefit from a particular drug combination, or as an early response biomarker specific for the combination response and intrinsically linked to expression patterns correlated with improved prognosis. Our approach in this study was enabled by the availability of high-quality, publicly available tumor gene expression datasets from large cohorts of myeloma patients that included either extensive survival annotation or comparisons of healthy vs. tumor tissues (GSE4581 and GSE6477)[21-25]. The schema depicted below illustrates the approach that we employed to understand the mechanism of drug synergy between the HDAC inhibitor, entinostat and an Mtor inhibitor, everolimus[19].
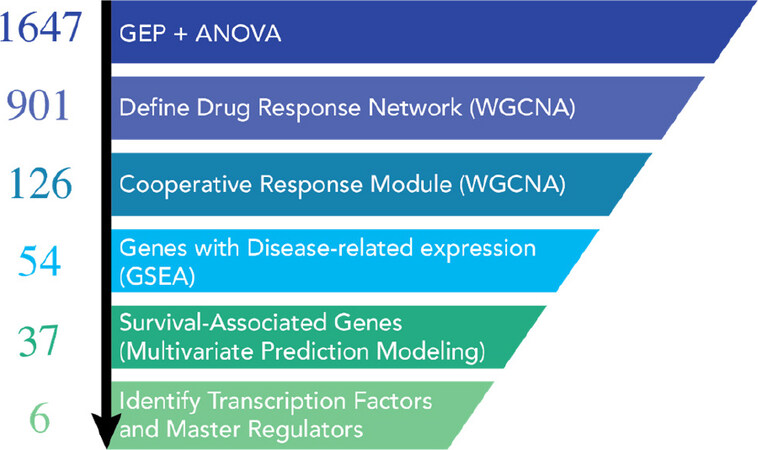
Figure 3. Graphical summary of the systems workflow used to dissect the mechanism of action for the mTOR inhibitor (mTORi) and HDAC inhibitor (HDACi) drug combination[19]. Initial ANOVA analysis from our gene expression profiling data started with 1647 differentially expressed genes. Weighted gene co-expression network analyses determined that there were 901 genes in the entire drug response network. Of these 901 genes, 126 genes could be assigned to the drug combination network. These genes were then evaluated for enrichment in myeloma vs. normal samples from the same patient (GEO databases) and by multivariate prediction modeling to assess their association with patient survival. 37 disease-specific genes were chosen for further analyses. When the data for the 37 genes (PatentUS2014357660-A1) was evaluated by IPA, 6 master regulators, including MYC, Rb, and Cdkn2a were identified. ANOVA: analysis of variance; GEO: gene expression omnibus; IPA: ingenuity pathway analysis
The upstream predictors identified as “activated” from the drug combination by ingenuity pathway analysis (IPA) from the 37 patient-survival associated genes included Cdkn2a (p16/p19), p53, and Rb. MYC, E2F, and TBX2 were predicted as “inhibited” by the combination. The combination worked cooperatively to lower MYC protein stability, partially through FBXW7-mediated degradation[19]. The combination also worked to increase the activity of the Rb1/Cdkn2a tumor suppressor pathways[20]. The drug combination enhanced the overall survival rate of tumor-bearing BALB-bclxl transgenic mice and lowered MYC protein levels in tumors of these immunocompetent mice. Our studies in the NCI-60 cell line panel found that most tumors, regardless of their tissue of origin, responded synergistically to the mTORi/HDACi combination. In early molecular classification schemes of multiple myeloma patients based on heirachical clustering of gene expression in myeloma samples, seven clusters were identifed as proliferation (PR), low bone, multiple myeloma SET domain (MMSET), hyperdiploid, cyclin D1 (CD-1), cyclin D2 (CD-2) and avian musculoaponeurotic fibrosarcoma (MAF)[21]. Using gene expression data from samples within these same subgroups, we determined a gene score for our 37 drug-responsive genes to predict how many patients would be expected to benefit from combination treatment. Roughly 50% in most subgroups were predicted to benefit; there were two exceptions: all patients in the PR group and only 17% in the CD-2 group were predicted to benefit from the drug combination based on their expression scores for the 37 gene signature[19]. This is of course, hypothetical and would need to be tested in a clinical trial. In addition, the drug combination did not have a direct effect on gene expression of genes involved in determining Zhan et al.[21]’s proliferation index. Cells with mutations in MYC residues required for its degradation did not respond to the drug combination[19].
