
The systemic hallmarks of cancer

Abstract
Cancer is not just a lump of cells that divide, invade, and spread randomly, but rather a multi-layered precisely tuned process that requires the participation of the whole organism. There is an urgent need to zoom-out from the cellular and the local stromal view and broaden our perspective by including the whole organism level. Geographically separated cancer tissues communicate between themselves, forming a system that interacts with the rest of the organism through cancer induced systemic pathogenic networks. In the present paper, I introduce six systemic hallmarks of cancer that emerge as a result of these interactions. I also describe several potential therapeutic approaches that can be developed using the cancer system concept. Overall, I argue that the tumoricentric paradigm should be replaced with a broader approach that brings into focus the “cancerized” organism.
Keywords
Introduction
The cancer system
From the systemic biology point of view, organisms are complex, embedded, multi-layered networks of interactions. At the cellular level, the networks are comprised of genes, metabolic intermediates, miRNA and signaling molecules (proteins, lipids, ions). At the tissular level, the networks are comprised of interactions between different cell types and between the cells and the supporting stroma. At the organismic level, the networks are comprised of interactions between different body systems (endocrine, nervous, immune, etc.). As described by the seminal work of Denis Noble[1-3] these three levels of networks (cellular, tissular, organismic) [Figure 1] are co-dependent and there is no priviledged level of causation. They interact and influence each other.
Figure 1. Three levels of organization
Cancer is also a multi-layered disease with multiple complex networks of interactions located at different levels, (i.e., cellular, tissular, organismic). The focus on the genome, the cancer cells, or even the cancer tissues, is too narrow and, in order to better understand the cancer process, there is an urgent need to zoom-out and broaden our perspective by including a broader, organismic level.
We define a system as a dynamic entity of several interacting components that are co-dependent and function in an integrated way. A single cell, an organ, the entire human body are all systems. In the present paper, I will focus on cancer at the macroscopic level. Macroscopically, experimental data accumulated over more than a decade, supports the concept of a cancer system formed by several geographically separated cancer tissues (the primary tumor, the local and the distant metastasis). The cancer system and the body systems are co-dependent and, through their interaction, new cancer induced pathologic systemic networks (CISPN) appear and the whole organism is “cancerized” to support cancer development [Figure 2].
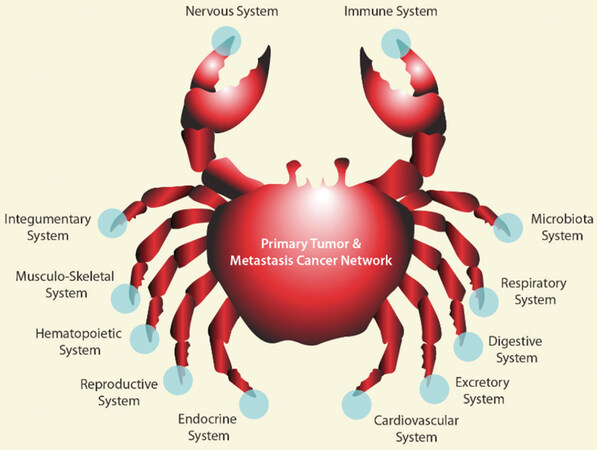
Figure 2. The cancer system and the body systems interact leading to novel cancer induced systemic pathologic networks
Cancer as a systemic disease
Over the last decade, several models of cancer as a systemic disease have been proposed[4-8]. In 2010, Mikala Egeblad and her collaborators introduced the model of the tumor as an organ that may influence the immunity, the metabolism and the coagulation status of the host[4]. In a paper published the same year, Sandra Mc Allister and Robert Weinberg suggested that tumor-host interactions extend well beyond the local tissue microenvironment and introduced the tumor “instigation” concept in which primary tumors perturb normal host organs and support the growth of metastatic tumors at distant anatomic sites[5]. They listed several factors secreted by the tumors with systemic effects: vascular endothelial growth factor (VEGF), interleukin (IL)-6, IL-8, stromal cell-derived factor-1 (SDF1), fibroblast growth factor, growth-related oncogene-α (CXCL1), platelet-derived growth factor (PDGF), angiopoietins, transforming growth factor β (TGF-β), hepatocyte growth factor (HGF), angiogenin, leptin, sonic hedgehog homolog, regulated upon activation normal T-cell expressed (CCL5), and osteopontin[5]. They also proposed a novel treatment of metastases by blocking their access to supporting stromal cells derived from the bone marrow. In a subsequent publication, the same authors[7], refined their original model bringing additional support to the complementary idea that tumors can be also significantly influenced by systemic processes. In 2014, a research team from Austria, introduced the concept of tumor macroenvironment and described mainly the global metabolic changes that tumors exert on the whole organism[6].
This systemic perspective should not be limited solely to clinically stage IV cancers. Often, systemic effects appear also when the tumors are localized and, in many instances, metastasis is present in a subclinical form even when the tumor appears localized. This may be the reason why even when detected very early, a significant proportion of cancers is incurable. For example, the 5 year survival of stage I non-small cell lung cancer disease is approximately 60% (American Cancer Society Statistics, 2020). In 2019, a neuroscientist, Jeremy Borniger, published two papers[8,9] focused on brain-tumor interactions, with special emphasis on the interaction of subcortical neural populations and cancer. Specifically, he and his team discovered that non-metastatic breast tumors may influence the endocrine and the immune systems of the hosts. Using a breast cancer mouse model, they demonstrated that by inhibiting the signaling of the lateral hypothalamic orexin/hypocretin neurons, the sleep quality and the metabolic dysregulations induced by the tumor were improved[9].
We have also proposed previously the need for a broader, systemic perspective on cancer and introduced briefly the idea of the cancer system[10]. The notion of the cancer system although closely related to the idea of the cancer-systemic disease is different in a fundamental way. The meaning of the word “systemic” refers to “affecting the body generally”. Metastatic and, sometimes, localized cancers, influence the whole organism and therefore they are classified as systemic diseases. On the other hand, macroscopically, cancer behaves like a sort of an organism within an organism and metastasis appears as a finely orchestrated process. The concept of a cancer system tries to capture precisely this deterministic behavior.
Cancer as a developmental disease
The idea that cancer represents an embryonal developmental program gone haywire has been around for more than four decades[11,12]. As shown by a recent review[13] there are some tissular and organismic genes that may play a role both in embryogenesis and cancer. It is important to point out that until approximately three weeks after fertilization, the embryo does not have a functioning circulatory system, and, therefore, the developmental programs involved in the embyo development are likely different from that of the metastatic process. A notable exception is the neural crest migration where epithelial-to-mesenchymal transition plays a critical role. Similar to the neural crest migration[14], the metastatic process represents a transformed cellular program that once activated leads to the development of disseminated tumors at distance from the original site. The fact that that the same genes (i.e., the nuclear hormone receptors, Hedgehog, Wnt, TGF-β, Notch) are involved in the generation and maintenance of multicellularity, and, they are also dysregulated in stem cells and metastasis[15], suggest the striking idea that cancer, in general, and the metastatic process, in particular, may represent the activation of a developmental program that leads to the creation of a novel, pervasive, multicellular entity. As previously suggested by Mark Vincent, cancer appears to be much more than a simple dysregulated growth and may represent a form of multicellular life, with symbiotic properties[16,17], that establishes a commensual relationship with the organism where it develops. In this process, the whole organism, “cancerized” through the development of CISPN, as we will argue in this paper, far from being a passive bystander, becomes an active enabler of cancer progression and spread.
Metastasis as a finely orchestrated deterministic process
As opposed to the apparition of malignant tumors that is, in general, related to genetic mutations, metastasis appears to be mainly an epigenetic process. In the 2011 updated version of their original hallmarks article[18], Hannahan and Weinberg, noted that the ability of cancer cells to invade and metastasize may not require additional genetic mutations in addition to those already present in the primary tumors[19]. Also, the majority of the genes proposed by Massagué and collaborators more than a decade ago[20,21] in their step by step model of metastasis are not mutated. In addition, Vogelstein et al.[22] noted that despite considerable effort, specific genetic alterations that distinguish cancers that metastasize from cancers that do not metastasize have not been yet identified. The immediate conclusion, drawn by the Vogelstein team[22], is that there are no specific metastasis genes.
This opinion of metastasis as a random, nondeterministic process has been challenged for 130 years since Paget asked the famous question: “What is what decides what organs suffer from disseminated cancer?” and launched the “seed and soil” hypothesis[23]. By the late 70’s, metastasis started to be understood more and more as the result of non-random tumor-host interactions[24], and the Paget’s “seed and soil” hypothesis has been strongly supported by the work of Fidler and Kripke[25] and Price et al.[26]. A recent study described widespread epigenetic reprogramming during the evolution of distant metastasis of pancreatic cancer in the absence of metastasis-specific driver mutations[27]. This manifested as global reprogramming of histone H3K9 and DNA methylation within large heterochromatin domains (LOCKs) as well as regional changes in gene regulatory modifications. Interestingly, the authors found that the epigenetic changes were controlled by an anabolic glucose metabolism enzyme 6-phosphogluconate dehydrogenase (PGD). Glucose deprivation, RNA interference (RNAi) against PGD, and, treatment with 6-aminonicotinamide, reprogrammed the chromatin state of the distant metastasis. In addition, cells treated with RNAi against PGD did not form distal metastasis[27]. Another recent study[28], found also that in prostate cancer the master regulator genes of metastasis are genes involved in epigenetic regulation. Silencing a particular histone methyltyransferase gene (Nuclear receptor binding SET Domain protein 2, NSD2) in vivo allografts, resulted in significant improvement in survival in the mice treated as well as a significant reduction in the metastatic burden without any effect on the primary tumor growth[28].
These observations suggest a different view of metastasis. The plethora of genetic abnormalities present in established malignant tumors may not be the main driver of metastasis. No genetic mutation or mutations have been unequivocally shown to be associated with progression from localized to metastatic disease[29]. As shown by several in vitro experiments, epigenetic factors present inside and outside tumor cells may control the metastatic process. The cytoplasm of human embryonic stem cells can epigenetically reprogram multipotent metastatic melanoma cells and made them to assume a melanocyte-like phenotype[30]. Adam Telerman and Robert Amson, two researchers from École Normale Supérieure from Paris, France, who have been modeling tumor reversion for more than 20 years, stated[31] that the “reversion process involves a reprogramming mechanism using epigenetic and probably genetic tools that will supersede the changes in cancer by assembling and triggering alternative ways leading to the suppression of tumorigenicity”. Some of the metastasis master regulators may not be even located inside the cancer cell. The work of Bissell and Radisky[32] and Orimo and Weinberg[33] demonstrated the crucial role of the tumor associated stroma in promoting tumor metastasis. Convincingly, a recent review[34] illustrated how hyaluronan, an integrated component of the extracellular matrix (ECM), may modulate several key hallmarks of cancer: sustaining of the proliferative signaling, evasion of apoptosis, angiogenesis, activation of invasion and metastasis, reprogramming of energy metabolism and evasion of the immune response.
Cancer cell programs
If cancer cells switch back and forth between different programs, cancer may represent a controllable cellular state that can rerouted to a non-neoplastic phenotype. The model of cancer as a potentially reversible cellular program[35] complements and refines the genetic model. It has been previously suggested that many of the properties associated with invasion and metastasis do not arise as purely cell autonomous processes[36]. In most of the cases, the metastatic process seems to be due to adaptation and not to selection of the cancer cells[37]. It is the secretion of factors such as TGF-β, HGF, tumor necrosis factor (TNF)-α, Wnt and PDGF by the surrounding tumor stroma, and, the activation in the tumor cells of several master regulators of embryogenesis, such as the transcription factors Twist, Snail, Slug, Zeb1 and Zeb2, regarded as the epithelial to mesenchymal transition (EMT) core regulators, that drive metastasis[13,29]. These processes may be mediated by miRNAs[38], that coordinate multiple genes at the same time, so it will be more appropriate to talk in terms of various cellular programs, i.e., a division program possibly controlling key mitotic genes[39], an invasion program possibly controlling key invasion genes[40,41], and, a metastatic program, possibly controlling key EMT genes[13][Figure 3]. The master regulators of these programs may or may not partially overlap. For example, miR-21 and miR-222 are involved in uncontrolled proliferation, miR-130 and miR-126 are involved in tumor angiogenesis and miR-373 and miR-155 are involved both in invasion and metastasis[38]. The existence of specific cellular programs activated during the cancer process, suggests the possibility that the transformations induced by cancer both at the level of the tissue where it originally appears, and at distant sites where metastasis are formed, are not simple random by-products of malignancy but represent a well orchestrated process of local and global “cancerization”. In this paper, I argue that the metastatic phenotype is initiated and maintained by non-random CISPN developed at the organismic level. The emergence of these CISPN may result mainly from the finely regulated secretion by the tumors and their stroma of specific exosomes[42,43]. Exosomes have been involved in the communication between the primary tumor and remote metastatic sites[44] and, cancer cell-derived exosomes contain miRNAs that may regulate all the systemic hallmarks of cancer described below. In spite of the presence of RNase in blood, miRNAs survive due to their presence in exosomes[45]. Both the nature of the proteins expressed on the surface of exosomes and the exosomes cargo are non-random as demonstrated by the work of David Lyden’s lab[46,47]. Recently his team analyzed the protein exosomes from plasma of patients with five cancer types (breast, colorectal, lung, pancreatic, mesothelioma) and found that in cancer patients, the circulating plasma exosomes proteins, derive not only from the tumor itself, but also from the tumor environment, distant organs (i.e., liver) and the immune cells, this data supporting the “cancerized” organism model[43].
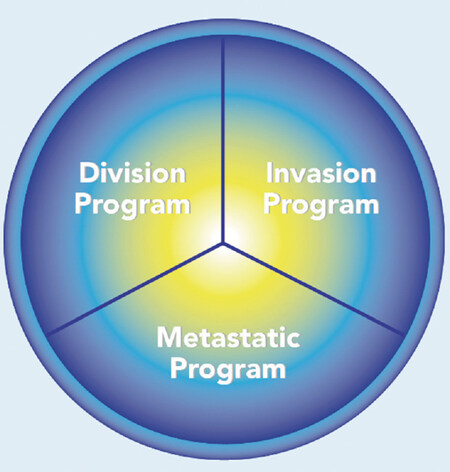
Figure 3. The cancer cell programs
Clinical implications of the model of cancer as a modified cellular program
The division, invasion and metastatic programs may be activated differently and in different order in various cancer types, and this may explain the fact that cancers arising in different tissues have different propensity to grow locally, to invade the surrounding stroma and to metastasize. This might be related to distinct modified cellular programs present in Cancer Stem Cells (CSCs). CSCs by definition are a small subpopulation of cells within tumors with capabilities of self-renewal, differentiation, and tumorigenicity when transplanted into an animal host. When we think of CSC we mainly think of their ability to grow and form colonies but CSCs present in different tissues may not be the same qualitatively, and even, quantitatively[48]. It is likely that due to these differences, programs for division, invasion and metastasis are differently activated in different CSCs. Some CSCs may “instigate” or “educate” the stromal cells by secreting signals that induce changes in these cells that facilitate local invasion of the tumor[49]. Other distinct population of stem cells, so called migrating cancer stem (MCS) cells, may be responsible for metastasis as proposed originally by Brabletz[50]. Recently, this population of tumor cells with MCS properties was identified in a study conducted at Memorial Sloan Kettering Cancer Institute (MSKCI)[51]. Using a colorectal cancer mouse model, the MSKCI investigators found two distinct population of stem cells: an adenoma forming stem cells population with oncogenic mutations and a L1CAM positive tumor-propagating metastasis-initiating stem cells without oncogenic mutations[51].
