
Recent advances in immunotherapy for pancreatic cancer

 , ...
, ... Abstract
Pancreatic ductal adenocarcinoma (PDAC) remains a disease with a dismal prognosis. Since 1996 there have only been two upfront regimens found to be superior to gemcitabine: FOLFIRINOX (5-fluorouracil, leucovorin, irinotecan and oxaliplatin), and gemcitabine plus nab-paclitaxel. Despite the improvement noted in these trials, PDAC is highly chemo-resistant and patients who respond will inevitably develop resistance. The unique immunosuppressive tumor microenvironment with extensive desmoplasia has posed challenges to developing new and effective treatments. Therapeutic vaccines, combination treatments, adoptive T cell transfer, as well as immunomodulators are being explored. With the emerging use of immunotherapy and immunomodulators, the scope of this review is to present the current data on these agents as well as focus on the advancements in the treatment of PDAC. Overall, results in this realm have been disappointing to date reflecting the non-immunogenic and complex tumor microenvironment of PDAC.
Keywords
Introduction
Pancreatic ductal adenocarcinoma (PDAC) has a grim prognosis and is now the third leading cause of cancer-related deaths in the United States[1]. Furthermore, it is estimated that pancreatic cancer will become the second leading cause of cancer-related deaths in the United States by 2030[2]. It is estimated that 57,600 Americans will be diagnosed with pancreatic cancer and more than 47,050 will die of the disease in 2020 making it the most lethal malignancy of all major cancers[1]. Most patients present with unresectable or metastatic disease leading to an abysmal 5-year-overall survival (OS) rate of only 7%. Even when surgery is feasible in 15%-20% of the patients, the 5-year survival remains only about 10%[3]. Current front-line treatment options include traditional chemotherapies that only provide modest survival benefits.
Gemcitabine was considered the backbone of management for metastatic PDAC as it provided a survival benefit as well as alleviation of symptoms compared to fluorouracil (5-FU)[4]. Many trials after this failed to show improvement from this integral study in 1997. Various combination treatments have been explored since then to see if any headway could be made in the treatment of this lethal disease. Notably, combination treatments with epidermal growth factor receptors (EGFR) as well as anti-vascular endothelial growth factors (VEGF) did not produce clinically meaningful results[4-6]. Additionally, combination treatments exploring gemcitabine with various cytotoxic agents including the addition of oxaliplatin, 5-FU, capecitabine, and irinotecan failed to demonstrate a statistically significant OS advantage[7-10]. Two chemotherapy combination regimens have shown superiority in patients with metastatic disease since then. In the PRODIGE/ACCORD and MPACT trials, FOLFIRINOX and gemcitabine plus nab-paclitaxel, respectively, showed an OS benefit at the cost of increased toxicity[11,12]. OS has improved with the addition of chemotherapy in the adjuvant setting as well, however, there is much room for improvement[13,14]. Despite the improvement noted in these trials, pancreatic cancer is highly chemo-resistant and patients who respond will inevitably develop resistance to these therapeutic modalities[15]. Based on this historical data, providers can attempt treatment with varying success, but there is still a dire need to develop newer agents and incorporate newer strategies to improve outcomes in pancreatic cancer.
Recently, immunotherapy has revolutionized cancer care and has garnered approval in many different solid tumors, including melanoma, lung cancer, and urothelial cancers, among others[16]. Therefore, it has been of great interest to explore the role of various immunotherapies in PDAC. Overall, results in this realm have been disappointing to date, reflecting the non-immunogenic and complex tumor microenvironment of PDAC. To overcome these challenges, therapeutic vaccines, combination treatments, adoptive T cell transfer, as well as immunomodulators are being explored. With the emerging use of immunotherapy and immunomodulators, the scope of this review is to present the current data on these agents as well as focus on the advancements in the treatment of PDAC.
Mechanisms of chemoresistance and tumorigenesis
Pancreatic cancer is highly resistant to chemotherapy and radiotherapy, making treatment less effective compared to other solid tumors. The dense desmoplasia surrounding the pancreatic tumor microenvironment (TME) plays a major role in immune modulation and chemotherapy resistance. Pancreatic TME is comprised of the tumor and its surrounding stroma. The development and progression of PDAC is associated with the interplay of inflammatory cells, mediators, pancreatic stellate cells (PSCs), and the extracellular matrix (ECM) that gives rise to the tumor micro environment favoring tumorigenesis[17,18][Figure 1].
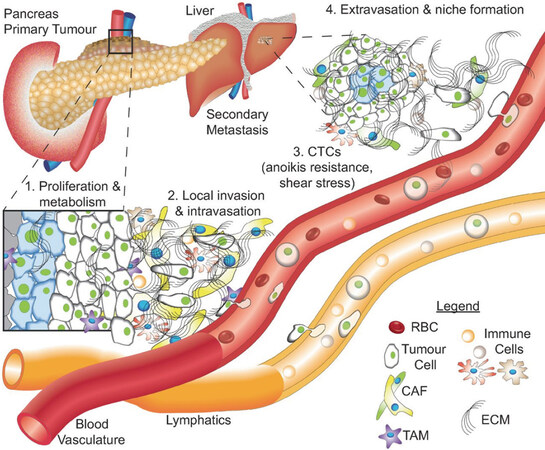
Figure 1. PDAC progression model and interaction with extracellular matrix (ECM). (1) Pancreatic cancer cells proliferate in the primary tumour, metabolising nutrients delivered by the blood vasculature and surrounding stroma; (2) cancer cells invade through the ECM, including cancer-associated fibroblasts (CAFs) and tumour-associated macrophages (TAMs), among other cancer-associated cell types, eventually intravasating or invading into the lymph and travelling to distant sites; (3) circulating tumour cells (CTCs) must develop resistance to anoikis, as well as shear stress, in order to survive in the circulation with red blood cells (RBCs) and leucocytes; (4) after travelling through the circulation, CTCs extravasate at secondary sites, commonly the liver, establishing a new niche. Re-use permitted under Creative Commons CC BY Attribution 4.0 Unported (CC BY 4.0) license, which permits others to copy, redistribute, remix, transform and build upon this work for any purpose, provided the original work is properly cited, a link to the license is given, and indication of whether changes were made. Link to license: https://creativecommons.org/licenses/by/4.0/). You are not required to obtain permission to reuse this article[18]
PSCs are part of normal pancreatic stroma and involved in vitamin A storage, exocrine/endocrine secretion, phagocytosis and maintenance of pancreatic stroma[19]. In PDAC, the malignant cells secrete transforming growth factor beta-1 (TGFβ-1), fibroblast growth factor-2 (FGF2), sonic hedgehog and platelet derive growth factor (PDGF) that drive PSC proliferation and increase ECM deposition leading to desmoplastic reaction[19,20]. The PSCs also contribute to the chemo resistance by actively entrapping gemcitabine in their cytoplasm lessening the effect of gemcitabine on pancreatic cancer cells[21]. Dense ECM deposition is thought to promote proliferation, chemoresistance, and limit T-cells accumulation near cancer cells[22,23]. ECM also increases intra-tumor interstitial fluid pressure, which leads to vascular dysfunction, limits accumulation of chemotherapeutic agents within the tumor and increases tumor hypoxia[24,25]. Hypoxic TME contributes to selection of more aggressive tumor cells that survive and proliferate[26]. These studies demonstrated the important role of desmoplastic reaction in the progression of PDAC making it an attractive target for therapeutic agents. However, studies with PDAC animal models lacking ability to form desmoplastic reaction demonstrated accelerated tumor progression, suggesting a more complex role of desmoplasia in PDAC. The recent HALO 301 trial using pegvorhyaluronidase alfa (PEGPH20), an enzyme that degrades hyaluronic acid in the ECM, in combination with gemcitabine and nab-paclitaxel, failed to improved survival further substantiating a more complex role of desmoplasia[27].