Targeting MYC transcription and degradation
Our systems analyses led us to explore a more direct approach to targeting MYC. We screened a small molecule microarray library for binders of the G-quadruplex located in the NHEIII region of the MYC promoter, and identified a benzofuran-containing molecule, D089, that could stabilize the G4 structure and inhibit MYC transcription[26]. We demonstrated that D089 inhibited MYC with greater affinity than other G4-containing genes (e.g., RAS, VEGF, BCL2, and Rb1). The small molecule was relatively potent in inhibiting multiple myeloma cell proliferation but was ineffective in tumor cell lines that had deleted the portion of the MYC promoter containing the G4 sequence[26]. In subsequent studies, we analyzed a series of analogs to find one, DC-34, that was more potent in its activity against myeloma cells. In these studies, we were able to define an nuclear magnetic resonance structure of the small molecule bound to the G4 structure which should allow structure-guided design of even more potent compounds[27]. The discovery of a drug that directly targets MYC has been elusive, and thus far there are no approved drugs for this indication. The development of a more direct approach for inhibiting MYC activity seems warranted given the overall importance of MYC to a wide range of tumor types[28].
Our early work involving retroviral induction studies of mouse PCTs suggest that inhibition of MYC alone may not be curative[29]. Early induction studies with retroviral vectors clearly showed that overexpression of MYC alone could not induce PCTs; but when MYC was paired with RAS or RAF, high incidences of plasma cell tumors could be induced even in PCT-resistant strains of mice[29]. These data are in agreement with recent studies indicating that MYC mutations are acquired secondary genetic events in myeloma progression[30]. A key aspect of the mTORi/HDACi combination is its ability to decrease MYC protein stability; however, in some myeloma cell lines, we have observed a “compensatory” MYCmRNA increase with combination treatment, although the steady-state protein level is decreased[19,20]. Thus, developing a combinational approach to MYC inhibition by inhibiting both transcription and post-translational activity [Figure 4] might be more effective in providing a longer treatment window. Our drug combination studies also highlighted the importance of not only inhibiting MYC, but also up-regulating Rb1/Cdkn2a pathways, again suggesting that a MYC G4 stabilizer may not be effective as a single agent[19,20]. Combining a MYC inhibitor with agents that can upregulate the RB1/CDKN2A pathways, such as CDK or HDAC inhibitors or other chromatin modifiers, may ultimately be more effective[31,32].
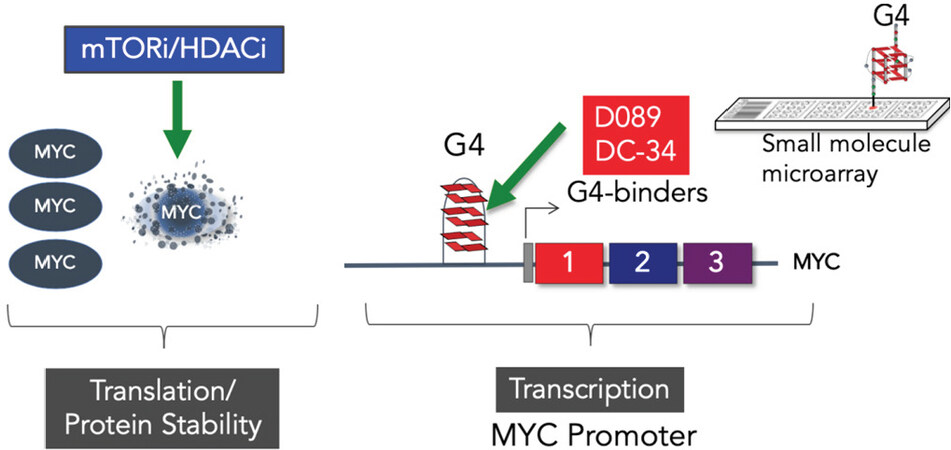
Figure 4. Drug combinations involving mTOR and HDAC inhibitors have a cooperative effect leading to MYC protein degradation. Small molecules targeting the G-quadruplex structure in the MYC promoter inhibit MYC transcription. HDAC: histone deacetylase; mTOR: mechanistic target of rapamycin
Conclusion
Since the initial sequencing of the human genome in 2001 and the myeloma genome in 2011, there has been a tremendous growth in the generation and availability of high-throughout MM omics datasets[33-35]. As a result of this, our knowledge and understanding of genetic underpinnings of MM tumor evolution has seen a similar expansion[36-38]. Tumor heterogeneity, both across different patients and between individual subclones within the same patient, has been shown to play an important role in MM disease progression, prognosis, and response to therapeutic treatments[36,39-41].
