
Bulbine frutescens phytochemicals as novel ABC-transporter inhibitor: a molecular docking and molecular dynamics simulation study

Abstract
Aim: The present in silico study aimed to evaluate the ATP-binding cassette (ABC) transporter inhibition potential of Bulbine frutescens (B. frutescens) phytochemicals.
Methods: Several previous studies and databases were used to retrieve the ligands and target protein structure. The molecular docking study was performed using the Auto Dock Tools, and the GROMACS package was applied to accomplish molecular dynamics simulation.
Results: Utilizing the molecular docking and simulation approach, ~25 phytochemicals were screened against the ABC transporter protein. Docking score analysis revealed that B. frutescens phytochemical 4’-Demethylknipholone 2’-β-D-glucopyranoside exhibited strong binding on the ABC transporter protein with a minimum binding score -9.8 kcal/mol in comparison to the standard ABC transporter inhibitor diltiazem (-6.86 kcal/mol). Furthermore, molecular dynamics simulation for 4’-Demethylknipholone 2’-β-D-glucopyranoside showed an acceptable root mean square deviation, radius of gyration, root mean square fluctuation, and hydrogen bond, in addition to other lead compounds.
Conclusion: The in-silico study demonstrated that B. frutescens phytochemical 4’-Demethylknipholone 2’-β-D-glucopyranoside possesses anti-drug resistance properties and requires further testing in preclinical settings.
Keywords
Introduction
Cancer is one of the major health problems worldwide. In addition to surgery, radiation, and hormonal therapy, chemotherapy is an important strategy in the management of cancer[1,2]. Cancer drug resistance is a well-known phenomenon that arises when the disease becomes refractory to chemotherapy[3]. One of the mechanisms related to drug resistance in cancer cells is the involvement of drug efflux transporters such as P-glycoprotein, also known as ABC (ATP-binding cassette) transporters. Besides their normal role, these proteins facilitate the movement of drugs and their metabolites across the cellular membrane. ABC transporters-mediated drug efflux is the cause of the relapse of disease in patients and decreases therapeutic output[4]. Therefore, ABC transporters remains an important target for therapeutic strategy against this devastating disease[4].
Phytochemicals are known to possess anticancer activity and play an important role in the management of the disease[5]. Various properties such as reducing side-effects, efficacy in chronic conditions, cost-effectiveness, and widespread usage of natural compounds give them a role as chemotherapeutic agent. Thus, currently, natural compounds are widely used in cancer chemotherapy and have a promising future[6]. Bulbine frutescens (B. frutescens) is a flowering plant that belongs to the Asphodelaceae family. It is a frequently used medicinal plant found in South Africa for the treatment of burns and skin wounds[7]. The potent anti-proliferative activity of B. frutescens was first reported in 2014[8]. There are few published articles on B. frutescens species which encompass the isolation of its phytochemicals and their biological activities. Aqueous extracts of B. frutescens can increase glucose utilization activity in vitro[9]. B. frutescens phytochemical isofuranonapthoquinone 1 is a known inhibitor of glutathione transferase P1-1 protein involved in anticancer drug detoxification-mediated drug resistance[10]. The ethanolic and aqueous extracts of the Bulbine genus inhibit HIV-1 protease activity. Recently, we reported that B. frutescens phytochemicals (extract) have strong potential to induce apoptosis and inhibit Notch signaling pathway in breast cancer cells[11].
The computational approach is a fast-growing stream that employs an integrative approach to characterize the selective target of a particular disease and explore the interactions involved with drugs or other molecules[12]. Computer-based drug discovery is a highly time- and cost-effective strategy. Various online and offline tools and programs are available which have been used in computer-based drug discovery and make the process easier and cost effective. In the present study, we screened the ABC transporter (PDB 3G60) inhibitory potential of B. frutescens phytochemicals and predicted their possible IC50 using an in silico approach.
Methods
Software, servers, and tools
The present study was performed on HP Workstation with 3.40 GHz processor, 16 GB RAM, and 1 TB hard disk running the Ubuntu 16.04.5 LTS and Windows 7 operating systems. Bioinformatics tools, such as AutoDock, PyRx, and GROMACS, were used for molecular docking and simulation studies. Pymol, VMD (visual molecular dynamics), Open Babel, LigPlot+ (v.1.4.5), Marvin Sketch 15.9.7.0 UCSF Chimera, and Avogadro were employed for visualization purposes. National Center for Biotechnology Information (NCBI), Protein data bank (PDB), PubChem, Swiss Model, Charmm-gui, RAMPAGE, etc. online servers/resources were used for the retrieval, evaluation, and analysis of the data.