The PSCs stimulated by PDAC cells in the TME can further differentiate into different populations of cancer associated fibroblasts (CAFs). Öhlund et al.[28] demonstrated that alpha-smooth muscle actin expressing CAFs is more prevalent in close proximity to PDAC cells whereas CAFs further away from the tumor cells have increased expression of fibroblast activating protein (FAP). The two populations of fibroblasts are mutually exclusive. FAP-positive CAFs secrete increased levels of cytokines [interleukin (IL)-1, IL-6, IL-8, IL-10] and growth factors such as VEGF, insulin-like-growth factor (IGF1), PDGF, connective tissue growth factor (CTGF) and FGF which stimulate angiogenesis, proliferation and metastasis[19,29]. The PSCs also promote immunosuppression by recruiting myeloid-derived suppressor cells (MDSCs) through the signal transducer and activator of transcription 3 (STAT3) pathway[30,31]. The MDSCs express programmed death-ligand 1 (PD-L1) and the cytotoxic T-lymphocyte antigen 4 (CTLA-4) receptors resulting in T cell tolerance. MDSCs also promote the development of regulatory T cells (Treg) through the CD40 engagement in presence of inteleukin-10 (IL-10) and TGFβ. Besides immune regulation, MDSCs also favor tumor progression via a non-immune mechanism by promoting tumor angiogenesis, cancer cell stemness, aggressiveness, and invasiveness[32].
The complex interactions among the components of the PDAC TME make the tumor highly inductive to angiogenesis, metastasis and treatment resistance.
The challenging immune environment in PDAC
Immunogenicity is related to the degree of epitope structural difference between the tumor and normal cells. Neoantigens, peptides generated from non-silent coding mutations in the cancer cell genome, are highly immunogenic[33]. Studies have shown that tumor mutation load is proportional to neoantigen burden, which positively correlates with response to immunotherapy[34,35]. However, PDAC is characterized by low tumor mutational burden (TMB) ranging from 10-60 encoded neoantigens in contrast to 100-1500 mutations per megabase expressed by other solid tumors that respond to immunotherapy[36]. The limited expression of neoantigens by PDAC leads to poor immune surveillance.
Additionally, the TME of PDAC is known to be immunosuppressive with reduced cytotoxic T cells (CD8+) and T helper cells (CD4+) with increased Tregs, tumor-associated macrophages (TAMs), and MDSCs[37]. The location of T cells in PDAC also has important implications in resistance mechanisms. For example, CD3+ T cells, which are either CD4+ or CD8+ T cells, have been identified more commonly at the invasive front of PDAC with fewer cells detected in the center thereby excluded by malignant cells. Moreover, the tumor infiltrating CD3+ T cells also cluster next to nests of malignant cells but are unable to interact with tumor cells since they are trapped within the stromal tissue[38]. Therefore, the overall regulatory immune population of cells with exclusion of cytotoxic T cells along with the physical barrier created by a dense stromal environment creates a non-immunogenic tumor that is often resistant to immune recognition and killing. Nevertheless, the impact of T-cell infiltration on prognosis has shown inconsistent results with some studies showing improved OS with increased intratumoral CD3+ T-cells, whereas other studies have not shown an association between T-cell density and patient survival[38,39].
The combination of an immunosuppressive environment and low TMB makes PDAC an “immune desert” that is resistant to immunotherapies. However, it also suggests a potential treatment strategy by overcoming low immunogenicity.
Immune check point inhibitors
The immune recruitment and response from T cells (CD4+ and CD8+) via antigen recognition in the presence of malignant cells are controlled by inhibitory and stimulatory signals called immune checkpoints. By expressing inhibitory ligands, tumor cells can evade immune surveillance[40,41]. The first immune checkpoints discovered were CTLA4 and its ligands B7-1 and B7-2, and programmed cell death receptor 1 (PD1) and its ligands PD-L1 and PD-L2[42,43]. Although immune checkpoint inhibitors (ICIs) such as ipilimumab (CTLA4 inhibitor), pembrolizumab (PD1 inhibitor) and durvalumab (PD-L1 inhibitors) have benefitted patients with a wide variety of solid tumors, they have shown limited success in patients with PDAC[44,45].
One of the first studies to test single agent immunotherapy for the treatment of PDAC was a phase II trial with ipilimumab, a CTLA4 inhibitor. It was given to 27 patients with PDAC including 20 patients with metastatic disease and 7 patients with locally advanced disease. There were no responders by response evaluation criteria in solid tumors criteria (RECIST ver.1.1) and three subjects experienced grade 3 or higher immune-mediated adverse events (colitis, encephalitis, and hypophysitis) leading to early discontinuation of the trial[45]. In a larger phase I study in 2012 with 207 patients, 14 of whom had PDAC, patients were treated with anti-PD-L1 antibody, and no responses were seen in patients with metastatic PDAC[46]. In 2015, another smaller phase I study of pembrolizumab (PD-1 inhibitor) in patients with advanced solid tumors showed similar results in 1 out of 30 patients with PDAC who did not respond[47].
ICIs have shown benefit in a small group of PDAC patients whose tumor harbors mismatch repair (MMR) deficiency, which comprise 1%-3% of PDAC diagnoses[48,49]. In a phase II trial, which included 8 patients with PDAC, who were MMR deficient and treated with pembrolizumab, there was a 62% objective response among PDAC patients[50]. There has been one case report for the benefit of pembrolizumab in MMR-proficient PDAC, but with high TMB potentially highlighting a new population that would benefit from immunotherapy[51]. Pembrolizumab is the first ICI to receive approval from FDA for tumor agnostic indication for use in patients with MMR-deficient malignancies[50]. Based on the success of pembrolizumab in patients with refractory, metastatic cancers with MMR deficiency, the National Comprehensive Cancer Network (NCCN) now recommends consideration of MSI or MMR testing in patients with locally advanced or metastatic PDAC.
Combination of chemotherapy, immunotherapy, and radiation
Given the lack of response with single agent ICIs in treatment of PDAC, combination with chemotherapy or with other immunotherapies has also been studied.
Phase I studies
A phase Ib dose finding study of ipilimumab and gemcitabine showed partial response in 15% (2 patients) and stable disease in 38% (5 patients)[52]. In a follow up phase Ib dose expansion study 2 out of 16 patients had partial response and 5 out of 16 had stable disease. Median PFS was 2.5 months (95%CI: 0.8-4.8 months) and medial OS of 8.5 months (95%CI: 2.2-10.3 months)[53].
Phase II studies
One of the largest phase II trials was a study comparing durvalumab (PD-L1 inhibitor) in combination with tremilimumab (CTLA-4 inhibitor) vs. durvalumab alone in 65 patients with metastatic PDAC. The objective response rate (ORR) was 3.1% and median OS of 3.1 months in the combination arm and ORR of 0% with median OS of 3.6 months in the monotherapy arm. Median PFS was 1.5 months in both arms[54]. Due to the low patient numbers, the trial was not powered to observe the association between treatment response and PD-L1 expression or MSI status. Therefore, combination ICIs thus far have shown minimal benefit in treatment of PDAC.
Combination with chemotherapy and radiation
The combination of radiation with chemotherapy (chemoXRT) and immunotherapy is also being explored in the treatment of PDAC. The rationale of combining radiation is based on a previously recognized phenomenon called the abscopal effect whereby a local treatment (i.e., ionizing radiation) results in systemic or off-target shrinkage of tumor. The abscopal effect is postulated to be induced by anti-tumor T cell response mediated by immunogenic cell death after radiation[55,56]. Therefore, it has been proposed that combining chemoXRT with immune therapy may increase tumor immunogenicity and increase pancreatic tumor response to immunotherapy[57]. This hypothesis is supported by various studies demonstrating that PDAC tumors treated with neoadjuvant chemoXRT had increased numbers of CD4+ and CD8+ T-lymphocytes compared to patients who did not receive neoadjuvant chemoXRT[58]. In another study, neoadjuvant chemoXRT showed significantly lower numbers of immunosuppressive regulatory T cells although there was no difference in the number of CD4+ and CD8+ T cells[57].
In pre-clinical studies, radiation has shown mixed results. In a mouse model of PDAC, a combination of radiation with dual blockade of PD-L1 and CTLA-4 resulted in improved survival and tumor responses compared to dual blockade without radiation or radiation alone[59]. Another mouse model suggested an immunosuppressive T cell effect where radiation exposure induced macrophage immunosuppressive phenotype with reduction in CD8+ T-cells and increased Tregs[60]. The role of radiation in clinical studies is still being explored. A phase Ib/II study with neoadjuvant pembrolizumab with chemoXRT is ongoing. The combination appears to be safe but efficacy data have not been reported[61]. Other ongoing clinical trials [Table 1] include a pilot study evaluating SBRT in combination with tremilimumab (anti-CTLA-4) and PD-L1 monoclonal antibody MEDI4736 (NCT02311361) and an open label phase II study combining radiation with nivolumab with or without ipilimumab (NCT02866383).