In a recent study by Maura et al.[42] serial whole genome sequencing (WGS) of 30 MM patients was collected and used to determine the chronological order of key driver events that occur during myeloma tumor evolution. In most patients, early driver events such as hyperdiploidy (including the characteristic trisomies of odd chromosomes), immunoglobulin translocation, and chromothripsis tended to precede whole genome duplication, chromoplexy, and point mutation events. In addition to these general patterns of driver event timings, Maura et al.[42] also found several examples of co-occurring or mutually exclusive events such as a co-occurrence between t(11;14) and t(14;16) chromosomal translocations and a mutually exclusive pattern of TRAF3 deletions with these same translocations. By combining the data from the 30 patients with an additional 804 patients from the MMRF CoMMpass trial[43], Maura et al.[42] were similarly able to detect important driver somatic mutations in MM, including well-known driver genes such as KRAS, NRAS, and DIS3, as well as novel putative driver mutations in genes encoding histone linkers (HIST1H1B, HIST1H1D, HIST1H1E, and HIST1H2BK), and mutations in or near genes involved in nucleosome binding.
Approximately 35%-40% of MM patients have IgH translocations (Chr 14), juxtaposed to an assortment of partners [MMSET (NSD2), FGFR2, MAF, CD-1 and D3 on other chromosomes (4, 6, 8, 11, 16 and 20)][44,45]. In contrast, about 80% of mouse PCTs carry translocations of the IgH locus on mouse Chr 12 juxtaposed to the MYC locus on Chr 15[13]. Many myeloma mouse genetically engineered models have focused on the dysregulation (overexpression or knock-out) of a particular gene or pathway, most notably, the dysregulation of MYC or BCL2[46,47], as well as the earlier spontaneous 5T models that have M spikes and develop bone lesions[48]. Adoptive B cell transfer mouse models also provide a novel approach to study MM pathogenesis[49]. Vlummens et al.[50] comprehensively reviewed numerous murine models, ranging from xenografts to immunocompetent spontaneous and transgenic models, for studying both the etiology and pathogenesis of MM. More recently, Rajagopalan et al.[51] generated a NrasLSL Q61R/+ mouse which takes advantage of crossing Vk*MYC mice to mice harboring a Q61R NRAS mutation (as found in WGS studies of myeloma)[35,42,46]. This rapid model also develops both bone lesions and M spikes.
In contrast, the focus of our studies has been on genetically inherited alleles of genes in immunocompetent strains of mice that predispose the mice to peritoneal plasmacytoma development. In the past several years, more than 17 risk loci for multiple myeloma susceptibility have now been mapped to unique regions of the human genome[52-57]. The one gene in common with our studies is the Cdkn2a locus; it is a tumor susceptibility gene in both mouse plasma cell tumors by genetic linkage studies[4,6,15] and in genome-wide association studies (GWAS) in human multiple myeloma[52]. Much progress has been made in understanding the omics of myeloma through GWAS[52-57], eQTL[58], and WGS[35,42] studies of myeloma patient samples. These studies have helped to identify new targets for intervention of myeloma disease progression and form the basis for developing companion diagnostics for drug treatments.
In our studies, we have viewed cancer treatment through the lens of cancer as a complex genetic trait by using pristane-induced mouse PCT as the model[4]. A goal in the molecular identification of these susceptibility/resistance genes has been to uncover the signaling pathways that are involved in promoting or controlling B cell neoplasia and to understand how these pathways may act in concert to contribute to or limit tumor progression. Tumor incidence data in congenic strains of mice, constructed to harbor different combinations of resistance alleles[5,9], led to the hypothesis that drug combinations affecting these pathways are likely to have at least an additive, if not synergistic effect in inhibiting tumor cell growth. We investigated experimental therapeutic approaches to target myeloma; that led to the twin goals of upregulating tumor suppressor activities and downregulating oncogenic processes simultaneously.
