
Primary malignant tumors of bone surface: a review with emphasis in differential diagnosis
 0
0Abstract
Surface tumors of the bone are broadly defined as a diverse group of osteogenic and chondrogenic benign and malignant neoplasms that arise adjacent to the outer surface of cortical bone. They may be a cause of diagnostic difficulty due to a degree of histological overlap, rarity, and nomenclature. In this review we summarize the different histological types of primary malignant tumors of bone surface, namely, secondary peripheral chondrosarcoma, periosteal chondrosarcoma, parosteal osteosarcoma, dedifferentiated parosteal osteosarcoma, periosteal osteosarcoma, and high-grade surface osteosarcoma. We provide a comprehensive updated review of their pathogenesis and highlight radiological, macroscopic, and histopathological features and recently available ancillary diagnostic tools that may aid in the differential diagnosis.
Keywords
Introduction
Primary malignant tumors of the bone surface are a group of osteogenic and chondrogenic neoplasms with different clinical behaviors, which depend greatly on their histologic type. Their overall incidence is very low, constituting approximately 3.5% of all primary bone tumors[1]. Their rarity, overlapping morphology, and the therapeutic implications of their correct identification make their diagnosis better rendered by expert hands in an integrated, multidisciplinary setting. The aims of this review are to outline the main clinicopathologic and radiologic features of malignant tumors of bone surface, to summarize their pathogenesis and to offer a useful framework for their differential diagnosis with emphasis on potential diagnostic difficulties.
Chondrogenic tumors
Secondary peripheral chondrosarcoma
Definition and clinical features
Secondary peripheral chondrosarcomas are cartilaginous malignant neoplasms arising from the chondroid cap of pre-existent osteochondromas. They are graded into grades 1 to 3, according to the World Health Organization (WHO) histological grading system used for central chondrosarcomas. Grade 1 neoplasms receive the name of secondary peripheral atypical cartilaginous tumor if they arise in the appendicular skeleton, and secondary peripheral chondrosarcoma grade 1 if they arise in axial locations[2].
They constitute approximately 12%-18% of all chondrosarcomas[1,3], and occur after puberty, with a peak incidence between 20 and 40 years. Males are affected more than females[1,4]. Secondary peripheral chondrosarcomas most commonly arise in the bones of the pelvis (40% of cases). Other common locations are the proximal and distal femur (19.5%), scapula (10%), vertebral column (9%), and ribs (5%)[1].
Rapid growth or onset of pain on a pre-existing osteochondroma after puberty, should raise suspicion of malignant transformation, which occurs in 1% of solitary osteochondroma and in up to 5% of patients with hereditary multiple osteochondromas. Depending on the location of the neoplasm, neurologic (e.g., numbness, weakness, radiating pain, paraplegia), urinary, or colonic symptoms may be present.
Imaging
Radiographically, secondary peripheral chondrosarcoma has the typical aspect of an osteochondroma with lytic areas and fuzzy margins. On CT, large, noncalcified tumoral lobules, ring-like or popcorn-like radiopacities, and a thick cartilaginous cap are evident features[1][Figure 1]. T1MRI images show a lobulated, ill-defined, inhomogeneous muscular signal intensity mass; whereas on T2MRI, heterogeneous signal intensity with foci of signal void due to the calcified areas and a thick peripheral layer of white signal due to the cap of the lesion are evident[1].
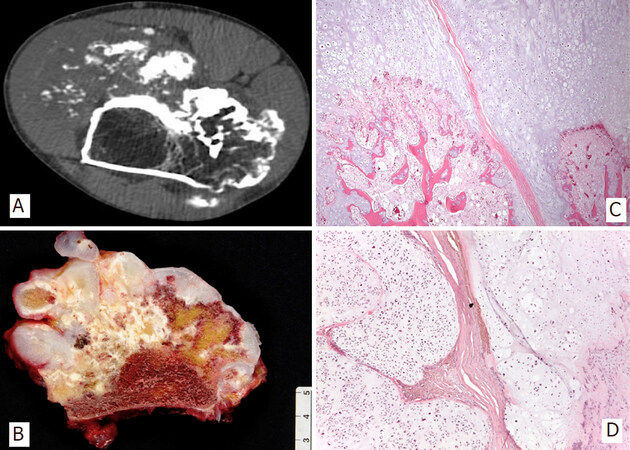
Figure 1. CT scan image of femoral peripheral secondary chondrosarcoma in patient with hereditary multiple osteochondromas with characteristic cortical flaring at the junction of the host bone with the stalk of the neoplasia (A); Macroscopic image of exophytic osteocartilaginous neoplasm containing a glistening cartilaginous cap more than 2 cm thick (B); Cartilaginous cap at the interphase with bone stalk, which is continuous with the host medullary canal (H&E; 25X) (C); Disorganized, hypercellular and atypical hyaline cartilage arranged in lobules separated by fibrous bands (H&E; 50X) (D)
Macroscopic features
Secondary peripheral chondrosarcomas are large, cauliflower-like masses covered by a thin pseudo-capsule and beneath it, a lobulated and chalky cartilaginous cap measuring more than 2 cm in its thickest portion (thickness mean 3.9 cm)[4][Figure 1].
Microscopic features
The overall appearance of the cap is that of disorganized cartilage arranged in lobules divided by fibrous trabecula. Grade 1 tumors (65% of the Rizzoli series of cases) are hyaline and mildly hypercellular, contain disorganized chondrocytes, and have areas of coarse calcifications. Grade 2 and 3 neoplasms (30% and 5% of Rizzoli series of cases, respectively) are more hypercellular, exhibit nuclear atypia and pleomorphism, and have a mostly myxoid cartilaginous matrix. Necrosis can be seen. Mitosis and spindling of peripheral chondrocytes are more easily found in grade 3 neoplasms[1].