A pervasive oncology dogma postulates that cancer develops in a linear way by initially growing locally, then subsequently invading the tissue where they appear and, finally, if given enough time, in the majority of cases, metastasize. Experimental data and clinical practice suggest that this assumption is incorrect. Some cancers, like breast or prostate, for example, sometimes behave as benign tumors that do not invade locally or metastasize and, maybe, this is why, the global, indiscriminately screening programs for breast and prostate cancers, may lead to over treatment of some patients. Another example is sarcoma, where roughly 50% of the sarcoma metastasize and 50% do not[52]. As demonstrated by the work of Ganesh et al.[51], the classical step by step genetic model of colorectal carcinogenesis of Fearon and Vogelstein[53] may not apply to the metastatic process who does not involve a specific set of mutated genes. The different activation of different cancer cells programs in tumors of different types might explain the striking difference in clinical stage presentations of different cancer locations. For example, as many as 55% of squamous head and neck cancer presents as stage 4 most frequently with lymph nodes metastasis[54], but only approximately 7% of thyroid cancers present as stage 4[55]. The presence of distinct cellular programs in cancer may also solve the enigma of the existence of carcinomas of unknown origin where the primary tumor is never found. It is conceivable that in metastasis of cancers of unknown origin the metastatic program is activated before the division and invasive programs.
The systemic hallmarks of cancer
The hallmarks of cancer described by Hanahan and Weinberg in their two articles[18,19], refer mainly to the cellular and tissular hallmarks of cancer. More recently[56], Welch and Hurst proposed four cancer hallmarks specifically associated with the metastatic process: motility and invasion, colonization, plasticity and modulation but these four hallmarks are practically identical with the succesive steps of the metastatic process described more than a decade ago by Joao Massagué[20,21] and his collaborators. In this paper, we describe six novel systemic cancer hallmarks, that appear as a result of the interaction between cancer and the organism at the macroscopic level. The first systemic hallmark is the cancer system itself established through the connections between the primary tumor, the bone marrow and the distal metastasis. The five other systemic hallmarks are as following: the global inflammation, the immunity inhibition, the metabolic changes leading to cachexia, the propensity to thrombosis, and the neuro-endocrine changes [Figure 4].
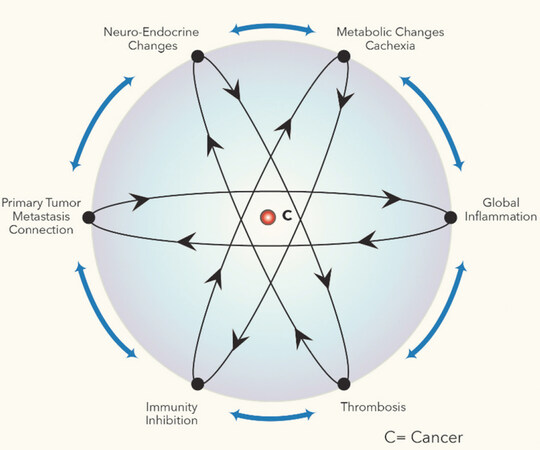
Figure 4. Systemic hallmarks of cancer
Each of these six hallmarks is established through a different CISPN. In the sections below, I will discuss one by one, the six CISPN. The accompanying figures [Figures 5-10] are raw sketches illustrating the salient components of the different CISPN. Apart from the nervous system, the connections between the CISPN components are made through exosomes, cytokines and other soluble factors, represented by the dotted lines.
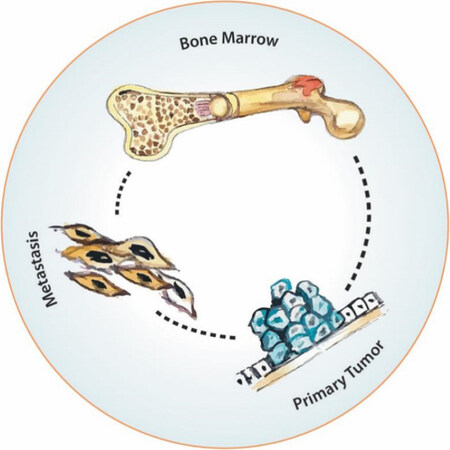
Figure 5. Primary tumor-metastasis-bone marrow network
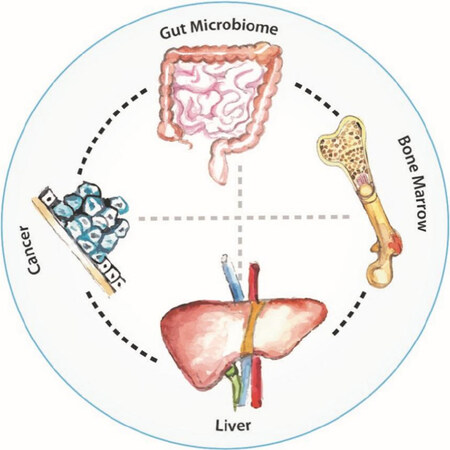
Figure 6. The systemic inflammation network
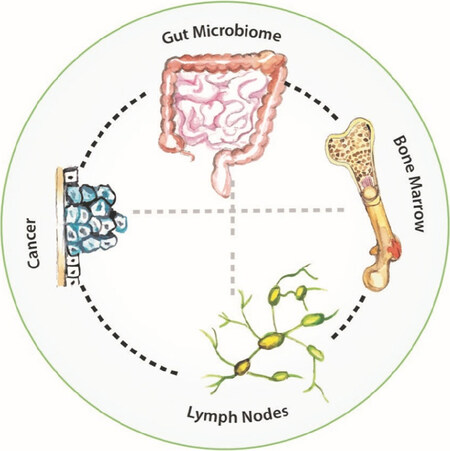
Figure 7. The immunity inhibition network
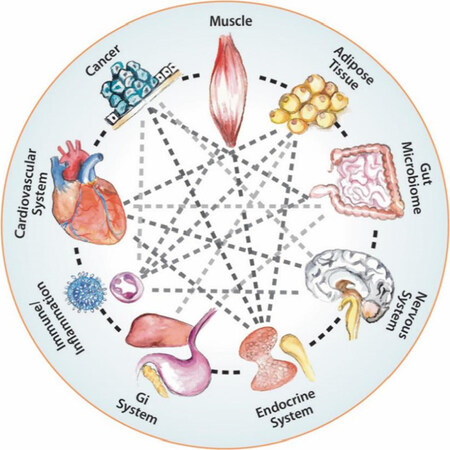
Figure 8. The global metabolism/cachexia network
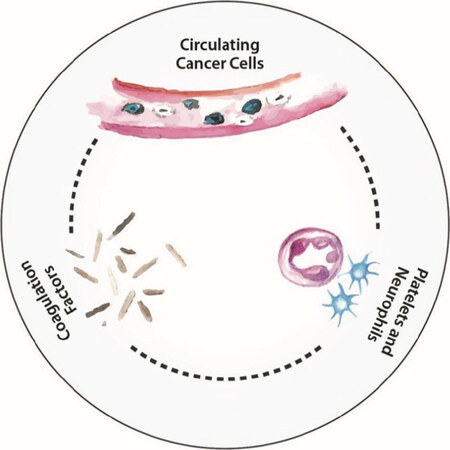
Figure 9. The thrombosis network
Figure 10. The neuro-endocrine network
The primary tumor-metastasis network
Clinically, it has been noticed for a long time that the primary tumor and the distal metastases are interconnected and co-dependent. In some cases of renal cell cancer, for example, resecting the primary tumor induces a regression in the distal metastasis[57]. On the contrary, in several experimental models, as shown by Folkman and his collaborators, resecting the primary tumor may accelerate the development of metastasis[58,59]. Over the last two decades, it has been demonstrated also that besides this primary tumor-metastasis influence, there is a permanent “trialogue” between the primary tumor, the metastatic sites and the bone marrow [Figure 5]. Bone marrow can function as a source of hematogenic progenitor cells that prepare the niche for metastasis[60,61] and, also possibly, as a source of malignant stem cells[62].
Bone marrow-derived cells (BMDCs), which are frequently recruited to sites of tissue injury and inflammation, are crucial for the malignant process. In an elegant experiment, Houghton et al.[63] demonstrated that in some cases, BMDCs may even represent the origin of malignant cells. These findings were subsequently confirmed by a different research team in a sarcoma mouse model[64].
Kaplan et al.[60] pioneered the work on the metastatic niche by demonstrating that BMDCs that express vascular endothelial growth factor receptor 1 (VEGFR-1) home to tumor-specific pre-metastatic sites and form cellular clusters before the arrival of tumor cells. Besides, BMDCs that facilitate the growth of tumor cells at distance from the site of origin and, cytokines and vesicles released into the circulation also contribute to the development of distal metastasis[65-68]. In a recent review[69], a comprehensive list of 34 primary-tumor, tumor stroma and myeloid derived factors that mobilize and recruit myeloid cells directly from the bone marrow to the pre-metastatic niche was compiled. The authors also proposed six characteristics of the pre-metastatic niche that empower the niche to favor tumor cell colonization and promote metastasis: angiogenesis and vascular permeability, lymphangiogenesis, inflammation, immunosuppression, organotropism and reprogramming. These characteristics determine whether the metastatic cells present in the blood circulation can colonize and survive or become dormant after arrival[69]. The best well described tumor secreted soluble molecules responsible for the niche formation and development are: VEGF-A, placental growth factor, and versican[69]. Besides the cancer cells, there are also factors secreted by the tumor stroma that play a similar role: TGF-β, TNF-α, hypoxia-inducible factor-1 (HIF-1), granulocyte colony-stimulating factor (G-CSF). Some of these factors are secreted by the BDMCs themselves: VLA-4 (integrin α4β1), matrix metalloproteinase (MMP)9 and ID3 protein (ID is a term that refers to its functional properties as both an inhibitor of DNA binding and an inhibitor of cell differentiation)[70].
The exosomes are key factors responsible for the preparation of the pre-metastatic niche and the communication between the tumor, the bone marrow and the distal metastatic site[43,71]. Different materials (proteins, mRNAs, DNA and miRNAs) carried inside the exosomes can be functionally delivered between different types of cells and transferred to distant locations, influencing the biological activities of tumor and non-tumor cells and promoting tumor growth, invasion, metastasis, angiogenesis, and drug resistance[43,71]. A comprehensive review highlighted the key role played by the ECM components like syndecan in regulating exosome biogenesis, protein composition, function, and docking to recipient cells[72]. Heparan sulphate chains of syndecans are essential for exosome formation within endosomal compartments, and trimming of heparan sulphate by heparanase activates the formation of an endosomal complex containing syndecan coupled to syntenin and ALIX[73]. Reversely, tumor exosomes expressing high CD44 expression bind to hyaluronic acid and modulate the ECM as demonstrated for degradation of collagens, laminins, and fibronectin[74]. Tumor-associated exosomes have been identified in biological (plasma, urine, saliva) and pathological (malignant effusions, pleural effusions, ascites) fluids from cancer patients. David Lyden’s team from Weill Cornell Medical Center (WCMC), was able to demonstrate that in melanoma the transfer of the MET oncoprotein from tumor-derived exosomes to BM progenitor cells promoted the metastatic process and to describe quantitative and qualitative exosome signatures, along with specific BM progenitor cell populations mobilized as representative hallmarks of metastatic disease[75]. Certain miRNAs are enriched in exosomes coming from the cancer cells, indicating that the exosomes composition seems to be controlled[76]. Exosomes from mutant KRAS colorectal cancer cells for example show a distinct miRNA profile compared to wild type cells[77]. The same WCMC team showed that in pancreatic cancer, the exosomes contained macrophage inhibitory factor (MIF) that is involved in the recruitment of bone marrow-derived macrophages and the blockade of MIF prevented liver pre-metastatic niche formation and metastasis. By releasing soluble factors, tumor cells were able to specifically direct bone marrow derived cells to the sites were they were supposed to go[68]. An “integrin code” present on the surface of the exosomes seems to be responsible for the homing of future metastasis to precise prespecified distal organs[46].
Redig and McAllister also included the participation of the bone marrow as a sine qua non component of the “instigation” model[78]. These authors demonstrated a process of “systemic instigation” by injecting cells coming from an aggressive breast tumor in the flank of a mouse and analyzing the effect of these cells on the growth in the opposite flank of tumor cells coming from an indolent breast cancer tumor. The explanation for the influence at distance of one cancer tissue on the growth of another was established to be bone marrow-derived cells that were recruited to the distal sites and instigated the previously indolent tumor cells to grow[78]. The importance of studying cancer as a systemic disease was underscored by another study done by a group of researchers from Harvard Medical School[79]. The authors described a systemic cross-talk between lung tumors and bones. In an elegant mouse model, lung adenocarcinomas were able to remotely activate osteoblasts in bones even in the absence of local metastasis. In turn, these osteoblasts supplied tumors with neutrophils, which fostered cancer progression[79].
Sometimes, the initiators of this “trialogue” are not the tumor cells themselves, but other stromal cells derived from the “cancerized” stroma. Cancer-associated fibroblasts have been shown, for example, to release cytokines[80] [i.e., SDF1] that once entered in the circulation may trigger the systemic release from the bone marrow of stem cells and haematopoietic progenitors that will support the formation of metastases[81]. A research team from MD Anderson have shown that the origin of these cancer-associated fibroblasts is primarily the bone marrow[82] but their origin and function within the tumor stroma varies[83]. The existence of these networks of communication between geographically separated sites, brings experimental evidence to the cancer system model, and, supports the idea that metastasis is a non-random, finely regulated process.
The systemic inflammation network
The link between local stromal inflammation and cancer progression is well known and the molecular pathways responsible for this link have been well characterized[84]. As any pathologist would certify, local inflammation is present in the stroma of many tumors and, inflammatory cells and molecules, may be involved in almost every aspect of cancer progression, including the tumour cells’ ability to metastasize. Colotta et al.[85] proposed that cancer related inflammation represents a seventh hallmark of cancer.
