
Targeting the JAK/STAT pathway in solid tumors

Abstract
Aberrant activation of signal transducer and activator of transcription (STAT) proteins is associated with the development and progression of solid tumors. However, as transcription factors, these proteins are difficult to target directly. In this review, we summarize the role of targeting Janus kinases (JAKs), upstream activators of STATs, as a strategy for decreasing STAT activation in solid tumors. Preclinical studies in solid tumor cell line models show that JAK inhibitors decrease STAT activation, cell proliferation, and cell survival; in in vivo models, they also inhibit tumor growth. JAK inhibitors, particularly the JAK1/2 inhibitor ruxolitinib, sensitize cell lines and murine models to chemotherapy, immunotherapy, and oncolytic viral therapy. Ten JAK inhibitors have been or are actively being tested in clinical trials as monotherapy or in combination with other agents in patients with solid tumors; two of these inhibitors are already Food and Drug Administration (FDA) approved for the treatment of myeloproliferative disorders and rheumatoid arthritis, making them attractive agents for use in patients with solid tumors as they are known to be well-tolerated. Four JAK inhibitors (two of which are FDA approved for other indications) have exhibited promising anti-cancer effects in preclinical studies; however, clinical studies specifically assessing their activity against the JAK/STAT pathway in solid tumors have not yet been conducted. In summary, JAK inhibition is a viable option for targeting the JAK/STAT pathway in solid tumors and merits further testing in clinical trials.
Keywords
Introduction
The Janus kinase/signal transducer and activator of transcription (JAK/STAT) signaling pathway is implicated in the development and progression of many cancers[1,2]. Hyperactivation of STAT transcription factors, has been reported in both hematologic malignancies and solid tumors, including cancers of the breast, lung, liver, head and neck, and stomach, among others[3-8]. For many of these cancers, increased activation of the JAK/STAT signaling pathway is associated with a worse prognosis, including increased recurrence and reduced overall survival[1,9,10]. Given the strong association between JAK/STAT hyperactivity and the development and prognosis of multiple cancers, STATs and their upstream activators, JAKs, are being extensively explored as targets for cancer therapy[1,11-13].
Certain hematologic malignancies such as myeloproliferative neoplasms are associated with specific JAK mutations that serve as predictive biomarkers for JAK-targeted therapy[14]. The majority of cases of polycythemia vera, essential thrombocytopenia, and myelofibrosis are characterized by an activating valine to phenylalanine mutation in JAK2 (JAK2 V617F) that drives the development of these neoplasms[15]. Clinical trials studying the impact of ruxolitinib, a selective JAK1/2 inhibitor, on polycythemia vera and myelofibrosis demonstrated significant improvement in patient outcomes, leading to Food and Drug Administration (FDA) approval and widespread use of this agent for these diseases[16-22]. However, mutations in the JAK/STAT pathway are rare in solid tumors, and the role of JAK and/or STAT inhibitors for the treatment of solid tumors is incompletely understood. In this review, we describe the rationale for targeting the JAK/STAT pathway in solid tumors and summarize preclinical studies and clinical trials to date that evaluate the impact of agents targeting this pathway.
JAK/STAT signaling
Ligands, particularly cytokines and growth factors, provide the initial stimulus for activating the JAK/STAT pathway[23]. Cytokines bind to their corresponding transmembrane receptor subunits, resulting in multimerization with other subunits and close physical interactions of receptor-associated JAKs[24]. The JAK family of tyrosine kinases consists of JAK1, JAK2, JAK3, and TYK2[25]. Once the receptor-associated JAKs are placed in close proximity, they become activated via trans-phosphorylation[24]. Activated JAKs phosphorylate tyrosine residues on the cytoplasmic region of the cytokine receptor to provide docking sites for the Src Homology 2 (SH2) domain of STAT proteins. The binding of a member of the STAT family of proteins (STAT1, STAT2, STAT3, STAT4, STAT5a, STAT5b, and STAT6) to the phosphorylated receptor intracellular domain results in JAK-mediated tyrosine phosphorylation and activation of the STAT protein [Figure 1][26]. In the case of receptors with intrinsic tyrosine kinase activity (e.g., epidermal growth factor receptor, EGFR), ligand binding results in receptor autophosphorylation of tyrosine residues which serve as the docking sites for STATs, and the bound STATs are directly phosphorylated/activated by the receptor tyrosine kinase. Activated STATs dimerize and translocate into the nucleus where they serve as transcription factors, inducing the expression of genes that regulate cellular proliferation, survival, and invasion, as well as the host immune response[14,24,27,28].
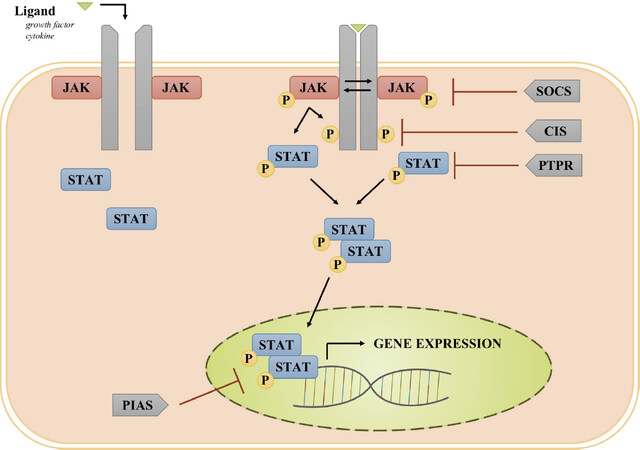
Figure 1. JAK/STAT pathway involving receptors lacking intrinsic tyrosine kinase activity: Upon ligand binding, transmembrane cytokine receptors multimerize, bringing receptor-associated JAKs into close physical proximity. Once activated via transphosphorylation, JAKs phosphorylate the cytoplasmic domain of the receptor to provide a docking site for STAT. The bound STATs are then phosphorylated and activated by JAKs. Activated STATs dimerize and translocate into the nucleus where act as transcription factors. Suppressors of cytokine signaling (SOCS) family of proteins inhibit JAK activation; cytokine-inducible SH2-containing protein (CIS) blocks the STAT docking site on the receptor; Protein inhibitor of activated STAT (PIAS) proteins inhibit STAT binding to promoter regions of target genes; and protein tyrosine phosphatase receptors (PTPRs) dephosphorylate STATs. JAKs: Janus kinases; STAT: signal transducer and activator of transcription
The JAK/STAT signaling pathway is modulated by several negative regulators[29]. Members of the suppressors of cytokine signaling (SOCS) family of proteins, such as SOCS1 and SOCS3, are inhibitory against JAKs, while cytokine-inducible SH2-containing protein (CIS) blocks STAT binding sites on receptor proteins[29,30]. Another family of inhibitory molecules, the protein inhibitor of activated STAT (PIAS) proteins, inhibit the binding of STATs to response elements in target genes[29,31]. Protein tyrosine phosphatase receptors (PTPRs), specifically PTPRT, PTPRD, and PTPRK, have been shown to dephosphorylate STAT3, resulting in STAT3 inactivation; a handful of non-receptor PTPs harbor a similar function[32-37]. Increased activity of JAKs and STATs coupled with decreased activity of negative regulators can lead to an upregulation of pro-proliferative, anti-apoptotic, and immunosuppressive proteins, potentially driving oncogenesis.
Dysregulation of the JAK/STAT pathway in solid tumors
Hyperactivation of STAT3
Hyperactivation of STATs, particularly STAT3, has been implicated in many cancers. Upstream JAK2 V617F mutations in myeloproliferative diseases and STAT3 mutations in T-cell large granular lymphocytic leukemia provide mechanisms for STAT3 hyperactivity in hematological malignancies[15,38,39]. JAK1 mutations have been identified in hepatocellular carcinoma (HCC) patient tumors; patient-derived xenografts with JAK1 S703I mutations had elevated levels of phosphorylated STAT3 and STAT5[40,41]. However, for most cases of solid tumors, activating mutations in this pathway have not been identified[42].
In most solid tumors associated with hyperactivation of STAT3, disease development and progression has been attributed to either increased cytokine signaling or inhibition of negative regulators of the JAK/STAT pathway[42,43]. In head and neck cancers (HNC), aberrant activation of STAT3, often through elevated IL-6 levels in the tumor microenvironment, is associated with increased tumor cell proliferation, survival, and metastasis, as well as immunosuppression of tumor-infiltrating immune cells[44-46]. As in HNC, gastric cancer cell lines exhibit IL-6-mediated STAT3 activation, which leads to increased cell survival and epithelial to mesenchymal transition in vitro[47,48]. Gastric cancer tumors were also found to have increased levels of phosphorylated STAT3 compared to healthy tissue[49]. In non-small cell lung cancer (NSCLC), secretion of oncostatin-M (OSM), a member of the IL-6 cytokine family, by cancer-associated fibroblasts increases STAT3 activity through activation of JAK1 and is a possible mechanism of resistance to targeted therapy such as EGFR and MEK inhibitors[50]. STAT3 hyperactivity seen in pancreatic cancers has been associated with increased IL-22-induced STAT3 signaling and SOCS3 suppression, leading to increased invasion, migration, and angiogenesis[51-53]. PTPRT, another negative regulator of the JAK/STAT pathway, is silenced via promoter hypermethylation in many cases of HNC and provides a likely mechanism for STAT3 hyperactivation in this cancer[54]. Loss-of-function mutations in PTPRD have also been implicated[55]. Hyperactivation of STAT3 has been reported in many other solid tumor malignancies, including breast cancer, HCC, and ovarian cancer, among others[56-58].
Hyperactivation of other STATs
While less common, hyperactivation of other members of the STAT protein family has been shown in some solid tumors. STAT1 drives aromatase inhibitor resistance in breast cancer, and is highly expressed in estrogen receptor-positive, tamoxifen-resistant breast cancer cell lines, indicating it may be a promising target in this malignancy[59]. STAT2 is not only highly expressed in ovarian cancer compared to normal ovarian tissue, but is also associated with metastasis and poor overall survival[60]. STAT2 is also associated with poor overall survival in NSCLC[61]. Hyperactivity of STAT5 is associated with enhanced cell viability, tumor growth, and recurrence in prostate cancers[62,63]. In colorectal cancer cell lines, elevated levels of activated STAT6 are correlated with metastasis and decreased apoptosis[64].
Collectively, there is ample evidence showing that increased JAK/STAT signaling is associated with increased cell proliferation, cell survival, immune evasion, recurrence, and drug resistance in solid tumors; this pathway therefore represents a promising target for therapeutic intervention.