Identification, retrieval, and preparation of ligands
The phytochemicals present in B. frutescens were searched in the published literature[13]. The 2D/3D structures of identified phytochemicals and standard inhibitor compounds of the test protein were downloaded from the NCBI PubChem database. Open Babel molecule format converter[14] and Marvin Sketch 15.9.7.0 software packages were utilized to perform 2D to 3D conformation and .sdf to .pdb format conversions, respectively. The force field mmff94 and conjugate gradients optimization algorithm were applied to minimize the test ligands energy using PyRx-Python prescription 0.8 for 200 steps[15].
Retrieval and preparation of the receptor
The 3D structure of ABC-transporter protein or P-glycoprotein (PDB ID 3G60) was retrieved from RCSB-PDB[16] with a resolution of 4.40 Å. The protein structure was loaded into UCSF Chimera for molecular docking preparation[17]. The protein model was cleaned and optimized by removing the ligands and heteroatoms such as water. The protein structure was minimized by using UCSF Chimera software with a 100-step steepest descent and a 10-step conjugate gradient method. The step size of both methods was 0.02 Å.
Molecular docking
Auto Dock Tools 1.5.6 (ADT) was used for the docking of the protein and ligands[18]. Gestgeiger partial charges were allocated and torsions were introduced to the test ligands. Using ADT, Kollman charges, polar hydrogen atoms, and solvation parameters were applied. The Lamarckian Genetic Algorithm was applied to discover the place for active binding. A maximum of 27,000 GA operations were made for each of the thirty independent runs on a single population of 150 individuals. Operator loads for cross overrate, elitism, and gene mutation rate were set as default parameters.
Molecular dynamics simulation
The GROMACS (version 2018) package was used to perform molecular dynamics simulation analysis of the dynamic behavior of membrane-embedded P-glycoprotein complexes with AMBER Lipid14 force field. The receptor topology was constructed by pdb2gmx. Online server CHARMM-gui was used to insert the dipalmitoylphosphatidylcholine (DPPC) membrane. The ligand potential energy function was derived using AMBER GAFF parameters. SPCe water model was applied to represent the water molecules explicitly. To neutralize the positive charge on the receptor, the respective number of negative ions was added. Bad contact inside the system was discarded by energy minimization through the steepest descent method, and the long-range interactions were computed by particle mesh Ewald at every step. The energy minimized system underwent 100 ps (NVT) and 1ns (NPT) equilibration run at constant T (323 K) and pressure (1 bar). We employed modified Berendsen thermostat and Parrinello-Rahman barostat for NVT and NPT equilibrations. In addition, to avoid the hot solvent-cold solute problem, temperature coupling was applied by the indexing system into water and non-water components. On the completion of two equilibration phases, the production run was conducted for 10 ns after taking away the position restraints. Throughout the data collection phase, the pressure and temperature were maintained with the same methods as in the NPT and NVT phases, respectively. Moreover, the P-glycoprotein ligand complex coordinates were saved every 1 ps. Structural analysis results were processed by using modules of GROMACS package. The appropriate GROMACS command was used for the root mean square fluctuation (RMSF), Root Mean Square Deviation (RMSD), radius of gyration, peptide-peptide hydrogen bond, peptide-water hydrogen bond, and SASA calculations. xmgrace software was used to visualize these GROMACS data calculations.
Molecular Mechanics/Generalized Born Model and Solvent Accessibility analysis
Prime Molecular Mechanics/Generalized Born Model and Solvent Accessibility (MM-GBSA) was performed to calculate the protein and protein-ligand binding energies. The analysis utilized the VSGB solvent model, OPLS_2005 force field, and rotamer search algorithms. The glide pose viewer file was utilized to compute the total binding free energy using the Prime MM-GBSA simulation aid. The MM/GBSA analysis provided relative binding affinity of the target protein and target protein-ligand complex (kcal/mol). High negative MM/GBSA binding energies indicate stronger binding[19].
Prediction of inhibitory concentration
The prediction of 50% inhibitory concentration (pIC50, Prediction of inhibitory concentration) for the lead compound was performed using the experimentally established IC50 of the standard inhibitors such as tariquidar (0.015 µM), elacridar (0.05 µM), verapamil (14.3 µM), felodipine (127.3 µM), diltiazem (11.7 µM), and quinidine (51.3 µM)[20,21]. Linear regression analysis was performed through GraphPad Prism software to calculate the pIC50.
