
Angiogenesis in acute myeloid leukemia

 , ...
, ... Abstract
Angiogenesis is a word that refers to new blood vessel formation, and this process is of fundamental importance for physiological development and tissue homeostasis, as well as the genesis of several diseases, including tumors. Thus, studies carried out in the last years have shown that angiogenesis is essential for the growth of many solid tumors. Angiogenesis is also important for the growth of many hematological malignancies, including acute myeloid leukemia (AML). Endothelial cells are essential constituents of the bone marrow vascular niches, structures essential for the survival and maintenance of normal hematopoietic stem/progenitor cells. Bone marrow endothelial cells play an essential role in leukemia development and there is growing evidence that a targeting of both leukemic and endothelial cells of the leukemic vascular niche may improve the efficacy of antileukemic therapies. Bone marrow angiogenesis is frequently increased in AML, is morphologically evidenced as increased microvascular density, and is typically associated with some AML subtypes. The molecular mechanisms underlying the increased angiogenesis in some AML subtypes have been defined. In conclusion, a better understanding of angiogenesis as well as the fundamental interactions between bone marrow endothelial cells and leukemic stem cells may contribute to improve antileukemia treatments.
Keywords
Introduction
Angiogenesis is a fundamental and vital process required for the generation of a functional vasculature essential for tissue and whole organism survival[1]. Blood vessels are not only simple conduits required to supply oxygen and nutrients to tissues, but are also involved in specialized functions to support tissue-specific homeostasis, through tissue-specific endothelial cells[1].
A deregulation of angiogenetic mechanisms is involved in many diseases, characterized by excessive angiogenetic processes, as observed in many malignant tumors or dysfunctional angiogenetic processes, such as in diabetic microvasculopathies[1]. Historically, the occurrence of a vascular phase of tumor development was first demonstrated for solid tumors, characterized by an angiogenic capability required to promote new vessel formation to sustain tumor growth[2,3]. Only more recently the occurrence of tumor-related angiogenic mechanisms was reported also for hematological malignancies, although some doubts were raised about the role of angiogenesis in the progression of hematological malignancies. However, numerous studies have shown that the degree of angiogenesis or the level of endothelial growth factors correlate with stage of disease, prognosis, or response to therapy, thus suggesting that angiogenesis induction in hematological malignancies has a physiological role in the mechanisms responsible for disease progression[4,5].
The terms angiogenesis and vasculogenesis underline two different biologic processes involving new blood vessel formation: the first one describes the formation of new blood vessels starting from pre-existing vessels, while the second one identifies the de novo formation of blood vessels and mostly occurs during embryonic and fetal life.
Blood vasculature forms a closed circulatory system involved in the circulation of blood from heart to all peripheral tissues; this complex function requires a highly articulated network composed of different types of vessels, including arteries, veins, and capillaries; the inner layer of vessels is constituted by endothelial cells, a unique and typical component of vessels. There is growing evidence that endothelial cells exert a number of complex biological functions not limited to the generation of a barrier between blood and tissues, but also consisting in specialized functions, such as the creation of a stem cell niche, a function particularly important at the level of bone marrow[6].
Endothelial cells originate from the mesoderm layer of the early embryo and subsequently a subset of endothelial cells, the hemogenic endothelium, gives rise to hematopoietic stem cells (HSCs)[7]. At the level of hemogenic endothelium, the hemangioblast, a mesoderm-derived stem cell, gives rise to both the angioblast, the progenitor of endothelial cells, and hematopoietic stem cells, the progenitors of hematopoietic cells. During embryogenesis, endothelial cells undergo a process of specification, necessary to generate the peculiar features of distinct vessel subtypes (arteries, veins, capillaries, and lymphatic vessels) to provide essential support to tissue morphogenesis[7]. During tissue and organ formation, endothelial cells undergo an additional process of tissue- and organ-characteristic specification.
However, recent studies indicate that the relationship between endothelial cells and hematopoietic lineage during embryonic life is more complex. During embryonic life, some mesoderm cells located in the extraembryonic organ called the yolk sac generate a population of HSCs, called erythro-myeloid progenitors (EMPs); EMPs migrate from the yolk sac into embryo and generate a population of primitive hematopoietic cells, but they also differentiate into endothelial cells, contributing to the vasculature of several organs[8]. Interestingly, the percentage of endothelial cells that originate from EMPs ranges from 30% in the brain to 60% in the liver[8]. Further studies will be required to demonstrate to which extent these observations made in mice apply also to humans; however, these studies strongly support the existence of a strong link between endothelial and hematopoietic cell lineages.
Recent studies support the existence of endothelial stem/progenitor cells located at the level of the endothelium of vessels of various tissues; under steady-state conditions, the endothelial stem cells are in a state of quiescence, but they can be activated by stimuli requiring a tissue regenerative response[9,10].
The mechanisms through which tumors stimulate angiogenesis are complex and heterogeneous in the various tumors implying different molecular and cellular processes; however, it is evident that in all tumors a major determinant of angiogenesis is related to the tumor microenvironment[11,12]. Tumor angiogenesis is also a key determinant of tumor heterogeneity in that the level of proximity of cancer cells to blood vessels in a tumor greatly influences phenotype and functional and metabolic properties of tumor cells[13].
The present paper analyzes the angiogenic mechanisms occurring in acute myeloid leukemia focusing on the analysis of changes in vessel density, architecture, and functional properties and on the direct contribution of bone marrow endothelial cells to the development of leukemia progression, promoting leukemic cell homing, survival and proliferation of leukemic cells, and resistance to therapy. As discussed below, a better understanding of the complex interactions occurring between leukemic cells and bone marrow vascular niches may contribute to the development of new therapeutic approaches, including the targeting of leukemic endothelium.
Role of endothelial cells in leukemia development
Role of bone marrow endothelial cells in the control of hematopoiesis
The bone marrow capillary network exhibits a complex structure and shows a linear columnar organization at the level of the metaphysis and endosteum and a sinusoidal organization forming a network of fenestrated, highly branched sinusoidal vessels at the level of the bone cavity; columnar and sinusoidal vessels are interconnected, thus generating a single capillary network, providing a unique and fundamental structure for supporting hematopoiesis[14]. The columnar and sinusoidal vessels can be distinguished according to the pattern of expression of some endothelial cell surface markers, such as endomucin and CD31: due to the high expression of these markers, columnar vessels are defined as type H vessels, whereas sinusoidal vessels are defined as type L vessels for the low expression of these markers[15]. These phenotypic differences between type H and type L vessels correspond also to important functional differences in that columnar vessels exhibit a higher oxygen pressure and blood flow than sinusoidal vessels[16]. The lower endothelial permeability of columnar vessels induces the generation of a microenvironment characterized by low reactive oxygen species (ROS)[17]. These differences between type H and type L vessels have important consequences at the tissue level, contributing to the generation of different microenvironments; thus, type H vessels connect to arterioles, are surrounded by osteoprogenitors, release factors that promote osteogenesis, and create a local microenvironment promoting the survival and quiescence of HSCs, while type L vessels lack arteriolar connections and association with osteo-progenitors and generate a local microenvironment more permissive of the differentiation of HSCs and Hemopoietic Progenitor Cells (HPCs)[14][Figure 1].
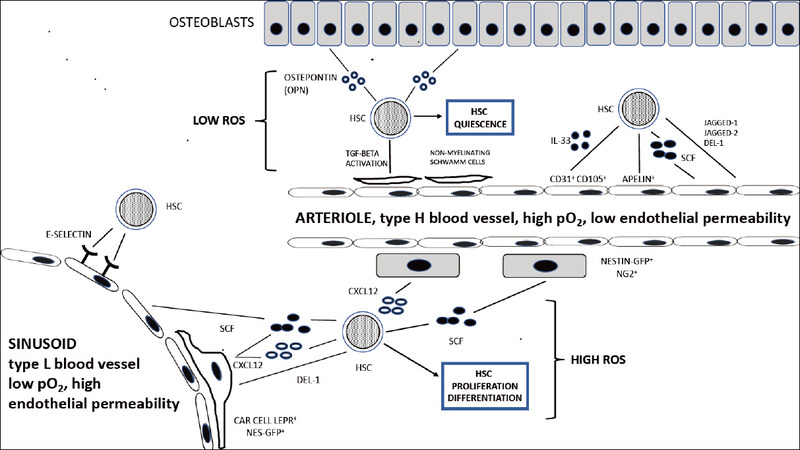
Figure 1. The HSC niche. Under normal conditions, HSCs reside near the bone marrow vessels, either endosteal arterioles or sinusoids. Endothelial cells, as well as mesenchymal stem cell populations (Nestin-GFP NG2+ cells and CAR cells) promote the maintenance of HSCs. The endosteal niche contributes to the creation of a microenvironment with higher pO2 levels; the type H vessels constituting the endosteal niche display low permeability and create an environment low in reactive oxygen species (ROS), promoting HSC quiescence. The sinusoidal niche contributes to the creation of a microenvironment with lower pO2 levels; the L vessels constituting the sinusoidal niche have a high permeability and create a microenvironment high in ROS, promoting HSC proliferation and differentiation. HSC: hematopoietic stem cell
Type H endothelial cells display high proliferation rate and mediate vascular growth in bone; regulators of vessel growth are highly expressed in type H endothelial cells, such as neuropilin1, plexin D1, and vascular endothelial growth factor-receptor 3 (VEGF-R3), compared to the low levels of these regulators observed in sinusoid type L endothelial cells. The generation of type H bone marrow vessels is promoted by NOTCH activation in endothelial cells, a surprising finding given the inhibitory effects on angiogenesis of NOTCH signaling exerted in other tissues[18].