JAK inhibitors
While hyperactivation of STATs, primarily STAT3, has been linked to the development and progression of solid tumors, STATs, similar to other transcription factors, have proven difficult to target directly. Therefore, upstream activators of STATs, such as JAKs, have been studied in preclinical and clinical settings as potential therapeutic targets. Several JAK inhibitors have been studied in solid tumors. Figure 2 depicts JAK inhibitors that are: (1) FDA approved and have been tested clinically in solid tumors [Figure 2A]; (2) not FDA approved, but have been tested clinically in solid tumors [Figure 2B]; and (3) have only been tested in solid tumor preclinical models. One multitarget agent (lestaurtinib) has been tested clinically for its activity against other targets [Figure 2C]. To date, there are 10 JAK inhibitors (two of which are FDA approved for other indications) that have been or are currently being investigated across 45 clinical trials in patients with solid tumors (excluding trials that have been withdrawn or in which JAK inhibitor was standard of care in studies investigating other agents) [Table 1]. Some compounds, a few of which are also FDA approved for other indications, have to date only been studied in solid tumor preclinical models.
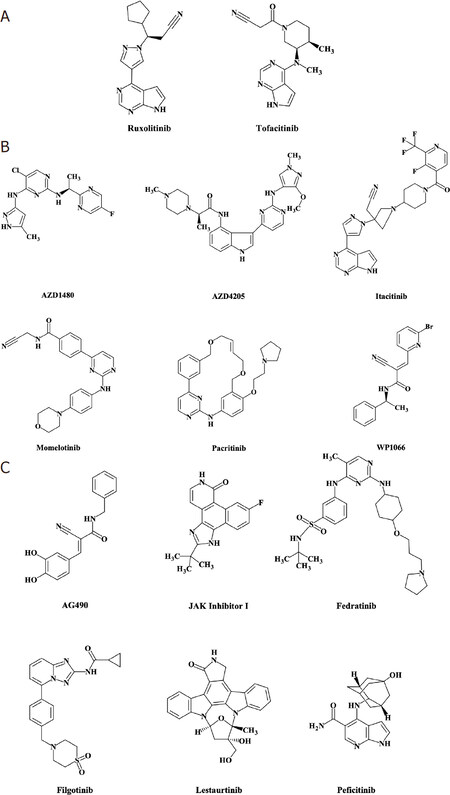
Figure 2. JAK inhibitors: chemical structures for JAK inhibitors described in this review were created using MarvinSketch software downloaded from ChemAxon (Budapest, Hungary)[115,171-183]. Food and Drug Administration (FDA)-approved JAK inhibitors that have been tested clinically in solid tumors (A); JAK inhibitors that are not FDA approved but have been tested clinically in solid tumors (B); JAK inhibitors that have only been tested in solid tumor preclinical models, and lestaurtinib, which has been tested clinically in solid tumors for its activity against other targets (C). Structures for INCB047986 and INCB052793 are not publicly available and therefore are not shown in this figure. *FDA approved for another indication; JAKs: Janus kinases
JAK inhibitors tested clinically in solid tumors
| Inhibitor | Direct Target(s) | NCT# | Type(s) of solid tumor | Status | Outcome |
|---|---|---|---|---|---|
| Ruxolitinib* | JAK1/2 | NCT01423604 | PDAC | Completed | Improved overall survival in subgroup of patients with inflammation[89] |
| NCT02117479 | PDAC | Terminated | No overall survival benefit[90] | ||
| NCT02119663 | PDAC | Terminated | No overall survival benefit[90] | ||
| NCT02120417 | BC | Terminated | Favorable HRQoL, no overall survival benefit[91] | ||
| NCT01562873 | BC | Terminated | No tumor response[92] | ||
| NCT01594216 | BC | Completed | None published | ||
| NCT02119676 | CRC | Terminated | No overall survival benefit[94] | ||
| NCT02119650 | NSCLC | Terminated | Unable to interpret efficacy[95] | ||
| NCT02145637 | NSCLC | Completed | 23.3% PR, 70.0% SD[96] | ||
| NCT02155465 | Lung adenocarcinoma | Completed | Lack of efficacy[97] | ||
| NCT01822756 | Advanced solid tumors | Terminated | Unable to interpret efficacy[98] | ||
| NCT00638378 | PC | Terminated | Lack of clinical response | ||
| NCT02955940 | PDAC, CRC, BC, NSCLC | Active | |||
| NCT03153982 | HNC | Active | |||
| NCT02928978 | Premalignant breast disease | Active | |||
| NCT03514069 | High-grade gliomas | Active | |||
| NCT04303403 | CRC, PDAC | Active | |||
| NCT03012230 | BC | Active | |||
| NCT02876302 | IBC | Active | |||
| NCT02041429 | IBC | Active | |||
| NCT02066532 | BC | Active | |||
| NCT02713386 | OC, fallopian tube cancer, peritoneal cancer | Active | |||
| NCT02788201 | UC | Completed | None published | ||
| Tofacitinib* | JAK1 | NCT04034238 | Epithelioid mesothelioma, cholangiocarcinoma, PDAC | Active | |
| AZD1480 | JAK1/2 | NCT01112397 | Advanced solid tumors, not specified | Terminated | pSTAT3 inhibition in granulocytes, neurotoxicity in patients[110] |
| NCT01219543 | HCC, NSCLC, GC | Terminated | None published | ||
| AZD4205 | JAK1 | NCT03450330 | NSCLC | Completed | None published |
| INCB047986 | JAK1 | NCT01929941 | PDAC, BC, non-specified advanced solid tumors | Terminated | None published |
| INCB052793 | JAK1 | NCT02265510 | Non-specified advanced solid tumors | Terminated | Lack of efficacy |
| Itacitinib | JAK1 | NCT01858883 | Variety (84% PDAC) | Completed | Lack of efficacy in JANUS 1 and JANUS 2 trials[114] |
| NCT04358185 | HCC | Active | |||
| NCT03425006 | NSCLC | Active | |||
| NCT02917993 | NSCLC | Active | |||
| NCT02646748 | CRC, endometrial cancer, HNC, lung cancer, BC, PDAC, RCC, UC | Active | |||
| NCT03670069 | Soft tissue sarcoma | Suspended | |||
| NCT02257619 | NSCLC | Terminated | None published | ||
| NCT02559492 | Non-specified advanced solid tumors | Terminated | None published | ||
| Momelotinib | JAK1/2
TBK1 | NCT02101021 | PDAC | Terminated | No overall survival benefit[122] |
| NCT02258607 | NSCLC | Terminated | No overall survival benefit[123] | ||
| NCT02206763 | NSCLC | Terminated | Neutropenia[124] | ||
| NCT02244489 | PDAC | Terminated | None published | ||
| Pacritinib | JAK2 | NCT02277093 | CRC | Terminated | Lack of clinical response |
| NCT02342353 | NSCLC | Terminated | None published | ||
| WP1066 | JAK2 | NCT04334863 | Medulloblastoma, brain metastases | Active | |
| NCT01904123 | Glioma, brain metastases | Active |
JAK inhibitors investigated in clinical trials
Ruxolitinib
The JAK1/2-selective inhibitor ruxolitinib is FDA approved for the treatment of polycythemia vera, myelofibrosis, and graft versus host disease, and it has been shown to decrease STAT3 activation in preclinical models of several solid tumors[18,22,65]. Ruxolitinib inhibited STAT3 activation and decreased cell growth in breast cancer[66,67], NSCLC[68], HNC[69], esophageal cancer[70], bladder cancer[71], HCC[72], cervical cancer[73], and colorectal cancer[74,75] cell lines. In pancreatic cancer cells, ruxolitinib treatment was also shown to decrease expression of pro-angiogenic genes and impede epithelial-to-mesenchymal transition[76,77]. In in vivo xenograft models of neuroblastoma[78,79], HCC (in which there was a JAK1 S703I mutation)[40], and KRAS-mutated lung adenocarcinoma[80], among others, ruxolitinib treatment significantly inhibited tumor growth. Ruxolitinib treatment was associated with an increase in CD8+ T cells in pancreatic cancer xenograft models and a decrease in myeloid-derived suppressor cells in KRAS-mutated lung adenocarcinoma models, indicating an impact on immune activity[52,80].
Ruxolitinib has also been shown to overcome drug resistance and increase sensitivity to several chemotherapeutic or targeted agents. In preclinical in vitro and in vivo models of cisplatin-resistant NSCLC, with increased JAK2 and STAT3 activation levels, the addition of ruxolitinib to cisplatin decreased STAT3 activation and cell growth, enhanced apoptosis, and inhibited tumor growth[81]. In myxoid liposarcoma cancer stem cells, which can be resistant to chemotherapy due to upregulated JAK/STAT signaling, ruxolitinib treatment inhibited phosphorylation of STAT3 and cell viability, overcoming chemotherapy resistance[82]. Ruxolitinib in combination with antibodies against cytokines such as IL-6 (tocilizumab) improved survival in mice bearing ovarian cancer tumors. Ruxolitinib in combination with paclitaxel reduced cell proliferation and colony formation in ovarian cancer cell lines and inhibited tumor growth in in vivo models[83,84]. Ruxolitinib has been shown to improve sensitivity to oncolytic viral therapy in HNC[85], pancreatic cancer[86], glioblastoma multiforme (GBM)[87], and NSCLC[88]. Collectively, the safety profile of ruxolitinib in conjunction with promising preclinical findings in a variety of tumor models make ruxolitinib an attractive therapeutic agent against solid tumors.
Several clinical trials have studied the impact of ruxolitinib in patients with solid tumors. In a Phase II study of ruxolitinib and capecitabine in patients with pancreatic cancer who failed to respond to gemcitabine, known as the RECAP trial, there was improved survival among a subgroup of patients with inflammation, defined by a C-reactive protein (CRP) greater than the population median of 13 mg/L (NCT01423604)[89]. Given these initial promising results, ruxolitinib was administered to patients with pancreatic cancer and an elevated CRP in two Phase III trials, JANUS 1 (NCT02117479) and JANUS 2 (NCT02119663). In both trials, patients were randomized to be treated with either ruxolitinib and capecitabine or placebo and capecitabine. However, these studies were terminated as there was no increase in overall or progression-free survival observed in the group receiving ruxolitinib compared with placebo[90]. The combination of ruxolitinib and capecitabine in breast cancer patients with elevated CRP was also investigated in a Phase II clinical trial (NCT02120417). While patients receiving ruxolitinib and capecitabine had a more favorable health-related quality of life outcome, this study was terminated because there was no improvement in overall survival compared to the group receiving placebo and capecitabine[91]. A Phase II trial of ruxolitinib in triple-negative breast cancer confirmed inhibition of STAT3 activation in patient tumor samples; however, no clinical response was observed, as evaluated by the RECIST criteria, and the study was terminated (NCT01562873)[92,93]. The most recently completed clinical trial (results not reported or published) included a Phase II study testing ruxolitinib in combination with exemestane in patients with estrogen receptor-positive breast cancer (NCT01594216). The addition of ruxolitinib to regorafenib in a Phase II trial in patients with colorectal cancer did not show a difference in overall survival or progression-free survival as compared to placebo and regorafenib; therefore, this study was terminated early (NCT02119676)[94]. Ruxolitinib was also tested in patients with lung cancers. A Phase II trial of ruxolitinib (or placebo), pemetrexed, and cisplatin in patients with stage IIIb/IV or recurrent NSCLC demonstrated that this combination was well-tolerated; the study was terminated without achieving an efficacy endpoint (NCT02119650)[95]. Partial responses were seen in 31% of patients who received ruxolitinib and in 35% of patients who received placebo. A Phase Ib study of ruxolitinib combined with afatinib, an inhibitor of mutant EGFR, in patients with NSCLC showed that this regimen was both well-tolerated and displayed activity against this malignancy, as 23.3% displayed a partial response and 80% had stable disease (NCT02145637)[96]. In a Phase I/II study, ruxolitinib combined with the EGFR inhibitor erlotinib in lung adenocarcinoma was shown to be well-tolerated but ineffective (NCT02155465)[97]. A Phase Ib study of ruxolitinib with gemcitabine or nab-paclitaxel in solid tumors showed that this combination was well-tolerated (NCT01822756). However, efficacy could not be evaluated due to early termination of the trial after results from JANUS 1 showed no benefit of ruxolitinib and capecitabine compared to placebo and capecitabine[98]. In a Phase II trial of ruxolitinib in metastatic prostate cancer, there was no significant clinical response and the trial was terminated (NCT00638378). There is currently a rollover study that is providing continued access to ruxolitinib for patients with pancreatic, colorectal, lung, and breast cancers enrolled in previous trials (NCT02955940).