Results
Binding mode analysis of ligands
The phytochemicals present in B. frutescens were searched from the published literature and their two-dimensional structures are depicted in Figure 1.
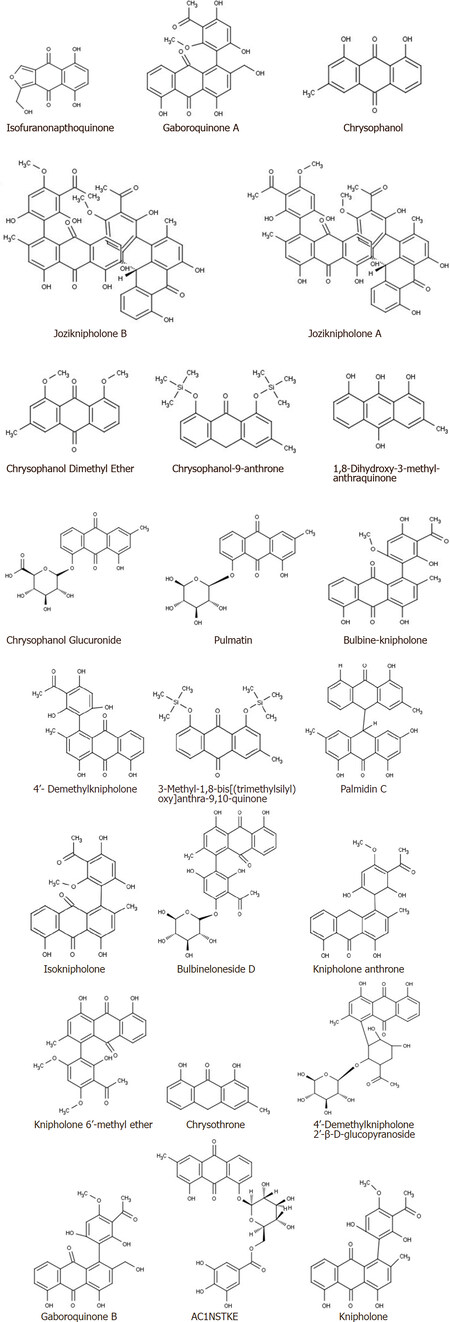
Figure 1. Structure of B. frutescens phytochemicals. B. frutescens: Bulbine frutescens
The results show that 4’-Demethylknipholone 2’-β-D-glucopyranoside acts as a potential P-glycoprotein inhibitor. The interaction poses of 4’-Demethylknipholone 2’-β-D-glucopyranoside with the target protein are depicted in Figure 2A. The phytochemical binds with the P-glycoprotein transporter with higher affinity (-9.8 kcal/mol) than the known standard diltiazem (-6.86 kcal/mol) inhibitor. Phe339, Ile336, Leu335, Phe728, Phe332, Ser725, Ala981, Val978, Ile977, Leu64, Tyr949, Phe974, Leu971, Met68, Phe71, Phe953, Gln128, and Glu871 of P-glycoprotein transporter showed hydrophobic and hydrogen interactions with the 4’-Demethylknipholone2’-β-D-glucopyranoside. However, amino acids Gln721 and Gln986, Gly985, Phe339, Met982, Ala981, Leu335, Phe332, Val978, Tyr303, Phe724, Leu758, Ser725, Ser975, Phe974, Phe728, Ser729, Leu972, and Leu971 of P-glycoprotein transporter form hydrogen and hydrophobic interactions, respectively, with standard inhibitor diltiazem. The binding energy score of other B. frutescens identified phytochemicals are shown in Figure 2B.