Blood flow was shown to be crucial for the formation of type H capillaries and angiogenic growth of the bone marrow vasculature; a pharmacologically-induced reduction of blood flow resulted in the inhibition of angiogenesis, osteogenesis, and NOTCH activity in the endothelium[19]. With aging, there is a decline of both blood flow and NOTCH endothelial activity[19].
Type H endothelial cells are essential for maintaining HSC number and vitality. A major determinant of this effect is represented by the production of the cytokine stem cell factor (SCF) selectively produced by type H endothelial cells but not by type L endothelial cells[6][Figure 1]. Genetic deletion of SCF in type H endothelial cells elicited a significant reduction of the number and activity of HSCs[20]. Importantly, lineage-tracing experiments have shown that type H and type L endothelial cells self-generate independently after a genotoxic insult, such as a radiation exposure; this finding suggests the existence of radioresistant endothelial progenitors separate for H type and L type endothelial cells[20].
Other cytokines or chemokines released by bone marrow endothelial cells, such as Chemokine (C-X-C motif) Ligand 12 (CXCL12)[21], Interleukin 33 (IL33)[22], and pleiotrophin[23], play an important role in the survival of HSCs. Particularly, endothelial cell-specific deletion of CXCL12 determines a decrease in the number and repopulating activity of HSCs[21]. Interestingly, in human bone marrow, IL33 is released from a subtype of endothelial cells expressing CD105 [Transforming Growth Factor β1(TGF-β1) co-receptor], involved in regeneration of endothelial cell after chemotherapy injury and displaying several similarities with murine type H endothelial cells[22].
Recent studies have reported the identification of an endothelial cell subpopulation, characterized by the production of the peptide Apelin: Apelin+ endothelial cells are distinct from other sinusoidal endothelial cells, express NOTCH ligands and pleiotrophin, and play a key role in HSC maintenance, hematopoiesis, and hematopoietic regeneration after irradiation[24]. Apelin receptor allows the identification in human fetal and adult bone marrow of a mesodermal-derived cell population with hemogenic potential[25].
Other studies have identified the secreted developmental endothelial locus-1 (Del-1) as a regulator of myelopoiesis in the HSC niche: this HSC niche factor interacts with β3 integrin on HSCs and is produced by bone marrow arteriolar endothelial cells and mesenchymal stromal cells (CAR cells)[26]. Del-1 regulates HSC proliferation and differentiation toward the myeloid lineage[26].
Endothelial permeability in acute myeloid leukemia
Vascular permeability is a peculiar property of blood vessels wall to be permissive to the flow of small molecules or even of whole cells in and out of the vessel. This flow occurs at the level of the cell junctions between endothelial cells that form microscopic gaps. Vascular permeability is a highly controlled process related to the intrinsic property of each single vessel and to the physiologic conditions of the tissue of residence of vessels.
A recent study by Passaro et al.[27] provided strong evidence that leukemic cells exert a disruptive effect on bone marrow permeability and vascular architecture; these events are essential for the development of the malignant phenotype.
To better understand these studies, it is important to analyze the studies on vascular permeability of vessels of normal bone marrow. In a fundamental study, Itkin and coworkers provided evidence that distinct bone marrow blood vessels, with different permeability properties, play a key role in the control of homeostasis of hematopoiesis through a control of hematopoietic stem cell/hematopoietic progenitor cell quiescence and differentiation. Thus, less-permeable arterial blood vessels maintain HSC in a state of metabolic quiescence, characterized by a low ROS state, whereas the more permeable sinusoids promote a state of metabolic activation of stem/progenitor cells, with high ROS production, triggering differentiation[28]. Sinusoids represent the site for immature and mature leukocyte trafficking to and from the bone marrow[28].
It is well known that there is a deterioration of bone and hematopoiesis with aging, and there is evidence for the progressive degeneration of arterial endothelial cells from endosteal regions of bone. Aging determines a reduction in HSC survival and an increase in bone marrow permeability[29].
The bone endothelial cell permeability is a physiologically very relevant process because it regulates the microenvironment and the hematopoietic stem and progenitor cell transmigration. Stem/progenitor cell transmigration is an essential and fundamental event in the process of homing of transfused HSCs in stem cell transplantation. This process implies first the firm adhesion of HSC/HPC to endothelial cells and subsequent transmigration across the endothelial lining.
The fundamental importance of vascular bone marrow permeabilization is highlighted by the procedure of mobilization of HSCs and HPCs from bone marrow into blood, utilized in clinical practice for the treatment of patients with hematological malignancies who undergo a treatment with HSC transplantation. In clinical studies, granulocyte-colony stimulating factor (G-CSF) is the preferred mobilizing agent. Several studies have in part clarified the mechanisms responsible for G-CSF-mediated HSC/HPC mobilization. The available evidence indicates that G-CSF-induced trafficking is mediated by bone marrow endothelial cells mainly through a mechanism involving a remodulation of the CXCL12-CXCR4 axis: (1) under steady-state conditions, the chemokine receptor CXCR4 expressed on bone marrow endothelial cells actively binds and internalizes CXCL12, its ligand, resulting in the translocation of this chemokine in the bone marrow with consequent activation of a homing signal for HSCs and HPCs in this tissue[30]; (2) G-CSF administration determines a decrease of both CXCL12 and CXCR4, determined by serine proteases able to cleave these molecules[31,32]; (3) among the various proteases, the dipeptidyl peptidase CD26 seems to play an essential role in the process of G-CSF-mediated mobilization, mediating CXCL12 cleavage[33]; and (4) G-CSF induces an increase of the expression of CD26 on the surface of bone marrow endothelial cells, promoting the cleavage of the neuropeptide Y (NPY) to its truncated form, which in turn binds with higher affinity to NPY receptors expressed on sinusoidal endothelial cells, triggering VE-cadherin internalization and degradation, an event that consistently enhances bone marrow vascular permeability[34].
Vascular bone marrow permeability is also affected by inflammatory and infectious processes. In fact, studies carried out in the context of infectious experimental models have shown a key role of endothelial bone marrow cells as mediators of stimuli enhancing granulopoiesis during acute infection[35]. Using a model of inflammatory response induced by interferon α (IFNα), a cytokine rapidly produced in response to infection, the induction of a rapid stimulation of endothelial cells bone marrow cells in vivo was shown, resulting in an increase of endothelium activation, vascular permeability, and vascularity[36]. This IFNα-mediated activation of bone marrow endothelial cells is in part dependent on an increased production of VEGF by bone marrow cells[36].
Passaro et al.[27] explored the bone marrow vasculature using intravital two-photon microscopy in acute myeloid leukemia (AML) patient-derived xenografts; this approach allowed defining changes in bone marrow vascularity since the first stages of leukemic cells engraftment in bone marrow of recipient animals. Using this approach, it was shown that: (1) AML engraftment induced a leukemic-specific expansion of the endothelial compartment among the non-hematopoietic stroma, associated with an increase of microvessel density (MVD) and an alteration of the architecture of bone marrow vasculature with loss of sinusoidal structures, reduction of the mean diameter of vessels, and an increase of tissue hypoxia; (2) increased vascular leakiness in bone marrow; (3) induction of remission with chemotherapy failing to induce a recovery of vascular architecture and permeability in bone marrow; (4) analysis of molecular signatures in vascular endothelial cells showed a consistent deregulation of various pathways involved in permeability regulation and cell adhesion; (5) importantly, among the genes hyperexpressed in vascular endothelial cells, there are the nicotinamide adenine dinucleotide phosphate (NADPH) oxidase 4 (NOX4) gene, encoding A NADPH, which, in response to hypoxia, increases ROS levels in the vascular cells and the endothelial nitric oxide synthetase (eNOS), responsible for the production of nitric oxide (NO) molecules; (6) as a consequence of the deregulated Nox4 and eNOS expression, all leukemic vascular niches express high ROS and NO levels; and (7) combined treatment with chemotherapy and genetic or pharmacological inhibitors of eNOS resulted in an improvement of the antileukemic effects and a restoration of bone marrow vasculature[27].