Several ongoing early-stage clinical trials are investigating ruxolitinib as monotherapy. There are two current window-of-opportunity trials: one testing neoadjuvant ruxolitinib in HNC (NCT03153982) and one examining ruxolitinib in premalignant breast disease (NCT02928978). Some trials are also investigating ruxolitinib in combination other agents. Among these are a Phase I study testing ruxolitinib in combination with temozolomide in patients with high-grade gliomas (NCT03514069), a Phase Ib study of ruxolitinib and trametinib (MEK inhibitor) in colon and pancreatic cancers with RAS mutations (NCT04303403), a Phase I study testing ruxolitinib with pembrolizumab (PD-L1 inhibitor) in triple-negative breast cancer (NCT03012230), two Phase II studies investigating ruxolitinib with chemotherapy in inflammatory breast cancer (NCT02876302, NCT02041429), a Phase I/II trial evaluating ruxolitinib with trastuzumab (HER2 inhibitor) in HER2+ breast cancer (NCT02066532), and a Phase I/II study of ruxolitinib with paclitaxel and carboplatin in ovarian, fallopian tube, and peritoneal cancers (NCT02713386). Ruxolitinib is one of 75 approved agents being tested in a trial that uses the Co-eXpression ExtrapolatioN (COXEN) model to identify biomarkers and to predict which drugs would provide the most benefit to patients with urothelial cancer (NCT02788201).
Tofacitinib
Tofacitinib is a JAK1/3 inhibitor that is FDA approved for treatment of rheumatoid arthritis and ulcerative colitis[99-102]. Tofacitinib treatment of breast cancer cells prevented activation and nuclear localization of STAT3[103]. In prostate cancer preclinical models, tofacitinib decreased STAT5 activation and epithelial-to-mesenchymal transition[104]. This JAK inhibitor is currently being tested in patients with solid tumors (mainly pancreatic adenocarcinoma and cholangiocarcinoma) in a Phase I trial (NCT04034238).
AZD1480
AZD1480 is a selective ATP-competitive JAK1/2 inhibitor that showed promising activity against many solid tumor preclinical models. AZD1480 treatment of cell lines and murine models, including but not limited to GBM[105], breast cancer[106,107], HNC[108], and ovarian cancer[109], inhibited STAT3 activation, cell viability, and tumor growth. Despite these encouraging preclinical findings, neurotoxicity was observed in a Phase I clinical trial of AZD1480 in solid tumors and halted the development of this agent, leading to the termination of this trial (NCT01112397) and a parallel Phase I study in patients with HCC, NSCLC, and gastric cancer (NCT01219543)[110].
AZD4205
AZD4205 is a selective JAK1 inhibitor[111]. In a preclinical NSCLC in vivo model, AZD4205 treatment inhibited tumor growth and STAT3 activation; these findings were more significant when AZD4205 was administered in combination with the EGFR inhibitor osimertinib. A Phase I/II clinical trial investigating AZD4205 combined with osimertinib was initiated in patients with NSCLC (NCT03450330).
INCB047986 and INCB052793
INCB047986 and INCB052793 are selective inhibitors of JAK1. INCB047986 was studied in a Phase I clinical trial in breast and pancreatic cancers, among other solid tumors, but the trial was terminated early (NCT01929941). INCB052793 has been studied in multiple myeloma (MM) preclinical models, but there are no reports using this agent in solid tumors. In combination with other anti-MM agents, INCB052793 decreased cell viability and inhibited tumor growth[112]. A Phase I/II trial was initiated investigating this agent in solid tumors but was terminated due to lack of efficacy (NCT02265510).
Itacitinib
Preclinical studies of the JAK1 inhibitor itacitinib have mostly been conducted in preclinical models of hematological malignancies. In conjunction with INCB054329, an inhibitor of bromodomain and extra-terminal motif proteins, itacitinib inhibited STAT3 activation and tumor growth in MM cell lines and murine models[113]. Given this effect of JAK1 inhibition on STAT3 activity, clinical studies with this agent were initiated in solid tumors. In a Phase Ib/II study of itacitinib in combination with nab-paclitaxel and gemcitabine in solid tumors (84% of which had pancreatic cancer), 24% of patients responded (all partial response) (NCT01858883). The therapeutic combination was well-tolerated after dose reduction of itacitinib; however, this study was terminated due to another Phase III clinical trial reporting no impact of the JAK1/2 inhibitor ruxolitinib on overall survival in pancreatic cancer[114]. Other ongoing clinical trials are studying itacitinib in patients with HCC (NCT04358185), NSCLC (NCT03425006 and NCT02917993), and a variety of advanced solid tumors (NCT02646748). A Phase II study in soft tissue sarcoma is currently suspended (NCT03670069); a Phase II study in NSCLC (NCT02257619) and a Phase Ib study in other solid tumors (NCT02559492) were terminated, with no published findings.
Momelotinib
Momelotinib is a JAK1/2 inhibitor that also has activity against TANK-binding kinase 1 (TBK1)[115,116]. Several preclinical studies in solid tumor models have investigated the impact of momelotinib on the JAK/STAT pathway. Momelotinib has been shown to increase sensitivity of ovarian cancer to chemotherapy in in vitro and in vivo preclinical models[117,118]. In combination with paclitaxel, momelotinib inhibited tumor growth, suppressed STAT3 activation, reduced expression of the stem cell marker OCT4, significantly increased the time to recurrence, and decreased tumor burden[117,118]. Similarly, in GBM preclinical models, momelotinib in combination with temozolomide inhibited STAT3 activation, decreased cell growth, increased apoptosis, and inhibited tumor growth compared to temozolomide monotherapy[119]. In colorectal cancer cells, momelotinib inhibited STAT5 activation, decreased cell growth, and increased cell death[120]. These promising preclinical results across several types of solid tumors support further investigation of momelotinib as a therapeutic agent.
Clinical use of momelotinib has been studied extensively in myeloproliferative diseases: in myelofibrosis, treatment with this agent was associated with a reduction in splenic volume that was non-inferior to ruxolitinib[121]. Its impact in solid tumors is under active clinical investigation. In a Phase I dose-escalation study in patients with untreated metastatic pancreatic cancer, momelotinib in combination with gemcitabine and nab-paclitaxel was well-tolerated; however, limited efficacy and no apparent association between efficacy and increasing dose led to the termination of this trial prior to the initiation of planned Phase III studies (NCT02101021)[122]. A Phase Ib study of momelotinib combined with trametinib in KRAS-mutated NSCLC showed no improvement in response compared with historic data with trametinib monotherapy (NCT02258607)[123]. In a Phase Ib study of momelotinib in combination with erlotinib in EGFR-mutated, metastatic NSCLC, patients experienced neutropenia as an adverse effect of this drug combination, and the trial was halted (NCT02206763)[124]. Another Phase Ib clinical trial of momelotinib with chemotherapeutic agents, capecitabine and oxaliplatin, in pancreatic adenocarcinoma was terminated (NCT02244489).
Pacritinib
Pacritinib is a selective JAK2 inhibitor currently being studied in a Phase III clinical trial for treatment of myelofibrosis (NCT02055781)[125,126]. In GBM cell lines, pacritinib, alone or in combination with afatinib, inhibited STAT3 activation, cell viability, and spheroid formation[127-129]. Pacritinib plus afatinib was also shown to decrease tumor burden in mice with GBM tumors[129]. Similar to momelotinib, pacritinib reduced resistance to temozolomide in GBM in vivo models[127,128]. Pacritinib has been shown to inhibit liver fibrosis and thus may be effective in preventing HCC[130]. A Phase II trial of pacritinib in refractory colorectal cancers is ongoing (NCT02277093)[131]. Pacritinib was also studied in combination with erlotinib in a Phase I/II trial in NSCLC, which was terminated (NCT02342353).
WP1066
WP1066 inhibits JAK2 phosphorylation and causes JAK2 degradation; it is an analog of the JAK2 inhibitor AG490, an agent which was widely tested in preclinical modes of solid tumors[132,133]. Preclinical studies have shown that WP1066 exhibits anti-cancer activity including inhibition of cell proliferation and survival, and/or inhibition of tumor growth in solid tumors including, but not limited to, bladder cancer, renal cell carcinoma (in which it was shown to inhibit angiogenesis), HNC, GBM, and NSCLC[134-139]. This agent also inhibited migration and invasion in bladder cancer, hepatocellular carcinoma, and GBM cell lines[134,139,140]. WP1066 treatment overcame STAT3-mediated cisplatin resistance in oral squamous cell carcinoma and ovarian cancer cell lines, and doxorubicin resistance in breast cancer cell lines[141-143]. Two current Phase I trials investigating the safety and efficacy of WP1066 are being conducted in pediatric medulloblastomas (NCT04334863) and adult malignant gliomas or brain metastases (NCT01904123).
JAK inhibitors with preclinical evidence supporting activity against solid tumors
While several JAK inhibitors have not yet been tested in patients with solid tumors, they have shown promising anti-cancer effects in preclinical models. Agents such as AG490, the compound from which WP1066 was derived, and JAK inhibitor I have been widely tested in preclinical in vitro and in vivo models. AG490 inhibited STAT3 activation and exhibited anti-cancer effects such as inhibition of cell growth and induction of apoptosis via targeting of JAK2 in preclinical models of breast cancer[4], gastric cancer[48], pancreatic cancer[144], and gallbladder cancer[145], among others. JAK inhibitor I is a JAK1/2/3 inhibitor that decreased cell proliferation in breast cancer cells[146], increased apoptosis in esophageal squamous cell carcinoma cancer stem cells[147], inhibited STAT3 phosphorylation in HCC cells[148], and, in combination with cisplatin, decreased PD-L1 expression in prostate cancer cells[149]. In addition to these agents, there are a handful of inhibitors that have either already been FDA approved or are being tested currently in clinical trials for other indications and have also shown promising findings in solid tumor preclinical models. The following inhibitors, therefore, are potential candidates for clinical testing and use in patients with solid tumors.
