
Genetic characteristics and molecular diagnostics of bone tumors

Abstract
The diagnosis of primary tumors of bone relies heavily on clinicopathological and radiological correlation and is often best performed in a multidisciplinary setting. Bone tumors comprise a heterogenous category of human lesions ranging from benign to malignant neoplasms. These tumors affect a wide age range and can become problematic for diagnosis when less common entities are encountered. Traditionally the pathological diagnosis of many bone tumors has been based primarily on the evaluation of hematoxylin and eosin-stained glass slides, sometimes combined with ancillary diagnostic techniques such as immunohistochemistry, conventional cytogenetics, fluorescence in situ hybridization, and polymerase chain reaction-based assays. More recently, the advent of massively parallel sequencing-based techniques has opened new avenues for diagnostic testing in bone tumors; however, these new testing modalities are sensitive to traditional decalcification procedures that are commonly used in the routine processing of bony specimens. Herein we provide a focused review concentrating on the molecular genetic features of bone tumors with specific, recurrent genetic alterations that make them appealing targets for directed ancillary testing by conventional or molecular techniques. In addition, specimen handling with regards to decalcification procedures are discussed and the different types of testing modalities available are reviewed.
Keywords
Introduction
Primary tumors of bone comprise a heterogeneous group of benign and malignant tumors that affect patients of a wide age range. While benign bone tumors are relatively common, malignant bone tumors are exceedingly rare, accounting for less than 1%-2% of all neoplastic disease[1-3]. While some of the more common bone lesions, such as osteochondroma, pose little problem for diagnosis, the less commonly occurring benign and malignant lesions can often lead to diagnostic difficulties. The histopathologic diagnosis of bone tumors is fraught with difficulty due to the histomorphologic overlap between many different types of tumors, including overlap between reactive, benign, and malignant lesions[4-5]. In addition, the diagnosis of bone lesions is heavily reliant on correlation with imaging characteristics to be able to localize lesions to specific locations within different bones and to sometimes assist in staging[6-8]. Thus, the diagnosis and treatment of patients with primary bone tumors is best accomplished with a multidisciplinary approach involving the coordination of surgeons, radiologists, pathologists, and oncologists to produce optimal patient care[9-10].
Given the inherent diagnostic difficulties associated with primary bone tumors, ancillary testing modalities that may assist in diagnosis are beneficial to patient care. Precision medicine for bone cancer has lagged behind soft tissue, epithelial, and hematologic neoplasms in part due to the special processing procedures required for bony tissues[11]. Traditionally, diagnosis has relied on histopathological assessment of tumor tissue combined with clinical and radiological correlation. This has been supplemented with cytogenetic analysis including karyotype analysis and fluorescence in situ hybridization (FISH)[12-16]. Immunohistochemical analysis has played little role in the diagnosis of bone tumors, although recent molecular advances have provided pathologists with specific targets amenable to antibody interrogation in some tumors such as giant cell tumor of bone and chondroblastoma[17]. Polymerase chain reaction (PCR)[18-20] and, more recently, massively parallel next generation sequencing (NGS) based assays have provided additional tools for assessing molecular alterations in bone tumors[21-23]. However, it is worth noting that many of these ancillary testing modalities are exquisitely sensitive to conventional processing techniques, particularly decalcification, and care must be taken when processing bony lesions with consideration for downstream testing that may take place after the histopathological examination. This review aims to discuss the molecular genetic landscape of many of the primary bone tumors, specimen handling, and the ancillary testing available for diagnosis.
Specimen handling
Processing and handling are critical components for the proper diagnosis of bony surgical pathology specimens. When sufficient material is available, receiving fresh tissue is preferred as it allows for a portion of the tissue to be preserved (often fresh in saline or snap frozen) for subsequent cytogenetic, molecular, and microbiological studies[24]. Priority however should be given to tissue that will be used for morphological analysis, as that remains the cornerstone of histopathologic diagnosis of bone lesions. Routine fixation using 10% formalin and paraffin embedding is acceptable for this purpose and generally does not interfere significantly with testing of antigen expression or genomic material[25,26]. It is also worth mentioning that numerous other pre-analytical variables can have large effects on nucleic acid retrieval including specimen collection techniques, storage practices, temperature, duration of processing, and dehydration protocols amongst others. However, an in-depth discussion of pre-analytical variables is beyond the scope of this review which will focus on decalcification as it pertains specifically to bone specimens. Bone specimens often require additional preparation in the form of decalcification to allow for proper processing of the bone sections and creation of glass slides that can then be reviewed microscopically; however, decalcification is known to have detrimental effects on subsequent molecular testing[27,28]. Decalcification is most often accomplished using strong (e.g., hydrochloric acid or nitric acid) or weak (e.g., acetic acid or formic acid) inorganic acidic solutions to aid in the demineralization of bony specimens [Table 1]. Although generally adequate for preserving histomorphology, decalcification with acidic solutions may lead to the degradation of genomic material and cause interference when molecular testing is performed[29,30]. Acid decalcification with formic acid (a weak acid) has been reported to preserve genomic material for sequencing analysis although it can cause severe damage to DNA and RNA when longer decalcification times are used[31]. Picric acid may also not be optimal for nucleic acid retrieval[32]. Alternative methods or modifications to standard decalcification that exist include the use of ethylenediaminetetraacetic acid (EDTA), microwave, ultrasonography, and other types of acid. Acids or EDTA may be used in bone decalcification and combined with microwave or ultrasonography to reduce the time needed to decalcify, and EDTA decalcification alone appears to provide some measure of genomic material preservation compared to stronger acid solutions[33-37]. Decalcification times vary for both acidic solutions and EDTA and can be modified with additional factors such as temperature or mechanical agitation[38]. When handling bone specimens it is important to be familiar with the tissue processing procedures employed by the laboratory so that consideration can be given to the types of subsequent testing performed.