Duarte et al.[37] analyzed by intravital microscopy (IVM) the changes in bone marrow vasculature induced by the engraftment of primary AML cells; the consistent advantage of this technique is related to its minimal invasiveness and its compatibility with longitudinal observations of cellular dynamics. These studies showed that following AML engraftment in bone marrow blood vessels appeared damaged: most vessels were narrower than those in control mice; limited and abnormal sprouting of bone marrow vessels; progressive decrease of vessels in the endosteum and metaphysis; endosteal vessels are progressively lost at intermediate and advanced disease stages; and the number of CD31high/Endomucinhigh was significantly reduced[37]. These changes in bone marrow (BM) vasculature are accompanied by a concomitant progressive depletion of bone marrow stroma. This endosteal remodeling correlated with a loss of normal hematopoiesis, further supporting the evidence that the vascular endosteal system is structurally and functionally damaged by the leukemic process[37]. Importantly, endosteal areas represent the major site for initiation of AML relapse. Finally, studies in genetically engineered mice provided evidence that rescue of endosteal vessels improves the efficacy of standard antileukemic chemotherapy[37].
Other components of the endothelial niche play a key role in the control of normal hematopoiesis and in AML development. Osteoblasts are an important component of the endosteal niche and regulate hematopoiesis through the secretion of various factors, such as osteopontin (OPN)[38]. OPN is a cytokine involved in many physiological processes, including angiogenesis. In adult bone marrow, OPN production is restricted to endosteal region, where this cytokine is required for HSC homing and quiescence[38]. In the endosteal niche, a truncated form of OPN, trOPN, interacts with α4β1 and α9β1 integrins expressed on HSCs and inhibits their proliferation and differentiation[39]. Interestingly, OPN expression is increased both in bone marrow leukemic blasts and in bone marrow serum; AML patients exhibiting high levels of bone marrow OPN showed a reduced overall survival[40]. The prognostic role of OPN was particularly evident at the level of the intermediate risk AMLs[40].
Targeting leukemic endothelium
Another set of recent studies has shown the key role of some adhesion molecules expressed on endothelial cells of the vascular niche in homing, survival, and chemoresistance of AML cells. E-selectin, also known as ELAM-1 (endothelial-leukocyte adhesion molecule-1) or CD62E (CD62 antigen-like family member E), is a selectin cell adhesion molecule selectively expressed on activated endothelial cells. E-selectin binds to different ligands expressed on various types of hematopoietic cells. E-selectin mediates the adhesion of some tumor cell types to endothelium through the interaction with ligands expressed on tumor cells.
E-selectin is constitutively expressed on bone marrow endothelium, where it plays a key role in allowing the homing and engraftment of HSCs/HPCs that express the correspondent ligands. Winkler et al.[41] showed that an increase of E-selectin at the vascular HSC niche in the bone marrow corresponds to a stimulation of proliferation and differentiation of dormant HSCs: following antileukemic chemotherapy or radiation treatment, E-selectin expression increases at the level of bone marrow by about 10-20-fold during the recovery phase of hematopoiesis in correspondence with the reparative proliferation and differentiation of HSCs; a genetic or pharmacologic inactivation of E-selectin reduces this response and increases the proportion of HSCs returning to quiescence [Figure 2]. Interestingly, deletion or blockade of E-selectin significantly enhanced the survival of normal HSCs after treatment of mice with chemotherapeutic drugs or irradiation[41]. These effects do not seem to be mediated by canonical E-selectin ligands, such as CD44 and CD162[41].
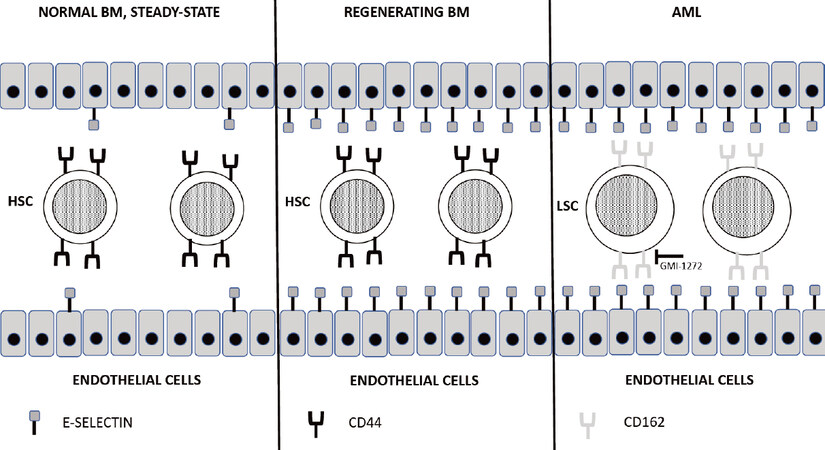
Figure 2. Role of endothelial E-selectin in the control of HSC and LSC. (Left) under steady-state conditions, few bone marrow endothelial cells express the adhesion molecule E-selectin on their surface, mediating in few instances the interaction with the CD44 receptor expressed on HSCs; (middle) under conditions of bone marrow regeneration following damage caused by radiotherapy or chemotherapy, E-selectin expression on bone marrow endothelial cells markedly increases and promotes HSC proliferation; (right) in AML patients, the inflammatory microenvironment promotes a marked increase of E-selectin expression on bone marrow endothelial cells, mediating its interaction with the CD162 receptor overexpressed on the surface of LSCs. GMI-1271, a E-selectin inhibitor, blocks the interaction between E-selectin and CD162, reducing the binding of LSCs to endothelium and their chemoresistance. HSC: hematopoietic stem cell; LSC: leukemic stem cell
Importantly, E-selectin expression was observed at the level of bone marrow areas where leukemia cells home at the moment of their engraftment[42]. Xenograft models of immune-deficient mice transplanted with AML blasts showed that a small number of CD34+ leukemic stem cells that survived antileukemia chemotherapy was detected under form of clusters located around endosteal vascular endothelium, a region where E-selectin is expressed on endothelial cells[43,44].
Recent studies have shown a direct role of bone marrow vascular E-selectin expression in homing and chemoresistance of leukemic cells. Thus, Winkler et al.[45] initially reported a remarkable up-modulation of E-selectin expression on the bone marrow vasculature in mice with AML; studies in the murine AML model generated by retroviral transduction of the MLL-AF9 fusion oncogene into HSCs showed that leukemic blasts rapidly upregulate E-selectin expression following oncogenic transformation. Experiments based on genetic or pharmacological inhibition of E-selectin expression provided evidence that E-selectin expression is important for retention of leukemic stem cells in bone marrow and protects leukemic stem cells from the cytotoxic effects of chemotherapy[45]. In a more recent study, the same authors showed that MLL-AF9 AML cells surviving cytarabine therapy display an increased E-selectin binding potential[46]. This is due to the capacity of these leukemic cells to interact with E-selectin-positive endothelial cells present in the vascular niche. This conclusion is supported by two lines of observations: (1) vascular niche E-selectin blockade by GMI-1271, a specific inhibitor of E-selectin binding, inhibits malignant AML reconstitution/survival potential in vivo; and (2) these effects occur through the inhibition of several pro-survival signals induced in leukemic cells[46]. According to these observations, it was suggested that E-selectin blockade may synergize with other pathway inhibitors to improve the therapeutic response of AML patients[46].
In a more recent study, the same investigators showed that AML cells generate an inflammatory state in the bone marrow at the level of the sites where they home, driving increased E-selectin expression on bone marrow endothelial cells[47][Figure 2]. This condition creates the basis for the interaction between leukemic cells and E-selectin-expressing endothelial cells, triggering the generation of pro-survival signaling (PI3K/AKT and mTOR pathways) that stimulates leukemic cell proliferation and promotes leukemic chemoresistance[47]. These observations support the clinical use of E-selectin inhibitors in combination with standard induction chemotherapy for AML patients. The preliminary results of a phase I clinical trial involving the administration of GMI-1271 in combination with induction chemotherapy to a group of 19 relapsed/refractory AML patients showed a high response rate (42% of complete responses), with a sufficient duration of response to allow five patients to proceed to salvage stem cell transplant. Phase II of this study is ongoing and implies two arms of treatment, one corresponding to an expansion cohort of phase I and the other involving treatment of newly diagnosed AML patients ≥ 60 years.