Ongoing clinical trials investigating immune check point inhibitors
| Study drug | Combination | Study phase | Treatment setting | Sponsor | Clinicaltrials.gov identifier |
|---|---|---|---|---|---|
| Anti-PD-1 antibodies | |||||
| Nivolumab | Nab-paclitaxel ± gemcitabine | I | Any | Celgene | NCT02309177 |
| ± Ipilimumab | I/II | Any | Bristol-Myers Squibb | NCT01928394 | |
| Radiotherapy ± ipilimumab | II | Second line | Herlev Hospital | NCT02866383 | |
| Pembrolizumab | Radiotherapy | I | Second line | University of Pennsylvania | NCT02303990 |
| None | I | Any | Merck Sharp & Dohme | NCT02054806 | |
| Paricalcitol | II | Any | Translational Genomics Research Institute | NCT03331562 | |
| BL-8040 (CXCR4 antagonist) | II | Second line | MD Anderson | NCT02907099 | |
| Azacitidine | II | Second line | Columbia University | NCT03264404 | |
| Anti-PD-L1 antibodies | |||||
| Atezolizumab | None | I | Any | Genentech | NCT01375842 |
| None | II | Any | Hoffmann La Roche | NCT02458638 | |
| Avelumab | Binimetinib (MEK inhibitor) and talazoparib | II | Second line | Pfizer | NCT03637491 |
| Durvalumab | Pexidartinib (CSF1R inhibitor) | I | Second line | Centre Leon Berard | NCT02777710 |
| Galunisertib (TGFβ antagonist) | I | Second line | Eli Lilly | NCT02734160 | |
| None | I/II | Any | MedImmune | NCT01693562 | |
| Radiotherapy ± tremelimumab | I/II | Second line | National Cancer Institute | NCT02311361 | |
Vaccines
Another strategy to overcome the immune desert of pancreatic cancer that is under investigation is the use of therapeutic vaccines [Table 2]. Vaccines may potentially turn PDAC into more immunogenic tumors by activating specific T cells with the ability to migrate into PDAC tumors[62]. Tumors harbor driver and passenger mutations that may lead to changes in amino acid sequences, which in turn produces mutant proteins that are expressed by the tumors. These mutant proteins are processed into short polypeptides and displayed on the cell surface by major histocompatibility complex (MHC) and are recognized by T cells as foreign antigens[63]. This distinguishing feature of a malignant cell from a normal cell can provide a method of targeting the tumor cells while sparing the normal cells by the immune system.
Ongoing clinical trials investigating vaccines
| Study drug | Combination | Study phase | Treatment setting | Sponsor | Clinicaltrials.gov identifier |
|---|---|---|---|---|---|
| Whole cell vaccines | |||||
| GVAX (+cyclophosphamide) | Nivolumab, Urelumab | I/II | Neoadjuvant and Adjuvant Treatment | Johns Hopkins | NCT02451982 |
| Nivolumab + SBRT | II | Neoadjuvant | Johns Hopkins | NCT03161379 | |
| Ipilimumab, nivolumab, CRS-207 | II | Second line | Johns Hopkins | NCT03190265 | |
| Pembrolizumab + SBRT | II | First line | Johns Hopkins | NCT02648282 | |
| Pembrolizumab, IMC-CS4 | early phase I | First line | Johns Hopkins | NCT03153410 | |
| Algenpantucel-L | None | n/a | Any | NewLink Genetics Corporation | NCT03165188 |
| Bacterial-based vaccines | |||||
| Listeria based vaccine CRS-207 | Ipilimumab, nivolumab with or without GVAX + cyclophasphamide | II | Second line | Johns Hopkins | NCT03190265 |
| Epacadostat, pembrolizumab ± GVAX and cyclophosphamide | II | Second line | Johns Hopkins | NCT03006302 | |
| Yeast-based vaccines | |||||
| GI-4000 | Aldoxorubicin, cyclophosphamide, oxaliplatin, capecitabine, fluorouracil, leucovorin, lovaza, nab-paclitaxel, bevacizumab, avelumab, ALT-803, aNK, ETBx-011, SBRT | I/II | Second line | NantKwest, Inc. | NCT03387098 |
| Cyclophosphamide, oxaliplatin, capecitabine, fluorouracil, leucovorin, nab-paclitaxel, bevacizumab, avelumab, ALT-803, aNK, ETBx-011, SBRT | I/II | Second line | NantKwest, Inc. | NCT03329248 | |
| Cyclophosphamide, oxaliplatin, capecitabine, fluorouracil, leucovorin, nab-paclitaxel, bevacizumab, avelumab, ALT-803, aNK and ETBx-011 | I/II | Second line | NantKwest, Inc. | NCT03136406 | |
| YE-NEO-001 | None | I | First line | NantBioScience, Inc. | NCT03552718 |
| Viral vector-based vaccines | |||||
| Ankara Vaccine Expressing p53 | Pembrolizumab | I | Second line | City of Hope Medical Center | NCT02432963 |
| Peptide vaccines | |||||
| Personalized mesothelin epitopes | Adjuvant chemotherapy + Polyinosinic-polycytidylic acid, and poly-L-lysine (poly-ICLC) | I | First line | Washington University School of Medicine | NCT03956056 |
| KRAS peptide vaccine | ipilimumab, nivolumab | I | First line | Johns Hopkins | NCT04117087 |
| iNeo-Vac-P01 | GMCSF | I | Second line | Zhejiang Provincial People’s Hospital | NCT03645148 |
| Personalized neoantigen vaccine | Adjuvant chemotherapy | I | First line | Changhai Hospital | NCT03558945 |
| DNA based vaccines | |||||
| Personalized neoantigen DNA vaccine | I | Washington University School of Medicine | NCT03122106 | ||
| Dandritic cell-based vaccines | |||||
| Autologous DC vaccine | None | I | First line | Baylor College of Medicine | NCT04157127 |
| mDC3/8-KRAS Vaccine | None | I | Any | University of Pennsylvania | NCT03592888 |
Antigens expressed by PDAC cells can be grouped into two categories: tumor-specific antigens (TSA), which are only expressed by the tumor cells, and tumor-associated antigens (TAA), which are mostly restricted to malignant cells with limited expression on normal cells[64]. TSAs are neoantigens unique to each patient. A vaccine that targets TSAs needs to be personalized to the individual patient. However, TSAs such as MUC1 or KRAS which is expressed by more than 90% of PDAC cells are an exception to this rule. Currently there are a number of vaccines targeting shared antigens and patient specific antigens that are under investigation. Examples of TAAs expressed by PDAC cells include EGFR family, carcinoembryonic antigen (CEA), mesothelin, VEGF family, and Wilms’ tumor 1 (WT-1); however, since they may also be expressed by normal cells, off-target toxicity can occur[65,66].
Whole cell vaccines
GVAX is the most extensively evaluated whole cell vaccine consisting of two human allogeneic pancreatic tumor cells lines irradiated to release antigens. It is also genetically engineered to release granulocyte-macrophage colony-stimulating factor (GM-CSF) at the vaccination site[67]. Dendritic cells are attracted to GM-CSF site where they phagocytose antigens released from apoptotic PDAC cells. These dendritic cells then travel to draining lymph nodes, present tumor antigens found in vaccine PDAC cell lines to effector T-cell and activate them.
In an early phase clinical trial, GVAX demonstrated improved disease-free survival (DFS) in PDAC patients who displayed delayed-type hypersensitivity responses to autologous tumor cells[68]. In a phase II trial of 60 patients with resected PDAC, GVAX was studied in combination with chemoXRT. Results demonstrated a DFS of 17.3 months with an OS of 24.8 months, and induction of mesothelin-specific CD8+ T cells was seen in those treated with GVAX, which correlated with DFS[69]. Another trial studied GVAX in the neoadjuvant setting where patients received cyclophosphamide and GVAX prior to pancreaticoduodenectomy. Surgical resection samples from these patients were noted to have novel vaccine-induced upregulation of PD-1/PD-L1 pathways in the intratumoral tertiary lymphoid aggregates[62]. In the metastatic setting, GVAX has also been studied with or without ipilimumab in patients that were refractory to gemcitabine-based therapy. This study enrolled 15 patients in each arm and results showed a median OS of 3.6 months vs. 5.7 months (P = 0.072) for single agent ipilimumab versus combination ipilimumab with GVAX[70]. Currently, there are ongoing phase I and II clinical trials of GVAX in various combinations with nivolumab and urelumab (NCT02451982), nivolumab and radiation therapy (NCT03161379), ipilimumab, nivolumab and CRS-207 (Listeria based vaccine) (NCT03190265), pembrolizumab and radiation therapy (NCT02648282), pembrolizumab and IMC-SC4 (NCT03153410).