Our initial preclinical studies focused on Mtor inhibition, through targeting Mtor kinase activity, coupled with HDAC inhibition, which inhibits histone deacetylation. HDAC inhibitors can target a broad spectrum of genes involved in chromatin modification, including those that regulate the Rb1 and p16 pathways. Our mechanistic analysis of the successful targeting of these two pathways, which induced synergistic anti-tumor activity in susceptible tumors, identified MYC as an important upstream driver regulated by the combined pathways through their cooperative effects on MYC protein degradation. Drug combinations targeting the two signaling pathways (Cyclin D/CDK/Cdkn2a/Rb and PI3K/AKT/Mtor) identified by our genetic analysis of PCT susceptibility, were indeed synergistic in their activity, not only for myeloma, but also a variety of tumor types as shown in their broad synergistic activity in the panel of NCI-60 cell lines. In fact, the only NCI-60 cell line for which this combination was antagonistic had a mutation in FBXW7 which is involved in MYC protein degradation[19].
MYC is often overexpressed and/or dysregulated in cancer, including mouse PCT, as well as human myeloma and Burkitt’s lymphoma[30,59,60]. MYC has often been considered an undruggable target, yet many researchers are pursuing a number of avenues to downregulate MYC, including drug combinations such as the one described above to target post-translational steps, such as protein stability. In addition, MYC’s transcription factor activity requires dimerization with its binding partner MAX (MYC-associated factor x), and many efforts have focused on interrupting this complex to downregulate its transcription factor activity[61]. Furthermore, JQ1, a BET (bromodomain and extra-terminal domain) inhibitor, can also inhibit MYC transcription, as well as other pathways[62]. We and others have focused on interfering with MYC gene transcription by an alternative inhibitory mechanism involving small molecules that stabilize complex DNA structures (G-quadruplexes) which form transiently in the MYC promoter[26,27,63,64].
Our studies to identify cooperative targets of mTORi/HDACi inhibition have: (1) provided a basic approach for broader application to identify potential biomarkers of drug combinations utilizing weighted gene coexpression network analyses combined with gene set enrichment analyses of survival annotated patient datasets; (2) identified upstream regulators/drivers of drug responses leading to a mechanistic understanding of how the combination is acting (upregulation of tumor suppressive pathways (Rb1 and p16) and downregulation of oncogenic pathways (MYC and E2F1), leading to MYC degradation; and (3) demonstrated that cell lines carrying mutations in FBXW7 or surrounding MYC residues Thr58,Ser62, involved in MYC degradation are not likely to respond to the combination of rapamycin/everolimus and entinostat. While MYC is known to be deregulated in a majority of cancers, its direct drug targeting has been elusive. We hope that our work to identify and develop a new class of compounds targeting MYC transcription will lead to new pharmacological agents for MYC inhibition. Our studies suggest that it would be clinically useful if these inhibitors were coupled with drugs that simultaneously upregulate tumor suppressors. Our studies are designed to provide the pre-clinical rationale and evidence of synergistic mechanisms required to advance candidate combinations for preclinical assessment in patient-derived cells and eventually in clinical study.
Declarations
AcknowledgmentsWe wish to thank our many collaborators and lab members for their contributions to the primary research referenced in this review: Douglas Lowy, Aleksandra Michalowski, Jyoti Patel, Patrick Sullivan, John S. Schneekloth, Jr., David Calabrese, Michael Kuehl, Ola Landgren, Robert Hawley, Anaisa Quintanilla-Arteaga, Patricia Wiley, Nana Gayabah-Kessie, Kenneth Felsenstein, Elena Leon, Zaw Phyo, Daniel Connors, Ben Gamache, Nick Watson, Max Eiden, Matt Steinsaltz, Maudeline Etienne, Joy Gary, Edward Ramsey, Marianne Krall, Cecilia Padlan, Danielle Holiday, Jenefer Dosik, Margaret Pruitt, Dena Tran, Siegfried Janz, Alexander Kovalchuk, Elizabeth Mushinski, Mari Hirano, Rania Salem, Jing Huang, and R. Mark Simpson. We also thank Richard Nordan, Michael Potter, J. Frederic Mushinski, and Valery Bliskovsky (in memoriam) for their many contributions to these studies.