Very recently, the membrane E-selectin receptor expressed on HSCs and responsible for induction of chemoresistance was identified[48]. E-selectin may interact with two different membrane receptors, CD44 and CD162, expressed on hematopoietic cells. To explore this issue, Erbani and coworkers used the CD34+ human myeloid cell line KG1a. This cell line express CD44 and CD162, and both of these receptors are functional in terms of E-selectin binding capacity. Only the silencing of both receptors abrogates the capacity of KG1a cells to bind E-selectin; however, only CD162 is critical for E-selectin-mediated chemoresistance in vitro. Importantly, CD162 expression on AML cells in vivo is a major determinant for E-selectin binding, bone marrow vascular niche retention, and leukemic progression[48][Figure 2]. Deletion of CD162 in AML cells induces a clear increase of the sensitivity of leukemia stem cells to therapy[48]. According to these findings, it was suggested that the binding of CD162 to E-selectin represents a potential therapeutic target to improve therapeutic outcomes through a potentiation of the efficacy of antileukemic chemotherapy.
Interestingly, recent studies carried out on chronic myeloid leukemia (CML) led to evidence similar to that observed in AML about a key role of interactions with E-selectin mediating leukemic homing and survival[49]. In fact, it was shown that binding of BCR-ABL1+ CML cells to E-selectin in the vascular niche stimulates cell cycle progression and response to imatinib treatment[49]. For CML cells, the E-selectin CD44 receptor and not CD162 seems to be involved in the mediation of E-selectin induced effects[50].
Angiogenesis in AML bone marrow: microvessel density
The technique currently used to evaluate the extent of angiogenesis at the level of bone marrow biopsies consists in staining of the histological sections with specific endothelial cell markers and then evaluation with a microscope of the density of microvessels present in these tissue sections (the number of vessels per millimeter length of bone marrow core biopsy is evaluated). This technique provides a score of MVD. Using this approach, two different groups of investigators in 2000 provided evidence that the MVD was increased (approximately doubled) in AML specimens compared to normal bone marrow[51,52]. Examination of AML bone marrow specimens after induction of disease remission was associated with a clear decrease of bone marrow MVD; in patients not achieving remission following chemotherapy treatment, no decrease of bone marrow MVD was observed[51,52]. Kini et al.[53] reported that the MVD and hot spot density were particularly increased in bone marrow biopsies of acute promyelocytic leukemia patients compared with normal bone marrow specimens; treatment with retinoic acid induced disease remission and a clear decrease of MVD. Other studies have evaluated whether the increased MVD observed in AML bone marrow was associated with an increased production of VEGF. Padrò et al.[54] showed, through the immunohistochemical analysis of 32 AML bone marrow samples, higher VEGF and VEGF-R2 expression than in control normal bone marrow: expression of VEGF and VEGF-R2 was clearly higher in patients with a high degree of MVD, compared to those observed with lower MVD. A direct correlation was observed between VEGF expression and MVD[54]. Ghannadan and coworkers explored VEGF expression in the bone marrow of 41 AMLs and observed positive expression in all AML subtypes classified according to the FAB classification, with the expression of most immature FAB M0 AMLs, expressing undetectable or only low levels of VEGF[55]. VEGF was detectable in immature elements, whereas it was undetectable or expressed at very low levels in mature hematopoietic elements[55].
Other studies confirmed the increased MVD in AML bone marrow and showed also a correlation between the increase of MVD and the proliferation index of leukemic blasts[56] and the association between increased MVD and increased bone marrow VEGF levels, both parameters decreasing in patients who achieved disease remission following induction therapy[57].
Kuzu et al.[58] showed a higher MVD in AML patients compared to controls, independent of cellularity or blast percentage; higher baseline MVD values in AML patients were associated with a shorter overall survival and thus are a negative prognostic factor.
Interestingly, Weidenaar and coworkers explored the vascular morphology within AML bone marrow biopsies[59]. The analysis of a pericyte marker (smooth muscle actin) provided evidence that in AML bone marrow at diagnosis only 35% of vessels were pericyte-coated, compared to 73% in the normal bone marrow and 55% in AML patients in remission. Furthermore, the percentage of pericyte-coated vessels was significantly higher in the group of AML patients with “low vessel count” compared with that observed in AML patients with “high vessel count”[59]. Two different patterns of vascular morphology were observed in AML bone marrow biopsies: high number of vessels with a large lumen and thin walls and a high vessel count with a network of small vessels with thin walls, narrow lumen, and branching. The first vessel pattern was associated with high secreted vascular endothelial growth factor A (VEGFA) protein levels[59].
Other investigators have reported a comparative analysis of bone marrow MVD in various hematological malignancies, confirming the higher values of MVD in AML compared to normal bone marrow. These studies also showed an increase of MVD in Acute Lymphoblastic Leukemia (ALL) and CML, at a level higher than that observed in AML[60,61]. These studies failed to demonstrate a significant increase in serum/plasma VEGF levels in AML patients[60,61].
As reported above, the analysis of plasma/serum VEGF levels in AML patients showed conflicting results as not all studies showed an increase of VEGF levels in these patients[59]. These discrepancies may be related to the variable sensitivities of the immunodetection assays used to quantify VEGF levels and the heterogeneity of AML patients (de novo or relapsed AMLs) included in these studies. However, the majority of these studies showed increased VEGF levels in plasma/serum of AML patients, a conclusion supported by a recent large meta-analysis performed by Song et al.[62]. A meta-analysis carried out by Guo et al.[63] showed that high VEGF expression in AML was associated with worse event-free survival and poor overall survival. In this context, two studies are particularly interesting. Aguayo et al.[64] showed that VEGF levels were similarly elevated in AML and MDS patients: in AMLs, but not in MDSs, elevated plasmatic VEGF levels were associated with reduced survival and reduced remission rate. De Bont et al.[65] evaluated the release of VEGF by leukemic blasts of pediatric AMLs, and the levels of this endothelial growth factor release by leukemic cells are an independent prognostic factor for relapse-free survival.
As mentioned above, some studies[59] have shown that the increased MVD in AML is a negative prognostic factor. This conclusion was confirmed by other studies. Rabitsch et al.[66] analyzed bone marrow MVD in 38 younger AML patients undergoing standard chemotherapy treatment and consecutive allogeneic bone marrow transplantation: at diagnosis, the MVD was markedly higher in AML patients than in normal controls (30/mm2vs. 7/mm2); in patients who failed to achieve a complete response following induction chemotherapy, the MVD was higher at diagnosis than in those achieving complete remission (41.5/mm2vs. 28.5/mm2); and patients with high MVD displayed a shorter overall survival and a higher risk of relapse than patients with lower MVD. Similarly, studies carried out in myelodysplastic syndromes, conditions frequently preceding AMLs, have shown that increased MVD was associated with a shorter survival time[67].
Myeloid sarcoma, also known as granulocytic sarcoma or chloroma, is a tissue extramedullary mass form of AML composed of myeloid blasts[68,69]. Myeloid sarcoma may occur de novo, may precede or coincide with AML, or may correspond to a blastic transformation of a preceding myeloproliferative neoplasm or myelodysplastic syndrome[68,69]. Myeloid sarcoma may occur as a manifestation of relapse in an AML patient, even after allogeneic stem cell transplantation[70]. Piccaluga et al.[71] explored the bone marrow MVD in 60 myeloid sarcomas and showed that these tumors have an increased MVD compared to normal bone marrow. The MVD observed in myeloid sarcomas was similar to that observed in AMLs. Among myeloid sarcomas, those with a monocytic morphology displayed a significantly higher MVD than those with blastic appearance[71]. In these patients, higher MVD was associated with a reduced overall survival in multivariate analysis[71].
Molecular mechanisms responsible for stimulation of angiogenesis
Recent studies have explored the molecular mechanisms involved in the stimulation of angiogenesis in some AML subtypes and have defined a link between leukemia-specific genetic abnormalities and enhanced angiogenesis.
Initial studies by Hiramatsu and coworkers showed that, among the various AML subtypes, AML M3, corresponding to APL, having the translocation t(15;17) generating the fusion protein Promyelocytic/Retinoic Acid Receptor Alfa (PML/RARA), showed the highest expression of VEGF and VEGF-R1[72]. The AML subtype characterized by the specific t(8;21) translocation, generating the fusion protein AML1/ETO (Eight-Twenty-One), displayed high expression of VEGF and VEGF-R2[72].