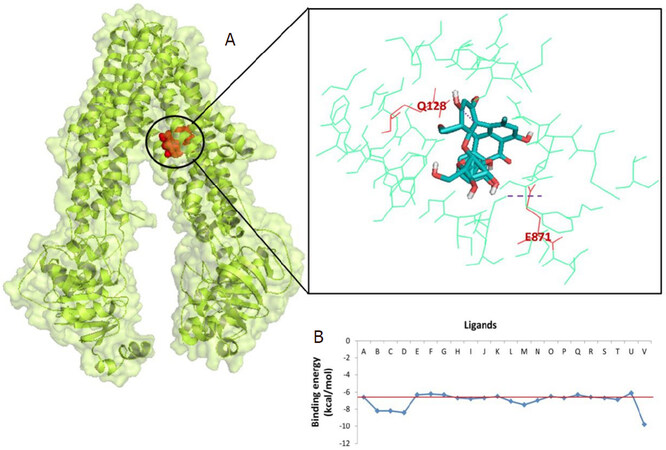
Figure 2. P-glycoprotein (PDB ID: 3G60) interaction with 4’-Demethylknipholone 2’-β-D-glucopyranoside: (A) representation of amino acid residues involved in hydrogen bond formation; and (B) binding energy score of the B. frutescens ligand and P-glycoprotein interaction. Ligand A represents standard inhibitor diltiazem while Ligands B-V represent B. frutescens phytoconstituents (Isofuranonapthoquinone, Joziknipholone, Joziknipholone, B,Chrysophanol, Chrysothrone, Chrysophanol Dimethyl Ether, 4’- Demethylknipholone, Pulmatin, Knipholone, Gaboroquinone B, AC1NSTKE, Palmidin C, Bulbineloneside D, Gaboroquinone A, Knipholone Anthrone, Knipholone 6’-methyl ether, Bulbine-Knipholone, Isoknipholone, Chrysophanol Glucuronide, 1,8-Dihydroxy-3-methyl-anthraquinone, and 4’-Demethylknipholone 2’-β-D-glucopyranoside, respectively). B. frutescens: Bulbine frutescens
Receptor-ligand energy calculation through MM-GBSA assay
The energy calculation for the unbound receptor and receptor-ligand complex was estimated by using MM-GBSA analysis, and the results are depicted in Figure 3A. The results show that binding free energy of the protein-ligand complex was highly negative (-41,338.2 kcal/mol) in comparison to the unbound protein (-41,241.2 kcal/mol).
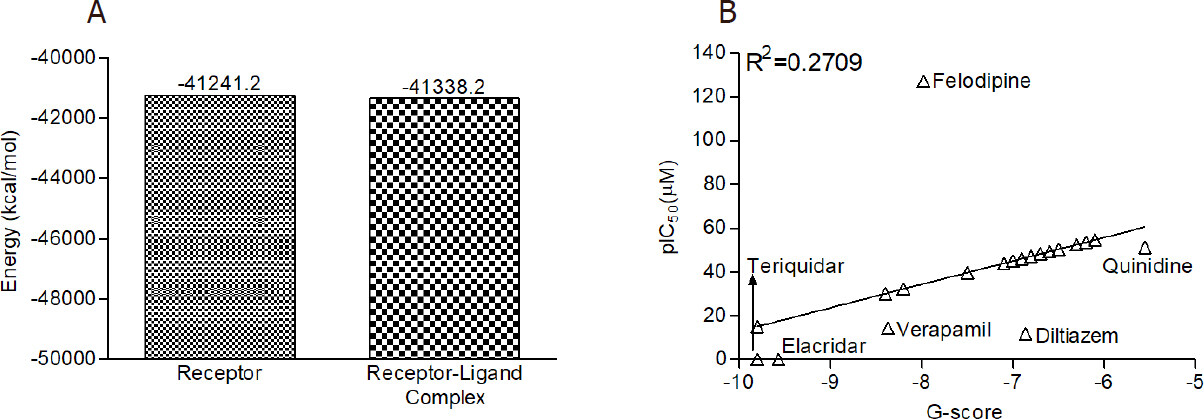
Figure 3. The MM-GBSA analysis and predicted IC50 of B. frutescens phytochemicals: (A) MM-GBSA analysis of unbound ABC transporter and lead phytochemical 4’-Demethylknipholone 2’-β-D-glucopyranoside-ABC transporter complex; and (B) regression analysis for the prediction of IC50 value of B. frutescens identified phytochemicals against P-glycoprotein. MM-GBSA: Molecular Mechanics/Generalized Born Model and Solvent Accessibility; ABC transporter: ATP-binding cassette transporter; pIC50: Prediction of inhibitory concentration; B. frutescens: Bulbine frutescens
Prediction of pIC50
The IC50 of lead B. frutescens phytochemicals were predicted on the basis of reported experimental IC50 of P-glycoprotein standard inhibitors. The predicted IC50 (pIC50) values for the lead phytochemicals were between 15.08 and 54.67 µM. The results are comparable to the range of IC50 of standard compounds (0.015-127.30 µM) [Figure 3B]. A weak positive correlation (R2 = 0.2709) was observed among the pIC50 and binding energy of the test compounds.