As described by Grivennikov et al.[86], there are several types of inflammation that can promote cancer development and progression, differing by cause, mechanism, outcome, and intensity. Briefly, there is chronic inflammation associated with infections or autoimmune disease, there is inflammation due to prolonged exposure to environmental irritants or obesity, there is another distinct type of inflammation related to the tumor, and, finally, there is inflammation related to cancer therapies themselves. Approximately 20% of human cancers may be related to chronic inflammation caused by infections, exposure to irritants, or autoimmune disease[87]. On the other hand, not only chronic inflammation may lead to cancer, but cancer may also cause local and systemic inflammation. Oncogene activation in cancer cells lead to expression of pro-inflammatory transcription factors within tumor cells [such as nuclear factor-kappaB (NF-κB), signal transducer and activator of transcription (STAT)3 or HIF-1α]. These activated transcription factors mediate the expression of key cytokines and chemokines as well as inflammatory enzymes within the tumor microenvironment. At the tissular level, different cytokines can either promote or inhibit tumor development and progression. Some of them may lead to tumor progression (IL-6, IL-17, IL-23), and also have direct effects on cancer cell growth and survival [TNF-related apoptosis-inducing ligand, Fas ligand, TNF-α, epidermal growth factor receptor (EGFR) ligands, TGF-β]. Others [IL-12, interferon (IFN)γ] may have an anti-tumor effect[86].
Cancer inflammation is not only a local phenomenon [Figure 6]. High serum concentrations of inflammatory cytokines, (i.e., IL-1, IL-6) are found in many advanced malignancies[88]. Circulating cytokines and small inflammatory molecules, such as chemokines and matrix-degrading proteins, are also involved in the systemic inflammation, and play a crucial role in the metastatic process[88]. IL-1, for example, is involved in invasion and angiogenesis. IL-1 may enhance the invasiveness of already existing tumor cells by the induction of inflammatory molecules, such as MMPs, VEGF, heparanase, chemokines, and integrins on the malignant cells and endothelial cells, or by switching on the angiogenesis leading to tumor dissemination and metastasis[89]. Systemic inhibition of IL-1 with anakinra (a recombinant derivative of IL-1RN) inhibits the growth and density of new vessels in IL-1-producing human tumor cell lines xenografted into immunodeficient mice, but not in their counterparts that do not produce IL-1[90]. A recent review[91], provided a comprehensive list of factors associated with colorectal systemic inflammation including cytokines, chemokines and growth factors (IL-6, C-C Motif Chemokine Ligand (CCL)2, CXCL (C-X-C Motif Chemokine Ligand)8, CSF1 [macrophage colony-stimulating factor (M-CSF)], and CSF2 [granulocyte-macrophage colony stimulating factor (GM-CSF)]. The authors noted that immune cells and fibroblasts are capable of producing many of these factors at much higher level than tumor cells and pointed out to the role of the stroma in systemic inflammation. Some of these immunomodulatory effects are modulated by exosomes that contain in their cargo a large variety of molecules[92,93]. Exosomes secreted by tumors contain IL-8, CCL2, CCL3, CCL4, CCL5, CCL20 and TGF-β[93].
The liver is a key player in the systemic inflammatory response. All the acute-phase proteins, including C-reactive protein (CRP), amyloid A, α1 antitrypsin, and α1 acid glycoprotein, are synthesised in the liver and secreted into the circulation[94]. Classical studies have shown the deleterious role played by the citokines secreted by the Kupfer cells in acute inflammation[95]. For example, complete elimination of liver macrophages, decreased the mortality of mice challenged with zimosan, a potent inflammatory agent, from 27% to 0%[96].
Another key component of the global inflammation network is the gut microbiota. The human body is in symbiosis with the gut microbiota, which outnumbers human cells by a 10-fold factor. As shown by two Science articles, gut microbiota modulates inflammation in both the tumor microenvironment and in the systemic circulation[97,98]. Microbiota also regulates steady-state myelopoiesis and neutrophil homeostasis[99]. Mouse models have shown that gut microbes promote the development of mammary carcinomas via a neutrophil-mediated mechanism[100], and, microbiota-driven mobilization of myeloid-derived suppressor cells, favors malignant progression through systemic tumor promoting inflammation[101].
Increase of the systemic markers of inflammation as neutrophils, lymphocytes and platelet counts and acute phase proteins, such as CRP and albumin or their combinations, computed in different scores, i.e., the neutrophil lymphocyte ratio, the platelet lymphocyte ratio and the Glasgow Prognostic Score, are associated with adverse prognosis in several malignancies[102].
A systemic immune-inflammation index (SII), which is calculated as platelet (P) × neutrophil (N)/lymphocyte (L) counts, has also been demonstrated to be closely associated with the prognosis of solid tumors especially lung cancer[103].
In most advanced cancers, systemic inflammation is caused by cancer itself and indicate the aggressiveness of the tumor[104]. Unfortunately, despite pre-clinical efficacy demonstrated in several animal studies, until present, agents used to manipulate systemic inflammation in the treatment of patients with advanced-stage cancer have only shown modest results[105]. The clinical trials that used inhibitors of primary inflammatory cytokines (e.g.,TNF-α, IL-6, IL-8), in the treatment of various types of human cancers (i.e., pancreas, renal) showed only limited benefit. This is not surprising as the function of cytokine varies with the clinical context and the same cytokine may promote or inhibit cancer progression. The same cytokine can be beneficial in some clinical context and detrimental in others, and the term yin-yang has been used for cytokine behavior[106]. Currently there are multiple clinical studies in progress using agents that target cytokines (i.e., IL-1, CXCR4/CXCL12), transcription factors (i.e., JAK-STAT pathway inhibitors) or local immune/inflammatory cells (i.e., macrophages M2) and the field of cancer inflammation is currently a very active area of research[105,107].
The immunity inhibition network
Tumor promoting inflammation and anti-tumor immunity are the two opposite factors that shape the evolution of tumors[108]. As illustrated in the above section, tumors actively induce a global inflammatory state. They also inhibit the immune system, both locally and systemically [Figure 7]. The local inhibition of the immune system by the tumor checkpoint molecules has been well characterized and the use of checkpoint inhibitors is currently approved in many types of cancers. Tumors may also have a global inhibitory effect on the immune system as recently shown by a team from the University of Pennsylvania. The researchers described the release of exosomes carrying programmed death-ligand 1 (PD-L1) on their surface by metastatic melanoma cells. Stimulation with IFN-γ increased the amount of PD-L1 on these vesicles, which suppressed the function of CD8 T cells and facilitated tumor growth[108]. Tumor cell-derived exosomes can also impair immunity through different mechanisms: exosomes containing miR-203 secreted by pancreatic cells may impair activation of the immune system through downregulation of toll-like receptor 4 and IL-12[109], exosomes can down regulate the functions of immune cells[110,111], may promote Tregs expansion[112], and inhibit the activity of natural killers (NK) cells[113].
As is the case with inflammation, there is a co-dependent relationship between the immune system and the gut microbiome. The immune system plays an important role in defining the composition of the microbiota and preserving the ecology of the microbiota. Reversely, the microbiota influences all aspects of the immune system. Gut microbiome plays an important role in the training and the functional tuning of the immune system and can be seen as one of the key modulators of the immune system[114]. In addition to influencing localized immune responses, microbiota also has broader effects contributing to innate and adaptive immunity at multiple levels[115]. Myeloid cells respond to microbial signals, and initiate innate and adaptive immune responses[99]. In 2013, it has been shown in two murine models that germfree or antibiotic-treated animals did not respond to chemotherapy, indicating that an intact microbiome was required for modulating the myeloid-derived immune cell responses in the tumor microenvironment[97,98]. Alterations in the gut microbiome can affect response to immunotherapy in several cancer types. Matson et al.[116] identified different bacterial species as being critical for response to therapy in their patients with advanced melanoma, with Bifidobacterium longum, Collinsella aerofaciens, and Enterococcus faecium, among others, found to be enriched in the feces of patients that responded to anti-PD-L1. Similar findings were reported by Routy et al.[117] in patients with advanced urothelial carcinoma, non-small cell lung cancer, and renal cell carcinoma. Patients who have been treated with antibiotics within several months before, during, or after treatment with PD-1/PD-L1 blockade had shorter progression-free survival and lower overall survival rates compared with patients who had not received antibiotics. After sequencing fecal samples from these patients, the genera Akkermansia and Alistipes were enriched, and, the bacterial species A. muciniphila, specifically, was found to be highly represented in patients that responded to checkpoint blockade.
The immune cells play a dual role in cancer[118,119]. Classically, some immune cells may promote cancer growth (M2 macrophages, T regs cells) and others fight cancer (M1 macrophages, CD8 cells). This is an over simplification as the same type of cells may play a pro, or anti-neoplastic role depending on the local and systemic context. For example, in the majority of cancers, an increased number of T regs in the tumor is associated with a poor prognostic, but in patients with colon or breast carcinomas, the presence and frequency of T reg in the tumor is correlated with an improved prognostic[120]. A similar phenomenon has been shown for tumor associated macrophages[121]. Like macrophages and T reg cells, tumor-associated neutrophils and NK cells may have both antitumoral and protumoral functions[122]. As shown in by Labelle et al.[123], platelets attract neutrophils into the tumor thrombi contributing to the metastatic niche development. Also, a high neutrophil to lymphocyte ratio, predicts poor outcome in several types of cancer including lung cancer, pancreatic cancer and colorectal cancer. There is new data showing direct involvement of neutrophils in different types of cancer and there is increasing evidence in preclinical models that granulocyte-CSF (G-CSF) can promote metastasis[124,125]. Also, as shown by several research teams, metastatic cancer cells can induce neutrophils to form metastasis-supporting neutrophil extracellular traps (NETs) and drugs that degrade NETs have been shown to have a profound inhibitory effect on the development of metastatic disease in preclinical models[126,127].
The global metabolism/cachexia network
In order to ensure sufficient biomass synthesis for their growth, cancer cells need to maintain high metabolic turnover rates. A large amount of energy is required to support this process. For example, an estimated ~17,700 kcal are required over 3 months to support metastatic colorectal cancer growth[128]. Since the seminal work of Warburg[129], it has been observed cancer cells have distinct metabolic programs than normal cells and metabolic reprogramming has been acknowledged as one of the classical hallmarks of cancer[19]. The most distinctive metabolic differences of cancer tissues are increased aerobic glycolysis, elevated glutaminolytic flux and enhanced amino acid and lipid metabolism. Some types of cancer cells utilize in excess glucose and, in some cases secrete lactate even in the presence of oxygen (the Warburg phenomenon). The propensity of cancer cells towards aerobic glycolysis does not seem to be related to an impairment of the respiration, as respiration is also needed for tumor growth[130,131]. In some cancer patients, lactate is converted back to glucose in the liver, a process known as the oncogenic Cori cycle[132-134] a process that is energetically very inefficient. Besides glucose and lactate, there are other nutriments needed for tumor growth for example, glutamine, glycine and aspartate for purine and pyrimidine synthesis, serine for membrane lipid component synthesis, branched aminoacids, lipids, acetate and others[135]. Not in all cancers the Warburg phenomenon is present, and, sometimes, high glycolytic rates in tumors and mitochondrial respiration often operate simultaneously in tumors[136]. A sort of metabolic parasitism has been described at the tissular level by a group of French researchers[137] who introduced the concept of the “reverse Warburg effect”[138,139]. These authors proposed that aggressive cancer cells are “parasites” that use oxidative stress as a “weapon” to extract nutrients from surrounding stromal cells, forced to undergo aerobic glycolysis, and produce energy-rich nutrients (such as lactate and ketones) to “feed” cancer cells. They suggested that stromal catabolism, via autophagy and mitophagy, fuels the anabolic growth of tumor cells, promoting tumor progression and metastasis.
What is also becoming apparent, is that cancer cells or tissues have an altered metabolism, but, they also induce systemic changes of the whole body metabolism by secreting humoral factors (i.e., TNF-α, IL-1 and IL-6) and pro-cachectic factors (i.e., proteolysis-inducing factor and lipid mobilization factor) that lead to a generalized catabolic state followed by significant and progressive energy loss from host tissue in the final stages of cancer[140,141]. A group of researchers from Taiwan metaphorically compared these influences of the tumor on the host’s metabolism as a “metabolic dictatorship”, the tumors imposing their high demands on the normal host these metabolic changes, ultimately, in some types of cancers (i.e., pancreatic or gastric cancer) leading to cachexia[142]. Basically, the metabolic parasitism described at the tissular level exists also at the level of the whole organism[143].
Cachexia is a multi-organ syndrome involving changes in many tissues and organs besides the muscle, the adipose tissue and the tumor itself, other organs including the liver, the pancreas, the brain and the gut[144][Figure 8]. It involves up-regulated tissue catabolism and impaired anabolism, release of tumor-derived catabolic factors and inflammatory cytokines, and neuroendocrine dysfunction[145]. As previously suggested by Al-Zoughbi and Porporato, the global metabolic changes that tumors exert on the whole organism are due to a precise reprogramming of the different key structures involved in the normal body energy expenditure balance and are not simply complications of tumor progression[6,134]. This is why these authors introduced new terminology to describe this phenomenon, i.e., “macroenvironment”[6] and “metabolic cancer syndrome”[134]. In addition to the direct effects of tumor-derived cytokines on individual organs, there is also an interplay between muscle, fat, and liver involving several signaling pathways and metabolites leading to pathologic networks formation resulting in disruption of key metabolic pathways[140][Figure 8].
The incidence of cachexia among cancer patients is very high, especially in gastric and pancreatic cancer where the incidence is more than 80%. One of the main causes of cancer cachexia is inflammation. The cytokines secreted by the tumor may lead to the symptoms commonly associated with cachexia (loss of apetite, pain, fever, fatigue, cachexia) but, ultimately, cachexia is dependent on the patient response to tumor progression and the activation of the inflammatory response[146]. One of the key cytokines involved in cachexia is IL-6, linked to both cachexia and metastasis events[86], but, also other cytokines - such as TNF-α, IL-1β, and TGF-β- are involved and they may induce inflammation and muscular and adipose tissue wasting[147,148]. Pro-inflammatory cytokines promote also a shift in liver protein synthesis towards the production of CRP instead of albumin - which contributes to sustaining chronic inflammation[149]. Inflammation is the key trigger of muscle wasting inducing alterations in protein and amino acid metabolism, together with activation of apoptosis and decreased regeneration[144]. Besides muscles wasting, adipose tissue wasting is also present in the majority of cachexia patients. Indeed, cachectic patients manifest high levels of circulating free fatty acids, glycerol and triacylglycerol[150] and the trigger of lipolysis may be also systemic inflammation[151]. Another characteristic of cancer cachexia is the progressive switch from white adipose tissue to brown adipose tissue-brown adipose tissue derives its name from the darker color associated with the enrichment in mitochondria[152]. Pro-inflammatory factors either derived from the host immune system or the tumor, contribute to this switch[152]. Browning strongly contributes to the increased energy expenditure common in cachectic patients and, interestingly, cachectic lipid wasting occurs mostly in tumors actively secreting parathyroid hormone-related protein[152,153]. Pro-inflammatory cytokines, such as IL-6, IL-1, TGF-β, and TNF-α, are common denominators both for metastasis and inflammation and for the metabolic reprogramming associated with cachexia[86,122,133] and their underlying molecular pathways might overlap[154]. One of these molecular pathways might be exosome secretion by tumour cells or adipose tissue. It has been shown recently that tumor related exosomes play a key role in activating the inflammatory process in cancer; thus, they might be involved in both host-wasting processes, as well as metastatic dissemination[155-158]. As a direct proof of this concept, a study[159] published in 2014, showed that cancer-derived microvesicles containing miR-21 induce apoptosis of skeletal muscle and lipolysis of the adipose tissue. Similarly, another study demonstrated that cancer associated microvesicles induce muscle wasting in mice through releasing extracellular Hsp70 and Hsp90[155].