In line with this initial study, Imai et al.[73] showed that t(8;21) AML cells are responsive to exogenous VEGF stimulation with activation of AKT pathway. Furthermore, t(8;21)-positive AML cell lines are inhibited in their proliferation by VEGF-R2 kinase inhibitors. The gene AML1/RUNX1 located on chromosome 21 is frequently involved in genetic alterations, such as chromosomal translocation events (AML1/ETO or AML1/EVI1) resulting in the formation of fusion proteins, associated with a loss of function of AML1/RUNX1. The analysis of a large dataset of gene expression arrays relative to AML samples showed the existence of an inverse correlation between expression of VEGF and AML1/RUNX1, with the highest VEGF levels being observed in leukemic blasts bearing t(8;21)[73]. Gene expression and transfection experiments provided evidence that AML1/RUNX1 acts as a repressor of VEGFA expression, through its direct binding at the level of three sites present on the promoter of the VEGF gene; AML1/ETO fails to exert this repressive effect on VEGF expression and results in a stimulation of its expression[72]. In line with this interpretation, silencing of AML1/ETO expression in t(8;21) leukemic cell lines resulted in an inhibition of VEGF expression[74].
Studies carried out in APLs have led to similar conclusions concerning the mechanisms through which the fusion protein observed in t(15;17) AMLs stimulates angiogenesis. Saulle et al.[75] through the analysis of a large TCGA dataset on gene expression in AMLs, confirmed that the highest expression of VEGF-A mRNA was observed in M3 t(15;17) AMLs. The molecular mechanism through which PML/RARA stimulates VEGF expression is related to its capacity to induce a significant downmodulation of Hematopoietically Expressed Homeobox (HHEX) expression, a homeobox transcription factor exerting a repressive effect on VEGF gene expression. The downmodulation of HHEX was also responsible for the stimulation of the expression of other genes involved in the control of angiogenesis[75].
Expression of endothliel growth factor receptors in AMLs
The receptors for various endothelial growth factors are frequently expressed on leukemic blasts and several studies have characterized the properties of AMLs expressing these receptors.
Padrò and coworkers explored VEGF-R1 and VEGF-R2 expression in AML bone marrow biopsies and observed that VEGF-R2 but not VEGF-R1 expression in AMLs was increased compared to normal bone marrow; VEGF-R2 expression was mostly increased in AMLs with an immature myeloid phenotype, classified as M1 and M2 in the FAB classification system or those with a monocytic phenotype (M5 AMLs)[54]. Studies in a model of mice xenotransplanted with human AML cells showed that inhibition of paracrine (dependent on endothelial cells present in the bone marrow microenvironment) and autocrine (dependent on leukemic cells) VEGF/VEGF-R2 signaling pathway was essential to induce long-term remission of these mice[76]. This study provided a rationale to investigate in humans the antileukemic potential of VEGF-R2 inhibitors. Other preclinical studies supported an inhibitory effect of anti-VEGF-R2 monoclonal antibodies on human AML development in leukemia animal models[77].
Zahiragic et al.[78] treated nine AML patients with refractory/relapsing disease with the anti-VEGF monoclonal antibody bevacizumab. Bevacizumab treatment reduced VEGF expression at the level of bone marrow but failed to show any significant clinical antileukemic activity[78]. It is interesting to note that, while bevacizumab reduced VEGF expression at the level of bone marrow, it was unable to modify bone marrow MVD[66]. Other clinical trials incorporating anti-VEGF or anti-VEGF-R2 antibodies have not produced results supporting a significant clinical benefit[79,80].
More recent studies have reevaluated the role of VEGF-R2 in leukemia development and its targeting with new VEGF-R2 inhibitors. Nobrega-Pereira provided evidence that VEGF-R2 signaling is involved in the mechanism of AML chemoresistance[81]. Treatment of chemoresistant AML cells with a VEGF-R2 inhibitor sensitized AML cells to chemotherapy[81]. These observations suggest the potential use of VEGF-R2 inhibitors in association with antileukemia chemotherapy.
Other recent studies have explored the effects of apatinib, a new VEGF-R2 inhibitor. Apatinib is an oral small-molecule tyrosine kinase inhibitor of VEGF-R2, showing in various experimental systems a marked inhibitory activity on angiogenesis. This inhibitor showed promising effects in several solid tumors[82]. In vitro studies have shown that apatinib exerts a consistent cytotoxicity toward AML by targeting VEGF-R2-mediated prosurvival signaling and angiogenetic effects. The sensitivity of AML blasts to apatinib was correlated with some molecular features, including presence of Nucleophosphomin 1 (NPM1) mutations and FAB M2 and M5 subtypes; importantly, AML blasts of relapsed/refractory patients displayed sensitivity to apatinib[83,84].
Sorafenib is a multi-kinase inhibitor exerting its effects by reducing the activity of various kinase receptors, including VEGF-R2, Fms Like Tyrosine Kinase 3 (FLT3), and Kinase Insert Tyrosine kinase (KIT). Sorafenib when used in monotherapy failed to exert a pronounced antileukemic activity in AML patients with refractory/relapsing disease[85]. However, a phase II study in untreated AML patients (aged £ 60 years) explored the antileukemic activity of sorafenib added to standard induction chemotherapy compared to placebo: sorafenib induced a significant prolongation of both event-free survival (EFS) and relapse-free survival (RFS) compared to placebo[86]. In an exploratory subgroup analysis, it was observed that no EFS prolongation was observed among AML patients with FLT3-ITD mutation, while AML patient FLT3-WT had significantly improved EFS and RFS[86]. This finding suggests that an anti-angiogenesis effect of sorafenib through inhibition of VEGF-R and platelet-derived growth factor receptors (PDGF-R) could mediate the effect of this drug on significant prolongation of EFS and RFS[86].
In addition to VEGF and its receptors, the endothelial growth factors angiopoietin1 (Ang1) and angiopoietin2 (Ang2) and their receptors Tie1 and Tie2 are also important regulators of physiologic and pathologic angiogenesis[87]. Peculiar is the function of the constitutively expressed Ang1 that acts as a stabilizer of blood vessels. In addition, Ang1 through the binding to Tie2 promotes endothelial cell survival and endothelial barrier function[88]. Ang2 acts as a context-dependent agonist or antagonist of the Ang1-Tie2 signaling axis[87]. Ang2 is expressed by endothelial cells. Its levels are increased by hypoxia and proinflammatory signals, and they are also increased in many types of cancers[69]. In several tumor models, there is evidence that Ang2 protects stressed endothelial cells from apoptosis[89] and limits the effects induced by VEGF inhibition[90].
The Ang-Tie system was explored in AMLs. In an initial study, Watarai et al.[91] showed that an AML subset, characterized by the inappropriate expression of the lymphoid membrane marker CD7, displays expression of Ang2, in association with an elevated expression of integrin-family adhesion molecules. The expression of Tie2 in these CD7+ AMLs was low[91].
Schliemann et al.[92] explored Ang1, Ang2, and Tie2 expression in 64 adult patients with newly diagnosed AML. They observed that: (1) expression of Ang2 was significantly higher in the bone marrow of AML patients than in healthy controls; (2) the expression of Ang1 in AML was similar to that observed in normal controls; and (3) Tie2 expression was often increased in AML samples compared to the levels observed in normal bone marrow[92]. Ang2 levels but not Ang1 or Tie2 levels had a prognostic impact: patients with high Ang2 levels exhibited a better overall survival compared to those with low Ang2 levels[92]. In a subsequent study, the same investigators analyzed the prognostic impact of plasmatic Ang1, Ang2, and soluble Tie2 (sTie2) in 68 AML patients[93]. Circulating levels of Ang2 and sTie2, but not of Ang1, were significantly elevated in AML patients as compared to controls[75]. Higher levels of Ang2 and sTie2 were predictive of poor survival in these patients; particularly, patients with elevated plasmatic Ang2 displayed a significantly reduced overall survival compared to that observed in patients with low plasmatic Ang2 levels[93]. The discrepancy observed in these two studies may be tentatively related to some relevant differences: the first study evaluated bone marrow expression of Ang2, while the second study evaluated plasmatic Ang2 levels and the source of circulating Ang2 is not only related to leukemic blasts, but also reflects the production by other cell types, including endothelial cells[93].
Loges et al.[94] explored VEGF-A, VEGF-C, Ang1, Ang2, and Tie2 mRNA levels in a cohort of 90 patients with de novo AML. This study showed that high Ang2 levels have a good prognostic impact on patient’s survival: sub-analysis according to the levels of other endothelial growth factors showed that the prognostic impact of Ang2 mRNA expression was most evident in AML subgroups with low VEGF-C and Ang1 levels[94].
Riccioni and coworkers reported a detailed immunophenotypic analysis of AMLs expressing high levels of Tie2[92]. In this study carried out in 111 de novo adult AML patients, 35% displayed high levels and 20% moderate levels of Tie2. Tie2 expression on leukemic blasts was associated with the expression of monocytic markers. Furthermore, Tie2 expression was associated with concomitant expression of other endothelial growth factors such as VEGF-R1, VEGF-R2, and VEGF-R3[95]. Highly-expressing Tie2 AMLs were characterized by high blast cell counts at diagnosis and frequent FLT3 mutations[95]. According to these findings, it was suggested that AMLs exhibiting high Tie2 expression resemble Tie2-expressing monocytes, a subpopulation of monocytes playing a role in promotion of tumor angiogenesis[96].