Secondary peripheral chondrosarcoma rarely grows in a truly infiltrative pattern. Instead, lobules of cartilage push into the soft tissues, sometimes in the form of satellite nodules. Soft tissue infiltration is a bona fide sign of malignancy.
There is continuity between the cortex and the medulla of the host bone and the bony stalk of the neoplasm [Figure 1].
Pathogenesis
The cartilaginous cap in osteochondromas is mosaic (genetically), and contains a subpopulation of chondrocytes harboring biallelic mutations of tumor-suppressor genes Exostosin-1 or 2 (EXT1 or EXT2), admixed with chondrocytes that are either wild-type or have monoallelic mutations[5-7]. EXT1 and EXT2 genes encode transmembrane glycosyltransferases that act in the polymerization of heparan sulfate chains[5].
The functional outcome of EXT1 and EXT2 mutations is the production of truncated proteins with loss of enzymatic activity[8] and impaired synthesis of heparan sulphate chains on cell surface and matrix proteoglycans. In the growth plates, heparan sulfate plays an important role in establishing and maintaining tissue polarity[6,9], in the diffusion of ligands, and in the binding of signaling molecules to receptors of intracellular signal transduction pathways[6,9,10] including Ihh, BMP, Wnt, and FGF[10,11]. The impaired elongation of heparan sulfate glycosaminoglycans disrupt cell polarity and the activity of these signaling pathways, which may result in ectopic growth of mutated cells and osteochondromagenesis.
Progression to secondary peripheral chondrosarcoma, a neoplasia with chromosomal instability and complex karyotypes[12], is hypothesized to happen upon acquisition of additional genetic changes by the chondrocytes within the cartilaginous cap that retain one or both functional copies of EXT genes[9,13]. The proliferative advantage and clonal outgrowth are likely driven by loss of cell cycle regulators CDKN2A and TP53[9,13,14].
Diagnostic ancillary techniques
Immunohistochemical or molecular testing are not relevant for diagnosis.
Main differential diagnosis
Osteochondroma develops in skeletally immature patients during stature growth. Peak incidence is in the first and second decades of life[1]. As opposed to secondary peripheral chondrosarcomas, the great majority of cases of osteochondromas occur in the growth plate area in the juxta-epiphyseal region of long tubular bones and less frequently involve flat bones. The most objective parameters to raise suspicion of a secondary peripheral atypical cartilaginous tumor/secondary peripheral chondrosarcoma grade 1 arising in an osteochondroma is the presence in the latter, of a cartilaginous cap greater than 2 cm (measured perpendicularly to the osteocartilaginous interphase). Nevertheless, this feature is in and of itself not enough to make a diagnosis of secondary peripheral chondrosarcoma in the absence of other signs of malignancy, such soft tissue invasion, as a very small percentage of osteochondromas may exhibits a cartilaginous cap greater than 2 cm[2,15]. Microscopic features can greatly overlap, thus a multidisciplinary approach to diagnosis is mandatory.
Periosteal chondrosarcoma lies directly over the cortex of the host bone and does not present corticomedullary continuity of the tumor with the bone of origin. A peripheral layer of metaplastic bone (i.e., pseudo-cortex) is present in nearly 50% of periosteal chondrosarcomas[16].
Periosteal osteosarcoma contains osteoid-producing primitive mesenchymal cells between the cartilage lobules, which are absent in secondary peripheral chondrosarcoma. Central chondrosarcoma is separated from secondary peripheral chondrosarcoma by its location in the medullary cavity. The former is molecularly characterized by IDH mutations, which are absent in secondary peripheral chondrosarcoma[17].
Treatment and prognosis
The treatment is surgery with wide margins, and will depend on the severity of symptoms, the size of the lesion, and its location[18,19]. Amputation is necessary when the neoplasm is otherwise inoperable. Radiotherapy and chemotherapy are not effective. Local recurrences can occur and are related to incomplete excision. Prognosis after treatment is excellent and metastasis is very rare.
Periosteal chondrosarcoma
Definition and clinical features
Periosteal chondrosarcoma is a low grade malignant cartilaginous neoplasm of the bone surface that, by definition, invades the underlying cortex and/or measures more than 5 cm[2]. It is approximately three times less frequent than periosteal chondroma[1,20] and represents 2.5% of all chondrosarcomas.
It occurs in adults, predominantly in males, with the peak incidence in the fourth decade[1]. It typically involves the metaphysis of long tubular bones, the distal femur being the most frequent location, followed by the proximal humerus.
Imaging
Radiographic findings of periosteal chondrosarcoma include a round, lobulated mass on the surface of bone with chondroid matrix mineralization in the form of “popcorn” opacities and sometimes metaplastic ossification. Occasionally, the neoplasm can exhibit the same radiopacity as soft tissues. The underlying cortex often appears thickened and sclerotic, slightly saucer-shaped and with a sharply defined border [Figure 2], although it may be thin and radiographically indistinct beneath tumors arising in infrequent sites, such as spongy bones[21].
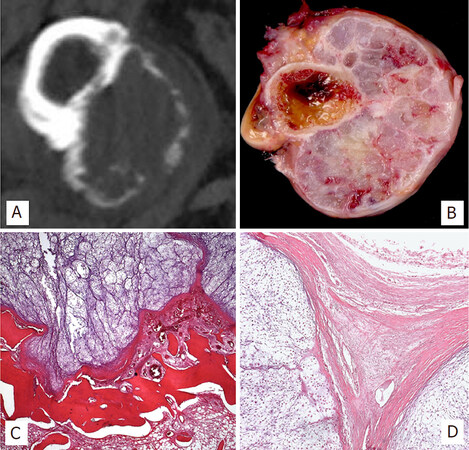
Figure 2. CT scan image of periosteal chondrosarcoma arising in the femur with cortex erosion in a well-limited way (A); Macroscopic image of periosteal chondrosarcoma showing a large, lobulated mass lying in direct apposition with the underlying outer cortex (B); Microscopic interphase of the neoplasm with the host bone cortex, showing erosion and focal thinning (H&E; 25X) (C); Outer surface of the tumor showing bosselated margins and hypercellular, myxoid neoplastic lobules (H&E; 50X) (D)
Macroscopic features
Periosteal chondrosarcoma presents as a large mass, usually measuring over 5 cm, located directly on the bone surface and associated with erosion or invasion of the subjacent cortical bone [Figure 2]. Grossly, it is lobulated, and its cut surface is greyish and glistening, frequently with gritty areas of calcification.