We will describe briefly below the contribution of different organs or body systems to cachexia. First, the liver plays a major role in cachexia. In the KRAS/P53 mouse model it has been shown that lung tumors act distally on the liver and reprograms hepatic metabolism through altered pro-inflammatory response via the STAT3-Socs3 pathway resulting in inhibition of hepatic insulin signaling, increased glucose production and a deregulated lipid synthesis[160]. The authors suggested that tumor-secreted ‘waste’ such as lactate is converted to pyruvate and shunted through gluconeogenesis to produce glucose, which can further satisfy the heightened energetic demand of cancer cells. A research team coordinated by Douglas Fearon also demonstrated in two mouse models of cancer-induced cachexia that in pre-cachectic mice, even before the onset of the weight-losing phase of the syndrome, tumor-induced IL-6 has altered the capacity of the liver to respond to caloric deprivation[141]. Tumors induce a reprogramming of the hepatic metabolism blocking the host’s capacity to make available endogenous sources of energy that compensate for decreased caloric intake. Through supressing ketogenesis, the tumor hampers the host’s capacity to produce endogenous sources of energy that compensate for decreased caloric intake. This energy deficit magnifies the host stress response and leads to increased glucocorticoid levels that suppress the tumor immunity[141]. Using an inducible lung cancer mouse model, a research group from WCMC, co-ordinated by Lewis Cantley, found that cachexia was associated with low ketones and increased glucocorticoid levels that suppresses tumor directed immunity[161]. The low ketones level associated with reduced expression of hepatic peroxisome proliferator-activated receptor-α (PPARα) targets that regulate fatty acid oxidation and ketogenesis. Treatment with fenofibrate, a PPARα agonist restored hepatic ketogenesis, prevented the reliance on hepatic gluconeogenesis, and skeletal muscle wasting. This model was consistent with the hypothesis that global inflammation induced by the tumor signals the brain that increases corticotropin-releasing hormone (CRH) leading to glucocorticoid production that will induce type 2-skeletal muscle fibers breakdown[161].
The pancreas also plays an important role in cachexia through secretion of insulin and glucagon. Insulin resistance is both a risk factor for cancer and is associated with cancer progression. The increase in insulin level driven by insulin resistance can drive cancer growth both directly through insulin receptors, and IGF-1 receptors present on the surface of cancer cells[162,163] and indirectly by promoting liver gluconeogenesis and muscular wasting. Also, the increased production of glucagon in the alpha islet of pancreas during cancer progression, may also increase liver gluconeogenesis[164]. Branched aminoacids released from the muscle will be used in the liver for gluconeogenesis or protein synthesis in lung tumors[165]. Interestingly, the increase of branched aminoacids blood levels may precede the clinical appearance of pancreatic cancer by several years[166].
The impact of gut on cachexia is mostly through the gut microbiota. Alteration of the gut flora due to undernutrition and chemotherapy ultimately affects specific metabolite availability and absorption, which in turn affects tumor growth and cachexia[167]. Host metabolism and energy balance are also influenced by an interplay between the intestinal microbiota, bile acids and nutrients that may have an impact on global inflammation, immune responses, gut hormone secretion and neuronal activity[168].
Several hormones including insulin, cathecolamines and atrial natriuretic peptide are involved in lipolysis[169]. Besides the endocrine system, the brain is also actively involved in the cachectic syndrome by controlling food intake through appetite, satiation, taste and smell of food. Receptors of TNF-α and IL-1 are found in the hypothalamic areas of the brain, which regulate food intake. Anorexia induced by both TNF-α and IL-6 can be blocked by inhibitors of cyclooxygenase, suggesting that a prostaglandin, such as PGE2, may be the direct mediator of appetite suppression[169,170]. Autonomic nervous system dysfunction has been also described in cancer patients with cachexia[171]. IL-6 was found to stimulate hypothalamic release of CRH, and increase glucocorticoid production[172].
Structural and functional heart changes similar to those found in cardiac failure are often associated with the cachexia syndrome. In addition to a loss of skeletal muscle mass and function, many patients with cancer cachexia also experience cardiac atrophy, remodeling, and dysfunction, which in the field of cancer cachexia is described as cardiac cachexia[173,174]. It has been shown for more than two decades that cardiac cachexia is linked to raised plasma levels of TNF-α and other inflammatory cytokines and that the degree of body wasting is strongly correlated with neurohormonal and immune abnormalities[175].
Israel and Schwartz[176] postulated that cancer cells have hybrid metabolic features that take advantage of the catabolic state that they also initially induce. The two French authors proposed a comprehensive model of the systemic metabolic changes induced by cancer that I will describe briefly. Normally, in starvation, when blood glucose level decreases, glucagon and epinephrine activate gluconeogenesis and ketogenesis to form nutriments, mobilizing body stores. On the contrary, when glycemia is elevated, the pancreas releases insulin, activating anabolism and oxidative glycolysis, energy being required to form new molecules or refill stores. Usually, these two opposite physiological states exclude each other; when anabolism is triggered by insulin, catabolism is blocked and the normal organism metabolic configuration is finely regulated by the state of key enzymes. Depending on the needs, enzymes function like switches and direct the metabolism towards different pathways that are open or closed depending on their phosphorylation state. In cancer, some of their enzymes are phosphorylated as normally observed when catabolic hormones stimulate Gs-coupled receptors, whereas other enzymes adopt a configuration normally found in anabolic situations, mediated via tyrosine kinase receptors. Basically, despite the fact that the organism as a whole is in a starvation-like state induced by cancer, tumor cells have their anabolic pathways turned ON through tyrosine kinase receptors, sometimes constitutively activated through genetic mutations or amplifications. The pyruvate kinase (PK) and pyruvate dehydrogenase (PDH) of cancer cells are OFF in a phosphorylated form but the citrate synthase is ON pulling the glucose flux in the glycolytic direction. So, on one hand, cancer cells, have their PKs and PDHs inhibited by phosphorylation, like in gluconeogenesis, on the other hand they have an increased glycolysis that will be used for the synthesis of new molecular building blocks for new mitotic daughter cells. As a result, cancer cells burn glucose and increase the tumor mass, at the same time consuming the muscle proteins and the lipid stores of the organism. The outcome of this hybrid rewired metabolism gives them a selective advantage over normal cells[176]. In subsequent publications, Israël proposed that the reason for this hybrid metabolism is an alteration of the GABA selection switch between anabolism and catabolism in the pancreas, leading to a concomitant release of catabolic glucagon and anabolic insulin[177]. According to him, the first cells that manifest a hybrid metabolism are the stem cells. Subsequently, this metabolic rewiring is stabilized through mutations or epigenetic changes selecting the most aggressive population and cancer cells arise. In his model, the pancreatic alteration is the primum movens of cancer followed by the metabolism switch of stem cells. The stem cells, initially committed to repair an organ, subsequently transform into cancer cells that use their metabolic advantage to compete for resources with the rest of the organism[177-179]. The practical value of Israël’s model is first the prediction that cancer could be detected several years before its clinical manifestations because of specific metabolome changes, and, second, the proposal of correcting the GABA switch pancreatic anomaly as a method for cancer prevention. In 2016, a group of German researchers analyzing data from the European Prospective Investigation into Cancer and Nutrition study found that abnormalities of two lipid metabolites (high levels of phosphatidylcholine PC ae C30:0 and low levels of lysophosphatidylcholines, C18:0, were consistently associated with increased risk of breast, prostate and colorectal cancer. These abnormalities were detected several years before the clinical apparition of cancer pointing to a global metabolic shift in phosphatidylcholine metabolism that may drive tumorigenesis[180].
The thrombosis network
The role of different blood components[181] and the lymphatic system[182] in the metastatic process has been coming more and more into focus. Therapies targeted against other blood and lymphatic factors involved in cancer are in development[181,182]. As many as 20% of cancer patients may have a thrombosis event during their lifetimes[183]. As shown in a recent review[184], a reciprocal connection exists between cancer and thrombosis, on one side cancer cells supporting clot formation, on the other side, clotting proteins support cancer growth and dissemination. Cancer is associated with a state of hypercoagulability, driven in part by the release of procoagulant factors, such as tissue factor (TF), released by the malignant tissue, as well as by inflammation-driven activation of endothelial cells, platelets, and leukocytes. Also, cancer cells are able to directly adhere to host cells (i.e., endothelial cells, monocytes, platelets, and neutrophils), thereby stimulating additional prothrombotic properties of the host thrombosis effector cells[184][Figure 9].
TF is considered to be the major molecular driver of cancer-associated coagulopathy and thromboembolic disorders. It is expressed either by cancer cells or its expression is induced by cancer cells in normal vascular tissues by both the release of soluble mediators and the direct cancer cell - host cell contact. Its expression is related to well defined oncogenic events: epidermal-to-mesenchymal transformation, TGF-β signaling, EGFR, phosphatase and tensin homolog (PTEN) and Src pathways, hypoxia induced signaling, etc. The majority of human epithelial cancers (lung, colorectal, prostate, breast, pancreatic, gastric, melanoma, etc.) are characterized by abundant levels of TF[185]. Also, as shown by a Canadian group in a glioma model, TF may also control the state of tumor dormancy by recruiting to the tumor niche myeloid and blood vessels forming cells[186]. Interestingly, the procoagulant and the signaling effect of TF in tumor biology can be targeted separately, and there are therapies under investigation that target solely the signaling effect of TF without affecting its homeostatic function[187].
In addition, cancer cell interactions with platelets and neutrophils contribute to cancer cell adhesion, extravasation, and the establishment of metastatic lesions[188]. Platelet-derived signals are required for the rapid intravascular recruitment of neutrophils to circulating tumor cells (CTCs) thrombi contributing to “early metastatic niches”[123]. Also, TGF-β secreted by degranulating platelets may contribute to the activation the NF-κB pathway in carcinoma cells, thereby inducing or sustaining the expression of EMT programs in the CTCs[189]. Selectins are carbohydrate-binding molecules that bind to glycan structures, present on endothelial cells, platelets and leukocytes. There are three members of the selectin family: P-selectin expressed on activated platelets and endothelial cells, L-selectin present on leukocytes and E-selectin expressed on activated endothelial cells[190]. P-selectin in particular seems to play a crucial role in several types of cancer metastasis by mediating the aggregation of platelets with tumor cells forming clots. A recent study showed that intravenous injection of melanoma cells into WT mice resulted in multiple lung metastases, while in P-selectin-deficient mice pulmonary tumor metastasis and trapping of tumor cells in the lung was significantly reduced[191]. Modulating the interaction between cancer cells and the circulating blood cells, and respectively, between cancer cells and the endothelial cells may represent novel therapeutic approaches. For example, there is experimental evidence that targeting specific types of the integrin receptors present on the surface of the platelets efficiently reduces tumor cell colonization into the lungs, suggesting that they could represent interesting targets for anti-metastatic drugs[192].
A team from France characterized the microparticulosome, the repertoire of plasma membrane vesicles produced by different types of cells and was able to differentiate a microparticle signature associated with pancreatic and colorectal cancer[193]. The same team showed in syngeneic ectopic and orthotopic mice models that treatment with the drug Clopidogrel prevented the binding of cancer cell-derived microparticles to fibrinogen-platelets aggregates at the site of thrombosis, and reduced the metastasis and the extent of thrombosis associated with cancer[194]. Procoagulant factors associated with exosomes from tumors have been described for almost four decades[195] Tissue factor associated with exosomes has been found to be responsible for the Trousseau syndrome in one patient with lung cancer[196] and there are several studies documenting the procoagulant effect of tumor exosomes[197].
The neuro-endocrine network
Both the central nervous system and the neurovegetative nervous system are intimately involved in cancer. One of the most studied links between central nervous system and cancer is stress. The neuroendocrine mediators reach the cells of the immune system either through the peripheral circulation or through direct innervation of lymphoid organs [Figure 10]. As suggested by Claire Magnon[198], a possible explanation for tumor formation associated with stress might rely on the activation of the sympathetic nervous system (SNS) through the sympathetic - adrenal - medullary axis, which controls the release of adrenergic neurotransmitters such as epinephrine or norepinephrine by the adrenals into the bloodstream in support of the fight-or-flight reflex. Catecholamine-mediated suppression of cellular immunity may play a role in increased growth of certain tumors[199]. Also, primary and secondary lymphoid organs are innervated by sympathetic nerve fibers. Lymphocytes and monocytes express receptors for several stress hormones, including CRH, adrenocorticotropic hormone (ACTH), cortisol, norepinephrine, and epinephrine. Therefore, it is possible that the neuroendocrine hormones released during a stressful event could alter immune function and subsequently alter the course of immune-based diseases[200]. It has been reported that mice living in an enriched housing environment (EE) show reduced tumor growth and increased remission[201]. This effect was described in melanoma and colon cancer models, and, it was proven that it was not caused by physical activity alone. Serum from animals held in an enriched environment (EE) inhibited cancer proliferation in vitro and was markedly lower in leptin. Hypothalamic brain derived neurotrophic factor (BDNF) was selectively upregulated by EE, its genetic overexpression reduced tumor burden, whereas BDNF knockdown blocked the effect of EE. The hypothalamic BDNF downregulated leptin production in adipocytes via sympathoneural β-adrenergic signaling[201].
A key central nervous system structure involved in cancer is the hypothalamus. In the context of systemic inflammation, the hypothalamus integrates signals from peripheral systems, translating them into neuroendocrine perturbations, altered neuronal signaling, and global metabolic derangements[202]. Cytokines, like IL-1β and TNF-α, for example, generated in the periphery during cancer progression are amplified and modified within the hypothalamus, leading to hypothalamic inflammation and aberrant activity of weight- and activity-modulating neurons that may induce muscle atrophy via activation of the hypothalamic-pituitary-adrenal axis[203,204]. Hypothalamic inflammation may be followed by dysregulation of homeostatic regulation of autonomic nerves (innervation of muscles, liver, fat tissue, endocrine glands and other organs) that may further potentiate dysregulation of metabolism and enhance peripheral, pro-inflammatory reactions[205]. Hypothalamus appears to be an important contributor in the development and maintenance of the cachectic state[202]. Lower hypothalamic activity has been demonstrated by functional magnetic resonance imaging scans in patients with cachexia associated with advanced lung cancer[206].