About 30% of adult AMLs display FLT3 mutations; in addition, 10%-15% of AMLs exhibit high FLT3 expression, in the absence of FLT3 mutations. This last group of AMLs is characterized by recurrent expression of receptors for endothelial growth factors, including Tie2[97].
The possible clinical efficacy of Tie2 inhibitors was not yet explored in AML patients. Pexmetinib, a dual inhibitor of Tie2 and p38 MAPK, showed antileukemic activity in preclinical models of AML[98].
Recent studies support a role for epidermal growth factor ligand 7 (EGFL7) as a pro-angiogenic factor promoting angiogenesis in AMLs. This secreted angiogenic factor possesses the unique property to be almost exclusively expressed by endothelial cells. EGFL7 is maximally expressed in proliferating endothelial cells and acts on endothelial cells[99]. Interestingly, microRNA-126, an endothelial cell-specific miRNA, is located in intron 7 of the EGFL7 gene. EGFL7 interacts with the extracellular domain of NOTCH, resulting in an antagonistic effect on NOTCH activation[99]. EGFL7 is a potent angiogenic factor, playing a key role in the control of vascular angiogenesis during embryogenesis[100]. EGFL7 is aberrantly overexpressed in solid tumors. A recent study showed that EGFL7 mRNA and EGFL7 protein levels are increased in blasts of AMLs compared to normal bone marrow cells: in AML patients with cytogenetically normal AMLs, high EGFL7 mRNA levels associate with decreased overall survival rates[101]. In vitro studies showed that EGFL7 stimulates the proliferation of leukemic blasts, whereas high EGFL7 expression predicts poor prognosis in AML patients undergoing allogeneic stem cell transplantation[102]. Functional studies have shown that EGFL7 inhibits NOTCH signaling in AML blasts antagonizing canonical NOTCH ligand binding; anti-EGF7L treatment resulted in reactivation of NOTCH signaling in AML cells, increased differentiation, and apoptosis, thus suggesting that it may represent a therapeutic strategy[103].
Conclusion
Endothelial cells are intimately associated with HSCs throughout the life of the stem cell, from peculiar endothelial cells (hemogenic endothelium) that give rise to HSCs, to the perivascular niche endothelial cells that regulate HSC homeostasis. Endothelial cells as constituents of bone marrow vascular niches play a key role in the control of HSC homing, migration, and maintenance. Bone marrow endothelial cells also play an essential role in the development of leukemia and growing evidence shows that targeting of both leukemic endothelium and leukemic stem cells represents a more effective antileukemic therapeutic strategy.
Bone marrow angiogenesis is enhanced in many patients with AML and several studies have shown a link between some leukemic-specific genetic alterations and stimulation of bone marrow angiogenesis. A better understanding of angiogenesis in AMLs may contribute both to a better understanding of the leukemogenetic process and to define an improved strategy for the treatment of these leukemias.
Declarations
Authors’ contributionsMade substantial contributions to conception, design of the study and analysis and interpretation of the literature data: Testa U, Castelli G, Pelosi E
Contributed to the preparation of the manuscript and to the supervision of all data included in this manuscript: Testa U
Availability of data and materialsNot applicable.
Financial support and sponsorshipNone.
Conflicts of interestAll authors declared that there are no conflicts of interest.
Ethical approval and consent to participateNot applicable.
Consent for publicationNot applicable.
Copyright©The Author(s) 2020.
REFERENCES
1. Eelen G, Treps L, Carmeliet P. Basic and therapeutic aspects of angiogenesis updated. Circ Res 2020;127:310-29.
2. Ribatti D, Vacca A, Dammacco F. The role of the vascular phase in solid tumor growth: a historical review. Neoplasia 1999;1:293-302.
3. Nguyen M, Watanabe H, Budson AE, Richie JP, Hayes DF, Folkman J. Elevated levels of an angiogenic peptide, basic fibroblast growth factor, in the urine of patients with a wide spectrum of cancers. J Natl Cancer Inst 1994;86:356-61.
4. Ribatti D. Is angiogenesis essential for the progression of hematological malignancies or is it an epiphenomenon? Leukemia 2009;23:433-4.
5. Testa U, Saulle E, Castelli G, Pelosi E. Endothelial progenitor cells in hematological malignancies. Stem Cell Invest 2016;3:26.
6. Marcu R, Choi YJ, Xue J, et al. Human organ-specific endothetial cell heterogeneity. IScience 2018;4:20-35.
7. Qiu J, Hirschi KK. Endothelial cell development and its application to regenerative medicine. Circulation Res 2019;125:489-501.
8. Plein A, Fantin A, Denti L, Pollard JW, Ruhberg C. Erythro-myeloid progenitors contribute endothelial cells to blood vessels. Nature 2018;562:223-8.
9. McDonald AI, Shirali AS, Aragon R, et al. Endothelial regeneration of large vessels is a biphasic process driven by local cells with distinct proliferative capacities. Cell Stem Cell 2018;23:210-25.
10. Wakabayashi T, Naito H, Suehiro JI, et al. CD157 marks tissue-resident endothelial stem cells with homeostatic and regenerative properties. Cell Stem Cell 2018;22:384-97.
11. Weis SM, Cheresh DA. Tumor angiogenesis: molecular pathways and therapeutic targets. Nature Med 2011;17:1359-69.
12. Testa U, Pelosi E, Castelli G. Endothelial progenitors in the tumor microenvironment. Adv Exp Med Biol 2020;1263:85-115.
13. Kumar S, Sharife H, Kreisel T, et al. Intra-tumoral metabolic zonation and resultant phenotypic diversification are dictated by blood vessel proximity. Cell Metab 2019;30:1-11.
14. Chen J, Hendriks M, Chatzis A, Ramasamy SK, Kusumbe AP. Bone vasculature and bone marrow vascular niches in health and disease. J Bone Min Res 2020;35:2103-20.
15. Kusumbe AP, Ramasamy SK, Adams RH. Coupling of angiogenesis and osteogenesis by a specific vessel subtype in bone. Nature 2014;507:323-8.
16. Spencer JA, Ferraro F, Roussakis E, et al. Direct measurement of local oxygen concentration in the bone marrow of live animals. Nature 2014;508:269-73.
17. Filipowska J, Tomaczewski KA, Niedzwiedzki L, Walocha JA, Niedzwedzki T. The role of vasculature in bone development, regeneration and proper systemic functioning. Angiogenesis 2017;20:291-302.
18. Ramasamu SK, Kusumbe AP, Wang L, Adams RH. Endothelial Notch activity promotes angiogenesis and osteogenesis in bone. Nature 2014;507:376-80.
19. Ramasamy SK, Kusumbe AP, Schiller M, et al. Blood flow controls bone vascular function and osteogenesis. Nat Commun 2016;7:13601.
20. Xu CL, Gao X, Wei QZ, Nakahara F, et al. Stem cell factor is selectively secreted by arterial endothelial cells in bone marrow. Nat Commun 2018;9:2449.
21. Ding L, Saunders TL, Enikolopov G, Morrison SJ. Endothelila and perivascular cells maintain hematopoietic stem eclls. Nature 2012;481:457-62.
22. Kenswil KJG, Jaramillo AC, Ping Z, et al. Characterization of endothelial cells associated with hematopoietic niche formation in humans identifies IL-33 as an anabolic factor. Cell Rep 2018;22:666-78.
23. Himburg HA, Termini CM, Schlussel L, et al. Distinct bone marrow sources of pleiotrophin control hematopoietic stem cell maintenance and regeneration. Cell Stem Cell 2018;23:370-81.
24. Chen Q, Liu Y, Jeong HW, et al. Apelin+ endothelial niche cells control hematopoiesis and mediate vascular regeneration after myeloablative injury. Cell Stem Cell 2019;25:768-83.
25. Mokhtari S, Colletti E, Yin WH, et al. A human bone marrow mesodermal-derived cell population with hemogenic potential. Leukemia 2018;32:1575-86.
26. Mitroulis I, Chen LS, Singh RP, et al. Secreted protein Del-1 regulates myelopoiesis in the hematopoietic stem cell niche. J Clin Invest 2017;127:3624-39.
27. Passaro D, Di Tullio A, Abarrategi A, et al. Increased vascular permeability in the bone marrow microenvironment contributes to disease progression and drug response in acute myeloid leukemia. Cancer Cell 2017;32:324-41.