Microscopic features
Periosteal chondrosarcoma is characterized by lobular growth of most often moderately cellular cartilage exhibiting myxoid matrix, frequent calcifications, and endochondral ossification. The neoplastic chondrocytes show variable degrees of cytological atypia, although marked nuclear pleomorphism is not a feature of periosteal chondrosarcoma. Osteoid or bone directly formed by the neoplastic chondrocytes is absent. Mitosis figures are very rare.
Invasion of the underlying cortex is present, even in tumors less than 5 cm. Also, involvement of medullary canal is seen in a proportion of cases, as is invasion of soft tissues. In a subset of cases (50%, in our experience) the periphery of the tumor is encased by a complete or incomplete layer of mature cortical bone[16,22].
Pathogenesis
A subset of periosteal chondrosarcomas harbors heterozygous, somatic IDH1 R132C mutations[17,22,23]. IDH1 is a metabolic enzyme of the tricarboxylic acid cycle that catalyzes the conversion of isocitrate to α-ketoglutarate.
Mutated IDH1 has reduced catalytic activity for the production of α-ketoglutarate and increased catalytic activity for the production of 2-hydroxyglutarate, due to an acquired neomorphic activity[24].
The elevated level of the oncometabolite 2-hydroxyglutarate epigenetically inhibits osteogenic differentiation of mesenchymal stem cells through histone hypermethylation in the promoter regions of alkaline phosphatase. It also promotes chondrogenic differentiation through histone modifications of promoter regions of master transcription factors for chondrogenesis, SOX9 and COL2A1[25,26].
Loss of protein expression of cell cycle regulator CDKN2A/P16/INK4A is found in 50% of cases of periosteal chondrosarcomas suggesting that the pRB pathway is involved in tumoral progression[22].
Diagnostic ancillary techniques
The commercially available IDH1 R132H antibody is a highly specific and sensitive marker for the detection of the R132H mutant allele, but does not detect other IDH mutants, including the R132C found in periosteal chondrosarcomas[17].
IDH mutational analysis can be useful in differentiating periosteal chondrosarcoma from the more aggressive periosteal osteosarcoma, but it does not discriminate between periosteal chondroma and periosteal chondrosarcoma.
Main differential diagnosis
The main differential diagnosis is periosteal chondroma, which occurs predominantly in younger adults and children (peak incidence in the second decade) in the proximal humerus and the short tubular bones of the hands[27]. Radiographically, it shows solid periosteal bone buttressing, which is absent in periosteal chondrosarcoma. Although some overlap in size might exist, periosteal chondromas are, on average, smaller than periosteal chondrosarcomas, the former only rarely exceeding 3 cm. Periosteal chondroma can erode and scallop the cortical bone, thus tumor nodules within the sclerotic cortex can be seen sometimes. They do not represent true invasion, but an artifact due to tangential sectioning. Real invasion of cortical bone and soft tissues is absent. Histologically, periosteal chondromas are usually hypocellular and devoid of cytological atypia.
Periosteal osteosarcoma is diaphyseal and has a pathognomonic radiological appearance. The cartilaginous areas in periosteal osteosarcoma show marked pleomorphism compared to the cartilage of periosteal chondrosarcoma. Also, the intervening bands of bone-producing primitive mesenchymal cells present in periosteal osteosarcoma are, by definition, absent in periosteal chondrosarcoma.
Secondary peripheral chondrosarcoma exhibits the characteristic corticomedullary continuity of the stalk of the tumor and the bone of origin, which is absent in periosteal chondrosarcoma [Figure 2].
Treatment and prognosis
Conservative surgery with wide margins is usually effective. The prognosis is very good, with local recurrence of 13%-28% and metastatic rates of 512.2%[20]. Metastasis primarily involve the lungs and, rarely, the lymph nodes[28]. In the Rizzoli case series of periosteal chondrosarcomas, no patient died of tumor[1,21].
Osteogenic tumors
Parosteal osteosarcoma
Definition and clinical features
Parosteal osteosarcoma is a low-grade osteosarcoma with low-metastatic potential that arises on the cortical surface of bone. It constitutes 4.3% of all osteosarcomas[1] and, although infrequent, is the most common osteosarcoma of the bone surface. Its peak of incidence is in young adults between 20 and 40 years old (mean age of 31 years) with a slight female predominance[29]. It almost exclusively affects the metaphysis of long bones of the extremities, with a marked predilection for the distal metaphysis of the femur (60% cases). Only exceptionally it occurs in flat bones, small bones of hands and feet and vertebrae. Parosteal osteosarcoma most commonly debuts as a long standing, painless and indurated mass.
Tumoral progression into a high-grade dedifferentiated parosteal osteosarcoma is seen in a percentage of cases and can occur either at presentation (primary/synchronous type) or at the time of recurrence (metachronous type). In primary dedifferentiated parosteal osteosarcomas the clinical history is sometimes more abrupt.
Imaging
Due to its indolent growth, parosteal osteosarcoma is usually discovered when it has achieved large dimensions. It is a circumferential or mushroom shaped mass of osseous radiodensity fused to the cortex [Figure 3]. Radiodensity is maximal near the implant base, while the outer margin tends to be blurred. Detailed radiographies show a mesh of trabecula (“steel-wool” pattern), ranging from ground glass to ivory density[1]. Areas of radiolucency on plain films or CT scans and hyperintense on fluid-sensitive MRI can be present in approximately 60% of dedifferentiated parostal osteosarcomas[30].