Fedratinib
Fedratinib is an orally bioavailable, small molecule, JAK2 inhibitor that is FDA approved for the treatment of myelofibrosis[150-153]. NSCLC cells have been shown to be sensitive to fedratinib; sensitivity was shown to be correlated with elevated JAK2 expression[154]. Two studies showed that fedratinib in combination with erlotinib (EGFR tyrosine kinase inhibitor) decreased STAT3 activation and increased apoptosis in erlotinib-resistant NSCLC cells and inhibited tumor growth in in vivo murine models[155,156]. This agent has also demonstrated cell-killing activity against ovarian and cervical cancer cells[157]. Fedratinib inhibited mammosphere formation and in combination with carboplatin, inhibited breast cancer tumor growth in mice[158]. In human papilloma virus (HPV)-positive cervical cancer cells, fedratinib treatment inhibited JAK2 and STAT3/5 activation, increased apoptosis, and reduced cyclin D1 expression, cell proliferation, and colony formation[73]. In HNC cells, treatment with fedratinib increased susceptibility to natural killer cell killing[159].
Filgotinib
Filgotinib is a selective JAK1 inhibitor currently being investigated in clinical trials for treatment of rheumatoid arthritis and inflammatory bowel disease; to date, this drug demonstrates a significant anti-inflammatory effect, as it reduces levels of cytokines such as IL-6[160-162]. Findings from preclinical studies in solid tumors have been reported. The OSM-JAK-STAT pathway has been implicated in progression of several cancers, including NSCLC; treatment of NSCLC cells with filgotinib resulted in inhibition of STAT3 activation and reduced OSM receptor expression[50]. Furthermore, treatment with filgotinib inhibited resistance to targeted therapy such as MEK, EGFR tyrosine kinase, and ALK inhibitors. In NCI-H889 lung cancer cells, derived from a metastatic site, filgotinib inhibited STAT3 activation[163]. In vivo, filgotinib treatment reduced metastatic seeding of NCI-H889-derived tumors. In a breast cancer cell line, filgotinib inhibited STAT3 phosphorylation; in combination with a histone deacetylase inhibitor, there was increased apoptosis in breast cancer cells as well as tumor growth inhibition in mice harboring breast cancer tumors[164].
Lestaurtinib
Lestaurtinib is a multitarget inhibitor that has activity against JAK2, in addition to fms-like tyrosine kinase tyrosine 3 (FLT3) and tropomyosin related kinase B (TrkB)[165]. Its impact on the JAK/STAT pathway has been studied clinically in myeloproliferative disorders, but trials in solid tumors such as neuroblastoma focus on its activity against other targets such as TrkB. One preclinical study showed that lestaurtinib treatment of anaplastic thyroid cancer cell lines inhibited STAT5 phosphorylation/activation, cell proliferation, cell survival, and cell migration, in addition to tumor growth in in vivo models[166].
Peficitinib
Peficitinib is a JAK1/2/3 and TYK2 inhibitor, approved in Japan in 2019 for rheumatoid arthritis after Phase III clinical trials demonstrated a reduction in symptoms and minimal toxicity compared to placebo in clinical trials[167,168]. Only one study to date has reported its potential use in solid tumors[169]. In ovarian cancer stem cells engineered to overexpress OCT4, peficitinib induced apoptosis and inhibited proliferation in conjunction with JAK1 inhibition.
Conclusion
Aberrant JAK/STAT signaling is associated with solid tumor development and progression. However, unlike hematopoietic malignancies which harbor activating JAK mutations that lead to increased JAK/STAT signaling, the majority of solid tumors that demonstrate increased JAK/STAT signaling lack somatic JAK mutations. Studies in preclinical cancer models of solid tumors collectively show that small molecule JAK inhibitors inhibit activation of STATs, particularly STAT3, in conjunction with inhibition of proliferation and tumor growth. The majority of JAK inhibitors tested in clinical trials, with the exception of AZD1480, were found to be safe and well-tolerated. Among these, ruxolitinib is the only inhibitor to date to demonstrate responses in early stage trials. While Phase II trial results in pancreatic cancer suggested an association between elevated CRP and response to ruxolitinib plus capecitabine, these findings were not seen in the Phase III trials[89,90]. In patients with elevated CRP, ruxolitinib combined with capecitabine was associated with improved health-related quality of life in breast cancer[91]. Additionally, treatment of patients with NSCLC with ruxolitinib plus afatinib resulted in partial responses and stable disease[96]. However, most trials testing ruxolitinib exhibited disappointing results, and several were terminated early; this could possibly be explained by JAK inhibition impeding immune cell function, which may counteract some of the drug’s other anti-cancer effects[170]. It is clear that only a subset of solid tumors is likely to be sensitive to JAK inhibition. Candidate predictive biomarkers to date include elevated CRP in pancreatic and breast cancers, PTPRT/D mutations in HNC, and a JAK1 S703I mutation in HCC, and assessments of biologically plausible biomarkers that predict clinical responses are needed[40,54,55,89,91]. The JAK inhibitors fedratinib, filgotinib, and peficitinib have been shown to abrogate JAK/STAT signaling and induce anti-tumor effects in solid tumor cell lines, but, to date, there are no clinical trials investigating these agents in solid tumors; lestaurtinib has been tested clinically in solid tumors for its activity against other targets not directly involved in the JAK/STAT pathway. Ruxolitinib, tofacitinib, fedratinib, and peficitinib are JAK inhibitors already approved for other indications, making them especially attractive options as they are known to be well-tolerated. Further investigation of JAK inhibitors in clinical trials is warranted to determine the therapeutic potential in solid tumors.
Declarations
Authors’ contributionsDrafted and edited manuscript, figures, and tables: Qureshy Z, Johnson DE, Grandis JR
Availability of data and materialsNot applicable.
Financial support and sponsorshipThis work was supported by NIH (R35CA231998) (to Grandis JR), (R01DE028289) (to Johnson DE, Grandis JR); and a Yearlong Research Fellowship awarded by the University of California San Francisco School of Medicine (to Qureshy Z).
Conflicts of interestJohnson DE and Grandis JR are co-inventors of cyclic STAT3 decoy and have financial interests in STAT3 Therapeutics. STAT3 Therapeutics holds an interest in cyclic STAT3 decoy. Qureshy Z declared that there are no conflicts of interest.
Ethical approval and consent to participateNot applicable.
Consent for publicationNot applicable.
Copyright© The Author(s) 2020.
REFERENCES
1. Thomas SJ, Snowden JA, Zeidler MP, Danson SJ. The role of JAK/STAT jnsignalling in the pathogenesis, prognosis and treatment of solid tumours. Br J Cancer 2015;113:365-71.
2. O’Shea JJ, Holland SM, Staudt LM. JAKs and STATs in Immunity, Immunodeficiency, and Cancer. N Engl J Med 2013;368:161-70.
3. O’Sullivan JM, Harrison CN. JAK-STAT signaling in the therapeutic landscape of myeloproliferative neoplas ms. Mol Cell Endocrinol 2017;451:71-9.
4. Khanna P, Lee JS, Sereemaspun A, Lee H, Baeg GH. GRAMD1B regulates cell migration in breast cancer cells through JAK/STAT and Akt signaling. Sci Rep 2018;8:9511.
5. Haque I, Ghosh A, Acup S, Banerjee S, Dhar K, et al. Leptin-induced ER-α-positive breast cancer cell viability and migration is mediated by suppressing CCN5-signaling via activating JAK/AKT/STAT-pathway. BMC Cancer 2018;18:99.
6. Lokau J, Schoeder V, Haybaeck J, Garbers C. Jak-Stat signaling induced by interleukin-6 family cytokines in hepatocellular carcinoma. Cancers (Basel) 2019;11:1704.
7. Grandis JR, Drenning SD, Zeng Q, Watkins SC, Melhem MF, et al. Constitutive activation of stat3 signaling abrogates apoptosis in squamous cell carcinogenesis in vivo. Proc Natl Acad Sci U S A 2000;97:4227-32.
8. Wu CS, Wei KL, Chou JL, Lu CK, Hsieh CC, et al. Aberrant JAK/STAT signaling suppresses TFF1 and TFF2 through epigenetic silencing of GATA6 in gastric cancer. Int J Mol Sci 2016;17:1467.
9. Shang AQ, Wu J, Bi F, Zhang YJ, Xu LR, et al. Relationship between HER2 and JAK/STAT-SOCS3 signaling pathway and clinicopathological features and prognosis of ovarian cancer. Cancer Biol Ther 2017;18:314-22.
10. Nishi M, Batsaikhan BE, Yoshikawa K, Higashijima J, Tokunaga T, et al. High STAT4 expression indicates better disease-free survival in patients with gastric cancer. Anticancer Res 2017;37:6723-9.
11. Nikitakis N, Siavash H, Sauk J. Targeting the STAT pathway in head and neck cancer: recent advances and future prospects. Curr Cancer Drug Targets 2005;4:637-51.
12. Khanna P, Chua PJ, Bay BH, Baeg GH. The JAK/STAT signaling cascade in gastric carcinoma (Review). Int J Oncol 2015;47:1617-26.
13. Tabassum S, Abbasi R, Ahmad N, Farooqi AA. Targeting of JAK-STAT signaling in breast cancer: therapeutic strategies to overcome drug resistance. Adv Exp Med Biol 2019;1152:271-81.
14. Vainchenker W, Constantinescu SN. JAK/STAT signaling in hematological malignancies. Oncogene 2013;32:2601-13.
15. Kralovics R, Passamonti F, Buser AS, Teo SS, Tiedt R, et al. A gain-of-function mutation of JAK2 in myeloproliferative disorders. N Engl J Med 2005;352:1779-90.
16. Plosker GL. Ruxolitinib: a review of its use in patients with myelofibrosis. Drugs 2015;75:297-308.
17. Verstovsek S, Mesa RA, Gotlib J, Levy RS, Gupta V, et al. A double-blind, placebo-controlled trial of ruxolitinib for myelofibrosis. N Engl J Med 2012;366:799-807.
18. Harrison CN, Vannucchi AM, Kiladjian JJ, Al-Ali HK, Gisslinger H, et al. Long-term findings from COMFORT-II, a phase 3 study of ruxolitinib vs best available therapy for myelofibrosis. Leukemia 2016;30:1701-7.
19. Vannucchi AM, Kiladjian JJ, Griesshammer M, Masszi T, Durrant S, et al. Ruxolitinib versus standard therapy for the treatment of polycythemia vera. N Engl J Med 2015;372:426-35.