A unique crosstalk between the central nervous system and prostate tumours was recently revealed. In a striking experiment[207], using a mouse model of prostate cancer, a French group demonstrated a process of tumour-associated neo-neurogenesis, in which neural progenitors leave the brain of the mouse and reach, through the systemic circulation, the primary tumour or the metastatic tissues. Once arrived there, they differentiated into new adrenergic neurons that are known to support the early stages of the development of cancer. The authors suggested the possibility that the tumour itself might deplete neurogenic niches in the brain by attracting neural progenitors to support its own development[207].
Recent experiments suggest a direct relationship between the neurovegetative nervous system and certain tumors. As reviewed by Cole[208], SNS activation modulates gene expression programs that promote metastasis of solid tumours by stimulating macrophage infiltration, inflammation, angiogenesis, epithelial-mesenchymal transition, and tumour invasion, and by inhibiting cellular immune responses and programmed cell death. SNS activation may also influence cancer progression via indirect pathways in which SNS innervation of distant tissues triggers secondary hormonal or cellular effects that subsequently affect the tumour microenvironment. For example, sympathetic innervation of bone marrow can stimulate the production of myeloid lineage immune cells which may infiltrate the tumoral microenvironment and promote metastasis[209-211]. In prostate cancer, sympathetic nerve fibers may help tumors grow by interacting with beta-adrenergic receptors on stromal cells[212]. Epidemiological studies showed that men with prostate adenocarcinoma who take non-selective beta-blockers have lower prostate cancer-specific mortality rates[213]. A similar activity of beta-blockers has been described in melanoma or breast cancer patients indicating that adrenergic signaling might be involved in various types of cancer[214,215].
If the role of the sympathetic system in cancer has been well documented, the contribution of the parasympathetic division of the autonomic nervous system is less clear. As shown by Kevin Tracey and his collaborators from the Feinstein Institute on Long Island, New York, the efferent vagus nerve-mediated cholinergic signaling controls immune function and pro-inflammatory responses via the inflammatory reflex[216]. T and B cells express most cholinergic system components - e.g., acetylcholine, choline acetyltransferase, acetylcholinesterase, and, both muscarinic and nicotinic acethycholine (Ach) receptors and the cholinergic signals generated by immune cells appear to be triggers of both the initiation and termination of cytokine synthesis (e.g., IL-2 in T cells and TNF-α in macrophages)[217]. A recent study from the University of Sichuan, China, suggested that parasympathetic innervation may contribute to stomach cancer development via acetylcholine-mediated activation of muscarinic acetylcholine receptors[218]. In a mouse model of stomach cancer, vagotomy suppressed gastric tumorigenesis[219]. Also in a prostate cancer mouse model cholinergic-induced tumor invasion and metastasis were inhibited by pharmacological blockade of the stromal type 1 muscarinic receptor, leading to improved survival of the mice[212]. However, as discussed by Cole et al.[208], cholinergic blockade may stimulate indirectly the SNS promotion of cancer. An alternative strategy would be to target neurotrophic growth factors in cancer as many cancers are associated with nerve infiltration. An antineurotrophic antibody (tanezumab) has been developed by Pfizer and is currently used as an analgesic[220].
As many as 8% of cancers might be associated with endocrine paraneoplastic syndromes[221], but, a detailed discussion on these syndromes, is beyond the scope of this article. In the context of our discussion on the systemic hallmarks of cancer, it is clear, however, that production of specific hormones by tumors of particular types is not a random event[222]. For example, squamous cell carcinomas typically produce parathyroid hormone - related protein and small cell carcinomas (SCC) of the lung typically produce calcitonin, adrenocorticotropin (ACTH), or gastrin releasing peptide (GRP). In some case for example, bombesin (BBS)-like neuropeptides secreted by SCC can act as autocrine growth factors[223]. Besides the involvement of cathecolamines in cancer described before, several other hormones (i.e., estrogens, androgens) are well known to promote cancer development and metastasis[224]. Also, the role of the thyroid hormones in promoting the metastatic process has been recently described[225]. Other hormones, like melatonin, for example, may inhibit cancer metastasis[226]. Patient with cancer have poor sleep and this may influence melatonin secretion. In a recent study done breast cancer women serum melatonin levels correlated significantly with self-reported sleep quality and psychometric profiles of depression[227].
Interogating the system: cancer induced systemic pathologic networks
In the recent years, liquid biopises and “omics” became useful tools of the developing field of precision oncology. Through liquid biopsies and “omics” we can interogate the global characteristics of the tumor itself, and obtain useful information that help us in the diagnostic, prognostic and treatment of cancer patients. In the near future, of great importance will be the characterization of the different CISPN through specific biomarkers designed to analyze the systemic cancer hallmarks. This information might be used to refine the staging and prognostic of patients with metastatic cancer currently lumped indiscriminatively under one large umbrella by the TNM staging and, also, design and monitor targeted interventions directed specifically against key CISPN that behave as master regulators of the metastatic process. Circulating miRNA present inside the exosomes are plausible CISPN master regulators[42]. Exosomal proteins isolated from plasma of cancer patients have been recently characterized not only as useful biomarkers associated with several cancer types but also for dissecting different CISPN involvement in the malignant process[43]. Potential systemic biomarkers might be also found analysing metabolomics data. In order for a tumor to develop and spread needs energy and, global metabolic reprogramming, might well be one of the key systemic cancer hallmarks driving cancer from its emergence through its progression and metastasis. The Consortium of Metabolomics Studies (COMETS) was established in 2014 to facilitate large-scale collaborative research on the human metabolome and its relationship with disease etiology, diagnosis, and prognosis[228]. Systemic metabolic changes in advanced cancers have been described in the past for several tumor types[229,230]. The essential role of metabolism at the cellular level in controlling cancer hallmarks was recently proven. Using molecular data of 9,125 patient samples from The Cancer Genome Atlas, a group of researchers identified distinct metabolic expression subtypes in 27 cancer types based on mRNA expression patterns of seven major metabolic processes (amino acid metabolism, carbohydrate metabolism, integration of energy, lipid metabolism, nucleotide metabolism, tricarboxylic acid cycle and vitamin & cofactor metabolism)[231]. The metabolic expression subtypes correlated with clinical outcomes: subtypes with upregulated carbohydrate, nucleotide, and vitamin/cofactor metabolism most consistently correlated with worse prognosis, whereas subtypes with upregulated lipid metabolism showed the opposite. The most interesting finding was that these metabolic subtypes were not related to specific genetic somatic drivers but were intrinsically coupled with cancer hallmark pathways (i.e., angiogenesis, cell division, etc.) and were modulated by highly recurrent master regulators across cancer types, ultimately leading to consistent survival patterns. As a proof-of-concept in vitro experiment, the authors also demonstrated that knockdown of two master regulators genes of carbohydrate metabolic subtypes (SNAI1 in a lung cancer cell line or RUNX1 in a sarcoma cell line) significantly decreased the concentrations of intracellular glucose. According to this model, the master metabolic regulators identified were key nodes with the greatest influence on systems-level metabolic activities and targeting these metabolic master regulators may inhibit tumor progression. Strikingly, all four master metabolism regulators genes identified in the 8 cancer types with significantly worse survival rates due to upregulated carbohydrate metabolism, SNAI1, RUNX1, RUNX2, and FOSL1[231], play also a key role in embryonal development and EMT[13,232,233] and might be also master regulators of the metastatic cellular program [Figure 3].
Novel therapeutic approaches using the cancer system model
Mark Vincent classified cancer treatments in two fundamentally different approaches: a “causality-inhibition” strategy, targeted towards the cancer cause, which at present is still a “moon shot”, remote from our current cancer treatment practices, and, an ”acausal” approach that target a specific cancer marker or signature[234]. At present, many aspects of cancer, in general, and, by large, the metastatic process are still incompletely charted territories, and, therefore, most our current cancer treatments are not directed towards the specific cause that triggeres the cancer process. In the near future, hopefully, once the mechanisms of the different cancer cellular programs are better described, we will be able to design effective causality-inhibition therapies.
For example, the immunomodulatory function of exosomes may be exploited for therapeutic effect. In 2008, a Chinese group from Guangxi University[235] reported the results of a Phase I study in which 40 patients with advanced colorectal cancer received four weekly intravenous injections of ascites derived exosomes plus or minus GM-CSF. Stable disease and a minor response were observed in two of the patients treated. More recently, another Chinese group[236], suggested that miRNA depleted pancreatic cancer exosomes might enhance the killing capacity of dendritic or cytokine-induced killer cells, and activate the immune system against pancreatic cancer. The exosomes packaging is closely regulated, and, different clones even from the same tumor may secret exosomes carrying a different cargo with different properties[237].
Dismantelling cancer networks at the organism level
Networks, composed of various nodes and edges may be described at different levels in an organism. In a cell, nodes may be amino acids of cancer-related proteins, where edges are their distances in the 3D protein structure or nodes may represent protein/RNA molecules or DNA-segments, where edges are their physical or signaling contacts. In metabolic networks, nodes are metabolites and edges are the enzymes, which catalyze the reactions to convert them to each other. At the tissular levels, nodes can be the cancer cells and the stromal cells and the edges the different molecules through which they communicate. At the level of the whole organism, nodes may represent the different components of the cancer system and the different components of the normal body systems [Figure 2] and, the edges, cellular, exosomal or proteic signals exchanged between them. Cancer is a robust system that is able to maintain stable functioning despite various perturbations. The essential robustness of cancer is maintained through heterogeneous redundancy, i.e., the cancer tissue contains a heterogeneous distribution of genetically different cancer cells maintained by genetic instability[238]. Communication is crucial for the development of the cancer system. In order to be able to dismantle such a complex multi-layered network as cancer, novel targeted multi-scale approaches are needed that target simultaneously key elements of the cellular, tissular and systemic cancer networks[239]. Targeting the master genetic regulators or the hubs at the cellular level led to promising results[28,240]. Also, a therapeutic approach based on game theory targeting the collaboration between cancer cells at the tissular level was recently proposed by Archetti and Pienta[241]. It is conceivable that using similar mathematical tools, treatments targeting specific CISPN elements at the organismic level can be designed. Evaluating the cancer system vulnerabilities through analysis of network topology and, especially, network dynamics can predict novel anti-cancer drug targets[242]. In general, therapeutic approaches targeting levels above the cellular level may be less affected by cancer genetic instability and heterogeneity than treatments targeting the cancer cells themselves. A suggestive example is the improvement in the long term survival associated with check-point inhibitors that target cancer tissue as opposed to cancer cells[243-245] as opposed to the almost universal development of acquired resistence associated to the use of tyrosine kinase inhibitors that target specific intra-cellular cancer networks[246]. An attractive top-down regulator of cancer is the nervous system and novel therapies could be designed stimulating or inhibiting some of its components[198]. As a proof of principle, amplyfing a single gene in the hypothalamus of obese mice through gene transfer of BDNF inhibited breast cancer progression and metastasis[247].
“Horizontal” vs. “vertical” approaches
Modalities to target cancer at the cellular level (i.e., tyrosine kinase inhibitors and antibodies directed to the antigens present on the surface of the cancer cells) have been already in place now for almost two decades. Immunotherapies with checkpoint inhibitors and CAR-T cell approaches have recently improved the quality and duration of life of many cancer patients. We envision that agents targeting the systemic hallmarks of cancer and interrupting the communication between the different components of the cancer system will represent the “new wave” of cancer treatments. In the multi-scale model of cancer a “horizontal” approach is considered targeting the cross-talk between the different components of the CISPN and blocking the communication between its parts and a “vertical” approach would use drugs that act simultaneously at the cellular, tissular and systemic level in a particular sub-component of the CISPN. An example of a “horizontal” approach is the finding that the blockade of the CXCL5/7 receptor CXCR2, or the transient depletion of either platelets or granulocytes, prevents the formation of early metastatic niches and significantly reduced metastatic seeding and progression[123]. Granulocyte recruitment depends on the secretion of CXCL5 and CXCL7 chemokines by platelets upon contact with tumor cells[123]. An example of a “vertical” approach using trans-level drugs acting simultaneously at the cellular, tissular and organismic levels simultaneously is the use of beta-blockers. Some beta-blockers, for example, like propranolol, at the tissular level have an immunomodulatory effect[248] and at the organismic level alter the metastatic potential of cancer cells[208]. A group of researchers at Penn State University found that melanoma patients who received immunotherapy while taking pan β-blockers lived longer than patients who received immunotherapy alone or patinets that received immunotherapy and β1-selective blockers[249]. In a follow-up experiment with mice, the researchers saw the same results[249]. Bisoprolol is another selective β1-blocker commonly used to treat hypertension, cardiac ischemia, and congestive heart failure. Bisoprolol improved survival, increased total heart mass, and other heart parameters and, importantly, improved food intake and activity levels in an AH-130 tumor-bearing rats model[250]. Clinical studies with Bisoprolol are planned in patients with cancer cachexia (Professor Anker, Charité Hospital, Berlin, personal communication).
Conclusion
Cancer is a multidimensional process with specific characteristics at the cellular, tissular and the organismic level. Basic research and clinical data obtained over the last decade suggests that, at the macroscopic level, cancer behaves like an evolving co-dependent system that interacts continuously through CISPN with the modified body systems. Cancer cells and cancer stroma secreted exosomes, cytokines and other soluble factors together with the modified, cancer-supporting body systems, are responsible for establishing the CISPN and the systemic hallmarks of cancer. Without taking into consideration this larger, organism-level picture, some of the current local treatments targeted towards the cancer cells or tissues may lead to cancer progression. For example, in some cases of head and neck cancer, (up to 29% in some series), checkpoint inhibitor treatments may induce cancer hyperprogression[251]. Treatments targeted towards the cancer system and the systemic hallmarks of cancer are urgently needed. Moving to the organismic level and targeting the systemic hallmarks of cancer in concerted therapeutic approaches with currently existing therapies may further improve our cancer armamentarium in the immediate future.
Declarations
Authors’ contributionsThe author solely contributed to the article.
Availability of data and materialsNot applicable.
Financial support and sponsorshipNot applicable.
Conflicts of interestThe author declared that there are no conflicts of interest.
Ethical approval and consent to participateNot applicable.
Consent for publicationA written informed consent for publication was provided.
Copyright© The Author(s) 2020.
REFERENCES
1. Noble D. A theory of biological relativity: no privileged level of causation. Interface Focus 2012;2:55-64.
2. Noble D. A biological relativity view of the relationships between genomes and phenotypes. Prog Biophys Mol Biol 2013;111:59-65.
3. Noble D. The music of life. Oxford; 2006.
4. Egeblad M, Nakasone ES, Werb Z. Tumors as organs: complex tissues that interface with the entire organism. Dev Cell 2010;18:884-901.