Authors’ contributionsMade conceptual contributions to the manuscript: Zhang S, DuBois W, Zhang K, Simmons JK, Hughitt VK, Gorjifard S, Gaikwad S, Peat TJ, Mock BA
Wrote the manuscript: Simmons JK, Hughitt VK, Gorjifard S, Gaikwad S, Peat TJ, Mock BA
Designed several of the figures: Gorjifard S
Availability of data and materialsNot applicable.
Financial support and sponsorshipThis work was supported by the Intramural Research Program of the National Institutes of Health, National Cancer Institute, Center for Cancer Research and the MMRF (Multiple Myeloma Research Foundation).
Conflicts of interestAll authors declared that there are no conflicts of interest.
Ethical approval and consent to participateNot applicable.
Consent for publicationNot applicable.
Copyright© The Author(s) 2020.
REFERENCES
1. Martincorena I, Campbell PJ. Somatic mutation in cancer and normal cells. Science 2015;349:1483-9.
2. Park S, Supek F, Lehner B. Systematic discovery of germline cancer predisposition genes through the identification of somatic second hits. Nat Commun 2018;9:2601.
3. Sud A, Kinnersley B, Houlston RS. Genome-wide association studies of cancer: current insights and future perspectives. Nat Rev Cancer 2017;17:692-704.
4. Mock BA, Krall MM, Dosik JK. Genetic mapping of tumor susceptibility genes involved in mouse plasmacytomagenesis. Proc Natl Acad Sci USA 1993;90:9499-503.
5. Mock BA, Hartley J, Le Tissier P, Wax JS, Potter M. The plasmacytoma resistance gene, Pctr2, delays the onset of tumorigenesis and resides in the telomeric region of chromosome 4. Blood 1997;90:4092-8.
6. Zhang S, Ramsay ES, Mock BA. Cdkn2a, the cyclin dependent kinase inhibitor encoding p16INK4a and p19ARF is a candidate for the plasmacytoma susceptibility locus, Pctr1. Proc Natl Acad Sci USA 1998;95:2429-34.
7. Zhang S, Mock BA. The role of p16INK4a (Cdkn2a) in mouse plasma cell tumors. Curr Top Microbiol Immunol 1999;246:363-7.
8. Potter M, Mushinski EB, Wax JS, Hartley J, Mock BA. Identification of two genes on chromosome 4 determine resistance to plasmacytoma induction in mice. Cancer Res 1994;54:969-75.
9. Mock BA, Zhang S, Ramsay ES, Bliskovski V, Zhang K, et al. Strategies for dissecting complex traits associated with cancer: lessons from plasma cell tumors. AACR Education Book 2005;2005:273-6.
10. Krall M, Ruff N, Zimmerman K, Aggarwal A, Dosik J, et al. Isolation and mapping of four new DNA markers from mouse chromosome 4. Mamm Genome 1992;3:653-5.
11. Bliskovsky V, Ramsay ES, Scott J, DuBois W, Shi W, et al. Frap, FKBP12 rapamycin-associated protein, is a candidate gene for the plasmacytoma resistance locus Pctr2 and can act as a tumor suppressor gene. Proc Natl Acad Sci USA 2003;100:14982-7.
12. Zhang K, Kagan D, DuBois W, Robinson R, Bliskovsky V, et al. Mndal, a new interferon-inducible family member, is highly polymorphic, suppresses cell growth, and may modify plasmacytoma susceptibility. Blood 2009;114:2952-60.
13. Janz S. Genetic and environmental cofactors of Myc translocations in plasma cell tumor development in mice. J Natl Cancer Inst Monogr 2008:37-40.
14. Dib A, Gabrea A, Glebov OK, Bergsagel PL, Kuehl WM. Characterization of MYC translocations in multiple myeloma cell lines. J Natl Cancer Inst Monogr 2008:25-31.