20. Vannucchi AM, Verstovsek S, Guglielmelli P, Griesshammer M, Burn TC, et al. Ruxolitinib reduces JAK2 p.V617F allele burden in patients with polycythemia vera enrolled in the RESPONSE study. Ann Hematol 2017;96:1113-20.
21. Verstovsek S, Passamonti F, Rambaldi A, Barosi G, Rosen PJ, et al. A phase 2 study of ruxolitinib, an oral JAK1 and JAK2 inhibitor, in patients with advanced polycythemia vera who are refractory or intolerant to hydroxyurea. Cancer 2014;120:513-20.
22. Verstovsek S, Vannucchi AM, Griesshammer M, Masszi T, Durrant S, et al. Ruxolitinib versus best available therapy in patients with polycythemia vera: 80-week follow-up from the RESPONSE trial. Haematologica 2016;101:821-9.
23. Morris R, Kershaw NJ, Babon JJ. The molecular details of cytokine signaling via the JAK/STAT pathway. Protein Sci 2018;27:1984-2009.
24. Rawlings JS, Rosler KM, Harrison DA. The JAK/STAT signaling pathway. J Cell Sci 2004;117:1281-3.
27. Alvarez JV, Frank DA, Alvarez J, Lab F. Genome-wide analysis of STAT target genes: Elucidating the mechanism of STAT-mediated oncogenesis. Cancer Biol Ther 2004;3:1045-50.
28. Wang Y, Shen Y, Wang S, Shen Q, Zhou X. The role of STAT3 in leading the crosstalk between human cancers and the immune system. Cancer Lett 2018;415:117-28.
29. Greenhalgh CJ, Hilton DJ. Negative regulation of cytokine signaling. J Leukoc Biol 2001;70:348-56.
30. Chikuma S, Kanamori M, Mise-Omata S, Yoshimura A. Suppressors of cytokine signaling: potential immune checkpoint molecules for cancer immunotherapy. Cancer Sci 2017;108:574-80.
31. Niu GJ, Xu JD, Yuan WJ, Sun JJ, Yang MC, et al. Protein inhibitor of activated STAT (PIAS) negatively regulates the JAK/STAT pathway by inhibiting STAT phosphorylation and translocation. Front Immunol 2018;9:2392.
32. Frankson R, Yu ZH, Bai Y, Li Q, Zhang RY, et al. Therapeutic targeting of oncogenic tyrosine phosphatases. Cancer Res 2017;77:5701-5.
33. Kim M, Morales LD, Jang IS, Cho YY, Kim DJ. Protein tyrosine phosphatases as potential regulators of STAT3 signaling. Int J Mol Sci 2018;19:2708.
34. Chen YW, Guo T, Shen L, Wong KY, Tao Q, et al. Receptor-type tyrosine-protein phosphatase κ directly targets STAT3 activation for tumor suppression in nasal NK/T-cell lymphoma. Blood 2015;125:1589-600.
35. Lee M, Hirpara JL, Eu JQ, Sethi G, Wang L, et al. Targeting STAT3 and oxidative phosphorylation in oncogene-addicted tumors. Redox Biol 2019;25:101073.
36. Zhang X, Guo A, Yu J, Possemato A, Chen Y, et al. Identification of STAT3 as a substrate of receptor protein tyrosine phosphatase T. Proc Natl Acad Sci U S A 2007;104:4060-4.
37. Veeriah S, Brennan C, Meng S, Singh B, Fagin JA, et al. The tyrosine phosphatase PTPRD is a tumor suppressor that is frequently inactivated and mutated in glioblastoma and other human cancers. Proc Natl Acad Sci U S A 2009;106:9435-40.
38. Shahmarvand N, Nagy A, Shahryari J, Ohgami RS. Mutations in the signal transducer and activator of transcription family of genes in cancer. Cancer Sci 2018;109:926-33.
39. Shi M, He R, Feldman AL, Viswanatha DS, Jevremovic D, et al. STAT3 mutation and its clinical and histopathologic correlation in T-cell large granular lymphocytic leukemia. Hum Pathol 2018;73:74-81.
40. Yang S, Luo C, Gu Q, Xu Q, Wang G, et al. Activating JAK1 mutation may predict the sensitivity of JAK-STAT inhibition in hepatocellular carcinoma. Oncotarget 2016;7:5461-9.
41. Kan Z, Zheng H, Liu X, Li S, Barber TD, et al. Whole-genome sequencing identifies recurrent mutations in hepatocellular carcinoma. Genome Res 2013;23:1422-33.
42. Igelmann S, Neubauer HA, Ferbeyre G. STAT3 and STAT5 activation in solid cancers. Cancers (Basel) 2019;11:1428.
43. Orlova A, Wagner C, De Araujo ED, Bajusz D, Neubauer HA, et al. Direct targeting options for STAT3 and STAT5 in cancer. Cancers (Basel) 2019;11:1930.
44. Wang Y, Wu C, Zhang C, Li Z, Zhu T, et al. TGF-β-induced STAT3 overexpression promotes human head and neck squamous cell carcinoma invasion and metastasis through malat1/miR-30a interactions. Cancer Lett 2018;436:52-62.
45. Kijima T, Niwa H, Steinman RA, Drenning SD, Gooding WE, et al. STAT3 activation abrogates growth factor dependence and contributes to head and neck squamous cell carcinoma tumor growth in vivo. Cell Growth Differ 2002;13:355-62.
46. Bu LL, Yu GT, Wu L, Mao L, Deng WW, et al. STAT3 Induces Immunosuppression by Upregulating PD-1/PD-L1 in HNSCC. J Dent Res 2017;96:1027-34.
47. Kanda N, Seno H, Konda Y, Marusawa H, Kanai M, et al. STAT3 is constitutively activated and supports cell survival in association with survivin expression in gastric cancer cells. Oncogene 2004;23:4921-9.
48. Wu X, Tao P, Zhou Q, Li J, Yu Z, et al. IL-6 secreted by cancer-associated fibroblasts promotes epithelial-mesenchymal transition and metastasis of gastric cancer via JAK2/STAT3 signaling pathway. Oncotarget 2017;8:20741-50.
49. Pan YM, Wang CG, Zhu M, Xing R, Cui JT, et al. STAT3 signaling drives EZH2 transcriptional activation and mediates poor prognosis in gastric cancer. Mol Cancer 2016;15:79.
50. Shien K, Papadimitrakopoulou VA, Ruder D, Behrens C, Shen L, et al. JAK1/STAT3 activation through a proinflammatory cytokine pathway leads to resistance to molecularly targeted therapy in non-small cell lung cancer. Mol Cancer Ther 2017;16:2234-45.
51. He W, Wu J, Shi J, Huo YM, Dai W, et al. IL22RA1/STAT3 signaling promotes stemness and tumorigenicity in pancreatic cancer. Cancer Res 2018;78:3293-305.
52. Lu C, Talukder A, Savage NM, Singh N, Liu K. JAK-STAT-mediated chronic inflammation impairs cytotoxic T lymphocyte activation to decrease anti-PD-1 immunotherapy efficacy in pancreatic cancer. Oncoimmunology 2017;6:e1291106.
53. Lin XM, Chen H, Zhan XL. MIR-203 regulates JAK-STAT pathway in affecting pancreatic cancer cells proliferation and apoptosis by targeting SOCS3. Eur Rev Med Pharmacol Sci 2019;23:6906-13.
54. Peyser ND, Freilino M, Wang L, Zeng Y, Li H, et al. Frequent promoter hypermethylation of PTPRT increases STAT3 activation and sensitivity to STAT3 inhibition in head and neck cancer. Oncogene 2016;35:1163-9.
55. Peyser ND, Du Y, Li H, Lui V, Xiao X, et al. Loss-of-function PTPRD mutations lead to increased STAT3 activation and sensitivity to STAT3 inhibition in head and neck cancer. PLoS One 2015;10:e0135750.
56. Gyamfi J, Lee YH, Eom M, Choi J. Interleukin-6/STAT3 signalling regulates adipocyte induced epithelial-mesenchymal transition in breast cancer cells. Sci Rep 2018;8:8859.
57. Jiang C, Long J, Liu B, Xu M, Wang W, et al. miR-500a-3p promotes cancer stem cells properties via STAT3 pathway in human hepatocellular carcinoma. J Exp Clin Cancer Res 2017;36:99.
58. Saini U, Naidu S, Elnaggar AC, Bid HK, Wallbillich JJ, et al. Elevated STAT3 expression in ovarian cancer ascites promotes invasion and metastasis: a potential therapeutic target. Oncogene 2017;36:168-81.
59. Escher TE, Lui AJ, Geanes ES, Walter KR, Tawfik O, et al. Interaction between MUC1 and STAT1 drives IFITM1 overexpression in aromatase inhibitor-resistant breast cancer cells and mediates estrogen-induced apoptosis. Mol Cancer Res 2019;17:1180-94.
60. Chen X, Huang J, Yuchun L. High expression of STAT2 in ovarian cancer and its effect on metastasis of ovarian cancer cells. Nan Fang Yi Ke Da Xue Xue Bao 2020;40:34-41. (in Chinese)
61. Yang M, Chen H, Zhou L, Chen K, Su F. Expression profile and prognostic values of STAT family members in non-small cell lung cancer. Am J Transl Res 2019;11:4866-80.
62. Haddad BR, Erickson A, Udhane V, LaViolette PS, Rone JD, et al. Positive STAT5 protein and locus amplification status predicts recurrence after radical prostatectomy to assist clinical precision management of prostate cancer. Cancer Epidemiol Biomarkers Prev 2019;28:1642-51.
63. Liao Z, Lutz J, Nevalainen MT. Transcription factor Stat5a/b as a therapeutic target protein for prostate cancer. Int J Biochem Cell Biol 2010;42:186-92.
64. Li BH, Yang XZ, Li PD, Yuan Q, Liu XH, et al. IL-4/Stat6 activities correlate with apoptosis and metastasis in colon cancer cells. Biochem Biophys Res Commun 2008;369:554-60.
65. von Bubnoff N, Ihorst G, Grishina O, Röthling N, Bertz H, et al. Ruxolitinib in GvHD (RIG) study: a multicenter, randomized phase 2 trial to determine the response rate of Ruxolitinib and best available treatment (BAT) versus BAT in steroid-refractory acute graft-versushost disease (aGvHD) (NCT02396628). BMC Cancer 2018;18:1132.
66. Lim ST, Jeon YW, Gwak H, Kim SY, Suh YJ. Synergistic anticancer effects of ruxolitinib and calcitriol in estrogen receptor-positive, human epidermal growth factor receptor 2-positive breast cancer cells. Mol Med Rep 2018;17:5581-8.