Molecular dynamics simulation
P-glycoprotein is a membrane-bound protein, thus the protein was incorporated in a DPPC membrane during the process of MD simulation run [Figure 4]. The Protein-ligand complex was subjected to MD simulation to analyze the dynamics of the binding mechanism of the ligand at the P-glycoprotein active site under clearly expressed water condition. The conformational changes analysis of the protein-ligand complex was analyzed during the last 5 ns of the 10-ns MD production run. RMSD is a fundamental method to analyze structural stability. A decreased RMSD of the complex with respect to the initial structure was observed [Figure 5A]. The RMSD plot revealed that binding of ligand stabilized the P-glycoprotein structure such that the complex showed less deviation and conformational changes compared to unbound protein [Figure 5A]. The radius of gyration (Rg) is used to analyze the compactness of the protein-ligand complex and unbound protein structure in a biological system. The results show that the Rg for P-glycoprotein-ligand complex decreased along the simulation time, showing an increase in the compactness of the structure [Figure 5B]. The RMSF of the P-glycoprotein and ligand complex was plotted to visualize the average fluctuation of all the amino acid residues during the simulation process [Figure 5C]. The RMSF plot indicates that amino acid residue fluctuations are present in the protein during ligand-bound state at several times. The results depict that interaction of ligand and protein brings both subunits (two arms of the protein) closer to each other and leads to the reduction in the distance between them [Figure 6A-D].
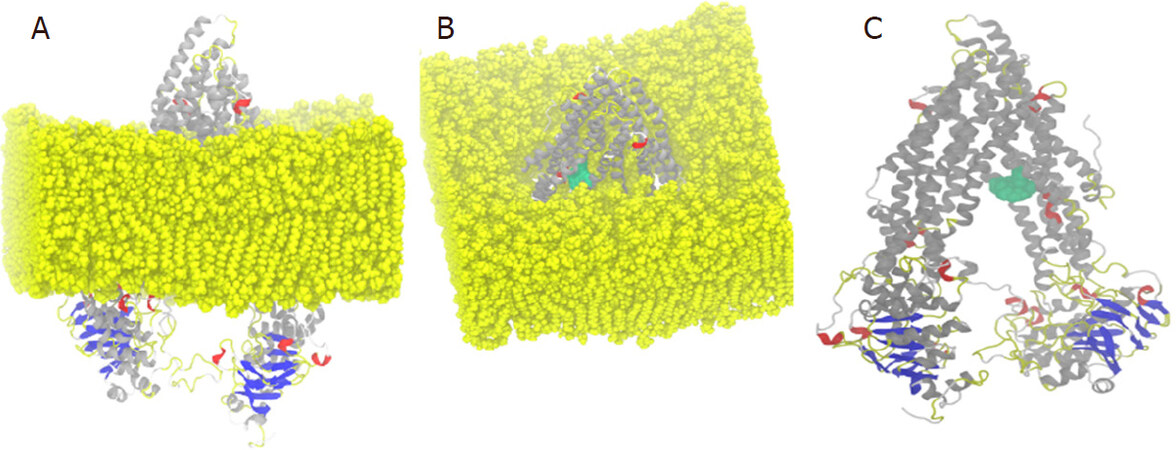
Figure 4. (A) Lateral view of protein-ligand complex incorporated in dipalmitoylphosphatidylcholine (DPPC) membrane; (B) upper view of protein-ligand complex incorporated in the DPPC membrane; and (C) ligand (green color) complex structure without DPPC membrane
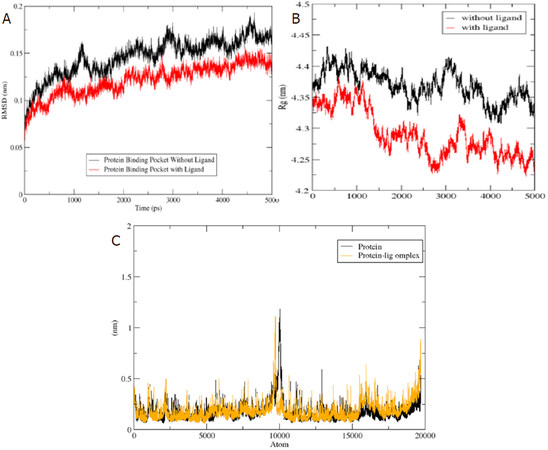
Figure 5. (A) The RMSD of unbound protein (ABC transporter) and bound protein-ligand (ABC transporter-4’-Demethylknipholone 2’-β-D-glucopyranoside) complex along the 5 ns of simulation time; (B) time evolution of radius of gyration value during 5 ns of MD simulation; and (C) RMSF plot during the 5 ns of simulation time. Black and yellow color peaks represent unbound protein and bound protein-ligand complex, respectively. RMSD: Root Mean Square Deviation; RMSF: root mean square fluctuation; ABC transporter: ATP-binding cassette transporter

Figure 6. (A) Distance measurement between two subunits of unbound protein; (B) distance measurement between two subunits of a bound protein-ligand complex; (C) yellow and red colored superimposed structure of unbound protein and protein-ligand complex, respectively; and (D) list of bond distances between residues of unbound protein and protein-ligand complex
Peptide-water hydrogen bond examination was conducted to determine the atomistic interaction between the protein and water in the presence and absence of the ligand (4’-Demethylknipholone 2’-β-D-glucopyranoside) during the simulation period. In the protein-ligand complex, there is a decrease in hydrogen bond number between protein and water compared to protein alone [Figure 7A]. Peptide-peptide hydrogen bond analysis was accomplished to find the interaction between the protein residues. During the analysis, overall, it was found that ligand-protein interaction significantly increased the number of hydrogen bonds [Figure 7B]. The SASA of the protein was calculated during the MD simulation in unbound and ligand-bound conditions. SASA values changed due to the binding of the ligand to the protein [Figure 7C]. The analysis indicates the folding states of protein and its stability upon ligand binding.