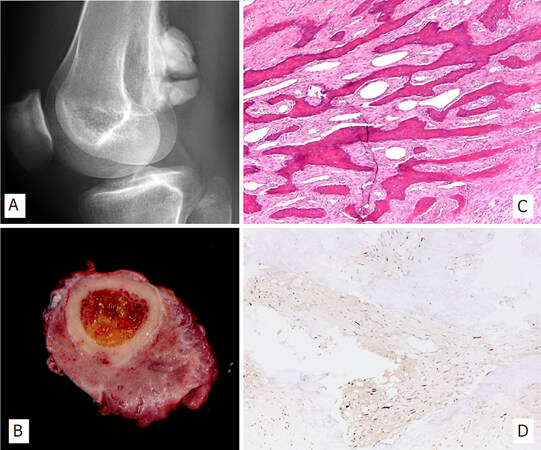
Figure 3. X-rays of distal femur exhibiting an exophytic and sclerotic mass projecting from the cortex in its popliteal fossa (A); Gross specimen of parosteal osteosarcoma, encircling the femur with a gritty cut surface (B); Characteristic microscopic arrangement of parallel bone trabecula and the intervening low-grade fibroblastic component (H&E; 50X) (C); MDM2 nuclear positivity (IHC; 50X) (D)
The combination of MRI and CT allows for high accuracy in the determination of the extent of intramedullary infiltration of the tumor, thus assisting in the planning of conservative surgeries that avoid surgical morbidity[31].
Macroscopic features
Grossly, parosteal osteosarcoma presents as a firm, exophytic and lobulated mass with a broad base attachment to the cortex of the host bone. It can grow either longitudinally along the long axis of the bone or encircle it [Figure 3]. Elevation of periosteum and reactive periosteal bone formation are typically absent. The cut surface is whitish, hard, or gritty and may contain areas of cartilage. When dedifferentiation is present, its occurrence is characterized by a tan and fleshy component not infrequently accompanied by areas of hemorrhage and fluid cavities that may correlate well with the lucent areas in imaging studies[2].
Microscopic features
It is composed of long fascicles of deceptively bland spindle cells embedded in a collagenous stroma and intervening, well-formed bone trabecula. The fibrous fascicles are hypocellular and the spindle cells exhibit minimal nuclear atypia. Mitotic figures are inconspicuous. The bone trabecula are typically arranged in parallel streamers but can also be more curvilinear and arranged in an anastomosing fashion [Figure 3]. Osteoblastic riming is inconsistently present.
A peripheral cartilaginous component is seen in 25-50% of cases[1,2] in the form of chondroid nodules. The presence of areas of higher histological grade, frank anaplasia and brisk mitotic activity within the tumor indicates dedifferentiation. The high-grade dedifferentiated component most commonly exhibits features of conventional osteosarcoma or undifferentiated pleomorphic sarcoma[30], although rhabdomyosarcomatous dedifferentiation has also been reported[32]. Infiltration of the soft tissue and medullary canal can be seen.
Pathogenesis
Supernumerary ring chromosomes containing high-level amplification of chromosomal regions 12q13-15, including MDM2 and CDK4 loci, is a consistent finding in parosteal osteosarcoma[33,34]. As a result, two major pathways of growth regulation may be inhibited: MDM2 may downregulate the p53- mediated growth control and CDK4 may affect the function of retinoblastoma tumor suppressor protein (pRb)[35], leading to deregulation of the cell cycle.
Diagnostic ancillary techniques
MDM2 gene amplification by in situ hybridization techniques, or surrogate immunohistochemical expression of MDM2 are useful and routinely used to confirm diagnosis[36].
Main differential diagnosis
Juxtacortical myositis ossificans is composed by a spindle-cell proliferation admixed with woven bone that exhibits a zonation pattern of progressive maturation toward the periphery of the lesion. The spindle cell component is similar to that seen in nodular fasciitis, characterized by non-cohesive, plump, spindle-shaped cells reminiscent of tissue culture; instead of the long fascicles present in parosteal osteosarcoma. MDM2 amplification is absent in myositis ossificans, while rearrangement of USP6 can be detected[37].
Desmoplastic fibroma and fibrous dysplasia are also characterized by hypocellular fascicles of bland spindle cells in a collagenous background but lack the bone trabecula seen in parosteal osteosarcoma. In ambiguous situations, they can be confidently separated from parosteal osteosarcoma by the absence of MDM2 amplification or MDM2 nuclear immunostaining.
Osteoma affects almost exclusively bones formed by membranous ossification, thus locations outside the craniofacial bones and jaws, where this entity might constitute a diagnostic differential, are extremely rare. Osteoma contains cortical-type bone and lacks the fascicles of spindle cells seen in parosteal osteosarcoma.
Up to a fourth of bizarre parosteal osteochondromatous proliferation (BPOP) may arise in long tubular bones[38-40], a location where it must be distinguished from parosteal osteosarcoma. The latter is exceedingly rare in small tubular bones of hands and feet, which are the sites of predilection of BPOP.
BPOP are smaller lesions measuring from a few millimeters up to 3 cm. BPOP has a zonal architecture, absent in parosteal osteosarcoma, comprised by an admixture of cancellous bone, aggregates of cartilage and fibrous tissue. Characteristically in BPOP, an irregular zone of endochondral ossification and calcification, characterized by the presence of so-called “blue bone” is present in the interface between cartilage and bone.
Dedifferentiated parosteal osteosarcoma can be distinguished from periosteal and high-grade surface osteosarcoma by the presence of a low-grade spindle-cell component and MDM2 amplification in dedifferentiated parosteal osteosarcoma.