28. Itkin T, Gur-Cohen S, Spencer JA, et al. Distinct bone marrow blood vessels differentially regulate hematopoiesis. Nature 2016;532:323-8.
29. Kusumbe AP, Ramasamy SK, Itkin T, et al. Age-dependent modulation of vascular niches for haematopoietic stem cells. Nature 2016;532:380-4.
30. Dar A, Goichberg P, Shinder V, et al. Chemokine receptor CXCR4-dependent internalization and resecretion of functional chemokine SDF-1 by bone marrow endothelial and stromal cells. Nat Immunol 2005;6:1038-47.
31. Lévesque JP, Hendy J, Takamatsu Y, Simmons PJ, Bendall LJ. Disruption of the CXCR4/CXCL12 chemotactic interaction during hematopoietic stem cell mobilization induced by GCSF or cyclophosphamide. J Clin Invest 2003;110:187-96.
32. Kim HK, Sierra M, Williams CK, Gulino AV, Tosato G. G-CSF down-regulation of CXCR4 expression identified as a mechanism for mobilization of myeloid cells. Blood 2006;108:812-20.
33. Christopherson KW, Cooper S, Hangoc G, Broxmeyer HE. CD26 is essential for normal G-CSF-induced progenitor cell mobilization as determined by CD26-/- mice. Exp Hematol 2003;31:1126-34.
34. Singh P, Hoggatt J, Kamocka M, et al. Neuropeptide Y regulates a vascular gateway for hematopoietic stem and progenitor cells. J Clin Invest 2017;127:4527-40.
35. Boettcher S, Gerosa RC, Redpour R, et al. Endothelial cells translate pathogen signals into G-CSF-driven emergency gran ulopoiesis. Blood 2014;124:1393-403.
36. Prendergast A, Kuck A, van Essen M, Haas S, Blaskiewicz S, Essers MAG. IFNα-mediated remodeling of endothelial cells in the bone marrow niche. Haematologica 2017;102:445-53.
37. Duarte D, Hawkins ED, Akinduro O, et al. Inhibition of endosteal vascular niche remodeling rescues hematopoietic stem cell loss in AML. Cell Stem Cell 2018;22:64-77.
38. Le PM, Andreef M, Battula VL. Ostogenic niche in the regulation of normal hematopoiesis and leukemogenesis. Haematologica 2018;103:1945-55.
39. Nilsson SK, Johnston HM, Whitty GA, et al. Osteopontin, a key component of the hematopoietic stem cell niche and regulator of primitive hematopoietic progenitor cells. Blood 2005;106:1232-9.
40. Liersch R, Gerss J, Schliemann C, et al. Osteopontin is a prognostic factor for survival of acute myeloid leukemia patients. Blood 2012;119:5215-20.
41. Winkler IG, Barbier V, Nowlan B, et al. Vascular niche E-selectin regulates hematopoietic stem cell dormancy, self-renewal and chemoresistance. Nat Med 2012;18:1651-61.
42. Spikins DA, Wei XB, Wei JW, et al. In vivo imaging of specialized bone marrow endothelial microdomains for tumour engraftment. Nature 2005;435:969-73.
43. Ishikawa F, Yoshida S, Saito Y, et al. Chemotherapy-resistant human AML stem cells home to and engraft within the bone-marrow endosteal region. Nat Biotechnol 2007;25:1315-21.
44. Ninomiya M, Abe A, Katsumi A, et al. Homing, proliferation and survival sites of human leukemia cells in vivo in immunodeficient mice. Leukemia 2007;21:136-42.
45. Winkler IG, Barbier V, Pattabiraman DR, Gonda TJ, Magani JL, Levesque JP. Vascular niche E-Selectin protects acute myeloid leukemia stem cells from chemotherapy. Blood 2014;124:620.
46. Winkler IG, Barbier V, Tay MJ, et al. Blocking vascular niche E-selectin dampens AML stem cell regeneration/survival potential in vivo by inhibiting MAPK/ERK and PI3K/AKT signaling pathways. Blood 2019;134:2657.
47. Barbier V, Erbani J, Fiveash C, et al. Endothelial E-selectin inhibition improves acute myeloid leukaemia therapy by disrupting vascular niche-mediated chemoresistance. Nast Commun 2020;11:2042.
48. Erbani J, Tay J, Barbier V, Levesque JP, Winkler IG. Acute myeloid leukemia chemo-resistance is mediated by E-selectin receptor CD162 in bone marrow niches. Front Cell Dev Biol 2020;8:668.
49. Godavarthy PS, Kumar R, Herkt SC, et al. The vascular bone marriw niche influences outcome in chronic myeloid leukemia via the E-selectin-SCL/TAL1-CD44 axis. Haematologica 2020;105:136-47.
50. Krause DS, Lazarides K, von Adrian UH, Van Etten RA. Requirement for CD44 in homing and engraftment of BCR-ABL-expressing leukemic stem cells. Nat Med 2006;12:1175-80.
51. Padrò T, Ruiz S, Bieker R, et al. Increased angiogenesis in the bone marrow of patients with acute myeloid leukemia. Blood 2000;95:2637-44.
52. Hussong JW, Rodgers GM, Shami PJ. Evidence of increased angiogenesis in patients with acute myeloid leukemia. Blood 2000;95:309-13.
53. Kini AR, Peterson LA, Tallman MS, Lingen MW. Angiogenesis in acute promyelocytic leukemia: induction by vascular endothelial growth factor and inhibition by all-trans retinoic acid. Blood 2001;97:3919-24.
54. Padrò T, Bieker R, Ruiz S, et al. Overexpression of vascular endothelial growth factor (VEGF) and its cellular receptor KDR (VEGFR2) in the bone marrow of patients with acute myeloid leukemia. Leukemia 2002;16:1302-10.
55. Ghannadan M, Wimazal F, Simonitsch I, et al. Immunohistochemical detection of VEGF in the bone marrow of patients with acute myeloid leukemia. Correlation between VEGF expression and the FAB category. Am J Clin Pathol 2003;119:663-71.
56. Jothlingam P, Basu D, Dutta TK. Angiogenesis and proliferation index in patients with acute myeloid leukemia: a prospective study. Bone Marrow Res 2014;2014:634874.
57. Song YQ, Tan Y, Liu LB, Wang Q, Zhu J, Liu M. Levels of bone marrow microvessel density are crucial for evaluating the status of acute myeloid leukemia. Oncology Lett 2015;10:211-5.
58. Kuzu I, Beksac M, Arat M, Celebi H, Elhan H, Erekul S. Bone marrow microvessel density (MVD) in adult acute myeloid leukemia (AML): therapy induced changes and effects on survival. Leuk Lymphoma 2004;45:1185-90.
59. Weidenaar AC, ter Elst A, Koopmans-Klein G, et al. High acute myeloid leukemia derived VEGFA levels are associated with a specific vascular morphology in the leukemic bone marrow. Cell Oncol 2011;34:289-96.
60. Aguayo A, Kantarjian H, Gidel C, et al. Angiogenesis in acute and chronic leukemias and myelodysplastic syndromes. Blood 2000;96:2240-5.
61. Chand R, Chandra H, Chandra S, Verma SK. Role of microvessel density and vascular endothelial growth factor in angiogenesis of hematological malignancies. Bone Marrow Res 2016;5043383.
62. Song MZ, Wang HP, Ye QL. Increased circulating vascular endothelial growth factor in acute myeloid leukemia patients: a systematic review and meta-analysis. Syst Rev 2020;9:103.
63. Guo BP, Liu Y, Tan XH, Cen H. Prognostic significance of vascular endothelial growth factor expression in adult patients with acute myeloid leukemia: a meta-analysis. Leuk Lymphoma 2013;54:1418-25.
64. Aguayo A, Kantarjian HM, Estey EH, et al. Plasma vascular endothelial growth factor levels have prognostic significance in patients with acute myeloid leukemia but not in patients with myelodysplastic syndromes. Cancer 2002;95:1923-30.
65. De Bont ES, Fidler V, Meeuwseen T, Scherpen F, Hahlen K, Kmaops WA. Vascular endothelial growth factor secretion is an independent prognostic factor for relapse-free survival in pediatric acute myeloid leukemia patients. Clin Cancer Res 2002;8:2856-61.
66. Rabitsch W, Sperr WR, Lechner K, et al. Bone marrow microvessel density and its prognostic significance in AML. Leuk Lymphoma 2004;45:1369-73.
67. Savic A, Cemericik-Martinovic V, Dovat S, et al. Angiogenesis and survival in patients with myelodysplastic syndrome. Pathol Oncol Res 2012;18:681-90.
69. Mohammadisl J, Khosravi A, Shahjahani M, Azizidoost S, Saki N. Molecular and cellular aspects of extramedullary manifestations of acute myeloid leukemia. J Cancer Metast Treat 2016;2:44-50.