67. Kim JW, Gautam J, Kim JE, Kim JA, Kang KW. Inhibition of tumor growth and angiogenesis of tamoxifen-resistant breast cancer cells by ruxolitinib, a selective JAK2 inhibitor. Oncol Lett 2019;17:3981-9.
68. Taverna JA, Hung CN, DeArmond DT, Chen M, Lin CL, et al. Single-cell proteomic profiling identifies combined AXL and JAK1 inhibition as a novel therapeutic strategy for lung cancer. Cancer Res 2020;80:1551-63.
69. Vallath S, Sage EK, Kolluri KK, Lourenco SN, Teixeira VS, et al. CADM1 inhibits squamous cell carcinoma progression by reducing STAT3 activity. Sci Rep 2016;6:24006.
70. Yang PW, Huang PM, Yong LS, Chang YH, Wu CW, et al. Circulating interleukin-6 is associated with prognosis and genetic polymorphisms of MIR608 in patients with esophageal squamous cell carcinoma. Ann Surg Oncol 2018;25:2449-56.
71. Ojha R, Singh SK, Bhattacharyya S. JAK-mediated autophagy regulates stemness and cell survival in cisplatin resistant bladder cancer cells. Biochim Biophys Acta - Gen Subj 2016;1860:2484-97.
72. Wilson GS, Tian A, Hebbard L, Duan W, George J, et al. Tumoricidal effects of the JAK inhibitor Ruxolitinib (INC424) on hepatocellular carcinoma in vitro. Cancer Lett 2013;341:224-30.
73. Morgan EL, Macdonald A. JAK2 inhibition impairs proliferation and sensitises cervical cancer cells to cisplatin-induced cell death. Cancers (Basel) 2019;11:1934.
74. An HJ, Choi EK, Kim JS, Hong SW, Moon JH, et al. INCB018424 induces apoptotic cell death through the suppression of pJAK1 in human colon cancer cells. Neoplasma 2014;61:56-62.
75. Radhakrishnan H, Ilm K, Walther W, Shirasawa S, Sasazuki T, et al. MACC1 regulates Fas mediated apoptosis through STAT1/3 - Mcl-1 signaling in solid cancers. Cancer Lett 2017;403:231-45.
76. Gore J, Craven KE, Wilson JL, Cote GA, Cheng M, et al. TCGA data and patient-derived orthotopic xenografts highlight pancreatic cancer-associated angiogenesis. Oncotarget 2015;6:7504-21.
77. Perusina Lanfranca M, Zhang Y, Girgis A, Kasselman S, Lazarus J, et al. Interleukin 22 signaling regulates acinar cell plasticity to promote pancreatic tumor development in mice. Gastroenterology 2020;158:1417-32.e11.
78. Borriello L, Nakata R, Sheard MA, Fernandez GE, Sposto R, et al. Cancer-associated fibroblasts share characteristics and protumorigenic activity with mesenchymal stromal cells. Cancer Res 2017;77:5142-57.
79. Hadjidaniel MD, Muthugounder S, Hung LT, Sheard MA, Shirinbak S, et al. Tumor-associated macrophages promote neuroblastoma via STAT3 phosphorylation and up-regulation of c-MYC. Oncotarget 2017;8:91516-29.
80. Mohrherr J, Haber M, Breitenecker K, Aigner P, Moritsch S, et al. JAK-STAT inhibition impairs K-RAS-driven lung adenocarcinoma progression. Int J Cancer 2019;145:3376-88.
81. Hu Y, Hong Y, Xu Y, Liu P, Guo DH, et al. Inhibition of the JAK/STAT pathway with ruxolitinib overcomes cisplatin resistance in nonsmall-cell lung cancer NSCLC. Apoptosis 2014;19:1627-36.
82. Dolatabadi S, Jonasson E, Lindén M, Fereydouni B, Bäcksten K, et al. JAK-STAT signalling controls cancer stem cell properties including chemotherapy resistance in myxoid liposarcoma. Int J Cancer 2019;145:435-49.
83. McLean K, Tan L, Bolland DE, Coffman LG, Peterson LF, et al. Leukemia inhibitory factor functions in parallel with interleukin-6 to promote ovarian cancer growth. Oncogene 2019;38:1576-84.
84. Reeves PM, Abbaslou MA, Kools FRW, Vutipongsatorn K, Tong X, et al. Ruxolitinib sensitizes ovarian cancer to reduced dose Taxol, limits tumor growth and improves survival in immune competent mice. Oncotarget 2017;8:94040-53.
85. Escobar-Zarate D, Liu YP, Suksanpaisan L, Russell SJ, Peng KW. Overcoming cancer cell resistance to VSV oncolysis with JAK1/2 inhibitors. Cancer Gene Ther 2013;20:582-9.
86. Cataldi M, Shah NR, Felt SA, Grdzelishvili VZ. Breaking resistance of pancreatic cancer cells to an attenuated vesicular stomatitis virus through a novel activity of IKK inhibitor TPCA-1. Virology 2015;485:340-54.
87. Kurokawa C, Iankov ID, Anderson SK, Aderca I, Leontovich AA, et al. Constitutive interferon pathway activation in tumors as an efficacy determinant following oncolytic virotherapy. J Natl Cancer Inst 2018;110:1123-32.
88. Patel MR, Dash A, Jacobson BA, Ji Y, Baumann D, et al. JAK/STAT inhibition with ruxolitinib enhances oncolytic virotherapy in nonsmall cell lung cancer models. Cancer Gene Ther 2019;26:411-8.
89. Hurwitz HI, Uppal N, Wagner SA, Bendell JC, Beck JT, et al. Randomized, double-blind, phase II study of ruxolitinib or placebo in combination with capecitabine in patients with metastatic pancreatic cancer for whom therapy with gemcitabine has failed. J Clin Oncol 2015;33:4039-47.
90. Hurwitz H, Van Cutsem E, Bendell J, Hidalgo M, Li CP, et al. Ruxolitinib + capecitabine in advanced/metastatic pancreatic cancer after disease progression/intolerance to first-line therapy: JANUS 1 and 2 randomized phase III studies. Invest New Drugs 2018;36:683-95.
91. O’Shaughnessy J, DeMichele A, Ma CX, Richards P, Yardley DA, et al. A randomized, double-blind, phase 2 study of ruxolitinib or placebo in combination with capecitabine in patients with advanced HER2-negative breast cancer and elevated C-reactive protein, a marker of systemic inflammation. Breast Cancer Res Treat 2018;170:547-57.
92. Stover DG, Gil Del Alcazar CR, Brock J, Guo H, Overmoyer B, et al. Phase II study of ruxolitinib, a selective JAK1/2 inhibitor, in patients with metastatic triple-negative breast cancer. NPJ Breast Cancer 2018;4:10.
93. Eisenhauer EA, Therasse P, Bogaerts J, Schwartz LH, Sargent D, et al. New response evaluation criteria in solid tumours: revised RECIST guideline (version 1.1). Eur J Cancer 2009;45:228-47.
94. Fogelman D, Cubillo A, García-Alfonso P, Mirón MLL, Nemunaitis J, et al. Randomized, double-blind, phase two study of ruxolitinib plus regorafenib in patients with relapsed/refractory metastatic colorectal cancer. Cancer Med 2018;7:5382-93.
95. Giaccone G, Sanborn RE, Waqar SN, Martinez-Marti A, Ponce S, et al. A placebo-controlled phase II study of ruxolitinib in combination with pemetrexed and cisplatin for first-line treatment of patients with advanced nonsquamous non-small-cell lung cancer and systemic inflammation. Clin Lung Cancer 2018;19:e567-74.
96. Park JS, Hong MH, Chun YJ, Kim HR, Cho BC. A phase Ib study of the combination of afatinib and ruxolitinib in EGFR mutant NSCLC with progression on EGFR-TKIs. Lung Cancer 2019;134:46-51.
97. Yu HA, Perez L, Chang Q, Gao SP, Kris MG, et al. A phase 1/2 trial of Ruxolitinib and Erlotinib in patients with EGFR-mutant lung adenocarcinomas with acquired resistance to Erlotinib. J Thoracic Oncol 2017;12:102-9.
98. Bauer TM, Rpatel M, Forero-Torres A, George TJ, Assad A, et al. Aphase Ib study of ruxolitinib + gemcitabine ± nab-paclitaxel in patients with advanced solid tumors. Onco Targets Ther 2018;11:2399-407.
99. Sandborn WJ, Su C, Sands BE, D’Haens GR, Vermeire S, et al. Tofacitinib as induction and maintenance therapy for ulcerative colitis. N Engl J Med 2017;376:1723-36.
100. Lee EB, Fleischmann R, Hall S, Wilkinson B, Bradley JD, et al. Tofacitinib versus methotrexate in rheumatoid arthritis. N Engl J Med 2014;370:2377-86.
101. Fleischmann R, Mysler E, Hall S, Kivitz AJ, Moots RJ, et al. Efficacy and safety of tofacitinib monotherapy, tofacitinib with methotrexate, and adalimumab with methotrexate in patients with rheumatoid arthritis (ORAL Strategy): a phase 3b/4, double-blind, head-to-head, randomised controlled trial. Lancet 2017;390:457-68.
102. Fleischmann R, Kremer J, Cush J, Schulze-Koops H, Connell CA, et al. Placebo-controlled trial of tofacitinib monotherapy in rheumatoid arthritis. N Engl J Med 2012;367:495-507.
103. Lapeire L, Hendrix A, Lambein K, Van Bockstal M, Braems G, et al. Cancer-associated adipose tissue promotes breast cancer progression by paracrine oncostatin M and Jak/STAT3 signaling. Cancer Res 2014;74:6806-19.
104. Seol MA, Kim JH, Oh K, Kim G, Seo MW, et al. Interleukin-7 Contributes to the invasiveness of prostate cancer cells by promoting epithelial-mesenchymal transition. Sci Rep 2019;9:6917.
105. McFarland BC, Ma JY, Langford CP, Gillespie GY, Yu H, et al. Therapeutic potential of AZD1480 for the treatment of human glioblastoma. Mol Cancer Ther 2011;10:2384-93. Page 16 of 18 Qureshy et al. J Cancer Metastasis Treat 2020;6:27 I http://dx.doi.org/10.20517/2394-4722.2020.58
106. Wang T, Fahrmann JF, Lee H, Li YJ, Tripathi SC, et al. JAK/STAT3-regulated fatty acid β-oxidation is critical for breast cancer stem cell self-renewal and chemoresistance. Cell Metab 2018;27:136-50.e5.
107. Chang Q, Bournazou E, Sansone P, Berishaj M, Gao SP, et al. The IL-6/JAK/Stat3 feed-forward loop drives tumorigenesis and metastasis. Neoplasia (United States) 2013;15:848-62.
108. Sen M, Pollock NI, Black J, DeGrave KA, Wheeler S, et al. JAK kinase inhibition abrogates STAT3 activation and head and neck squamous cell carcinoma tumor growth. Neoplasia 2015;17:256-64.