Treatment and prognosis
Parosteal osteosarcoma is a locally aggressive tumor with limited potential for distant spread. Hence, surgical resection with adequate margins is the treatment of choice. Chemotherapy is reserved for dedifferentiated paraosteal osteosarcomas. The incidence of local recurrences has been demonstrated to be strictly related to the adequacy of surgical margins[41].
The prognosis of parosteal osteosarcoma is excellent. Long-term survival rate for this entity is at least 90%. Nevertheless, it possesses a risk of transformation into an intermediate or high-grade sarcoma (dedifferentiation), either at first diagnosis or after recurrences, estimated to occur in between 14%[42] and 24% of cases[29,30]. Dedifferentiation confers a significantly worse prognosis to parosteal osteosarcomas, diminishing overall survival and equating it to that of conventional high-grade osteosarcomas[29,43].
Periosteal osteosarcoma
Definition and clinical features
Periosteal osteosarcoma is an intermediate grade, mainly chondroblastic, bone-producing sarcoma that arises on the surface of bone beneath the periosteal layer[2]. It is rare, accounting for less than 2% of all osteosarcomas[1,27,44,45]. It mostly affects children, adolescents and young adults with a peak incidence in the second decade of life[1]. The most common sites of involvement are the diaphysis of the tibia and the femur[1,27,44,46], but other long bones of extremities and much less frequently, flat bones, can be involved[1,44].
Periosteal osteosarcomas present as swellings of relatively short duration, with or without accompanying pain.
Imaging
Periosteal osteosarcoma is a fusiform, mainly lucent mass with well-defined borders [Figure 4]. It usually contains perpendicular linear striae that radiate from the underlying host bone. Sometimes a Codman triangle can be seen[1,3]. CT and MRI are helpful in ruling out neoplastic infiltration to the bone marrow, which is rarely present.
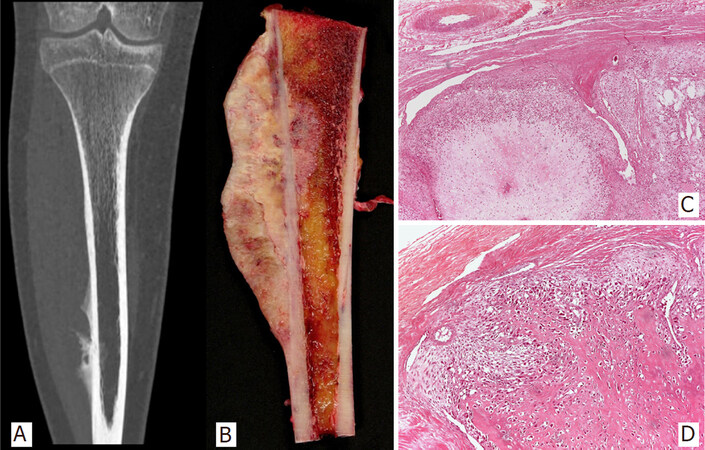
Figure 4. CT scan image of periosteal osteosarcoma originating in tibial diaphysis, forming bone, with thick periosteal bone formation. The neoplasm is elongated, mainly lucent (A); Subperiosteal, fusiform mass with broad base of attachment to outer cortex of host bone. The external surface is covered by fibrous tissue. The cut surface is mostly cartilaginous, but also contains perpendicular bands of ossification (B); Chondroblastic lobules separated by bands of undifferentiated spindle cells (H&E; 25X) (C); Osteoid-producing undifferentiated neoplastic cells (H&E, 50X) (D)
Macroscopic features
Periosteal osteosarcoma is a subperiosteal, fusiform mass with a broad base attached to the outer cortex. Periosteal elevation and reactive bone formation are present. The external surface is covered by a layer of fibrous tissue derived from periosteum. Scalloping and thickening of outer cortex is characteristic. Intramedullary extension may be present. The cut surface consists of lobules of glistening cartilaginous areas, separated by bands of ossification perpendicular to the cortical surface of the host bone [Figure 4].
Microscopic features
Ill-defined lobules of atypical cartilage dominate the histological picture. The cartilaginous areas are separated by sarcomatous bands of undifferentiated spindle cells with primitive appearance. Within these bands, osteoid and immature bone produced by neoplastic cells are seen [Figure 4]. Perpendicular streaks of reactive bone with osteoblastic rimming traverse the neoplasm.
Pathogenesis
No consistent genetic abnormality has been reported for periosteal osteosarcoma[2].
Diagnostic ancillary techniques
Compared to low-grade central osteosarcoma and parosteal osteosarcomas, MDM2 and CDK4 are very rarely expressed in periosteal osteosarcoma and therefore they do not seem to be molecules central to the control of cancer development, growth, and progression in this rare subtype of osteosarcoma[47].
Main differential diagnosis
BPOP arising in long bones may be mistaken for a periosteal osteosarcoma, due to its rapid growth, high rate of local recurrences and atypical histology[38,39]. BPOP does not exhibit the brisk mitotic activity nor the level of cytological atypia observed in periosteal osteosarcomas. In addition, blue bone is a feature of BPOP absent in periosteal osteosarcoma.
Parosteal osteosarcoma arises in metaphysis of long bones in slightly older population. Their radiological growth patterns are dissimilar. Microscopically their recognition is usually straightforward due to the fact that parosteal osteosarcoma is predominantly fibroblastic, is typically low-grade, and contains abundant tumor bone; whereas periosteal osteosarcoma is predominantly chondroblastic and of intermediate to high grade.