15. Zhang SL, DuBois W, Ramsay ES, Bliskovski V, Morse HC 3rd, et al. Efficiency alleles of the Pctr1 modifier locus for plasmacytoma susceptibility. Mol Cell Biol 2001;21:310-8.
16. Zhang S, Qian X, Redman C, Bliskovski V, Ramsay ES, et al. p16 INK4a gene promoter variation and differential binding of a repressor, the ras-responsive zinc-finger transcription factor, RREB. Oncogene 2003;22:2285-95.
17. Zhang S, Shi W, Ramsay ES, Bliskovsky V, Eiden AM, et al. The transcription factor MZF1 differentially regulates murine Mtor promoter variants linked to tumor susceptibility. J Biol Chem 2019;294:16756-64.
18. Boyle EM, Davies FE, Leleu X, Morgan GJ. Understanding the multiple biological aspects leading to myeloma. Haematologica 2014;99:605-12.
19. Simmons JK, Michalowski AM, Gamache BJ, DuBois W, Patel J, et al. Cooperative targets of combined mTOR/HDAC inhibition promote MYC degradation. Mol Cancer Ther 2017;16:2008-21.
20. Simmons JK, Patel J, Michalowski A, Zhang S, Wei BR, et al. TORC1 and class I HDAC inhibitors synergize to suppress mature B cell neoplasms. Mol Oncol 2014;8:261-72.
21. Zhan F, Huang Y, Colla S, Stewart JP, Hanamura I, et al. The molecular classification of multiple myeloma. Blood 2006;108:2020-8.
22. Chng WJ, Kumar S, Vanwier S, Ahmann G, Price-Troska T, et al. Molecular dissection of hyperdiploid multiple myeloma by gene expression profiling. Cancer Res 2007;67:2982-9.
23. Tiedemann RE, Zhu YX, Schmidt J, Yin H, Shi CX, et al. Kinome-wide RNAi studies in human multiple myeloma identify vulnerable kinase targets, including a lymphoid-restricted kinase, GRK6. Blood 2010;115:1594-604.
24. Chng WJ, Gertz MA, Chung TH, Van Wier S, Keats JJ, et al. Correlation between array-comparative genomic hybridization-defined genomic gains and losses and survival: identification of 1p31-32 deletion as a prognostic factor in myeloma. Leukemia 2010;24:833-42.
25. Chng WJ, Huang GF, Chung TH, Ng SB, Gonzalez-Paz N, et al. Clinical and biological implications of MYC activation: a common difference between MGUS and newly diagnosed multiple myeloma. Leukemia 2011;25:1026-35.
26. Felsenstein KM, Saunders LB, Simmons JK, Leon E, Calabrese DR, et al. Small molecule microarrays enable the identification of a selective, quadruplex-binding inhibitor of MYC expression. ACS Chem Biol 2016;11:139-48.
27. Calabrese DR, Chen X, Leon EC, Gaikwad SM, Phyo Z, et al. Chemical and structural studies provide a mechanistic basis for recognition of the MYC G-quadruplex. Nat Commun 2018;9:4229.
28. Grandori C, Kemp CJ. Personalized cancer models for target discovery and precision Medicine. Trends Cancer 2018;4:634-42.
29. Mock B, Wax J, Clynes R, Marcu KB, Potter M. The genetics of susceptibility to RIM-induced plasmacytomagenesis. Curr Top Microbiol Immunol 1988;141:125-7.
30. Misund K, Keane N, Stein CK, Asmann YW, Day G, et al. MYC dysregulation in the progression of multiple myeloma. Leukemia 2020;34:322-6.
31. Kadoch C, Copeland RA, Keilhack H. PRC2 and SWI/SNF chromatin remodeling complexes in health and disease. Biochemistry 2016;55:1600-14.
32. Orlando KA, Nguyen V, Raab JR, Walhart T, Weissman BE. Remodeling the cancer epigenome: mutations in the SWI/SNF complex offer new therapeutic opportunities. Expert Rev Anticancer Ther 2019;19:375-91.
33. Lander ES, Linton LM, Birren B, Nusbaum C, Zody MC, et al. Initial sequencing and analysis of the human genome. Nature 2001;409:860-921.