Another whole cell vaccine is Algenpantucel-L which contains two irradiated allogeneic PDAC tumor cell lines transfected to express alpha-1, 3-galactosyltransferase epitopes, a cell surface carbohydrate. A phase III IMPRESS trial comparing chemoXRT + algenpantucel-L vs. chemoXRT alone in the adjuvant setting showed no improvement in OS. Unfortunately, no further trials are planned at this time due to limited clinical benefit[71].
Bacterial-based vaccines
Vectors based on bacteria, including Listeria monocytogens, salmonella, Lactobacillus Plantarum, and Bacillus Calmette-Guerin, have been studied as they can elicit both innate and adaptive immune responses to TAAs. The bacterial vectors are modified to express specific antigens and elicit potent CD8+ and CD4+ responses via MHC I and MHC II antigen processing pathways. CRS-207, a live attenuated listeria vaccine genetically modified to secrete mesothelin was studied in combination with GVAX[72,73]. In a phase II trial with metastatic PDAC patients, GVAX with cyclophosphamide (Cy/GVAX) was compared to Cy/GVAX followed by CRS-207. The Cy/GVAX plus CRS-207 arm showed an OS benefit of 6.1 months vs. 3.9 months in the Cy/GVAX alone arm[74]. It was also noted that patients who elicited mesothelin specific CD8+ T-cell responses also had longer OS. However, a more recent phase IIb trial in which previously treated patients with metastatic PDAC who failed > 2 lines of chemotherapy were randomized 1:1:1 to receive Cy/GVAX + CRS 207 (arm A), CRS-207 (arm B), or a physician’s choice of singe-agent chemotherapy (arm C), showed Cy/GVAX + CRS-207 did not improve OS compared to chemotherapy[75]. CRS-207 is now being studied in combination with indoleamine 2, 3 dioxygenase-1 (IDO1) inhibitor and ICIs (NCT03190265, NCT03006302).
Yeast-based vaccines
Yeast cells have also been used as vaccine vectors given their ability to elicit robust T cell responses. Yeast cells can be genetically engineered to express TSAs. GI-4000 is a combination of four vaccines made up of heat-inactivated S. cerevisiae expressing the three most common Ras mutations of human cancers. A phase II randomized trial in which GI-4000 was given together with gemcitabine in PDAC showed a modest OS benefit of 524 days vs. 444 days[76]. Another phase I trial (NCT03552718) is evaluating a personalized neoepitope yeast vaccine in patients with resected pancreas, liver and lung cancers.
Viral vector based vaccines
Another strategy to elicit T-cell response is using viral vectors modified to encode TAAs/TSAs. These vectors include adenovirus (AdV), adeno-associated virus (AAV), vaccinia virus (VV), and alphavirus. Phase I results were encouraging. However, a phase III trial with PANVAC-VF, a recombinant attenuated vaccinia and fowlpox vector expressing CEA, MUC-1and immunostimulatory molecules ICAM-1, B7.1 LFA-3, did not show statistical significance in overall survival[77]. A phase I trial (NCT02432963) is evaluating a combination of vaccinia virus expressing p53 (MVA-p54) and pembrolizumab in PDAC and other solid tumors.
Peptide vaccines
Antigenic peptide vaccines are phagocytosed by dendritic cells and presented to T-cells to trigger a response. Theses vaccines have the potential to be personalized to individual patients with improved selection of immunogenic epitopes and have been shown to have some benefit in high risk melanoma patients[78]. In PDAC, despite demonstration of T-cell immune response by several peptide vaccines (elpamotide-VEGFR2 vaccine, KIF202A-66-member of kinesin super family protein 20A, and RAS peptide) in clinical trials, no OS benefit was observed[79-81].
DNA-based vaccines
DNA vaccines contain a DNA plasmid that encodes for highly immunogenic TAA that are specific to a patient’s PDAC including epitopes of mesothelin. After administration, the DNA plasmid is electroporated into cells. The expressed neoantigens are taken up by antigen presenting dendritic cells to elicit T-cells response. A phase I double blind, placebo controlled trial of VXM01, a DNA vaccine, in 71 patients with advanced PDAC showed that it was well tolerated and led to activation of VEGFR2-specific cytotoxic T-cells[82]. A phase I DNA vaccine trial (NCT03122106) of resected PDAC patients is ongoing.
Dendritic cell-based vaccines
Dendritic cells are specialized antigen presenting cells that process antigens to prime T cells. They can be loaded with tumor cell lysate and antigens, expanded ex-vivo and administered into patients[83,84]. Two phase I trials with autologous dendritic cells pulsed with mutant KRAS peptides (NCT03592888) and dendritic cells loaded with tumor cell lysate and RNA (NCT04157127) corresponding to the patient’s specific tumor mutation and HLA type in resected PDAC patients are currently ongoing.
Adoptive T-cell therapy
Adoptive T-cell therapy is another area of interest in treatment of PDAC given its success in number of hematologic malignancies[63]. T-cells are genetically engineered to express chimeric antigen receptor (CAR) which targets specific tumor antigens. The receptors are made up of an extracellular antigen recognition domain to an intracellular signaling domain of CD28, 4-1BB, and other co-stimulatory molecules which activate the T cell when an antigen is bound. These cells are expanded ex-vivo and re-infused into patients. In a phase I trial in which mesothelin-specific CAR-T cells were given to 6 patients with chemotherapy-refractory metastatic PDAC, 2 patients had stable disease with PFS of 3.8 and 5.4 months. Additionally, one patient had 69.2% decrease in metabolic activity on PET (positron emission tomography) scan with complete reduction in FDG (F-2-fluoro-2-deoxy-D-glucose) uptake in all liver lesions at 1 month but no effect was seen on primary PDAC[85]. Only small numbers of patients have been treated with adoptive T-cell therapy and there have been variable results in metastatic sites versus primary tumor. There are currently various other preclinical and phase I/II trials with CAR T cells targeting various tumor antigens such as Nectin4/FAP, EpCAM, CD70, CLD18, TnMUC1, PSCA (NCT03932565, NCT03013712, NCT02830724, NCT03302403, NCT04025216, NCT02744287) [Table 3].
Ongoing clinical trials investigating CART cells immunotherapy
| Study drug | Combination | Study phase | Treatment setting | Sponsor | Clinicaltrials.gov identifier |
|---|---|---|---|---|---|
| CART-meso | None | I | Second line | University of Pennsylvania | NCT03323944 |
| None | N/A | Second line | Shenzhen BinDeBio | NCT03638193 | |
| None | I/II | Second line | National Cancer Institute | NCT01583686 | |
| CART-Nectin4 | None | I | Second line | The Sixth Affiliated Hospital of Wenzhou Medical University | NCT03932565 |
| CART-EpCAM | None | I/II | Any | First Affiliated Hospital of Chengdu Medical College | NCT03013712 |
| CART-CD70 | Cyclophosphamide, Fludarabine, Aldesleukin | I/II | Second line | National Cancer Institute (NCI) | NCT02830724 |
| CART-CLD18 | Cylophosphamide, Fludarabine | N/A | Any | Kang YU, First Affiliated Hospital of Wenzhou Medical University | NCT03302403 |
| CART-TnMUC1 | Cylophosphamide, Fludarabine | I | Second line | Tmunity Therapeutics | NCT04025216 |
| CART-PSCA | Rimiducid | I/II | Second line | Bellicum Pharmaceuticals | NCT02744287 |
Although clinical data suggests that vaccines and CAR T-cell therapy can elicit anti-tumor T cell responses in PDAC with suggestion of clinical benefit, no clinically meaningful responses have been reported with CAR T-cell treatment of PDAC. These approaches have the potential to target the PDAC specifically; however, the tumor fighting cells may not be able to reach their target due to the desmoplasia and immunosuppressive tumor microenvironment. There may be also be other barriers to effective anti-tumor immune therapy. Further studies are needed to better understand and target these barriers.