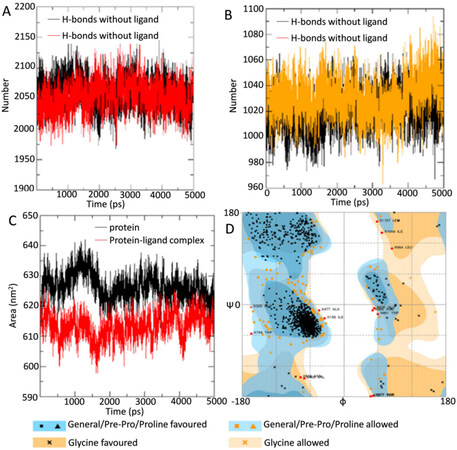
Figure 7. (A) H-bonds between protein and water in unbound protein and protein-ligand complex; (B) intramolecular H-bonds in unbound protein and bound protein-ligand complex; (C) solvent accessible surface area analysis for unbound protein and bound protein-ligand complex; and (D) assessment of Ramachandran plot of protein using the online server RAMPAGE
Discussion
The failure of cancer therapy largely arises due to the multidrug resistance potential of the cancer cells which occurs through the overexpression and/or hyperactivation of the ABC transporters. The alteration in molecular mechanisms and gene regulation often promotes overexpression of P-glycoprotein (ABC transporter) in tumors. Overexpression of ABC transporters limit the anticancer drug response in cancer patients by reducing the accumulation of the chemotherapeutic drug in cancer cell/tumor through an efflux mechanism. Plasma membrane glycoprotein (Pgp) has been known to employ resistance to a variety of cytotoxic drugs including anticancer drugs such as vinblastine, doxorubicin, indinavir, ritonavir, and paclitaxel[4]. Chemotherapy failure in different types of cancer occurs due to MDR, which enables failure of cancer treatment in ~90% of cancer patients[22]. Development of ABC transporter inhibitors might be one tactic to overcome multidrug resistance through sensitization of cancer cells for chemotherapeutic drugs. P-glycoprotein inhibition may also increase the drug load in the cells and increase excessive toxicity.
In the present in silico study, we employed B. frutescens phytochemicals against ABC transporter protein. Following the in-silico analysis, compound 4’-Demethylknipholone 2’-β-D-glucopyranoside was identified as a potent natural inhibitor of P-glycoprotein transporter. This is the first report on the Knipholone group of compounds acting as potent inhibitors for ABC transporter. Aller et al.[23] revealed that hydrophobic, aromatic, polar, and charged residues of the drug binding pocket provide poly-specificity to the P-glycoprotein toward the drug. They also showed that amino acid residues Phe332, Ile336, Phe339, Tyr949, Phe974, Val978, Ala981, Leu64, Leu335, Leu971, and Phe728 play a crucial role in the substrate/inhibitor-transporter interaction. They showed that V978 plays an important role in binding with the standard inhibitors (verapamil and QZ59) of the protein. Interestingly, in the present study, we found that the lead molecule (4’-Demethylknipholone 2’-β-D-glucopyranoside) showed interaction with key amino acids including V978. The IC50 of the identified B. frutescens phytochemical inhibitors were also predicted. The results show that compound 4’-Demethylknipholone 2’-β-D-glucopyranoside possesses higher activity at a lower concentration (15.08 µM). Jouan et al.[21] demonstrated that felodipine possess ABC transporter inhibitory activity at a concentration of 127.3 µM, which is comparatively much higher than the tested compound 4’-Demethylknipholone 2’-β-D-glucopyranoside.