34. Venter JC, Adams MD, Myers EW, Li PW, Mural RJ, et al. The sequence of the human genome. Science 2001;291:1304-51.
35. Chapman MA, Lawrence MS, Keats JJ, Cibulskis K, Sougnez C, et al. Initial genome sequencing and analysis of multiple myeloma. Nature 2011;471:467-72.
36. Schürch CM, Rasche L, Frauenfeld L, Weinhold N, Fend F. A review on tumor heterogeneity and evolution in multiple myeloma: pathological, radiological, molecular genetics, and clinical integration. Virchows Arch 2020;476:337-51.
37. Rustad EH, Yellapantula V, Leongamornlert D, Bolli N, Ledergor G, et al. Timing the initiation of multiple myeloma. Nat Commun 2020;11:1917.
38. Maura F, Rustad EH, Boyle EM, Morgan GJ. Reconstructing the evolutionary history of multiple myeloma. Best Pract Res Clin Haematol 2020;33:101145.
39. Rasche L, Kortüm KM, Raab MS, Weinhold N. The impact of tumor heterogeneity on diagnostics and novel therapeutic strategies in multiple myeloma. Int J Mol Sci 2019;20:1248.
40. Merz M, Jauch A, Hielscher T, Bochtler T, Schönland SO, et al. Prognostic significance of cytogenetic heterogeneity in patients with newly diagnosed multiple myeloma. Blood Adv 2017;2:1-9.
41. Ledergor G, Weiner A, Zada M, Wang SY, Cohen YC, et al. Single cell dissection of plasma cell heterogeneity in symptomatic and asymptomatic myeloma. Nat Med 2018;24:1867-76.
42. Maura F, Bolli N, Angelopoulos N, Dawson KJ, Leongamornlert D, et al. Genomic landscape and chronological reconstruction of driver events in multiple myeloma. Nat Commun 2019;10:3835.
43. Lonial S, Yellapantula VD, Liang W, Kurdoglu A, Aldrich J, et al. Interim analysis of the mmrf commpass trial: identification of novel rearrangements potentially associated with disease initiation and progression. Blood 2014;124:722.
44. Kuehl WM, Bergsagel PL. Multiple myeloma: evolving genetic events and host interactions. Nat Rev Cancer 2002;2:175-87.
45. Stewart AK, Bergsagel PL, Greipp PR, Dispenzieri A, Gertz MA, et al. A practical guide to defining high-risk myeloma for clinical trials, patient counseling and choice of therapy. Leukemia 2007;21:529-34.
46. Chesi M, Robbiani DF, Sebag M, Chng WJ, Affer M, et al. AID-dependent activation of a MYC transgene induces multiple myeloma in a conditional mouse model of post-germinal center malignancies. Cancer Cell 2008;13:167-80.
47. Cheung WC, Kim JS, Linden M, Peng L, Van Ness B, et al. Novel targeted deregulation of c-Myc cooperates with Bcl-X(L) to cause plasma cell neoplasms in mice. J Clin Invest 2004;113:1763-73.
48. Radl J, Croese JW, Zurcher C, Van den Enden-Vieveen MH, de Leeuw AM. Animal model of human disease. Multiple myeloma. Am J Pathol 1988;132:593-7.
49. Tompkins VS, Rosean TR, Holman CJ, DeHoedt C, Olivier AK, et al. Adoptive B-cell transfer mouse model of human myeloma. Leukemia 2016;30:962-6.
50. Vlummens P, De Veirman K, Menu E, De Bruyne E, Offner F, et al. The use of murine models for studying mechanistic insights of genomic instability in multiple myeloma. Front Genet 2019;10:740.
51. Rajagopalan A, Wen Z, Furumo Q, Ranheim E, Finn R, et al. Mice expressing MYC and NrasQ61R in germinal venter B vells fevelop highly aggressive multiple myeloma. Blood 2018;132:1006.
52. Mitchell JS, Li N, Weinhold N, Forsti A, Ali M, et al. Genome-wide association study identifies multiple susceptibility loci for multiple myeloma. Nat Commun 2016;7:12050.
53. Morgan GJ, Johnson DC, Weinhold N, Goldschmidt H, Landgren O, et al. Inherited genetic susceptibility to multiple myeloma. Leukemia 2014;28:518-24.