5. McAllister SS, Weinberg RA. Tumor-host interactions: a far-reaching relationship. J Clin Oncol 2010;28:4022-8.
6. Al-Zoughbi W, Huang J, Paramasivan GS, Till H, Pichler M, et al. Tumor macroenvironment and metabolism. Semin Oncol 2014;41:281-95.
7. McAllister SS, Weinberg RA. The tumour-induced systemic environment as a critical regulator of cancer progression and metastasis. Nat Cell Biol 2014;16:717-27.
8. Borniger JC. Central regulation of breast cancer growth and metastasis. J Cancer Metastasis Treat 2019;5.
9. Borniger JC, Walker Ii WH, Surbhi, Emmer KM, Zhang N, Zalenski AA, et al. A role for hypocretin/orexin in metabolic and sleep abnormalities in a mouse model of non-metastatic breast cancer. Cell Metab 2018;28:118-29.e5.
10. Paul D. Cancer the big picture: seeing the forest beyond the trees. Oncolog-Hematolog 2015;1:28-30.
11. Udrişte O. Gena ancestrală şi originea cancerului (in Romanian). Bucharest, Romania: Editura ştiinţifică şi enciclopedică; 1978.
12. Arechaga J. On the boundary between development and neoplasia. An interview with Professor G. Barry Pierce. Int J Dev Biol 1993;37:5-16.
13. Dongre A, Weinberg RA. New insights into the mechanisms of epithelial-mesenchymal transition and implications for cancer. Nat Rev Mol Cell Biol 2019;20:69-84.
14. Gallik KL, Treffy RW, Nacke LM, Ahsan K, Rocha M, et al. Neural crest and cancer: divergent travelers on similar paths. Mech Dev 2017;148:89-99.
15. Vincent MD. The animal within: carcinogenesis and the clonal evolution of cancer cells are speciation events sensu stricto. Evolution 2010;64:1173-83.
17. Vincent M. Cancer: a de-repression of a default survival program common to all cells? a life-history perspective on the nature of cancer. Bioessays 2012;34:72-82.
22. Vogelstein B, Papadopoulos N, Velculescu VE, Zhou S, Diaz LA Jr, et al. Cancer genome landscapes. Science 2013;339:1546-58.
24. Sugarbaker EV. Cancer metastasis: a product of tumor-host interactions. Curr Probl Cancer 1979;3:1-59.
25. Fidler IJ, Kripke ML. Metastasis results from preexisting variant cells within a malignant tumor. Science 1977;197:893-5.
26. Price JE, Naito S, Fidler IJ. Growth in an organ microenvironment as a selective process in metastasis. Clin Exp Metastasis 1988;96:91-102.
27. McDonald OG, Li X, Saunders T, Tryggvadottir R, Mentch SJ, et al. Epigenomic reprogramming during pancreatic cancer progression links anabolic glucose metabolism to distant metastasis. Nat Genet 2017;49:367-76.
28. Aytes A, Giacobbe A, Mitrofanova A, Ruggero K, Cyrta J, et al. NSD2 is a conserved driver of metastatic prostate cancer progression. Nat Commun 2018;9:5201.
29. Lambert AW, Pattabiraman DR, Weinberg RA. Emerging biological principles of metastasis. Cell 2017;168:670-91.
30. Hendrix MJC, Seftor EA, Seftor REB, Kasemeier-Kulesa J, Kulesa PM, et al. Reprogramming metastatic tumour cells with embryonic microenvironments. Nat Rev Cancer 2007;7:246-55.
31. Telerman A, Amson R. The molecular programme of tumour reversion: the steps beyond malignant transformation. Nat Rev Cancer 2009;9:206-16.
33. Orimo A, Weinberg RA. Stromal fibroblasts in cancer: a novel tumor-promoting cell type. Cell Cycle 2006;5:1597-601.
34. Caon I, Bartolini B, Parnigoni A, Carava E, Moretto P, et al. Revisiting the hallmarks of cancer: the role of hyaluronan. Semin Cancer Biol 2020;62:9-19.
35. Amson R, Karp JE, Telerman A. Lessons from tumor reversion for cancer treatment. Curr Opin Oncol 2013;25:59-65.
36. Sun Y, Ma L. The emerging molecular machinery and therapeutic targets of metastasis. Trends Pharmacol Sci 2015;36:349-59.
37. Scheel C, Onder T, Karnoub A, Weinberg RA. Adaptation versus selection: the origins of metastatic behavior. Cancer Res 2007;67:11476-9. discussion 9-80
38. Ruan K, Fang X, Ouyang G. MicroRNAs: novel regulators in the hallmarks of human cancer. Cancer Lett 2009;285:116-26.
39. Nath S, Ghatak D, Das P, Roychoudhury S. Transcriptional control of mitosis: deregulation and cancer. Front Endocrinol (Lausanne) 2015;6:60.
40. Chiaretti S, de Curtis I. Role of liprins in the regulation of tumor cell motility and invasion. Curr Cancer Drug Targets 2016;16:238-48.
42. Anfossi S, Fu X, Nagvekar R, Calin GA. MicroRNAs, regulatory messengers inside and outside cancer cells. Adv Exp Med Biol 2018;1056:87-108.
43. Hoshino A, Kim HS, Bojmar L, Gyan KE, Cioffi C, et al. Extracellular vesicle and particle biomarkers define multiple human cancers. Cell 2020;182:1-18.
44. Wortzel I, Dror S, Kenific CM, Lyden D. Exosome-mediated metastasis: communication from a distance. Dev Cell 2019;49:347-60.
45. Schwarzenbach H, Gahan PB. MicroRNA shuttle from cell-to-cell by exosomes and its impact in cancer. Noncoding RNA 2019;5.
46. Hoshino A, Costa-Silva B, Shen TL, Rodrigues G, Hashimoto A, et al. Tumour exosome integrins determine organotropic metastasis. Nature 2015;527:329-35.
47. Rodrigues G, Hoshino A, Kenific CM, Matei IR, Steiner L, et al. Tumour exosomal CEMIP protein promotes cancer cell colonization in brain metastasis. Nat Cell Biol 2019;21:1403-12.
48. Tomasetti C, Vogelstein B. Cancer etiology. Variation in cancer risk among tissues can be explained by the number of stem cell divisions. Science 2015;347:78-81.
49. Lau EY, Ho NP, Lee TK. Cancer stem cells and their microenvironment: biology and therapeutic implications. Stem Cells Int 2017;2017:3714190.
50. Brabletz T, Jung A, Spaderna S, Hlubek F, Kirchner T. Opinion: migrating cancer stem cells - an integrated concept of malignant tumour progression. Nat Rev Cancer 2005;5:744-9.
51. Ganesh K, Basnet H, Kaygusuz Y, Laughney AM, He L, et al. L1CAM defines the regenerative origin of metastasis-initiating cells in colorectal cancer. Nature Cancer 2020:20-45.
52. Brennan MF, Antonescu CR, Moraco N, Singer S. Lessons learned from the study of 10,000 patients with soft tissue sarcoma. Ann Surg 2014;260:416-21. discussion 21-2
54. Guizard AN, Dejardin OJ, Launay LC, Bara S, Lapotre-Ledoux BM, et al. Diagnosis and management of head and neck cancers in a high-incidence area in France: a population-based study. Medicine (Baltimore) 2017;96:e7285.
55. Olson E, Wintheiser G, Wolfe KM, Droessler J, Silberstein PT. Epidemiology of thyroid cancer: a review of the national cancer database, 2000-2013. Cureus 2019;11:e4127.
57. Flanigan RC, Yonover PM. The role of radical nephrectomy in metastatic renal cell carcinoma. Semin Urol Oncol 2001;19:98-102.
58. O’Reilly MS, Boehm T, Shing Y, Fukai N, Vasios G, et al. Endostatin: an endogenous inhibitor of angiogenesis and tumor growth. Cell 1997;88:277-85.
59. Kisker O, Onizuka S, Banyard J, Komiyama T, Becker CM, et al. Generation of multiple angiogenesis inhibitors by human pancreatic cancer. Cancer Res 2001;61:7298-304.
60. Kaplan RN, Riba RD, Zacharoulis S, Bramley AH, Vincent L, et al. VEGFR1-positive haematopoietic bone marrow progenitors initiate the pre-metastatic niche. Nature 2005;438:820-7.
61. Mareel M, Oliveira MJ, Madani I. Cancer invasion and metastasis: interacting ecosystems. Virchows Arch 2009;454:599-622.
62. Kano Y, Ishii H, Konno M, Yamasaki M, Miyata H, et al. Cells of origin of squamous epithelium, dysplasia and cancer in the head and neck region after bone marrow transplantation. Int J Oncol 2014;44:443-50.
63. Houghton J, Stoicov C, Nomura S, Rogers AB, Carlson J, et al. Gastric cancer originating from bone marrow-derived cells. Science 2004;306:1568-71.
64. Guest I, Ilic Z, Ma J, Grant D, Glinsky G, et al. Direct and indirect contribution of bone marrow-derived cells to cancer. Int J Cancer 2010;126:2308-18.
65. Kaplan RN, Rafii S, Lyden D. Preparing the "soil": the premetastatic niche. Cancer Res 2006;66:11089-93.
66. Kim S, Takahashi H, Lin WW, Descargues P, Grivennikov S, et al. Carcinoma-produced factors activate myeloid cells through TLR2 to stimulate metastasis. Nature 2009;457:102-6.
67. Spano D, Zollo M. Tumor microenvironment: a main actor in the metastasis process. Clin Exp Metastasis 2012;29:381-95.
68. Costa-Silva B, Aiello NM, Ocean AJ, Singh S, Zhang H, et al. Pancreatic cancer exosomes initiate pre-metastatic niche formation in the liver. Nat Cell Biol 2015;17:816-26.
69. Liu Y, Cao X. Characteristics and significance of the pre-metastatic niche. Cancer Cell 2016;30:668-81.
70. Norton JD, Deed RW, Craggs G, Sablitzky F. Id helix-loop-helix proteins in cell growth and differentiation. Trends Cell Biol 1998;8:58-65.
71. Guo L, Guo N. Exosomes: potent regulators of tumor malignancy and potential bio-tools in clinical application. Crit Rev Oncol Hematol 2015;95:346-58.
72. Karamanos NK, Piperigkou Z, Theocharis AD, Watanabe H, Franchi M, et al. Proteoglycan chemical diversity drives multifunctional cell regulation and therapeutics. Chem Rev 2018;118:9152-232.
73. Baietti MF, Zhang Z, Mortier E, Melchior A, Degeest G, et al. Syndecan-syntenin-ALIX regulates the biogenesis of exosomes. Nat Cell Biol 2012;14:677-85.
74. Mu W, Rana S, Zoller M. Host matrix modulation by tumor exosomes promotes motility and invasiveness. Neoplasia 2013;15:875-87.
75. Peinado H, Aleckovic M, Lavotshkin S, Matei I, Costa-Silva B, et al. Melanoma exosomes educate bone marrow progenitor cells toward a pro-metastatic phenotype through MET. Nat Med 2012;18:883-91.
76. Hessvik NP, Llorente A. Current knowledge on exosome biogenesis and release. Cell Mol Life Sci 2018;75:193-208.
77. Cha DJ, Franklin JL, Dou Y, Liu Q, Higginbotham JN, et al. KRAS-dependent sorting of miRNA to exosomes. Elife 2015;4:e07197.
78. Redig AJ, McAllister SS. Breast cancer as a systemic disease: a view of metastasis. J Intern Med 2013;274:113-26.
79. Engblom C, Pfirschke C, Zilionis R, Da Silva Martins J, Bos SA, et al. Osteoblasts remotely supply lung tumors with cancer-promoting SiglecF(high) neutrophils. Science 2017;358.
80. Orimo A, Gupta PB, Sgroi DC, Arenzana-Seisdedos F, Delaunay T, et al. Stromal fibroblasts present in invasive human breast carcinomas promote tumor growth and angiogenesis through elevated SDF-1/CXCL12 secretion. Cell 2005;121:335-48.
81. Hattori K, Heissig B, Tashiro K, Honjo T, Tateno M, et al. Plasma elevation of stromal cell-derived factor-1 induces mobilization of mature and immature hematopoietic progenitor and stem cells. Blood 2001;97:3354-60.
82. Kidd S, Spaeth E, Watson K, Burks J, Lu H, et al. Origins of the tumor microenvironment: quantitative assessment of adipose-derived and bone marrow-derived stroma. PLoS One 2012;7:e30563.
83. LeBleu VS, Kalluri R. A peek into cancer-associated fibroblasts: origins, functions and translational impact. Dis Model Mech 2018;11.
84. Allavena P, Germano G, Mantovani A. Molecular links between inflammation and cancer. Systems Biology of Cancer: Cambridge University Press; 2015. pp. 273-81.
85. Colotta F, Allavena P, Sica A, Garlanda C, Mantovani A. Cancer-related inflammation, the seventh hallmark of cancer: links to genetic instability. Carcinogenesis 2009;30:1073-81.
88. Terlizzi M, Casolaro V, Pinto A, Sorrentino R. Inflammasome: cancer's friend or foe? Pharmacol Ther 2014;143:24-33.
89. Setrerrahmane S, Xu H. Tumor-related interleukins: old validated targets for new anti-cancer drug development. Mol Cancer 2017;16:153.
90. Elaraj DM, Weinreich DM, Varghese S, Puhlmann M, Hewitt SM, et al. The role of interleukin 1 in growth and metastasis of human cancer xenografts. Clin Cancer Res 2006;12:1088-96.
91. Tuomisto AE, Makinen MJ, Vayrynen JP. Systemic inflammation in colorectal cancer: Underlying factors, effects, and prognostic significance. World J Gastroenterol 2019;25:4383-404.
92. Becker A, Thakur BK, Weiss JM, Kim HS, Peinado H, et al. Extracellular vesicles in cancer: cell-to-cell mediators of metastasis. Cancer Cell 2016;30:836-48.
93. Manning S, Danielson KM. The immunomodulatory role of tumor-derived extracellular vesicles in colorectal cancer. Immunol Cell Biol 2018; doi: 10.1111/imcb.12038.
94. Jain S, Gautam V, Naseem S. Acute-phase proteins: as diagnostic tool. J Pharm Bioallied Sci 2011;3:118-27.
95. Bankey PE. Hepatic reagulation of systemic inflammation following acute injury. Curr Opin Crit Care 1996;2:280-6.
96. Nieuwenhuijzen GA, Haskel Y, Lu Q, Berg RD, van Rooijen N, et al. Macrophage elimination increases bacterial translocation and gut-origin septicemia but attenuates symptoms and mortality rate in a model of systemic inflammation. Ann Surg 1993;218:791-9.
97. Iida N, Dzutsev A, Stewart CA, Smith L, Bouladoux N, et al. Commensal bacteria control cancer response to therapy by modulating the tumor microenvironment. Science 2013;342:967-70.