MM-GBSA analysis also revealed that lead B. frutescens phytoconstituent 4’-Demethylknipholone 2’-β-D-glucopyranoside forms a tight complex with ABC transporter and was stable in comparison to unbound ABC transporter protein. Previous studies have also utilized MMGBSA analysis to evaluate the ligand binding potential with the ABC transporter[24]. The stabilization of ligand-protein complex can be analyzed thorough MD simulations In addition, this approach evaluates conformational changes and stability of the three-dimensional structure of the protein. The present MD simulation study showed tight binding potential of the ligand, which provided stability to the ABC transporter-ligand complex. Our study corroborates the previous study performed by Shahraki et al.[25], which showed that 1,4-Dihydropyridines inhibits ABC transporter by providing stability to the ABC transporter-ligand complex. The major limitations of the study include the isolation and characterization of the lead molecule 4’-Demethylknipholone 2’-β-D-glucopyranoside from the B. frutescens plant. In conclusion, the present study highlighted that the compound 4’-Demethylknipholone 2’-β-D-glucopyranoside possesses remarkable inhibitory potential against ABC transporter. Further, preclinical in vitro and in vivo validation is required to establish the B. frutescens phytoconstituents as natural ABC transporter inhibitors. Altogether, the identified lead inhibitor in the present study might serve as a starting molecule in the chemotherapy resistance drug discovery in the future.
Declarations
Authors’ contributionsConceptualized and designed the study and prepared the draft manuscript: Kumar S
Performed the experiments and analyzed data: Kushwaha PP, Maurya SK, Singh A
Prepared manuscript: Prajapati KS, Singh AK, Shuaib M
Availability of data and materialsNot applicable.
Financial support and sponsorshipPPK acknowledges financial support from the Indian Council of Medical Research (ICMR), India in the form of ICMR Senior Research fellowship. SK acknowledges the University Grants Commission (UGC), India and Department of Science and Technology (DST), India for providing financial support in the form of UGC-BSR Research Start-Up-Grant [No. F.30–372/2017 (BSR)] and DST-SERB Grant (EEQ/2016/000350) respectively. SK also acknowledges DST, India for providing the Departmental DST-FIST grant to the Department of Biochemistry, Central University of Punjab, India. AKS and MS acknowledges CSIR and ICMR, India funding agencies respectively for providing financial assistance in the form of a Senior Research Fellowship. KSP acknowledge DBT, India funding agencies for providing financial assistance in the form of Junior Research Fellowship.
Conflicts of interestAll authors declared that there are no conflicts of interest.
Ethical approval and consent to participateThis study does not contain any studies with human participants or animals performed by any of the authors.
Consent for publicationNot applicable.
Copyright© The Author(s) 2021.
REFERENCES
1. Kaliberov SA, Buchsbaum DJ. Cancer treatment with gene therapy and radiation therapy. Adv Cancer Res 2012;115:221-63.
2. Kushwaha PP, Rapalli KC, Kumar S. Geminin a multi task protein involved in cancer pathophysiology and developmental process: a review. Biochimie 2016;131:115-27.
3. Kumar S, Kushwaha PP, Gupta S. Emerging targets in cancer drug resistance. Cancer Drug Resist 2019;2:161-77.
4. Falasca M, Linton KJ. Investigational ABC transporter inhibitors. Expert OpinInvestig Drugs 2012;21:657-66.
5. Kushwaha PP, Singh AK, Shuaib M, et al. 3-O-(E)-p-Coumaroylbetulinic acid possess anticancer activity and inhibit notch signaling pathway in breast cancer cells and mammosphere. ChemBiol Interact 2020;328:1-9.
6. Kinghorn AD, Chin YW, Swanson SM. Discovery of natural product anticancer agents from biodiverse organisms. Curr Opinion Drug Dis Dev 2009;12:189-96.
7. Pinar R. To investigate knowledge of Turkish people about cancer. Cancer Agenda 1998;12:66-73.
8. Tambama P, Abegaz B, Mukanganyama S. Antiproliferative activity of the isofuranonaphthoquinone isolated from Bulbinefrutescens against jurkat T cells. BioMed Res Int 2014;2014:1-14.
9. van Huyssteen M, Milne PJ, Campbell EE, van de Venter M. Antidiabetic and cytotoxicity screening of five medicinal plants used by traditional African health practitioners in the Nelson Mandela Metropole, South Africa. Afr J Tradit Complement Altern Med 2011;8:150-8.