Other targeted agents
There are also strategies using agents that target DNA repair, pathway inhibitors, metabolism and extracellular matrix. Some of the notable ones are discussed below [Table 4].
Ongoing clinical trials investigating novel targeting agents
| Study drug | Combination | Study phase | Treatment setting | Sponsor | Clinicaltrials.gov identifier |
|---|---|---|---|---|---|
| PARP inhibitors | |||||
| Olaparib | None | II | Any | AstraZeneca | NCT01078662 |
| None | III | Maintenance | AstraZeneca | NCT02184195 | |
| None | II | Second line | AstraZeneca | NCT02677038 | |
| Cediranib (VEGF inhibitor) | II | Second line | National Cancer Institute | NCT02498613 | |
| Rucaparib | None | II | Maintenance | Abramson Cancer Center, UPenn | NCT03140670 |
| Liposomal irinotecan, 5-fluorouracil and leucovorin | I/II | Any | Academic and Community Cancer Research United | NCT03337087 | |
| Niraparib | Ipilimumab or nivolumab | I/II | Second line | Upenn | NCT03404960 |
| None | III | Any | Anup Kasi | NCT03553004 | |
| Veliparib | FOLFIRI or mFOLFIRI | II | Second line | National Cancer Institute | NCT02890355 |
| mFOLFOX6 | I/II | Any | Georgetown University | NCT01489865 | |
| ± Cisplatin and gemcitabine | II | Any | National Cancer Institute | NCT01585805 | |
| Talazoparib | Avelumab and binimetinib | Ib/II | Any | Pfizer | NCT03637491 |
| NTRK inhibitors | |||||
| Larotrectinib | None | II | Any | Bayer | NCT 02576431 |
| Entrectinib | None | II | Any | Hoffmann-La Roche | NCT02568267 |
| Mitochondrial inhibitor | |||||
| Devimistat | Gemcitabine plus nab-paclitaxel | I | First line | Atlantic Health System | NCT03435289 |
| mFOLFIRINOX | III | Any | Rafael Pharmaceuticals | NCT03504423 | |
| FAK inhibitor | |||||
| PF-00562271 | None | I | Second line | Verastem | NCT00666926 |
| Connective tissue growth factor inhibitor | |||||
| Pamrevlumab | Gemcitabine plus nab-paclitaxel | I/II | Neoadjuvant | FibroGen | NCT02210559 |
| Gemcitabine plus nab-paclitaxel | III | First line | FibroGen | NCT03941093 | |
PARP inhibitors
Poly (ADP-ribose) polymerase (PARP) enzymes repair single stranded DNA breaks and play crucial roles in DNA damage repair (DDR). PARP inhibitors are small molecules that trap PARP enzymes on DNA and prevent the process of DDR. Cancer cells with deficient in DNA repair via homologous recombination due to mutations in BRCA 1/2 are very sensitive to PARP inhibitors[86]. The presence of PARP inhibitors lead to death of BRCA 1/2 mutated cancer cells due to a markedly reduced capacity for DDR. The first randomized, phase III trial, POLO found that maintenance therapy with a PARP inhibitor, Olaparib, significantly prolonged the progression of disease in advanced PDAC with germline BRCA gene mutations compared to placebo (PFS 7.3 months vs. 3.8 months)[87]. Currently, there are several clinical trials at varying phases studying the efficacy of different PARP inhibitors in advanced PDAC.
NTRK inhibitors
Topomyosin receptor kinase (TRK) transmembrane proteins plays an important role in neural development. When different partners fuse with TRK encoding genes (NTRK1, NTRK2 or NTRK3) the resulting gene encode for oncogenic proteins that constitutively activate the RAS-MEK-ERK, PI3k-AKT, and diacyleglycerol-inositol-1,4,5-triphosphate signaling pathways[88]. NTRK fusions are found in < 1% of PDAC. In the STARTRK-2 trial (NCT02568267) with entrectinib, a TRK A/B/C inhibitor, 3 patients with PDAC had PR based on RECIST criteria. Another ongoing trial NAVIGATE (NCT 02576431) showed response rate was as high as 75% among 55 patients with NTRK fusion positive solid tumors treated with larorectinib. However, only one patient with PDAC was included in the cohort. Currently, entrectinib and larorectinib are approved by FDA for a tissue-agnostic indication in solid tumors that harbor NRTK fusion gene.
Devimistat
Devimistat selectively inhibits the tricarboxylic acid cycle in the mitochondria and is hypothesized to work synergistically with cytotoxic agents by decreasing mitochondria metabolism, impairing production of intermediates required for DNA damage repair. Devimistat was studied in a phase I trial in combination with mFOLFIRINOX in 20 patients with metastatic PDAC. The response rate was 61% with 3 patients achieving CR[89]. Currently Devimistat is being studied in a phase III trial in combination with mFOLFIRINOX (NCT03504423) and in a phase I trial in combination with gemcitabine and nab-paclitaxel (NCT03435289).
Focal adhesion kinase inhibitors
Focal adhesion kinase (FAK) is a tyrosine kinase that promotes tumor growth, invasion and metastasis by interacting with tumor cells and stromal cells in various types of cancer[90]. In preclinical research, FAK is shown to be overexpressed in pancreatic tumor tissues. FAK promotes metastasis by regulating focal adhesions, matrix metalloproteinase surface expressions and enhancing tumor growth by promoting anti-apoptotic functions. The combination of FAK inhibitors together with gemcitabine and nab-paclitaxel has been shown to delay tumor growth in patient derived xenograph models, relative to chemotherapy alone[90]. Currently, a phase I clinical trial with FAK inhibitor PF00562271 is under investigation in patients with advanced pancreatic cancer (NCT00666926).
Connective tissue growth factor inhibitors
CTGF is a protein is highly expressed in PDAC that contributes to the dense desmoplasia. It is also thought to protect the cells from hypoxia-mediated apoptosis and drive more aggressive tumor cells selection[91]. In preclinical study with mouse model of PDAC, pamrevlumab, a monoclonal antibody against CTGF in combination with gemcitabine has been shown to prolonged survival[92]. A phase I/II trial involving patients with unresectable PDAC with pamrevlumab and gemcitabine/nab-paclitaxel (NCT02210559) showed higher resection rate in the arm with patients who received pamrevlumab. Currently, FDA granted fast track designation to pamrevlumab for the treatment of unresectable locally advanced PDAC[93]. A phase III trial with this agent is recruiting (NCT03941093).
Expert opinion
Surgery, chemotherapy and radiation therapy are three of the most commonly used modalities to treat pancreatic cancer. However, even with intense chemotherapy regimens, such as FOLFIRINOX, outcomes for these patients remains dismal with conventional therapies. Cancer cells prevent immune cells from recognizing them as a threat, thereby, allowing cancer cells to evade the immune system resulting in continued growth and metastasis. Immunotherapy restores the ability of the immune system to detect and destroy cancer cells by overcoming mechanisms by which tumors evade and suppress the immune response.
Recent negative findings from studies evaluating immune checkpoint inhibitors, whether alone or in combination, have suggested that they may not solely be ideal agents for treatment of pancreatic cancer. Instead, additional agents that prime the immune microenvironment may be needed to see efficacy. This may be attributed to the fact that immune checkpoint inhibitor combination therapy has shown efficacy for immunogenic, “hot” tumors, such as melanoma and lung cancer, while pancreatic cancer on the other hand, is not an immunogenic tumor, commonly called a “cold” tumor.
It is now known that pancreatic cancer does not contain a lot of immune cells and actually also has immunosuppressive signals. Therefore, we may have to adopt a “multi-pronged approach”. Such an approach may include different types of agents, including oncolytic viruses, or vaccines that can prime the immune microenvironment so that the checkpoint inhibitors can then impart their efficacy. Investigators have reported feasibility and clinical activity of T-cell therapy in patients with pancreatic cancer. The question is how do we facilitate bringing in these immune cells?
Pancreatic cancer tumors are well known for the presence of dense desmoplastic stroma that is made up of pancreatic stellate cells, fibroblasts, immune cells, and extracellular matrix proteins. The tumor microenvironment is immunosuppressive and dominated by myeloid-derived suppressor cells and regulatory T cells. Though recent data has shown disappointing results with agents that promised to deplete the stroma, such as agents that inhibit the sonic Hedgehog signaling pathway and pegylated hyaluronic acid, it is important to note that the tumor microenvironment does play a significant role in modulating the immune recognition of PDAC. It is now postulated that extracellular matrix depletion may actually lead to tumor progression further substantiates a more complex role of desmoplasia in tumor biology.