98. Viaud S, Saccheri F, Mignot G, Yamazaki T, Daillere R, et al. The intestinal microbiota modulates the anticancer immune effects of cyclophosphamide. Science 2013;342:971-6.
99. Gorjifard S, Goldszmid RS. Microbiota-myeloid cell crosstalk beyond the gut. J Leukoc Biol 2016;100:865-79.
100. Kostic AD, Chun E, Meyerson M, Garrett WS. Microbes and inflammation in colorectal cancer. Cancer Immunol Res 2013;1:150-7.
101. Rutkowski MR, Conejo-Garcia JR. TLR5 signaling, commensal microbiota and systemic tumor promoting inflammation: the three parcae of malignant progression. Oncoimmunology 2015;4:e1021542.
102. Hui D. Prognostication of survival in patients with advanced cancer: predicting the unpredictable? Cancer Control 2015;22:489-97.
103. Wang C, Jin S, Xu S, Cao S. High systemic immune-inflammation index (SII) represents an unfavorable prognostic factor for small cell lung cancer treated with etoposide and platinum-based chemotherapy. Lung 2020;198:405-14.
104. Dupréa A, Malika HZ. Inflammation and cancer: what a surgical oncologist should know. Eur J Surg Oncol 2018;44:566-70.
105. Nakamura K, Smyth MJ. Targeting cancer-related inflammation in the era of immunotherapy. Immunol Cell Biol 2017;95:325-32.
106. Dinarello CA. Why not treat human cancer with interleukin-1 blockade? Cancer Metastasis Rev 2010;29:317-29.
107. Crusz SM, Balkwill FR. Inflammation and cancer: advances and new agents. Nat Rev Clin Oncol 2015;12:584-96.
108. Chen G, Huang AC, Zhang W, Zhang G, Wu M, et al. Exosomal PD-L1 contributes to immunosuppression and is associated with anti-PD-1 response. Nature 2018;560:382-6.
109. Zhou M, Chen J, Zhou L, Chen W, Ding G, et al. Pancreatic cancer derived exosomes regulate the expression of TLR4 in dendritic cells via miR-203. Cell Immunol 2014;292:65-9.
112. Yin Y, Cai X, Chen X, Liang H, Zhang Y, et al. Tumor-secreted miR-214 induces regulatory T cells: a major link between immune evasion and tumor growth. Cell Res 2014;24:1164-80.
113. Berchem G, Noman MZ, Bosseler M, Paggetti J, Baconnais S, et al. Hypoxic tumor-derived microvesicles negatively regulate NK cell function by a mechanism involving TGF-beta and miR23a transfer. Oncoimmunology 2016;5:e1062968.
115. Honda K, Littman DR. The microbiota in adaptive immune homeostasis and disease. Nature 2016;535:75-84.
116. Matson V, Fessler J, Bao R, Chongsuwat T, Zha Y, et al. The commensal microbiome is associated with anti-PD-1 efficacy in metastatic melanoma patients. Science 2018;359:104-8.
117. Routy B, Le Chatelier E, Derosa L, Duong CPM, Alou MT, et al. Gut microbiome influences efficacy of PD-1-based immunotherapy against epithelial tumors. Science 2018;359:91-7.
118. Tamura R, Tanaka T, Yamamoto Y, Akasaki Y, Sasaki H. Dual role of macrophage in tumor immunity. Immunotherapy 2018;10:899-909.
119. Shitara K, Nishikawa H. Regulatory T cells: a potential target in cancer immunotherapy. Ann N Y Acad Sci 2018;1417:104-15.
120. Whiteside TL. The role of regulatory T cells in cancer immunology. Immunotargets Ther 2015;4:159-71.
121. Zamarron BF, Chen W. Dual roles of immune cells and their factors in cancer development and progression. Int J Biol Sci 2011;7:651-8.
122. Shalapour S, Karin M. Immunity, inflammation, and cancer: an eternal fight between good and evil. J Clin Invest 2015;125:3347-55.
123. Labelle M, Begum S, Hynes RO. Platelets guide the formation of early metastatic niches. Proc Natl Acad Sci U S A 2014;111:E3053-61.
124. Ocana A, Nieto-Jimenez C, Pandiella A, Templeton AJ. Neutrophils in cancer: prognostic role and therapeutic strategies. Mol Cancer 2017;16:137.
125. Mouchemore KA, Anderson RL, Hamilton JA. Neutrophils, G-CSF and their contribution to breast cancer metastasis. FEBS J 2018;285:665-79.
126. Cools-Lartigue J, Spicer J, Najmeh S, Ferri L. Neutrophil extracellular traps in cancer progression. Cell Mol Life Sci 2014;71:4179-94.
127. Park J, Wysocki RW, Amoozgar Z, Maiorino L, Fein MR, et al. Cancer cells induce metastasis-supporting neutrophil extracellular DNA traps. Sci Transl Med 2016;8:361ra138.
128. Lieffers JR, Mourtzakis M, Hall KD, McCargar LJ, Prado CM, et al. A viscerally driven cachexia syndrome in patients with advanced colorectal cancer: contributions of organ and tumor mass to whole-body energy demands. Am J Clin Nutr 2009;89:1173-9.
130. Vaupel P, Mayer A. Availability, not respiratory capacity governs oxygen consumption of solid tumors. Int J Biochem Cell Biol 2012;44:1477-81.
131. Davidson SM, Papagiannakopoulos T, Olenchock BA, Heyman JE, Keibler MA, et al. Environment impacts the metabolic dependencies of ras-driven non-small cell lung cancer. Cell Metab 2016;23:517-28.
133. Fearon KC, Glass DJ, Guttridge DC. Cancer cachexia: mediators, signaling, and metabolic pathways. Cell Metab 2012;16:153-66.
135. Vander Heiden MG, DeBerardinis RJ. Understanding the Intersections between Metabolism and Cancer Biology. Cell 2017;168:657-69.
136. Alam MM, Lal S, FitzGerald KE, Zhang L. A holistic view of cancer bioenergetics: mitochondrial function and respiration play fundamental roles in the development and progression of diverse tumors. Clin Transl Med 2016;5:3.
137. Martinez-Outschoorn UE, Pavlides S, Howell A, Pestell RG, Tanowitz HB, et al. Stromal-epithelial metabolic coupling in cancer: integrating autophagy and metabolism in the tumor microenvironment. Int J Biochem Cell Biol 2011;43:1045-51.
138. Pavlides S, Tsirigos A, Migneco G, Whitaker-Menezes D, Chiavarina B, et al. The autophagic tumor stroma model of cancer: role of oxidative stress and ketone production in fueling tumor cell metabolism. Cell Cycle 2010;9:3485-505.
139. Pavlides S, Tsirigos A, Vera I, Flomenberg N, Frank PG, et al. Loss of stromal caveolin-1 leads to oxidative stress, mimics hypoxia and drives inflammation in the tumor microenvironment, conferring the “reverse Warburg effect”: a transcriptional informatics analysis with validation. Cell Cycle 2010;9:2201-19.
140. Tsoli M, Robertson G. Cancer cachexia: malignant inflammation, tumorkines, and metabolic mayhem. Trends Endocrinol Metab 2013;24:174-83.
141. Flint TR, Janowitz T, Connell CM, Roberts EW, Denton AE, et al. Tumor-induced IL-6 reprograms host metabolism to suppress anti-tumor immunity. Cell Metab 2016;24:672-84.
142. Lee YM, Chang WC, Ma WL. Hypothesis: solid tumours behave as systemic metabolic dictators. J Cell Mol Med 2016;20:1076-85.
143. Argiles JM, Stemmler B, Lopez-Soriano FJ, Busquets S. Inter-tissue communication in cancer cachexia. Nat Rev Endocrinol 2018;15:9-20.
144. Argiles JM, Busquets S, Stemmler B, Lopez-Soriano FJ. Cancer cachexia: understanding the molecular basis. Nat Rev Cancer 2014;14:754-62.
145. George J, Cannon T, Lai V, Richey L, Zanation A, et al. Cancer cachexia syndrome in head and neck cancer patients: Part II. pathophysiology. Head Neck 2007;29:497-507.
146. Roxburgh CS, McMillan DC. Cancer and systemic inflammation: treat the tumour and treat the host. Br J Cancer 2014;110:1409-12.
147. Payen VL, Porporato PE, Baselet B, Sonveaux P. Metabolic changes associated with tumor metastasis, part 1: tumor pH, glycolysis and the pentose phosphate pathway. Cell Mol Life Sci 2016;73:1333-48.
148. Porporato PE, Payen VL, Baselet B, Sonveaux P. Metabolic changes associated with tumor metastasis, part 2: Mitochondria, lipid and amino acid metabolism. Cell Mol Life Sci 2016;73:1349-63.
149. Chasen M, Bhargava R, Hirschman S. Immunomodulatory agents for the treatment of cachexia. Curr Opin Support Palliat Care 2014;8:328-33.
150. Das SK, Eder S, Schauer S, Diwoky C, Temmel H, et al. Adipose triglyceride lipase contributes to cancer-associated cachexia. Science 2011;333:233-8.
151. Mantovani G, Maccio A, Mura L, Massa E, Mudu MC, et al. Serum levels of leptin and proinflammatory cytokines in patients with advanced-stage cancer at different sites. J Mol Med (Berl) 2000;78:554-61.
152. Kir S, White JP, Kleiner S, Kazak L, Cohen P, et al. Tumour-derived PTH-related protein triggers adipose tissue browning and cancer cachexia. Nature 2014;513:100-4.
153. Petruzzelli M, Schweiger M, Schreiber R, Campos-Olivas R, Tsoli M, et al. A switch from white to brown fat increases energy expenditure in cancer-associated cachexia. Cell Metab 2014;20:433-47.
154. Tomasin R, Martin A, Cominetti MR. Metastasis and cachexia: alongside in clinics, but not so in animal models. J Cachexia Sarcopenia Muscle 2019;10:1183-94.
155. Zhang G, Liu Z, Ding H, Zhou Y, Doan HA, et al. Tumor induces muscle wasting in mice through releasing extracellular Hsp70 and Hsp90. Nat Commun 2017;8:589.
156. Lazar I, Clement E, Dauvillier S, Milhas D, Ducoux-Petit M, et al. Adipocyte exosomes promote melanoma aggressiveness through fatty acid oxidation: a novel mechanism linking obesity and cancer. Cancer Res 2016;76:4051-7.
157. Tomasetti M, Lee W, Santarelli L, Neuzil J. Exosome-derived microRNAs in cancer metabolism: possible implications in cancer diagnostics and therapy. Exp Mol Med 2017;49:e285.
158. Marinho R, Alcantara PSM, Ottoch JP, Seelaender M. Role of exosomal microRNAs and myomiRs in the development of cancer cachexia-associated muscle wasting. Front Nutr 2017;4:69.
159. He WA, Calore F, Londhe P, Canella A, Guttridge DC, et al. Microvesicles containing miRNAs promote muscle cell death in cancer cachexia via TLR7. Proc Natl Acad Sci U S A 2014;111:4525-9.
160. Masri S, Papagiannakopoulos T, Kinouchi K, Liu Y, Cervantes M, et al. Lung adenocarcinoma distally rewires hepatic circadian homeostasis. Cell 2016;165:896-909.
161. Goncalves MD, Hwang SK, Pauli C, Murphy CJ, Cheng Z, et al. Fenofibrate prevents skeletal muscle loss in mice with lung cancer. Proc Natl Acad Sci U S A 2018;115:E743-52.
162. Bruning PF, Bonfrer JM, van Noord PA, Hart AA, de Jong-Bakker M, et al. Insulin resistance and breast-cancer risk. Int J Cancer 1992;52:511-6.
163. Maloney EK, McLaughlin JL, Dagdigian NE, Garrett LM, Connors KM, et al. An anti-insulin-like growth factor I receptor antibody that is a potent inhibitor of cancer cell proliferation. Cancer Res 2003;63:5073-83.
164. Hartl WH, Demmelmair H, Jauch KW, Koletzko B, Schildberg FW. Effect of glucagon on protein synthesis in human rectal cancer in situ. Ann Surgery 1998;227:390-7.
165. Mayers JR, Torrence ME, Danai LV, Papagiannakopoulos T, Davidson SM, et al. Tissue of origin dictates branched-chain amino acid metabolism in mutant Kras-driven cancers. Science 2016;353:1161-5.
166. Mayers JR, Wu C, Clish CB, Kraft P, Torrence ME, et al. Elevation of circulating branched-chain amino acids is an early event in human pancreatic adenocarcinoma development. Nat Med 2014;20:1193-8.
167. Bindels LB, Delzenne NM. Muscle wasting: the gut microbiota as a new therapeutic target? Int J Biochem Cell Biol 2013;45:2186-90.
168. Nieuwdorp M, Gilijamse PW, Pai N, Kaplan LM. Role of the microbiome in energy regulation and metabolism. Gastroenterology 2014;146:1525-33.
169. Argiles JM, Lopez-Soriano J, Almendro V, Busquets S, Lopez-Soriano FJ. Cross-talk between skeletal muscle and adipose tissue: a link with obesity? Med Res Rev 2005;25:49-65.
170. Hellerstein MK, Meydani SN, Meydani M, Wu K, Dinarello CA. Interleukin-1-induced anorexia in the Rat - influence of prostaglandins. J Clin Invest 1989;84:228-35.
171. Chauhan A, Sequeria A, Manderson C, Maddocks M, Wasley D, et al. Exploring autonomic nervous system dysfunction in patients with cancer cachexia: a pilot study. Auton Neurosci 2012;166:93-5.
172. Chrousos GP. The hypothalamic-pituitary-adrenal axis and immune-mediated inflammation. N Engl J Med 1995;332:1351-62.
173. Murphy KT. The pathogenesis and treatment of cardiac atrophy in cancer cachexia. Am J Physiol Heart Circ Physiol 2016;310:H466-77.
174. Belloum Y, Rannou-Bekono F, Favier FB. Cancer-induced cardiac cachexia: Pathogenesis and impact of physical activity (Review). Oncol Rep 2017;37:2543-52.
177. Israël M. A primary cause of cancer: GABA deficiency in endocrine pancreas. Cancer Therapy 2012;8:171-83.
178. Cancer metabolism: an alteration of the anabolic–catabolic selection switch. OA Cancer 2014;2:1.
179. Israël M. Metabolic rewiring of stem cells and differentiated cells in cancer: the hypothetical consequences of a GABA deficiency in endocrine pancreas. J Cancer Metastasis Treat 2019;5.
180. Kuhn T, Floegel A, Sookthai D, Johnson T, Rolle-Kampczyk U, et al. Higher plasma levels of lysophosphatidylcholine 18:0 are related to a lower risk of common cancers in a prospective metabolomics study. BMC Med 2016;14:13.
181. Beleva E, Grudeva-Popova J. From Virchow's triad to metastasis: circulating hemostatic factors as predictors of risk for metastasis in solid tumors. J BUON 2013;18:25-33.