10. Mukanganyama S, Bezabih M, Robert M, et al. The evaluation of novel natural products as inhibitors of human glutathione transferase P1-1. J Enzyme Inhib Med Chem 2011;26:460-7.
11. Kushwaha PP, Vardhan PS, Kapewangolo P, et al.
12. Ezebuo FC, Kushwaha PP, Singh AK, Kumar S, Singh P. In silico methods of drug design: molecular simulations and free energy calculations. In: Kumar S, Chukwuebuka E, editors. Phytochemistry: An in-silico and in-vitro Update. Singapore: Springer; 2019. pp. 521-33.
13. Kushwaha PP, Kumar A, Maurya S, Singh AK, Sharma AK, Kumar S. Bulbine frutescens phytochemicals as a promising anti-cancer drug discovery source: a computational study. In: Phytochemistry: An in-silico and in-vitro Update. Singapore: Springer; 2019. pp. 491-510.
14. O’Boyle NM, Banck M, James CA, et al. Open babel: an open chemical toolbox. J Cheminformatics 2011;3:1-14.
15. Dallakyan S. PyRx-python prescription v. 0.8. The Scripps Research Institute 2010. Available from: https://pyrx.sourceforge.io/. [Last accessed on 31 Dec 2020].
16. Berman H, Henrick K, Nakamura H, Markley JL. The worldwide Protein Data Bank (wwPDB): ensuring a single, uniform archive of PDB data. Nucleic Acids Res 2007;35:301-3.
17. Pettersen EF, Goddard TD, Huang CC, et al. UCSF Chimera - a visualization system for exploratory research and analysis. J ComputChem 2004;25:1605-12.
18. Trott O, Olson AJ. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem 2010;31:455-461.
19. Gupta S, Singh AK, Kushwaha PP, et al. Identification of potential natural inhibitors of SARS-CoV2 main protease by molecular docking and simulation studies. J Biomol Struct Dyn 2020:1-9.
20. Fox E, Bates SE. Tariquidar (XR9576): a P-glycoprotein drug efflux pump inhibitor. Expert Rev Anticancer Ther 2007;7:447-59.
21. Jouan E, Le Vée M, Mayati A, et al. Evaluation of P-glycoprotein inhibitory potential using a rhodamine 123 accumulation assay. Pharmaceutics 2016;82:1-13.
23. Aller SG, Yu J, Ward A, et al. Structure of P-glycoprotein reveals a molecular basis for poly-specific drug binding. Science 2009;323:1718-22.
24. Di Meo F, Fabre G, Berka K, et al. In silico pharmacology: drug membrane partitioning and crossing. Pharmacol Res 2016;111:471-86.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Kushwaha PP, Maurya SK, Singh A, Prajapati KS, Singh AK, Shuaib M, Kumar S. Bulbine frutescens phytochemicals as novel ABC-transporter inhibitor: a molecular docking and molecular dynamics simulation study. J Cancer Metastasis Treat 2021;7:2. http://dx.doi.org/10.20517/2394-4722.2020.92
AMA Style
Kushwaha PP, Maurya SK, Singh A, Prajapati KS, Singh AK, Shuaib M, Kumar S. Bulbine frutescens phytochemicals as novel ABC-transporter inhibitor: a molecular docking and molecular dynamics simulation study. Journal of Cancer Metastasis and Treatment. 2021; 7: 2. http://dx.doi.org/10.20517/2394-4722.2020.92
Chicago/Turabian Style
Kushwaha, Prem Prakash, Santosh Kumar Maurya, Amit Singh, Kumari Sunita Prajapati, Atul Kumar Singh, Mohd Shuaib, Shashank Kumar. 2021. "Bulbine frutescens phytochemicals as novel ABC-transporter inhibitor: a molecular docking and molecular dynamics simulation study" Journal of Cancer Metastasis and Treatment. 7: 2. http://dx.doi.org/10.20517/2394-4722.2020.92
ACS Style
Kushwaha, PP.; Maurya SK.; Singh A.; Prajapati KS.; Singh AK.; Shuaib M.; Kumar S. Bulbine frutescens phytochemicals as novel ABC-transporter inhibitor: a molecular docking and molecular dynamics simulation study. J. Cancer. Metastasis. Treat. 2021, 7, 2. http://dx.doi.org/10.20517/2394-4722.2020.92
About This Article
Copyright

Data & Comments
Data


 Cite This Article 41 clicks
Cite This Article 41 clicks

 Like This Article 31
likes
Like This Article 31
likes






Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.