109. Wen W, Wu J, Liu L, Tian Y, Buettner R, et al. Synergistic anti-tumor effect of combined inhibition of EGFR and JAK/STAT3 pathways in human ovarian cancer. Mol Cancer 2015;14:100.
110. Plimack ER, LoRusso PM, McCoon P, Tang W, Krebs AD, et al. AZD1480: a phase I study of a novel JAK2 inhibitor in solid tumors. Oncologist 2013;18:819-20.
111. Su Q, Banks E, Bebernitz G, Bell K, Borenstein CF, et al. Discovery of (2R)-N-[3-[2-[(3-methoxy-1-methyl-pyrazol-4-yl)amino] pyrimidin-4-yl]-1H-indol-7-yl]-2-(4-methylpiperazin-1-yl)propenamide (AZD4205) as a potent and selective Janus Kinase 1 (JAK1) inhibitor. J Med Chem 2020;63:4517-27.
112. Sanchez E, Li M, Patil S, Soof CM, Nosrati JD, et al. The anti-myeloma effects of the selective JAK1 inhibitor (INCB052793) alone and in combination in vitro and in vivo. Ann Hematol 2019;98:691-703.
113. Stubbs MC, Burn TC, Sparks R, Maduskuie T, Diamond S, et al. The novel bromodomain and extraterminal domain inhibitor INCB054329 induces vulnerabilities in myeloma cells that inform rational combination strategies. Clin Cancer Res 2019;25:300-11.
114. Beatty GL, Shahda S, Beck T, Uppal N, Cohen SJ, et al. A phase Ib/II study of the JAK1 inhibitor, itacitinib, plus nab -paclitaxel and gemcitabine in advanced solid tumors. Oncologist 2019;24:14.
115. Xu L, Feng J, Gao G, Tang H. Momelotinib for the treatment of myelofibrosis. Expert Opin Pharmacother 2019;20:1943-51.
116. Burns CJ, Bourke DG, Andrau L, Bu X, Charman SA, et al. Phenylaminopyrimidines as inhibitors of Janus kinases (JAKs). Bioorganic Med Chem Lett 2009;19:5887-92.
117. Chan E, Luwor R, Burns C, Kannourakis G, Findlay JK, et al. Momelotinib decreased cancer stem cell associated tumor burden and prolonged disease-free remission period in a mouse model of human ovarian cancer. Oncotarget 2018;9:16599-618.
118. Abubaker K, Luwor RB, Escalona R, McNally O, Quinn MA, et al. Targeted disruption of the JAK2/STAT3 pathway in combination with systemic administration of paclitaxel inhibits the priming of ovarian cancer stem cells leading to a reduced tumor burden. Front Oncol 2014;4:75.
119. Liu T, Li A, Xu Y, Xin Y. Momelotinib sensitizes glioblastoma cells to temozolomide by enhancement of autophagy via JAK2/STAT3 inhibition. Oncol Rep 2019;41:1883-92.
120. Giordano G, Parcesepe P, D’Andrea MR, Coppola L, Di Raimo T, et al. JAK/Stat5-mediated subtype-specific lymphocyte antigen 6 complex, locus G6D (LY6G6D) expression drives mismatch repair proficient colorectal cancer. J Exp Clin Cancer Res 2019;38:28.
121. Mesa RA, Kiladjian JJ, Catalano JV, Devos T, Egyed M, et al. Simplify-1: a phase III randomized trial of momelotinib versus ruxolitinib in janus kinase inhibitor-naïve patients with myelofibrosis. J Clin Oncol 2017;35:3844-50.
122. Ng K, Hendifar A, Starodub A, Chaves J, Yang Y, et al. Phase 1 dose-escalation study of momelotinib, a Janus kinase 1/2 inhibitor, combined with gemcitabine and nab-paclitaxel in patients with previously untreated metastatic pancreatic ductal adenocarcinoma. Invest New Drugs 2019;37:159-65.
123. Barbie DA, Spira A, Kelly K, Humeniuk R, Kawashima J, et al. Phase 1B study of momelotinib combined with trametinib in metastatic, kirsten rat sarcoma viral oncogene homolog-mutated non-small-cell lung cancer after platinum-based chemotherapy treatment failure. Clin Lung Cancer 2018;19:e853-9.
124. Padda S, Reckamp K, Koczywas M, Neal J, Kawashima J, et al. P2.03-043 a phase 1b study of erlotinib and momelotinib for TKI-Naïve EGFR-mutated metastatic non-small cell lung cancer. J Thorac Oncol 2017;12:S2143-4.
125. Mascarenhas J, Hoffman R, Talpaz M, Gerds AT, Stein B, et al. Pacritinib vs best available therapy, including ruxolitinib, in patients with myelofibrosis: a randomized clinical trial. JAMA Oncol 2018;4:652-9.
126. Verstovsek S, Odenike O, Singer JW, Granston T, Al-Fayoumi S, et al. Phase 1/2 study of pacritinib, a next generation JAK2/FLT3 inhibitor, in myelofibrosis or other myeloid malignancies. J Hematol Oncol 2016;9:137.
127. Jensen KV, Cseh O, Aman A, Weiss S, Luchman HA. The JAK2/STAT3 inhibitor pacritinib effectively inhibits patient-derived GBM brain tumor initiating cells in vitro and when used in combination with temozolomide increases survival in an orthotopic xenograft model. PLoS One 2017;12:e0189670.
128. Chuang HY, Su Y, Liu HW, Chen CH, Chiu SC, et al. Preclinical evidence of STAT3 inhibitor pacritinib overcoming temozolomide resistance via downregulating miR-21-enriched exosomes from M2 glioblastoma-associated macrophages. J Clin Med 2019;8:959.
129. Jensen KV, Hao X, Aman A, Luchman HA, Weiss S. EGFR blockade in GBM brain tumor stem cells synergizes with JAK2/STAT3 pathway inhibition to abrogate compensatory mechanisms in vitro and in vivo. Neuro-Oncology Adv 2020;2:vdaa020.
130. Hin Tang JJ, Hao Thng DK, Lim JJ, Toh TB. JAK/STAT signaling in hepatocellular carcinoma. Hepatic Oncol 2020;7:HEP18.
131. Regenbogen T, Chen L, Trinkaus K, Wang-Gillam A, Tan BR, et al. Pacritinib to inhibit JAK/STAT signaling in refractory metastatic colon and rectal cancer. J Gastrointest Oncol 2017;8:985-9.
132. Iwamaru A, Szymanski S, Iwado E, Aoki H, Yokoyama T, et al. A novel inhibitor of the STAT3 pathway induces apoptosis in malignant glioma cells both in vitro and in vivo. Oncogene 2007;26:2435-44.
133. Ferrajoli A, Faderl S, Van Q, Koch P, Harris D, et al. WP1066 disrupts janus kinase-2 and induces caspase-dependent apoptosis in acute myelogenous leukemia cells. Cancer Res 2007;67:11291-9.
134. Tsujita Y, Horiguchi A, Tasaki S, Isono M, Asano T, et al. STAT3 inhibition by WP1066 suppresses the growth and invasiveness of Qureshy et al. J Cancer Metastasis Treat 2020;6:27. I http://dx.doi.org/10.20517/2394-4722.2020.58 Page 17 of 18 bladder cancer cells. Oncol Rep 2017;38:2197-204
135. Horiguchi A, Asano T, Kuroda K, Sato A, Asakuma J, et al. STAT3 inhibitor WP1066 as a novel therapeutic agent for renal cell carcinoma. Br J Cancer 2010;102:1592-9.
136. Huang Y, Zhou X, Liu A, Li S, Wang X, et al. Signal transducer and activator of transcription-3 inhibitor WP1066 affects human tongue squamous cell carcinoma proliferation and apoptosis in vitro and in vivo. Zhonghua Kou Qiang Yi Xue Za Zhi 2014;49:308-13. (in Chinese)
137. Zhou X, Ren Y, Liu A, Han L, Zhang K, et al. STAT3 inhibitor WP1066 attenuates miRNA-21 to suppress human oral squamous cell carcinoma growth in vitro and in vivo. Oncol Rep 2014;31:2173-80.
138. Bai H, Wang C, Qi Y, Xu J, Li N, et al. Major vault protein suppresses lung cancer cell proliferation by inhibiting STAT3 signaling pathway. BMC Cancer 2019;19:454.
139. Hou J, Lv A, Deng Q, Zhang G, Hu X, et al. TROP2 promotes the proliferation and metastasis of glioblastoma cells by activating the JAK2/STAT3 signaling pathway. Oncol Rep 2019;41:753-64.
140. Zhou Q, Jiang H, Zhang J, Yu W, Zhou Z, et al. Uridine-cytidine kinase 2 promotes metastasis of hepatocellular carcinoma cells via the stat3 pathway. Cancer Manag Res 2018;10:6339-55.
141. Zhou X, Ren Y, Liu A, Jin R, Jiang Q, et al. WP1066 sensitizes oral squamous cell carcinoma cells to cisplatin by targeting STAT3/miR21 axis. Sci Rep 2014;4:7461.
142. Cheng CC, Shi LH, Wang XJ, Wang SX, Wan XQ, et al. Stat3/Oct-4/c-Myc signal circuit for regulating stemness-mediated doxorubicin resistance of triple-negative breast cancer cells and inhibitory effects of WP1066. Int J Oncol 2018;53:339-48.
143. Tang YJ, Sun ZL, Wu WG, Xing J, He YF, et al. Inhibitor of signal transducer and activator of transcription 3 (STAT3) suppresses ovarian cancer growth, migration and invasion and enhances the effect of cisplatin in vitro. Genet Mol Res 2015;14:2450-60.
144. Palagani V, Bozko P, El Khatib M, Belahmer H, Giese N, et al. Combined inhibition of Notch and JAK/STAT is superior to monotherapies and impairs pancreatic cancer progression. Carcinogenesis 2013;35:859-66.
145. Fu LX, Lian QW, Pan JD, Xu ZL, Zhou TM, et al. JAK2 tyrosine kinase inhibitor AG490 suppresses cell growth and invasion of gallbladder cancer cells via inhibition of JAK2/STAT3 signaling. J Biol Regul Homeost Agents 2017;31:51-8.
146. Faouzi M, Chopin V, Ahidouch A, Ouadid-Ahidouch H. Intermediate Ca2+-sensitive K+ channels are necessary for prolactin-induced proliferation in breast cancer cells. J Membr Biol 2010;234:47-56.
147. Kano Y, Konno M, Kawamoto K, Tamari K, Hayashi K, et al. Novel drug discovery system for cancer stem cells in human squamous cell carcinoma of the esophagus. Oncol Rep 2014;31:1133-8.
148. Xie L, Zeng Y, Dai Z, He W, Ke H, et al. Chemical and genetic inhibition of STAT3 sensitizes hepatocellular carcinoma cells to sorafenib induced cell death. Int J Biol Sci 2018;14:577-85.