In summary, PDAC responds poorly to immune checkpoint blockade with both anti-CTLA-4 and anti-PD1/anti-PD-L1 immunotherapies. This could be due to the presence of dense desmoplasia and tumor microenvironment made up of extracellular matrix proteins, fibroblasts, endothelial cells, and immune cells. It has been clearly indicated by preclinical studies that the tumor microenvironment has abundant immunosuppressive cell types and few effector T cells.
In addition, cancer associated fibroblasts (CAF) especially FAP positive CAF have been shown to be important in mediating immunosuppression in the PDAC tumor microenvironment. Although treating PDAC with immunotherapy alone has not been very successful, various combination strategies of immunotherapies (including CAR-cells and immune check point inhibitors) with therapeutic vaccine and/or radiation therapies to improve immunogenicity of PDAC are ongoing. The multipronged approach to treating PDAC has led to various trials targeting tumor DNA repair (PARP inhibitors), and tumor metabolism (mitochondrial inhibitor). Novel agents such as FAK inhibitors, CTGFs are also being studied in early phase trials. The results of these studies are highly anticipated and some trials hold promise to advance into later-phase clinical trials. It is hoped that the results from these studies will eventually improve the outcome for pancreatic cancer patients.
Take home points
Patients with pancreatic cancer show poor response to chemotherapies and ICI due to its unique immunosuppressive tumor microenvironment with extensive desmoplasia.
The complex interactions among the components of PDAC TME (fibroblasts, endothelial cells, and extracellular matrix proteins, tumor-associated macrophages, MDSC, and Treg cells) lead to tumorignesis, chemo-resistance, and immune suppression although some recent studies also show evidence of tumor-restraining role of the desmoplasia.
PDAC is characterized by low TMB due to limited expression of neoantigens and leads to poor immune surveillance and poor response to ICIs. However, a small subset (1%-3%) of PDAC patients with dMMR or MSI-H tumors showed high response rate to single agent pembrolizumab.
ChemoXRT may increase immunogenicity in PDAC, which can sensitize tumors to immunotherapy. Multiple phase I/II trials with combination chemoXRT and immunotherapy are ongoing.
GVAX, vaccine developed from PDAC tumor associated antigens, showed T-cell mediated antitumor activity in preclinical and early phase clinical trials. It is now under investigation in combinations with ICIs. There are various ongoing early phase trials using viral vector, peptide based vaccines, DNA based vaccines, yeast-based vaccines and dendritic cell-based vaccines.
Strategies with various targeted therapies are being explored in clinical trials targeting tumor DNA repair (PARP inhibitors), and tumor metabolism (mitochondrial inhibitors).
Novel agents such as FAK inhibitors and CTGF are also being studied in early phase trials.
Declarations
Authors’ contributionsMade substantial contributions to conception and design of the study and performed data analysis and interpretation: Chi J, Patel R, Rehman H, Goyal S, Saif MW
Performed data acquisition, as well as provided administrative, technical, and material support: Chi J, Patel R, Rehman H, Goyal S, Saif MW
Availability of data and materialsNot applicable.
Financial support and sponsorshipNone.
Conflicts of interestAll authors declared that there are no conflicts of interest.
Ethical approval and consent to participateNot applicable.
Consent for publicationNot applicable.
Copyright© The Author(s) 2020.
REFERENCES
1. American Cancer Society. Cancer Facts & Statistics. Available from: http://cancerstatisticscenter.cancer.org/. [Last accessed on 30 Oct 2020].
2. Rahib L, Smith BD, Aizenberg R, Rosenzweig AB, Fleshman JM, Matrisian LM. Projecting cancer incidence and deaths to 2030: the unexpected burden of thyroid, liver, and pancreas cancers in the United States. Cancer Res 2014;74:2913-21.
3. SEER Cancer Statistics Review (CSR) 1975-2017. Available from: https://seer.cancer.gov/csr/1975_2017/index.html. [Last accessed on 30 Oct 2020].
4. Burris HA, Moore MJ, Andersen J, et al. Improvements in survival and clinical benefit with gemcitabine as first-line therapy for patients with advanced pancreas cancer: a randomized trial. J Clin Oncol 1997;15:2403-13.
5. Philip PA, Benedetti J, Corless CL, et al. Phase III study comparing gemcitabine plus cetuximab versus gemcitabine in patients with advanced pancreatic adenocarcinoma: Southwest Oncology Group-directed intergroup trial S0205. J Clin Oncol 2010;28:3605-10.
6. Kindler HL, Niedzwiecki D, Hollis D, et al. Gemcitabine plus bevacizumab compared with gemcitabine plus placebo in patients with advanced pancreatic cancer: phase III trial of the Cancer and Leukemia Group B (CALGB 80303). J Clin Oncol 2010;28:3617-22.
7. Louvet C, Labianca R, Hammel P, et al. Gemcitabine in combination with oxaliplatin compared with gemcitabine alone in locally advanced or metastatic pancreatic cancer: results of a GERCOR and GISCAD phase III trial. J Clin Oncol 2005;23:3509-16.
8. Berlin JD, Catalano P, Thomas JP, Kugler JW, Haller DG, Benson AB. Phase III study of gemcitabine in combination with fluorouracil versus gemcitabine alone in patients with advanced pancreatic carcinoma: Eastern Cooperative Oncology Group Trial E2297. J Clin Oncol 2002;20:3270-5.
9. Herrmann R, Bodoky G, Ruhstaller T, et al. Gemcitabine plus capecitabine compared with gemcitabine alone in advanced pancreatic cancer: a randomized, multicenter, phase III trial of the Swiss Group for Clinical Cancer Research and the Central European Cooperative Oncology Group. J Clin Oncol 2007;25:2212-7.
10. Rocha Lima CM, Green MR, Rotche R, et al. Irinotecan plus gemcitabine results in no survival advantage compared with gemcitabine monotherapy in patients with locally advanced or metastatic pancreatic cancer despite increased tumor response rate. J Clin Oncol 2004;22:3776-83.
11. Conroy T, Desseigne F, Ychou M, et al. FOLFIRINOX versus gemcitabine for metastatic pancreatic cancer. N Engl J Med 2011;364:1817-25.
12. Von Hoff DD, Ervin T, Arena FP, et al. Increased survival in pancreatic cancer with nab-paclitaxel plus gemcitabine. N Engl J Med 2013;369:1691-703.
13. Neoptolemos JP, Palmer DH, Ghaneh P, et al. Comparison of adjuvant gemcitabine and capecitabine with gemcitabine monotherapy in patients with resected pancreatic cancer (ESPAC-4): a multicentre, open-label, randomised, phase 3 trial. Lancet 2017;389:1011-24.
14. Conroy T, Hammel P, Hebbar M, et al. FOLFIRINOX or Gemcitabine as adjuvant therapy for pancreatic cancer. N Engl J Med 2018;379:2395-406.
15. Chand S, O’Hayer K, Blanco FF, Winter JM, Brody JR. The landscape of pancreatic cancer therapeutic resistance mechanisms. Int J Biol Sci 2016;12:273-82.
16. Nixon NA, Blais N, Ernst S, et al. Current landscape of immunotherapy in the treatment of solid tumours, with future opportunities and challenges. Curr Oncol 2018;25:e373-84.
17. Kessenbrock K, Plaks V, Werb Z. Matrix metalloproteinases: regulators of the tumor microenvironment. Cell 2010;141:52-67.
18. Conway JR, Herrmann D, Evans TJ, Morton JP, Timpson P. Combating pancreatic cancer with PI3K pathway inhibitors in the era of personalised medicine. Gut 2019;68:742-58.
19. Apte MV, Wilson JS, Lugea A, Pandol SJ. A starring role for stellate cells in the pancreatic cancer microenvironment. Gastroenterology 2013;144:1210-9.
20. Löhr M, Schmidt C, Ringel J, et al. Transforming growth factor-beta1 induces desmoplasia in an experimental model of human pancreatic carcinoma. Cancer Res 2001;61:550-5.
21. Hessmann E, Patzak MS, Klein L, et al. Fibroblast drug scavenging increases intratumoural gemcitabine accumulation in murine pancreas cancer. Gut 2018;67:497-507.