Differentiating a periosteal osteosarcoma from a high-grade surface osteosarcoma is of prognostic and therapeutic importance, as high-grade surface osteosarcoma behaves in a much more aggressive manner. High-grade surface osteosarcomas most frequently arise in metaphysis of long bones of extremities. Histologically, periosteal osteosarcoma shows less anaplasia and more prominent chondroid differentiation than high-grade surface osteosarcoma. High-grade surface osteosarcoma is a high-grade tumor (grades 3 or 4), whereas periosteal osteosarcoma is an intermediate grade tumor (grades 2 or 3)[2].
Periosteal chondrosarcoma arises predominantly in metaphyseal locations and in older patients. The neoplastic cartilage lacks marked nuclear pleomorphism, mitotic figures are rare, and osteoid or bone formed by the neoplastic chondrocytes are absent.
Treatment and prognosis
Treatment consists of en bloc resection with wide margins without chemotherapy. Periosteal osteosarcoma has less aggressive behavior compared to high-grade osteosarcoma, including a lower propensity for systemic recurrence which is reported to be around 15% in periosteal osteosarcoma[21,48]. The 10-year overall survival rate is 77.1%-84%[44-46].
High-grade surface osteosarcoma
Definition and clinical features
High-grade surface osteosarcoma is a high-grade, malignant, bone-forming neoplasm arising in the bony surface with minimal or no involvement of the underlying cortical bone. It is the least common type of surface osteosarcoma, representing approximately 1% of all osteosarcoma types. It mostly affects adolescents and young adults with a peak incidence in the second decade of life and a striking male predominance[1]. The skeletal distribution is similar to that of conventional osteosarcoma, most frequently involving the metaphysis of the distal femur, proximal tibia, and proximal humerus.
Imaging
X-rays show mixed non-mineralized and mineralized matrix with fine cloud-like areas of opacity[1]. Periosteal reaction is uncommon, and the margins are indistinct [Figure 5]. Up to one third of the tumors show minimal medullary involvement in CT and MRI images[3]. MRI facilitates the identification of cartilage that is scattered throughout the mass.
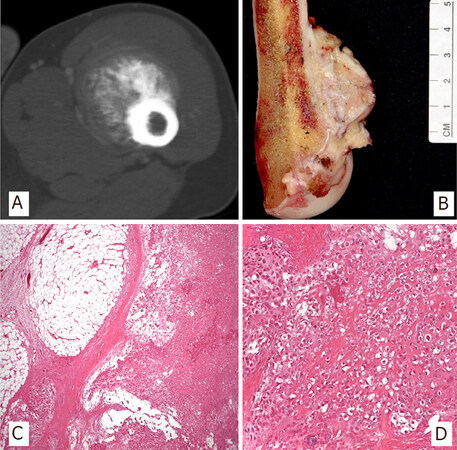
Figure 5. CT scan shows a tumor that develops in the soft tissue but starts from the cortex (A); High-grade surface osteosarcoma of distal femur with cortical erosion (B); Infiltrating neoplasm in the surrounding soft tissues (H&E;25X) (C); Highly atypical features and osteoid production (H&E; 50X) (D)
Macroscopic features
Lobulated, relatively well-circumscribed, broad-base mass on the periosteal surface of the bone. It frequently extends to the periosteum and can erode the cortex, but the vast majority of the tumor remains outside the bone [Figure 5]. Any significant penetration beyond the endosteum exclude the diagnosis of high- grade surface osteosarcoma. The cut surface may be dominated by cartilaginous tissue or composed of hard, tan-white areas admixed with others with fish flesh consistency.
Microscopic features
The microscopic features are identical to high-grade conventional osteosarcoma. They exhibit anaplasia, necrosis, osteoid, and immature bone and/or cartilage formation [Figure 5]. Mitotic activity is brisk, and atypical mitotic figures are easily found. Microscopic evidence of medullary extension is either absent or minimal.
Pathogenesis
The pathogenesis remains unknown.
Diagnostic ancillary techniques
The diagnostic ancillary techniques are not clinically relevant for high-grade surface osteosarcoma.
Main differential diagnosis
The distinction between a high-grade surface osteosarcoma and dedifferentiated parosteal osteosarcoma or extramedullary extension from a conventional central osteosarcoma does not carry prognostic or therapeutic implications. Areas of low-grade fibroblastic osteosarcoma are found in dedifferentiated parosteal osteosarcoma and absent in high-grade surface osteosarcoma. A considerable intramedullary component argues in favor of a central conventional osteosarcoma.
Due to therapeutic and prognostic implications, the most important differential diagnosis is with periosteal osteosarcoma as indicated in the paragraph on periosteal osteosarcoma.
Treatment and prognosis
Treatment and prognosis do not differ from those of central conventional osteosarcoma. Treatment is preoperative chemotherapy followed by surgical resection. The overall 5-year survival rate ranges between 46% to 62%[49,50]. A poor response to neoadjuvant chemotherapy is associated with a poor outcome[3].
Conclusion
There are a variety of bone and, to a lesser extent, soft tissue entities that should be considered in the differential diagnosis of primary tumors of the bone surface. These comprise a wide clinical spectrum of neoplasms that can impose diagnostic challenges outside the setting of large referral centers, due to their extremely low incidence and unawareness of their clinical, radiological, and often subtle microscopic differences. A multidisciplinary and expert approach to diagnosis is mandatory.
Declarations
AcknowledgmentsWe would like to thank Fundación Martha Stella C. de Vallarino for financial support through a medical educational scholarship to one of the authors (MP).
Authors’ contributionsPreparing the manuscript draft and capturing the microscopic images: Pacheco M
Reviewing the manuscript: Righi A
Writing, producing the illustrations, reading and approving the final manuscrip: Pacheco M, Righi A
Availability of data and materialsNot applicable.
Financial support and sponsorshipNot applicable.
Conflicts of interestAll authors declared that there are no conflicts of interest.
Ethical approval and consent to participateNot applicable.
Consent for publicationNot applicable.
Copyright©The Author(s) 2020.