70. Frietschj JJ, Huntstig F, Wittke C, et al. Extra-medullary recurrence of myeloid sarcoma after allogeneic stem cell transplantation: impact of conditioning intensity. Bone Marrow Transplant 2020. in press
71. Piccaluga PP, Paolini S, Navari M, Etebari M, Visani G, Ascani S. Increased angiogenesis seems to correlate with inferior overall survival. Pol J Pathol 2018;69:254-65.
72. Hiramatsu A, Miwa H, Shikami M, et al. Disease-specific expression of VEGF and its receptors in AML cells: possible autocrine pathway of VEGF/type1 receptor if VEGF in t(15;17) AML and VEGF/type 2 receptor of VEGF in t(8;21) AML. Leuk Lymphoma 2006;47:89-95.
73. Imai N, Shikami M, Miwa H, Suganuma K. T(8;21) acute myeloid leukaemia cells are dependent on vascular endothelial growth factor (VEGF)/VEGF receptor type 2 pathway and phosphorylation of Akt. Brit J Haematol 2006;135:673-82.
74. Ter Elst A, Ma B, Scherpen F, et al. Repression of vascular endothelial growth factor expression by the runt-related transcription factor 1 in acute myeloid leukemia. Cancer Res 2011;71:2761-71.
75. Saulle E, Petronelli A, Pelosi E, et al. PML-RAR alpha induces the downmodulation of HHEX: a kay event responsible for the induction of an angiogenetic response. J Hematol Oncol 2016;9:33.
76. Dias S, Hattori K, Heissig B, et al. Inhibition of both paracrine and autocrine VEGF /VEGF-R2 signaling pathways is essential to induce long-term remission of xenotransplanted human leukemias. Proc Natl Acad Sci U S A 2001;98:10857-62.
77. Zhu Z, Hattori K, Zhang H, et al. Inhibition of human leukemia in an animal model with human antibodies directed against vascular endothelial growth factor receptor 2. Correlation between antibody affinity and biological activity. Luekemia 2003;17:604-11.
78. Zahiragic L, Schliemann C, Bieker R, et al. Bevacizumab reduces VEGF expression in patients with relapsed and refractory acute myeloid leukemia without clinical antileukemic activity. Leukemia 2007;21:1310-2.
79. Fiedler W, Mesters R, Tinnefeld H, et al. A phase 2 clinical study of SU5416 in patients with refractory acute myeloid leukemia. Blood 2003;102:2763-7.
80. Fiedler W, Serve H, Dohner H, et al. A phase 1 study of SU11248 in the treatment of patients with refractory or resistant acute myeloid leukemia (AML) or not amenable to conventional therapy for the disease. Blood 2005;105:986-93.
81. Nobrega-Pereeira S, Caiado F, et al. VEGFR2-mediated reprogramming of mitochondrial metabolism regulates the sensitivity of acute myeloid leukemia to chemotherapy. Cancer Res 2018;78:731-41.
82. Zhao D, Hou H, Zhang XC. Progress in the treatment of solid tumors with apatinib: a systematic review. OncoTargets Ther 2018;11:4137-47.
83. Yu L, Deng MM, Li ZF, Fang ZH, Dai Y, Xu B. Apatinib exhibits cytotoxicity to acute myeloid leukemia cell via targeting VEGFR2-mediated pro-survival signaling and angiogenesis. Blood 2019;134:51548.
84. Deng MM, Zha J, Zhao HJ, et al. Apatinib exhibits cytotoxicity toward leukemia cells by targeting VEGFR2-nediated prosurvival signaling and angiogenesis. Exp Cell Res 2020;390:111934.
85. Borthakur G, Kantarjian H, Ravandi F, et al. Phase I study of sorafenib in patients with refractory or relapsed acute leukemias. Haematologica 2011;96:62-8.
86. Rollig C, Serve H, Huttmann A, et al. Addition of sorafenib versus placebo to standard therapy in patients aged 60 years or younger with newly diagnosed acute myeloid leukemia (SORAML): a multicentre, phase 2, randomized controlled trial. Lancet Oncol 2015;16:1691-9.
87. Sharinen P, Eklund L, Alitalo K. Therapeutic targeting of angiopoietin-TIE pathway. Nat Rev Drug Discov 2017;16:635-51.
88. Jeansson M, Gawlik A, Anderson G, et al. Angiopoietin-1 is essential in mouse vasculature during development and in response to injury. J Clin Invest 2011;121:2278-89.
89. Daly C, Pasnikowski E, Burova E, et al. Angiopoietin-2 functions as an autocrine protective factor in stressed endothelial cells. Proc Natl Acad Sci U S A 2006;103:15491-96.
90. Daly C, Eichten A, Castanero C, et al. Angiopoietin-2 functions as a Tie2 agonist in tumor models, where it limits the effects of VEGF inhibition. Cancer Res 2013;73:108-18.
91. Watarai M, Miwa H, Shikami M, et al. Expression of endothelial cell-associated molecules in AML cells. Leukemia 2002;16:112-9.
92. Schliemann C, Bieker R, Padro T, et al. Expression of angiopoietins and their receptor Tie2 in the bone marrow of patients with acute myeloid leukemia. Haematologica 2006;91:1203-11.
93. Schliemann C, Bieker R, Thoennissen N, et al. Circulating angiopoietin-2 is a strong prognostic factor in acute myeloid leukemia. Leukemia 2007;21:1901-6.
94. Loges S, Heil G, Bruweleit M, et al. Analysis of concerted expression in acute myeloid leukemia: expression of angiopoietin-2 represents an independent prognostic factor for overall survival. J Clin Oncol 2005;23:1109-17.
95. Riccioni R, Diverio D, Mariani G, et al. Expression of Tie-2 and other receptors for endothelial growth factors in acute myeloid leukemias is associated with monocytic features of leukemic blasts. Stem Cells 2007;25:1862-71.
96. Lewis CE, De Palma M, Naldini L. Tie-2 expressing monocytes and tumor angiogenesis: regulation by hypoxia and angiopoietin-2. Cancer Res 2009;67:8429-32.
97. Riccioni R, Pelosi E, Riti V, Castelli G, Lo-Coco F, Testa U. Immunophenotypic features of acute myeloid leukaemia patients exhibiting high FLT3 expression not associated with mutations. Br J Haematol 2011;153:33-42.
98. Bchegowda L, Morrone K, Winski SL, et al. Pexmetinib: a novel dual inhibitor of Tie2 and p38 MAPK with efficacy in preclinical models of myelodysplastic syndromes and acute myeloid leukemia. Cancer Res 2016;76:4841-9.
99. Nichol D, Stuhlmann H. EGFL7: a unique angiogenic signaling factor in vascular development and disease. Blood 2012;119:1345-52.
100. Hong G, Kuek V, Shi JX, et al. EGFL7: master regulator of cancer pathogenesis, angiogenesis and an emerging mediator of bone homeostasis. J Cell Physiol 2018;233:8526-37.
101. Papaioiannou D, Shen CX, Nicolet D, et al. Prognostic and biological significance of the proangiogenic factor EGFL7 in acute myeloid leukemia. Proc Natl Acad Sci U S A 2017;114:E4641-7.
102. Chen ZH, Dai YF, Pang YF, et al. High EGFL7 expression may predict poor prognosis in acute myeloid leukemia patients undergoing allogeneic hematopoietic stem cell transplantation. Cancer Biol Ther 2019;20:1314-8.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Testa U, Castelli G, Pelosi E. Angiogenesis in acute myeloid leukemia. J Cancer Metastasis Treat 2020;6:53. http://dx.doi.org/10.20517/2394-4722.2020.111
AMA Style
Testa U, Castelli G, Pelosi E. Angiogenesis in acute myeloid leukemia. Journal of Cancer Metastasis and Treatment. 2020; 6: 53. http://dx.doi.org/10.20517/2394-4722.2020.111
Chicago/Turabian Style
Testa, Ugo, Germana Castelli, Elvira Pelosi. 2020. "Angiogenesis in acute myeloid leukemia" Journal of Cancer Metastasis and Treatment. 6: 53. http://dx.doi.org/10.20517/2394-4722.2020.111
ACS Style
Testa, U.; Castelli G.; Pelosi E. Angiogenesis in acute myeloid leukemia. J. Cancer. Metastasis. Treat. 2020, 6, 53. http://dx.doi.org/10.20517/2394-4722.2020.111
About This Article
Special Issue
Copyright

Data & Comments
Data


 Cite This Article 18 clicks
Cite This Article 18 clicks

 Like This Article 39
likes
Like This Article 39
likes






Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.