22. Hartmann N, Giese NA, Giese T, et al. Prevailing role of contact guidance in intrastromal T-cell trapping in human pancreatic cancer. Clin Cancer Res 2014;20:3422-33.
23. Armstrong T, Packham G, Murphy LB, et al. Type I collagen promotes the malignant phenotype of pancreatic ductal adenocarcinoma. Clin Cancer Res 2004;10:7427-37.
24. Provenzano PP, Cuevas C, Chang AE, Goel VK, Von Hoff DD, Hingorani SR. Enzymatic targeting of the stroma ablates physical barriers to treatment of pancreatic ductal adenocarcinoma. Cancer Cell ;21:418-29.
25. Jacobetz MA, Chan DS, Neesse A, et al. Hyaluronan impairs vascular function and drug delivery in a mouse model of pancreatic cancer. Gut 2013;62:112-20.
26. Erkan M, Kurtoglu M, Kleeff J. The role of hypoxia in pancreatic cancer: a potential therapeutic target? Expert Rev Gastroenterol Hepatol 2016;10:301-16.
27. Halozyme Announces HALO-301 Phase 3 Study Fails To Meet Primary Endpoint. Available from: https://www.halozyme.com/investors/news-releases/news-release-details/2019/Halozyme-Announces-HALO-301-Phase-3-Study-Fails-To-Meet-Primary-Endpoint/default.aspx. [Last accessed on 30 Oct 2020].
28. Öhlund D, Handly-Santana A, Biffi G, et al. Distinct populations of inflammatory fibroblasts and myofibroblasts in pancreatic cancer. J Exp Med 2017;214:579-96.
29. Neesse A, Algül H, Tuveson DA, Gress TM. Stromal biology and therapy in pancreatic cancer: a changing paradigm. Gut 2015;64:1476-84.
30. Mace TA, Bloomston M, Lesinski GB. Pancreatic cancer-associated stellate cells: a viable target for reducing immunosuppression in the tumor microenvironment. Oncoimmunology 2013;2:e24891.
31. Nagathihalli NS, Castellanos JA, VanSaun MN, et al. Pancreatic stellate cell secreted IL-6 stimulates STAT3 dependent invasiveness of pancreatic intraepithelial neoplasia and cancer cells. Oncotarget 2016;7:65982-92.
32. Ugel S, De Sanctis F, Mandruzzato S, Bronte V. Tumor-induced myeloid deviation: when myeloid-derived suppressor cells meet tumor-associated macrophages. J Clin Invest 2015;125:3365-76.
34. Brown SD, Warren RL, Gibb EA, et al. Neo-antigens predicted by tumor genome meta-analysis correlate with increased patient survival. Genome Res 2014;24:743-50.
35. Rooney MS, Shukla SA, Wu CJ, Getz G, Hacohen N. Molecular and genetic properties of tumors associated with local immune cytolytic activity. Cell 2015;160:48-61.
36. Yarchoan M, Hopkins A, Jaffee EM. Tumor mutational burden and response rate to PD-1 inhibition. N Engl J Med 2017;377:2500-1.
37. Evans A, Costello E. The role of inflammatory cells in fostering pancreatic cancer cell growth and invasion. Front Physiol 2012;3:270.
38. Ryschich E, Nötzel T, Hinz U, et al. Control of T-cell-mediated immune response by HLA class I in human pancreatic carcinoma. Clin Cancer Res 2005;11:498-504.
39. Tewari N, Zaitoun AM, Arora A, Madhusudan S, Ilyas M, Lobo DN. The presence of tumour-associated lymphocytes confers a good prognosis in pancreatic ductal adenocarcinoma: an immunohistochemical study of tissue microarrays. BMC Cancer 2013;13:436.
40. Rosenberg SA, Yang JC, Restifo NP. Cancer immunotherapy: moving beyond current vaccines. Nat Med 2004;10:909-15.
41. Ito A, Kondo S, Tada K, Kitano S. Clinical development of immune checkpoint inhibitors. Biomed Res Int 2015;2015:605478.
42. Brunet JF, Denizot F, Luciani MF, et al. A new member of the immunoglobulin superfamily--CTLA-4. Nature 1987;328:267-70.
43. Ishida Y, Agata Y, Shibahara K, Honjo T. Induced expression of PD-1, a novel member of the immunoglobulin gene superfamily, upon programmed cell death. EMBO J 1992;11:3887-95.
44. Taube JM, Klein A, Brahmer JR, et al. Association of PD-1, PD-1 ligands, and other features of the tumor immune microenvironment with response to anti-PD-1 therapy. Clin Cancer Res 2014;20:5064-74.
45. Royal RE, Levy C, Turner K, et al. Phase 2 trial of single agent Ipilimumab (anti-CTLA-4) for locally advanced or metastatic pancreatic adenocarcinoma. J Immunother Hagerstown Md 1997 2010;33:828-33.
46. Brahmer JR, Tykodi SS, Chow LQM, et al. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N Engl J Med 2012;366:2455-65.
47. Patnaik A, Kang SP, Rasco D, et al. Phase I study of Pembrolizumab (MK-3475; Anti-PD-1 Monoclonal Antibody) in patients with advanced solid tumors. Clin Cancer Res 2015;21:4286-93.
48. Le DT, Durham JN, Smith KN, et al. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science 2017;357:409-13.
49. Hu ZI, Shia J, Stadler ZK, et al. Evaluating mismatch repair deficiency in pancreatic adenocarcinoma: challenges and recommendations. Clin Cancer Res 2018;24:1326-36.
50. Le DT, Uram JN, Wang H, et al. PD-1 blockade in tumors with mismatch-repair deficiency. N Engl J Med 2015;372:2509-20.
51. Chen M, Yang S, Fan L, et al. Combined antiangiogenic therapy and immunotherapy is effective for pancreatic cancer with mismatch repair proficiency but high tumor mutation burden: a case report. Pancreas 2019;48:1232-6.
52. Mohindra N, Kircher S, Nimeiri H, et al. Results of the phase Ib study of ipilimumab and gemcitabine for advanced pancreas cancer. J Clin Oncol 2015;33:e15281.
53. Kalyan A, Kircher SM, Mohindra NA, et al. Ipilimumab and gemcitabine for advanced pancreas cancer: a phase Ib study. J Clin Oncol 2016;34:e15747.
54. O’Reilly EM, Oh DY, Dhani N, et al. Durvalumab with or without tremelimumab for patients with metastatic pancreatic ductal adenocarcinoma: a phase 2 randomized clinical trial. JAMA Oncol 2019;5:1431-8.
55. Ng J, Dai T. Radiation therapy and the abscopal effect: a concept comes of age. Ann Transl Med 2016;4:118.
56. Park B, Yee C, Lee KM. The effect of radiation on the immune response to cancers. Int J Mol Sci 2014;15:927-43.
57. Tsuchikawa T, Hirano S, Tanaka E, et al. Novel aspects of preoperative chemoradiation therapy improving anti-tumor immunity in pancreatic cancer. Cancer Sci 2013;104:531-5.
58. Homma Y, Taniguchi K, Murakami T, et al. Immunological impact of neoadjuvant chemoradiotherapy in patients with borderline resectable pancreatic ductal adenocarcinoma. Ann Surg Oncol 2014;21:670-6.
59. Twyman-Saint Victor C, Rech AJ, Maity A, et al. Radiation and dual checkpoint blockade activate non-redundant immune mechanisms in cancer. Nature 2015;520:373-7.
60. Seifert L, Werba G, Tiwari S, et al. Radiation therapy induces macrophages to suppress T-cell responses against pancreatic tumors in mice. Gastroenterology 2016;150:1659-72.e5.
61. Katz MHG, Varadhachary GR, Bauer TW, et al. Preliminary safety data from a randomized multicenter phase Ib/II study of neoadjuvant chemoradiation therapy (CRT) alone or in combination with pembrolizumab in patients with resectable or borderline resectable pancreatic cancer. J Clin Oncol 2017;35:4125.
62. Lutz ER, Wu AA, Bigelow E, et al. Immunotherapy converts nonimmunogenic pancreatic tumors into immunogenic foci of immune regulation. Cancer Immunol Res 2014;2:616-31.
63. Wu AA, Jaffee E, Lee V. Current status of immunotherapies for treating pancreatic cancer. Curr Oncol Rep 2019;21:60.
64. Ward JP, Gubin MM, Schreiber RD. The role of neoantigens in naturally occurring and therapeutically induced immune responses to cancer. Adv Immunol 2016;130:25-74.