REFERENCES
1. Picci P, Manfrini M, Donati DM, et al. Diagnosis of musculoskeletal tumors and tumor-like conditions 2nd edition. Switzerland: Springer Nature; 2020.
2. Bovée JVMG, Bloem JL, Flanagan AM, et al. WHO Classification of Tumours of Soft Tissue and Bone. 5th edition. Lyon: International Agency for research on cancer; 2020.
3. Unni KK, Inwards CY, Bridge JA, Kindblom LG, Wold LE. AFIP atlas of tumor pathology. Tumors of the bones and joints. 4th series. Washington, DC: The American Registry of Pathology; 2005.
4. Ahmed AR, Tan TS, Unni KK, Collins MS, Wenger DE, Sim FH. Secondary chondrosarcoma in osteochondroma: report of 107 patients. Clin Orthop Relat Res 2003:193-206.
5. Jones KB, Piombo V, Searby C, et al. A mouse model of osteochondromagenesis from clonal inactivation of Ext1 in chondrocytes. Proc Natl Acad Sci U S A 2010;107:2054-9.
6. de Andrea CE, Wiweger M, Prins F, Bovée JV, Romeo S, Hogendoorn PC. Primary cilia organization reflects polarity in the growth plate and implies loss of polarity and mosaicism in osteochondroma. Lab Invest 2010;90:1091-101.
7. Matsumoto K, Irie F, Mackem S, Yamaguchi Y. A mouse model of chondrocyte-specific somatic mutation reveals a role for Ext1 loss of heterozygosity in multiple hereditary exostoses. Proc Natl Acad Sci U S A 2010;107:10932-7.
8. Jennes I, Pedrini E, Zuntini M, et al. Multiple osteochondromas: mutation update and description of the multiple osteochondromas mutation database (MOdb). Hum Mutat 2009;30:1620-7.
9. de Andrea CE, Reijnders CM, Kroon HM, et al. Secondary peripheral chondrosarcoma evolving from osteochondroma as a result of outgrowth of cells with functional EXT. Oncogene 2012;31:1095-104.
10. Häcker U, Nybakken K, Perrimon N. Heparan sulphate proteoglycans: the sweet side of development. Nat Rev Mol Cell Biol 2005;6:530-41.
11. Pacifici M. The pathogenic roles of heparan sulfate deficiency in hereditary multiple exostoses. Matrix Biol 2018;71-72:28-39.
12. Hallor KH, Staaf J, Bovée JV, et al. Genomic profiling of chondrosarcoma: chromosomal patterns in central and peripheral tumors. Clin Cancer Res 2009;15:2685-94.
13. de Andrea CE, Hogendoorn PC. Epiphyseal growth plate and secondary peripheral chondrosarcoma: the neighbours matter. J Pathol 2012;226:219-28.
14. de Andrea CE, Zhu JF, Jin H, Bovée JV, Jones KB. Cell cycle deregulation and mosaic loss of Ext1 drive peripheral chondrosarcomagenesis in the mouse and reveal an intrinsic cilia deficiency. J Pathol 2015;236:210-8.
15. Nielsen GP, Rosenberg AE, Deshpande V, Hornicek FJ, Kattapuram SV, Rosenthal DI. Diagnostic Pathology: Bone. 2nd ed. Philadelphia, PA: Elsevier; 2017.
16. Vanel D, De Paolis M, Monti C, Mercuri M, Picci P. Radiological features of 24 periosteal chondrosarcomas. Skeletal Radiol 2001;30:208-12.
17. Amary MF, Bacsi K, Maggiani F, et al. IDH1 and IDH2 mutations are frequent events in central chondrosarcoma and central and periosteal chondromas but not in other mesenchymal tumours. J Pathol 2011;224:334-43.
18. Gelderblom H, Hogendoorn PC, Dijkstra SD, et al. The clinical approach towards chondrosarcoma. Oncologist 2008;13:320-29.
19. van Maldegem AM, Gelderblom H, Palmerini E, et al. Outcome of advanced, unresectable conventional central chondrosarcoma. Cancer 2014;120:3159-64.
20. Papagelopoulos PJ, Galanis EC, Mavrogenis AF, et al. Survivorship analysis in patients with periosteal chondrosarcoma. Clin OrthopRelat Res 2006;448:199-207.
21. Bertoni F, Boriani S, Laus M, Campanacci M. Periosteal chondrosarcoma and periosteal osteosarcoma. Two distinct entities. J Bone Joint Surg Br 1982;64:370-6.
22. Cleven AH, Zwartkruis E, Hogendoorn PC, et al. Periosteal chondrosarcoma: a histopathological and molecular analysis of a rare chondrosarcoma subtype. Histopathology 2015;67:483-90.
23. Pansuriya TC, van Eijk R, d’Adamo P, et al. Somatic mosaic IDH1 and IDH2 mutations are associated with enchondroma and spindle cell hemangioma in Ollier disease and Maffucci syndrome. Nat Genet 2011;43:1256-61.
24. Dang L, White DW, Gross S, et al. Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature 2009;462:739-44.
25. Suijker J, Baelde HJ, Roelofs H, Cleton-Jansen AM, Bovée JV. The oncometabolite D-2-hydroxyglutarate induced by mutant IDH1 or -2 blocks osteoblast differentiation in vitro and in vivo. Oncotarget 2015;6:14832-42.
26. Jin Y, Elalaf H, Watanabe M, et al. Mutant IDH1 dysregulates the differentiation of mesenchymal stem cells in association with gene-specific histone modifications to cartilage- and bone-related genes. PLoS One 2015;10:e0131998.
27. Czerniak B. Dorfman and Czerniak’s Bone Tumors. 2nd edition. Philadelphia: Saunders; 2016.
28. Goedhart LM, Ploegmakers JJ, Kroon HM, Zwartkruis EC, Jutte PC. The presentation, treatment and outcome of periosteal chondrosarcoma in the Netherlands. Bone Joint J 2014;96:823-8.