54. Swaminathan B, Thorleifsson G, Jöud M, Ali M, Johnsson E, et al. Variants in ELL2 influencing immunoglobulin levels associate with multiple myeloma. Nat Commun 2015;6:7213.
55. Chubb D, Weinhold N, Broderick P, Chen B, Johnson DC, et al. Common variation at 3q26.2, 6p21.33, 17p11.2 and 22q13.1 influences multiple myeloma risk. Nat Genet 2013;45:1221-5.
56. Weinhold N, Johnson DC, Chubb D, Chen B, Försti A, et al. The CCND1.c.870G>A polymorphism is a risk factor for t(11;14)(q13;q32) multiple myeloma. Nat Genet 2013;45:522-5.
57. Broderick P, Chubb D, Johnson DC, Weinhold N, Försti A, et al. Common variation at 3p22.1 and 7p15.3 influences multiple myeloma risk. Nat Genet 2011;44:58-61.
58. Chattopadhyay S, Thomsen H, Yadav P, da Silva Filho MI, Weinhold N, et al. Genome-wide interaction and pathway-based identification of key regulators in multiple myeloma. Commun Biol 2019;2:89.
59. Walker BA, Wardell CP, Brioli A, Boyle E, Kaiser MF, et al. Translocations at 8q24 juxtapose MYC with genes that harbor superenhancers resulting in overexpression and poor prognosis in myeloma patients. Blood Cancer J 2014;4:e191.
60. Li ZR, Van Calcar S, Qu C, Cavenee WK, Zhang MQ, et al. A global transcriptional regulatory role for c-Myc in Burkitt’s lymphoma cells. Proc Natl Acad Sci USA 2003;100:8164-9.
61. Mathsyaraja H, Freie B, Cheng PF, Babaeva E, Catchpole JT, et al. Max deletion destabilizes MYC protein and abrogates Emicro-Myc lymphomagenesis. Genes Dev 2019;33:1252-64.
62. Filippakopoulos P, Qi J, Picaud S, Shen Y, Smith WB, et al. Selective inhibition of BET bromodomains. Nature 2010;468:1067-73.
63. Balasubramanian S, Hurley LH, Neidle S. Targeting G-quadruplexes in gene promoters: a novel anticancer strategy? Nat Rev Drug Discov 2011;10:261-75.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Zhang S, DuBois W, Zhang K, Simmons JK, Hughitt VK, Gorjifard S, Gaikwad S, Peat TJ, Mock BA. Mouse tumor susceptibility genes identify drug combinations for multiple myeloma. J Cancer Metastasis Treat 2020;6:21. http://dx.doi.org/10.20517/2394-4722.2020.40
AMA Style
Zhang S, DuBois W, Zhang K, Simmons JK, Hughitt VK, Gorjifard S, Gaikwad S, Peat TJ, Mock BA. Mouse tumor susceptibility genes identify drug combinations for multiple myeloma. Journal of Cancer Metastasis and Treatment. 2020; 6: 21. http://dx.doi.org/10.20517/2394-4722.2020.40
Chicago/Turabian Style
Zhang, Shuling, Wendy DuBois, Ke Zhang, John K. Simmons, V. Keith Hughitt, Sayeh Gorjifard, Snehal Gaikwad, Tyler J. Peat, Beverly A. Mock. 2020. "Mouse tumor susceptibility genes identify drug combinations for multiple myeloma" Journal of Cancer Metastasis and Treatment. 6: 21. http://dx.doi.org/10.20517/2394-4722.2020.40
ACS Style
Zhang, S.; DuBois W.; Zhang K.; Simmons JK.; Hughitt VK.; Gorjifard S.; Gaikwad S.; Peat TJ.; Mock BA. Mouse tumor susceptibility genes identify drug combinations for multiple myeloma. J. Cancer. Metastasis. Treat. 2020, 6, 21. http://dx.doi.org/10.20517/2394-4722.2020.40
About This Article
Copyright

Data & Comments
Data


 Cite This Article 6 clicks
Cite This Article 6 clicks

 Like This Article 38
likes
Like This Article 38
likes






Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.