65. Johnson BA, Yarchoan M, Lee V, Laheru DA, Jaffee EM. Strategies for Increasing Pancreatic Tumor Immunogenicity. Clin Cancer Res 2017;23:1656-69.
66. Morgan RA, Yang JC, Kitano M, Dudley ME, Laurencot CM, Rosenberg SA. Case report of a serious adverse event following the administration of T cells transduced with a chimeric antigen receptor recognizing ERBB2. Mol Ther 2010;18:843-51.
67. Beatty GL, Gladney WL. Immune escape mechanisms as a guide for cancer immunotherapy. Clin Cancer Res 2015;21:687-92.
68. Jaffee EM, Hruban RH, Biedrzycki B, et al. Novel allogeneic granulocyte-macrophage colony-stimulating factor-secreting tumor vaccine for pancreatic cancer: a phase I trial of safety and immune activation. J Clin Oncol 2001;19:145-56.
69. Lutz E, Yeo CJ, Lillemoe KD, et al. A lethally irradiated allogeneic granulocyte-macrophage colony stimulating factor-secreting tumor vaccine for pancreatic adenocarcinoma. A Phase II trial of safety, efficacy, and immune activation. Ann Surg 2011;253:328-35.
70. Le DT, Lutz E, Uram JN, et al. Evaluation of ipilimumab in combination with allogeneic pancreatic tumor cells transfected with a GM-CSF gene in previously treated pancreatic cancer. J Immunother Hagerstown Md 1997 2013;36:382-9.
71. Inc LP. NewLink genetics announces results from phase 3 IMPRESS trial of Algenpantucel-L for patients with resected pancreatic cancer. GlobeNewswire News Room. 2016. Available from: http://www.globenewswire.com/news-release/2016/05/09/837878/0/en/NewLink-Genetics-Announces-Results-from-Phase-3-IMPRESS-Trial-of-Algenpantucel-L-for-Patients-with-Resected-Pancreatic-Cancer.html. [Last accessed on 30 Oct 2020].
72. Lauer P, Chow MYN, Loessner MJ, Portnoy DA, Calendar R. Construction, characterization, and use of two Listeria monocytogenes site-specific phage integration vectors. J Bacteriol 2002;184:4177-86.
73. Brockstedt DG, Giedlin MA, Leong ML, et al. Listeria-based cancer vaccines that segregate immunogenicity from toxicity. Proc Natl Acad Sci U S A 2004;101:13832-7.
74. Le DT, Wang-Gillam A, Picozzi V, et al. Safety and survival with GVAX pancreas prime and Listeria Monocytogenes-expressing mesothelin (CRS-207) boost vaccines for metastatic pancreatic cancer. J Clin Oncol Off 2015;33:1325-33.
75. Le DT, Picozzi VJ, Ko AH, et al. Results from a phase IIb, randomized, multicenter study of GVAX pancreas and CRS-207 compared with chemotherapy in adults with previously treated metastatic pancreatic adenocarcinoma (ECLIPSE study). Clin Cancer Res 2019;25:5493-502.
76. A randomized, placebo-controlled, double blind, multicenter phase II adjuvant trial of the efficacy, immunogenicity, and safety of GI-4000 plus gem versus gem alone in patients with resected pancreas cancer with activating RAS mutations/survival and immunology analysis of the R1 subgroup. Available from: https://ascopubs.org/doi/abs/10.1200/jco.2012.30.15_suppl.e14501. [Last accessed on 30 Oct 2020].
77. Finke LH, Wentworth K, Blumenstein B, Rudolph NS, Levitsky H, Hoos A. Lessons from randomized phase III studies with active cancer immunotherapies - outcomes from the 2006 Meeting of the Cancer Vaccine Consortium (CVC). Vaccine 2007;25:B97-109.
78. Ott PA, Hu Z, Keskin DB, et al. An immunogenic personal neoantigen vaccine for patients with melanoma. Nature 2017;547:217-21.
79. Yamaue H, Tsunoda T, Tani M, et al. Randomized phase II/III clinical trial of elpamotide for patients with advanced pancreatic cancer: PEGASUS-PC study. Cancer Sci 2015;106:883-90.
80. Asahara S, Takeda K, Yamao K, Maguchi H, Yamaue H. Phase I/II clinical trial using HLA-A24-restricted peptide vaccine derived from KIF20A for patients with advanced pancreatic cancer. J Transl Med 2013;11:291.
81. Gjertsen MK, Buanes T, Rosseland AR, et al. Intradermal ras peptide vaccination with granulocyte-macrophage colony-stimulating factor as adjuvant: Clinical and immunological responses in patients with pancreatic adenocarcinoma. Int J Cancer 2001;92:441-50.
82. VXM01, an oral T-cell vaccine targeting the tumor vasculature: results from a randomized, controlled, first-in-man study in pancreatic cancer patients. Available from: https://meetinglibrary.asco.org/record/84680/abstract. [Last accessed on 30 Oct 2020].
83. Kondo H, Hazama S, Kawaoka T, et al. Adoptive immunotherapy for pancreatic cancer using MUC1 peptide-pulsed dendritic cells and activated T lymphocytes. Anticancer Res 2008;28:379-87.
84. Tanyi JL, Bobisse S, Ophir E, et al. Personalized cancer vaccine effectively mobilizes antitumor T cell immunity in ovarian cancer. Sci Transl Med 2018;10.
85. Beatty GL, O’Hara MH, Lacey SF, et al. Activity of mesothelin-specific chimeric antigen receptor T cells against pancreatic carcinoma metastases in a phase 1 trial. Gastroenterology 2018;155:29-32.
87. Golan T, Hammel P, Reni M, et al. Maintenance olaparib for germline BRCA-mutated metastatic pancreatic cancer. N Engl J Med 2019;381:317-27.
88. Kheder ES, Hong DS. Emerging targeted therapy for tumors with NTRK fusion proteins. Clin Cancer Res 2018;24:5807-14.
90. Sulzmaier FJ, Jean C, Schlaepfer DD. FAK in cancer: mechanistic findings and clinical applications. Nat Rev Cancer 2014;14:598-610.
91. Bennewith KL, Huang X, Ham CM, et al. The role of tumor cell-derived connective tissue growth factor (CTGF/CCN2) in pancreatic tumor growth. Cancer Res 2009;69:775-84.
92. Neesse A, Frese KK, Bapiro TE, et al. CTGF antagonism with mAb FG-3019 enhances chemotherapy response without increasing drug delivery in murine ductal pancreas cancer. Proc Natl Acad Sci 2013;110:12325-30.
93. Picozzi VJ, Pishvaian MJ, Mody K, et al. Effect of anti-CTGF human recombinant monoclonal antibody pamrevlumab on resectability and resection rate when combined with gemcitabine/nab-paclitaxel in phase 1/2 clinical study for the treatment of locally advanced pancreatic cancer patients. J Clin Oncol 2018;36:4016.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Chi J, Patel R, Rehman H, Goyal S, Saif MW. Recent advances in immunotherapy for pancreatic cancer. J Cancer Metastasis Treat 2020;6:43. http://dx.doi.org/10.20517/2394-4722.2020.90
AMA Style
Chi J, Patel R, Rehman H, Goyal S, Saif MW. Recent advances in immunotherapy for pancreatic cancer. Journal of Cancer Metastasis and Treatment. 2020; 6: 43. http://dx.doi.org/10.20517/2394-4722.2020.90
Chicago/Turabian Style
Chi, Jeffrey, Rajvi Patel, Hasan Rehman, Shreya Goyal, Muhammad Wasif Saif. 2020. "Recent advances in immunotherapy for pancreatic cancer" Journal of Cancer Metastasis and Treatment. 6: 43. http://dx.doi.org/10.20517/2394-4722.2020.90
ACS Style
Chi, J.; Patel R.; Rehman H.; Goyal S.; Saif MW. Recent advances in immunotherapy for pancreatic cancer. J. Cancer. Metastasis. Treat. 2020, 6, 43. http://dx.doi.org/10.20517/2394-4722.2020.90
About This Article
Special Issue
Copyright

Data & Comments
Data


 Cite This Article 77 clicks
Cite This Article 77 clicks

 Like This Article 26
likes
Like This Article 26
likes






Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.