29. Ruengwanichayakun P, Gambarotti M, Frisoni T, et al. Parosteal osteosarcoma: a monocentric retrospective analysis of 195 patients. Hum Pathol 2019;91:11-8.
30. Bertoni F, Bacchini P, Staals EL, Davidovitz P. Dedifferentiated parosteal osteosarcoma: the experience of the Rizzoli Institute. Cancer 2005;10:2373-82.
31. Aparisi Gómez MP, Righi A, Errani C, et al. Inflammation and infiltration: can the radiologist draw a line? MRI versus CT to accurately assess medullary involvement in parosteal osteosarcoma. Int J Biol Markers 2020;35:31-6.
32. Reith JD, Donahue FI, Hornicek FJ. Dedifferentiated parosteal osteosarcoma with rhabdomyosarcomatous differentiation. Skeletal Radiol 1999;28:527-31.
33. Szymanska J, Mandahl N, Mertens F, et al. Ring chromosomes in parosteal osteosarcoma contain sequences from 12q13-15: a combined cytogenetic and comparative genomic hybridization study. Genes Chromosomes Cancer 1996;16:31-4.
34. Dujardin F, Binh MB, Bouvier C, et al. MDM2 and CDK4 immunohistochemistry is a valuable tool in the differential diagnosis of low-grade osteosarcomas and other primary fibro-osseous lesions of the bone. Mod Pathol 2011;24:624-37.
35. Duhamel LA, Ye H, Halai D, et al. Frequency of Mouse Double Minute 2 (MDM2) and Mouse Double Minute 4 (MDM4) amplification in parosteal and conventional osteosarcoma subtypes. Histopathology 2012;60:357-9.
36. Baumhoer D, Amary F, Flanagan AM. An update of molecular pathology of bone tumors. Lessons learned from investigating samples by next generation sequencing. Genes Chromosomes Cancer 2019;58:88-99.
37. Bekers EM, Eijkelenboom A, Grünberg K, et al. Myositis ossificans - another condition with USP6 rearrangement, providing evidence of a relationship with nodular fasciitis and aneurysmal bone cyst. Ann Diagn Pathol 2018;34:56-9.
38. Meneses MF, Unni KK, Swee RG. Bizarre parostealosteochondromatous proliferation of bone (Nora’s lesion). Am J Surg Pathol 1993;17:691-7.
39. Berber O, Dawson-Bowling S, Jalgaonkar A, et al. Bizarre parostealosteochondromatous proliferation of bone: clinical management of a series of 22 cases. J Bone Joint Surg Br 2011;93:1118-21.
40. Abramovici L, Steiner GC. Bizarre parostealosteochondromatous proliferation (Nora’s lesion): a retrospective study of 12 cases, 2 arising in long bones. Hum Pathol 2002;3:1205-10.
41. Lewis VO, Gebhardt MC, Springfield DS. Parosteal osteosarcoma of the posterior aspect of the distal part of the femur. Oncological and functional results following a new resection technique. J Bone Joint Surg Am 2000;82:1083-8.
42. Han I, Oh JH, Na YG, Moon KC, Kim HS. Clinical outcome of parosteal osteosarcoma. J Surg Oncol 2008;97:146-9.
43. Laitinen M, Parry M, Albergo JI, et al. The prognostic and therapeutic factors which influence the oncological outcome of parosteal osteosarcoma. Bone Joint J 2015;97:1698-703.
44. Cesari M, Alberghini M, Vanel D, et al. Periosteal osteosarcoma: a single-institution experience. Cancer 2011;117:1731-5.
45. Chan CM, Lindsay AD, Spiguel ARV, Gibbs CP Jr, Scarborough MT. Periosteal Osteosarcoma: a single-institutional study of factors related to oncologic outcomes. Sarcoma 2018;2018:8631237.
46. Grimer RJ, Bielack S, Flege S, et al. Periosteal osteosarcoma:a European review of outcome. Eur J Cancer 2005;41:2806-11.
47. Righi A, Gambarotti M, Benini S, et al. MDM2 and CDK4 expression in periosteal osteosarcoma. Hum Pathol 2015;46:549-53.
48. Ritts GD, Pritchard DJ, Unni KK, Beabout JW, Eckardt JJ. Periosteal osteosarcoma. Clin Orthop Relat Res 1987;219:299-307.
49. Okada K, Unni KK, Swee RG, Sim FH. High grade surface osteosarcoma: a clinicopathologic study of 46 cases. Cancer 1999;85:1044-54.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Pacheco M, Righi A. Primary malignant tumors of bone surface: a review with emphasis in differential diagnosis. J Cancer Metastasis Treat 2020;6:49. http://dx.doi.org/10.20517/2394-4722.2020.85
AMA Style
Pacheco M, Righi A. Primary malignant tumors of bone surface: a review with emphasis in differential diagnosis. Journal of Cancer Metastasis and Treatment. 2020; 6: 49. http://dx.doi.org/10.20517/2394-4722.2020.85
Chicago/Turabian Style
Pacheco, Marina, Alberto Righi. 2020. "Primary malignant tumors of bone surface: a review with emphasis in differential diagnosis" Journal of Cancer Metastasis and Treatment. 6: 49. http://dx.doi.org/10.20517/2394-4722.2020.85
ACS Style
Pacheco, M.; Righi A. Primary malignant tumors of bone surface: a review with emphasis in differential diagnosis. J. Cancer. Metastasis. Treat. 2020, 6, 49. http://dx.doi.org/10.20517/2394-4722.2020.85
About This Article
Special Issue
Copyright

Data & Comments
Data
0

 Cite This Article 33 clicks
Cite This Article 33 clicks

 Like This Article 10
likes
Like This Article 10
likes






Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.