182. Karikoski M, Marttila-Ichihara F, Elima K, Rantakari P, Hollmen M, et al. Clever-1/stabilin-1 controls cancer growth and metastasis. Clin Cancer Res 2014;20:6452-64.
183. Khorana AA, Francis CW, Culakova E, Kuderer NM, Lyman GH. Thromboembolism is a leading cause of death in cancer patients receiving outpatient chemotherapy. J Thromb Haemost 2007;5:632-4.
184. Falanga A, Russo L, Milesi V, Vignoli A. Mechanisms and risk factors of thrombosis in cancer. Crit Rev Oncol Hematol 2017;118:79-83.
185. van den Berg YW, Osanto S, Reitsma PH, Versteeg HH. The relationship between tissue factor and cancer progression: insights from bench and bedside. Blood 2012;119:924-32.
186. Magnus N, Garnier D, Meehan B, McGraw S, Lee TH, et al. Tissue factor expression provokes escape from tumor dormancy and leads to genomic alterations. Proc Natl Acad Sci U S A 2014;111:3544-9.
187. Versteeg HH, Schaffner F, Kerver M, Petersen HH, Ahamed J, et al. Inhibition of tissue factor signaling suppresses tumor growth. Blood 2008;111:190-9.
188. Labelle M, Hynes RO. The initial hours of metastasis: the importance of cooperative host-tumor cell interactions during hematogenous dissemination. Cancer Discov 2012;2:1091-9.
189. Labelle M, Begum S, Hynes RO. Direct signaling between platelets and cancer cells induces an epithelial-mesenchymal-like transition and promotes metastasis. Cancer Cell 2011;20:576-90.
191. Becker KA, Beckmann N, Adams C, Hessler G, Kramer M, et al. Melanoma cell metastasis via P-selectin-mediated activation of acid sphingomyelinase in platelets. Clin Exp Metastasis 2017;34:25-35.
192. Lavergne M, Janus-Bell E, Schaff M, Gachet C, Mangin PH. Platelet integrins in tumor metastasis: do they represent a therapeutic target? Cancers (Basel) 2017;9.
193. Mege D, Panicot-Dubois L, Ouaissi M, Robert S, Sielezneff I, et al. The origin and concentration of circulating microparticles differ according to cancer type and evolution: a prospective single-center study. Int J Cancer 2016;138:939-48.
194. Mezouar S, Frere C, Darbousset R, Mege D, Crescence L, et al. Role of platelets in cancer and cancer-associated thrombosis: experimental and clinical evidences. Thromb Res 2016;139:65-76.
195. Dvorak HF, Quay SC, Orenstein NS, Dvorak AM, Hahn P, et al. Tumor shedding and coagulation. Science 1981;212:923-4.
196. Del Conde I, Bharwani LD, Dietzen DJ, Pendurthi U, Thiagarajan P, et al. Microvesicle-associated tissue factor and Trousseau’s syndrome. J Thromb Haemost 2007;5:70-4.
197. Geddings JE, Mackman N. Tumor-derived tissue factor-positive microparticles and venous thrombosis in cancer patients. Blood 2013;122:1873-80.
198. Magnon C. Role of the autonomic nervous system in tumorigenesis and metastasis. Mol Cell Oncol 2015;2:e975643.
199. Elenkov IJ, Wilder RL, Chrousos GP, Vizi ES. The sympathetic nerve--an integrative interface between two supersystems: the brain and the immune system. Pharmacol Rev 2000;52:595-638.
200. Li T, Harada M, Tamada K, Abe K, Nomoto K. Repeated restraint stress impairs the antitumor T cell response through its suppressive effect on Th1-type CD4+ T cells. Anticancer Res 1997;17:4259-68.
201. Cao L, Liu X, Lin EJ, Wang C, Choi EY, et al. Environmental and genetic activation of a brain-adipocyte BDNF/leptin axis causes cancer remission and inhibition. Cell 2010;142:52-64.
202. Burfeind KG, Michaelis KA, Marks DL. The central role of hypothalamic inflammation in the acute illness response and cachexia. Semin Cell Dev Biol 2016;54:42-52.
203. Braun TP, Zhu X, Szumowski M, Scott GD, Grossberg AJ, et al. Central nervous system inflammation induces muscle atrophy via activation of the hypothalamic-pituitary-adrenal axis. J Exp Med 2011;208:2449-63.
204. Burfeind KG, Zhu X, Levasseur PR, Michaelis KA, Norgard MA, et al. TRIF is a key inflammatory mediator of acute sickness behavior and cancer cachexia. Brain Behav Immun 2018;73:364-74.
205. Mravec B, Horvathova L, Cernackova A. Hypothalamic inflammation at a crossroad of somatic diseases. Cell Mol Neurobiol 2019;39:11-29.
206. Molfino A, Iannace A, Colaiacomo MC, Farcomeni A, Emiliani A, et al. Cancer anorexia: hypothalamic activity and its association with inflammation and appetite-regulating peptides in lung cancer. J Cachexia Sarcopenia Muscle 2017;8:40-7.
207. Mauffrey P, Tchitchek N, Barroca V, Bemelmans AP, Firlej V, et al. Progenitors from the central nervous system drive neurogenesis in cancer. Nature 2019;569:672-8.
208. Cole SW, Nagaraja AS, Lutgendorf SK, Green PA, Sood AK. Sympathetic nervous system regulation of the tumour microenvironment. Nat Rev Cancer 2015;15:563-72.
209. Jones DH, Nakashima T, Sanchez OH, Kozieradzki I, Komarova SV, et al. Regulation of cancer cell migration and bone metastasis by RANKL. Nature 2006;440:692-6.
210. Katayama Y, Battista M, Kao WM, Hidalgo A, Peired AJ, et al. Signals from the sympathetic nervous system regulate hematopoietic stem cell egress from bone marrow. Cell 2006;124:407-21.
211. Elefteriou F. Role of sympathetic nerves in the establishment of metastatic breast cancer cells in bone. J Bone Oncol 2016;5:132-4.
212. Magnon C, Hall SJ, Lin J, Xue X, Gerber L, et al. Autonomic nerve development contributes to prostate cancer progression. Science 2013;341:1236361.
213. Grytli HH, Fagerland MW, Fossa SD, Tasken KA, Haheim LL. Use of beta-blockers is associated with prostate cancer-specific survival in prostate cancer patients on androgen deprivation therapy. Prostate 2013;73:250-60.
214. Lemeshow S, Sorensen HT, Phillips G, Yang EV, Antonsen S, et al. Beta-blockers and survival among Danish patients with malignant melanoma: a population-based cohort study. Cancer Epidemiol Biomarkers Prev 2011;20:2273-9.
215. Melhem-Bertrandt A, Chavez-Macgregor M, Lei X, Brown EN, Lee RT, et al. Beta-blocker use is associated with improved relapse-free survival in patients with triple-negative breast cancer. J Clin Oncol 2011;29:2645-52.
216. Pavlov VA, Tracey KJ. The vagus nerve and the inflammatory reflex--linking immunity and metabolism. Nat Rev Endocrinol 2012;8:743-54.
217. Fujii T, Mashimo M, Moriwaki Y, Misawa H, Ono S, et al. Expression and function of the cholinergic system in immune cells. Front Immunol 2017;8:1085.
218. Yu H, Xia H, Tang Q, Xu H, Wei G, et al. Acetylcholine acts through M3 muscarinic receptor to activate the EGFR signaling and promotes gastric cancer cell proliferation. Sci Rep 2017;7:40802.
219. Zhao CM, Hayakawa Y, Kodama Y, Muthupalani S, Westphalen CB, et al. Denervation suppresses gastric tumorigenesis. Sci Transl Med 2014;6:250ra115.
220. Jobling P, Pundavela J, Oliveira SM, Roselli S, Walker MM, et al. Nerve-cancer cell cross-talk: a novel promoter of tumor progression. Cancer Res 2015;75:1777-81.
221. Pelosof LC, Gerber DE. Paraneoplastic syndromes: an approach to diagnosis and treatment. Mayo Clin Proc 2010;85:838-54.
222. DeLellis RA, Xia L. Paraneoplastic endocrine syndromes: a review. Endocr Pathol 2003;14:303-17.
223. Castellone MD, Laukkanen MO, Teramoto H, Bellelli R, Ali G, et al. Cross talk between the bombesin neuropeptide receptor and Sonic hedgehog pathways in small cell lung carcinoma. Oncogene 2015;34:1679-87.
224. Sherbet GV. Hormonal influences on cancer progression and prognosis. Vitam Horm 2005;71:147-200.
225. Mousa SA, Glinsky GV, Lin HY, Ashur-Fabian O, Hercbergs A, et al. Contributions of thyroid hormone to cancer metastasis. Biomedicines 2018;6:89.
226. Su SC, Hsieh MJ, Yang WE, Chung WH, Reiter RJ, et al. Cancer metastasis: mechanisms of inhibition by melatonin. J Pineal Res 2017;62.
227. Zaki NF, Sabri YM, Farouk O, Abdelfatah A, Spence DW, et al. Depressive symptoms, sleep profiles and serum melatonin levels in a sample of breast cancer patients. Nat Sci Sleep 2020;12:135-49.
228. Yu B, Zanetti KA, Temprosa M, Albanes D, Appel N, et al. The consortium of metabolomics studies (COMETS): metabolomics in 47 prospective cohort studies. Am J Epidemiol 2019;188:991-1012.
229. Dean DC, Shen S, Hornicek FJ, Duan Z. From genomics to metabolomics: emerging metastatic biomarkers in osteosarcoma. Cancer Metastasis Rev 2018;37:719-31.
230. Xiao S, Zhou L. Gastric cancer: metabolic and metabolomics perspectives (Review). Int J Oncol 2017;51:5-17.
231. Peng X, Chen Z, Farshidfar F, Xu X, Lorenzi PL, et al. Molecular characterization and clinical relevance of metabolic expression subtypes in human cancers. Cell Rep 2018;23:255-69.e4.
232. Cieslik M, Hoang SA, Baranova N, Chodaparambil S, Kumar M, et al. Epigenetic coordination of signaling pathways during the epithelial-mesenchymal transition. Epigenetics Chromatin 2013;6:28.
233. Hong D, Messier TL, Tye CE, Dobson JR, Fritz AJ, et al. Runx1 stabilizes the mammary epithelial cell phenotype and prevents epithelial to mesenchymal transition. Oncotarget 2017;8:17610-27.
234. Vincent MD. Cancer: towards a general theory of the target: all successful cancer therapies, actual or potential, are reducible to either (or both) of two fundamental strategies. Bioessays 2017;39.
235. Dai S, Wei D, Wu Z, Zhou X, Wei X, et al. Phase I clinical trial of autologous ascites-derived exosomes combined with GM-CSF for colorectal cancer. Mol Ther 2008;16:782-90.
236. Que RS, Lin C, Ding GP, Wu ZR, Cao LP. Increasing the immune activity of exosomes: the effect of miRNA-depleted exosome proteins on activating dendritic cell/cytokine-induced killer cells against pancreatic cancer. J Zhejiang Univ Sci B 2016;17:352-60.
237. Tauro BJ, Greening DW, Mathias RA, Mathivanan S, Ji H, et al. Two distinct populations of exosomes are released from LIM1863 colon carcinoma cell-derived organoids. Mol Cell Proteomics 2013;12:587-98.
239. Tyson JJ, Chen KC, Novak B. Sniffers, buzzers, toggles and blinkers: dynamics of regulatory and signaling pathways in the cell. Curr Opin Cell Biol 2003;15:221-31.
240. Chen JC, Alvarez MJ, Talos F, Dhruv H, Rieckhof GE, et al. Identification of causal genetic drivers of human disease through systems-level analysis of regulatory networks. Cell 2014;159:402-14.
241. Archetti M, Pienta KJ. Cooperation among cancer cells: applying game theory to cancer. Nat Rev Cancer 2019;19:110-7.
242. Csermely P, Korcsmaros T. Cancer-related networks: a help to understand, predict and change malignant transformation. Semin Cancer Biol 2013;23:209-12.
243. Antonia SJ, Borghaei H, Ramalingam SS, Horn L, De Castro Carpeno J, et al. Four-year survival with nivolumab in patients with previously treated advanced non-small-cell lung cancer: a pooled analysis. Lancet Oncol 2019;20:1395-408.
244. Garon EB, Hellmann MD, Rizvi NA, Carcereny E, Leighl NB, et al. Five-year overall survival for patients with advanced nonsmall-cell lung cancer treated with pembrolizumab: results from the phase I keynote-001 study. J Clin Oncol 2019;37:2518-27.
245. Larkin J, Chiarion-Sileni V, Gonzalez R, Grob JJ, Rutkowski P, et al. Five-year survival with combined nivolumab and ipilimumab in advanced melanoma. N Engl J Med 2019;381:1535-46.
246. Rosenzweig SA. Acquired resistance to drugs targeting tyrosine kinases. Adv Cancer Res 2018;138:71-98.
247. Liu X, McMurphy T, Xiao R, Slater A, Huang W, et al. Hypothalamic gene transfer of BDNF inhibits breast cancer progression and metastasis in middle age obese mice. Mol Ther 2014;22:1275-84.
248. Bucsek MJ, Qiao G, MacDonald CR, Giridharan T, Evans L, et al. Beta-adrenergic signaling in mice housed at standard temperatures suppresses an effector phenotype in CD8(+) T cells and undermines checkpoint inhibitor therapy. Cancer Res 2017;77:5639-51.
249. Kokolus KM, Zhang Y, Sivik JM, Schmeck C, Zhu J, et al. Beta blocker use correlates with better overall survival in metastatic melanoma patients and improves the efficacy of immunotherapies in mice. Oncoimmunology 2018;7:e1405205.
250. Springer J, Tschirner A, Haghikia A, von Haehling S, Lal H, et al. Prevention of liver cancer cachexia-induced cardiac wasting and heart failure. Eur Heart J 2014;35:932-41.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Paul D. The systemic hallmarks of cancer. J Cancer Metastasis Treat 2020;6:29. http://dx.doi.org/10.20517/2394-4722.2020.63
AMA Style
Paul D. The systemic hallmarks of cancer. Journal of Cancer Metastasis and Treatment. 2020; 6: 29. http://dx.doi.org/10.20517/2394-4722.2020.63
Chicago/Turabian Style
Paul, Doru. 2020. "The systemic hallmarks of cancer" Journal of Cancer Metastasis and Treatment. 6: 29. http://dx.doi.org/10.20517/2394-4722.2020.63
ACS Style
Paul, D. The systemic hallmarks of cancer. J. Cancer. Metastasis. Treat. 2020, 6, 29. http://dx.doi.org/10.20517/2394-4722.2020.63
About This Article
Copyright

Data & Comments
Data


 Cite This Article 179 clicks
Cite This Article 179 clicks

 Like This Article 37
likes
Like This Article 37
likes




Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.