149. Zhang L, Xu LJ, Zhu J, Li J, Xue BX, et al. ATM-JAK-PD-L1 signaling pathway inhibition decreases EMT and metastasis of androgenindependent prostate cancer. Mol Med Rep 2018;17:7045-54.
150. Wernig G, Kharas MG, Okabe R, Moore SA, Leeman DS, et al. Efficacy of TG101348, a selective JAK2 inhibitor, in treatment of a murine model of JAK2V617F-induced polycythemia vera. Cancer Cell 2008;13:311-20.
151. Pardanani A, Gotlib JR, Jamieson C, Cortes JE, Talpaz M, et al. Safety and efficacy of TG101348, a selective JAK2 inhibitor, in myelofibrosis. J Clin Oncol 2011;29:789-96.
152. Pardanani A, Harrison C, Cortes JE, Cervantes F, Mesa RA, et al. Safety and efficacy of fedratinib in patients with primary or secondary myelofibrosis: a randomized clinical trial. JAMA Oncol 2015;1:643-51.
153. Harrison CN, Schaap N, Vannucchi AM, Kiladjian JJ, Tiu RV, et al. Janus kinase-2 inhibitor fedratinib in patients with myelofibrosis previously treated with ruxolitinib (JAKARTA-2): a single-arm, open-label, non-randomised, phase 2, multicentre study. Lancet Haematol 2017;4:e317-24.
154. Li SD, Ma M, Li H, Waluszko A, Sidorenko T, et al. Cancer gene profiling in non-small cell lung cancers reveals activating mutations in JAK2 and JAK3 with therapeutic implications. Genome Med 2017;9:89.
155. Zhang FQ, Yang WT, Duan SZ, Xia YC, Zhu RY, et al. JAK2 inhibitor TG101348 overcomes erlotinib-resistance in nonsmall cell lung carcinoma cells with mutated EGF receptor. Oncotarget 2015;6:14329-43.
156. Chen D, Zhang F, Wang J, He H, Duan S, et al. Biodegradable nanoparticles mediated Co-delivery of erlotinib (ELTN) and fedratinib (FDTN) toward the treatment of ELTN-resistant non-small cell lung cancer (NSCLC) via suppression of the JAK2/STAT3 signaling pathway. Front Pharmacol 2018;9:1214.
157. Gorshkov K, Sima N, Sun W, Lu B, Huang W, et al. Quantitative chemotherapeutic profiling of gynecologic cancer cell lines using approved drugs and bioactive compounds. Transl Oncol 2019;12:441-52.
158. Zhou B, Damrauer JS, Bailey ST, Hadzic T, Jeong Y, et al. Erythropoietin promotes breast tumorigenesis through tumor-initiating cell self-renewal. J Clin Invest 2014;124:553-63.
159. Lin C, Cao W, Ren Z, Tang Y, Zhang C, et al. GDNF secreted by nerves enhances PD-L1 expression via JAK2-STAT1 signaling activation in HNSCC. Oncoimmunology 2017;6:e1353860.
160. Genovese MC, Kalunian K, Gottenberg JE, Mozaffarian N, Bartok B, et al. Effect of filgotinib vs placebo on clinical response in patients with moderate to severe rheumatoid arthritis refractory to disease-modifying antirheumatic drug therapy: the FINCH 2 randomized clinical trial. JAMA 2019;322:315-25.
161. Tarrant JM, Galien R, Li W, Goyal L, Pan Y, et al. Filgotinib, a JAK1 inhibitor, modulates disease-related biomarkers in rheumatoid arthritis: results from two randomized, controlled phase 2b trials. Rheumatol Ther 2020;7:173-90.
162. Van Rompaey L, Galien R, van der Aar EM, Clement-Lacroix P, Nelles L, et al. Preclinical characterization of GLPG0634, a selective Page 18 of 18 Qureshy et al. J Cancer Metastasis Treat 2020;6:27. I http://dx.doi.org/10.20517/2394-4722.2020.58 inhibitor of JAK1, for the treatment of inflammatory diseases. J Immunol 2013;191:3568-77.
163. Chuang CH, Greenside PG, Rogers ZN, Brady JJ, Yang D, et al. Molecular definition of a metastatic lung cancer state reveals a targetable CD109-Janus kinase-Stat axis. Nat Med 2017;23:291-300.
164. Huang Z, Zhou W, Li Y, Cao M, Wang T, et al. Novel hybrid molecule overcomes the limited response of solid tumours to HDAC inhibitors via suppressing JAK1-STAT3-BCL2 signalling. Theranostics 2018;8:4995-5011.
165. Hexner EO, Serdikoff C, Jan M, Swider CR, Robinson C, et al. Lestaurtinib (CEP701) is a JAK2 inhibitor that suppresses JAK2/STAT5 signaling and the proliferation of primary erythroid cells from patients with myeloproliferative disorders. Blood 2008;111:5663-71.
166. Pinto N, Prokopec SD, Vizeacoumar F, Searle K, Lowerison M, et al. Lestaurtinib is a potent inhibitor of anaplastic thyroid cancer cell line models. PLoS One 2018;13:e0207152.
167. Takeuchi T, Tanaka Y, Iwasaki M, Ishikura H, Saeki S, et al. Efficacy and safety of the oral Janus kinase inhibitor peficitinib (ASP015K) monotherapy in patients with moderate to severe rheumatoid arthritis in Japan: a 12-week, randomised, double-blind, placebo-controlled phase IIb study. Ann Rheum Dis 2016;75:1057-64.
168. Kivitz AJ, Gutierrez-Ureña SR, Poiley J, Genovese MC, Kristy R, et al. Peficitinib, a JAK inhibitor, in the treatment of moderate-tosevere rheumatoid arthritis in patients with an inadequate response to methotrexate. Arthritis Rheumatol 2017;69:709-19.
169. Ruan Z, Yang X, Cheng W. OCT4 accelerates tumorigenesis through activating JAK/STAT signaling in ovarian cancer side population cells. Cancer Manag Res 2019;11:389-99.
170. Schwartz DM, Kanno Y, Villarino A, Ward M, Gadina M, et al. JAK inhibition as a therapeutic strategy for immune and inflammatory diseases. Nat Rev Drug Discov 2017;17:78.
171. Levitzki A. Tyrphostins: tyrosine kinase blockers as novel antiproliferative agents and dissectors of signal transduction. FASEB J 1992;6:3275-82.
172. Ioannidis S, Lamb ML, Wang T, Almeida L, Block MH, et al. Discovery of 5-chloro- N 2-[(1 S)-1-(5-fluoropyrimidin-2-yl) ethyl]- N 4-(5-methyl-1 H -pyrazol-3-yl)pyrimidine-2,4-diamine (AZD1480) as a novel inhibitor of the jak/stat pathway. J Med Chem 2011;54:262-76.
173. Cao JJ, Hood J, Lohse D, Mak CC, McPherson A, et al. WO2007053452A1 - Bi-aryl meta-pyrimidine inhibitors of kinases. Accessed from: https://patents.google.com/patent/WO2007053452A1/el. [Accessed on 12 Aug 2020].
174. Menet CJ, Fletcher SR, Van Lommen G, Geney R, Blanc J, et al. Triazolopyridines as selective JAK1 inhibitors: from hit identification to GLPG0634. J Med Chem 2014;57:9323-42.
175. Huang T, Xue CB, Wang A, Kong L, Ye HF, et al. WO2011112662A1 Piperidin-4-yl azetidine derivatives as jak1 inhibitors. Accessed from: https://patents.google.com/patent/WO2011112662A1/en. [Accessed on 12 Aug 2020].
176. Thompson JE, Cubbon RM, Cummings RT, Wicker LS, Frankshun R, et al. Photochemical preparation of a pyridone containing tetracycle: a Jak protein kinase inhibitor. Bioorganic Med Chem Lett 2002;12:1219-23.
177. Gingrich DE, Hudkins RL. Synthesis and kinase inhibitory activity of 3ʹ-(S)-epi-K-252a. Bioorganic Med Chem Lett 2002;12:2829-31.
178. Tyner JW, Bumm TG, Deininger J, Wood L, Aichberger KJ, et al. CYT387, a novel JAK2 inhibitor, induces hematologic responses and normalizes inflammatory cytokines in murine myeloproliferative neoplasms. Blood 2010;115:5232-40.
179. William AD, Lee ACH, Blanchard S, Poulsen A, Teo EL, et al. Discovery of the macrocycle 11-(2-pyrrolidin-1-yl-ethoxy)-14,19- dioxa-5,7, 26-triaza-tetracyclo[19.3.1.1(2,6).1(8,12)]heptacosa-1(25),2(26),3,5,8,10,12(27),16,21,23-decaene (SB1518), a potent Janus Kinase 2/Fms-like tyrosine kinase-3 (JAK2/FLT3) inhibitor for the treatment of myelofibrosis and lymphoma. J Med Chem 2011;54:4638-58.
180. Hamaguchi H, Amano Y, Moritomo A, Shirakami S, Nakajima Y, et al. Discovery and structural characterization of peficitinib (ASP015K) as a novel and potent JAK inhibitor. Bioorganic Med Chem 2018;26:4971-83.
181. Fridman J, Nussenzveig R, Liu P, Rodgers J, Burn T, et al. Discovery and preclinical characterization of INCB018424, a selective JAK2 inhibitor for the treatment of myeloproliferative disorders. Blood 2007;110:3538.
182. Flanagan ME, Blumenkopf TA, Brissette WH, Brown MF, Casavant JM, et al. Discovery of CP-690,550: a potent and selective janus kinase (JAK) inhibitor for the treatment of autoimmune diseases and organ transplant rejection. J Med Chem 2010;53:8468-84.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Qureshy Z, Johnson DE, Grandis JR. Targeting the JAK/STAT pathway in solid tumors. J Cancer Metastasis Treat 2020;6:27. http://dx.doi.org/10.20517/2394-4722.2020.58
AMA Style
Qureshy Z, Johnson DE, Grandis JR. Targeting the JAK/STAT pathway in solid tumors. Journal of Cancer Metastasis and Treatment. 2020; 6: 27. http://dx.doi.org/10.20517/2394-4722.2020.58
Chicago/Turabian Style
Qureshy, Zoya, Daniel E. Johnson, Jennifer R. Grandis. 2020. "Targeting the JAK/STAT pathway in solid tumors" Journal of Cancer Metastasis and Treatment. 6: 27. http://dx.doi.org/10.20517/2394-4722.2020.58
ACS Style
Qureshy, Z.; Johnson DE.; Grandis JR. Targeting the JAK/STAT pathway in solid tumors. J. Cancer. Metastasis. Treat. 2020, 6, 27. http://dx.doi.org/10.20517/2394-4722.2020.58
About This Article
Copyright

Data & Comments
Data


 Cite This Article 77 clicks
Cite This Article 77 clicks

 Like This Article 7
likes
Like This Article 7
likes






Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.