Common methods of bone decalcification
| Agent | Speed | Mechanism of Action | Effect on Histomorphology and Antigen Expression | Effect on Genomic Material (DNA/RNA) |
| Strong Acids (Hydrochloric, Nitric, etc.) | Rapid decalcification (minutes to hours) | Calcium dissolution | Preserved with short decalcifications | Cause degradation of genomic material |
| Weak Acids (Acetic, Formic, Picric, etc.) | Slow decalcification (2-3 days) | Calcium dissolution | Preserved | May cause less degradation of genomic material although long decalcification times can still cause damage and formic acid may cause severe damage |
| EDTA | Slow decalcification (1-2 days) | Calcium chelation | Preserved | Generally better at preserving genomic material, although molecular tests may still fail |
| Microwave/Ultrasonography/Electricity^ | Generally, increase rate of decalcification | Heat/bone cavitation/electrolytic removal of ions | Can cause distortion of histomorphology | Can cause degradation of genomic material |
Diagnostic testing modalties
Karyotype, FISH, and chromosomal microarrays (CMA) such as array comparative genomic hybridization (aCGH)/single nucleotide polymorphism (SNP) arrays have all traditionally constituted the backbone of molecular diagnostics in bone and soft tissue tumors. In more recent decades, PCR based assays, first-generation sequencing technologies and the development of massively parallel targeted sequencing have provided new options for molecular testing of bone tumors[22,39-41]. In general, there are certain advantages and disadvantages to each type of testing modality with some special considerations with regards to tissue processing. Karyotype and CMA, while less commonly used these days for bone tumors, require fresh tissue or perform better with fresh tissue and may still provide some valuable information when used. FISH, PCR, and NGS can be performed on formalin fixed paraffin embedded tissue; however, they suffer from degradation of genomic material during decalcification procedures particularly when strong acids are used.
The type of testing offered by different laboratories also varies widely making it important for pathologists and clinicians to understand the capabilities of each different type of assay and the information being returned. For example, FISH may be used to identify specific translocations or amplification of certain genes in a case where a particular diagnosis is suspected and can be done at a relatively low price with a rapid turnaround time; however, it is less sensitive than PCR or NGS and may lead to false negative results, particularly in bone tumors where neoplastic cells are present in the background of numerous benign cells[42]. PCR and NGS based sequencing assays may be better diagnostic options for tumors where there is a broad differential such as a primary malignant small round blue cell tumor of bone, however these assays are exquisitely sensitive to decalcification and require proper triaging of pre-analytical variables such as tissue fixation and decalcification. In addition, NGS is expensive compared to older techniques and may not be available outside of larger academic institutions or referral centers. Table 2 provides a summary of some of the more common testing modalities that are available for diagnostic testing of bone tumors.
Cytogenetic and molecular diagnostic tests available for bone tumors
| Test | Use | Effects of Processing | Advantages | Limitations |
|---|---|---|---|---|
| FISH | Useful for any bone tumor displaying amplification or a specific chromosomal rearrangement | FFPE material works well for FISH, decalcification can cause interference with FISH probes if genomic material is sufficiently degraded | -Available in most academic centers
-Can be performed on FFPE samples -Rapid turnaround time (48-72 h) | -Technically difficult to interpret signals and requires trained personnel
-Requires a fluorescence microscope and imaging capability -Intermediate resolution: 200 kb -Misses CN-LOH -Provides no mutational information -May provide false negatives on hypocellular tumor material |
| PCR | PCR can provide rapid diagnosis of point mutations and gene fusions in bone tumors, although it is being replaced by other testing methods | Very sensitive to degradation of DNA and RNA due to decalcification procedures, some literature exists showing sequencing works with weak acid or EDTA decalcification however assays may still fail | -Easy assay to set up and perform
-Available at most academic centers -Fast turnaround time (2-5 days) -Works well on FFPE samples -Highly sensitive | -Can only interrogate specific suspected alterations†
-Primers must be designed to cover specific areas of interest -RT-PCR assays require RNA which can be difficult to work with and degrade easily |
| Sequencing | Can be used to analyze various types of bone tumors depending on type of sequencing panel used, including identifying point mutations, rearrangements, and copy number alterations | Very sensitive to degradation of DNA and RNA due to decalcification procedures, some literature exists showing sequencing works with weak acid or EDTA decalcification however assays may still fail | -NGS can interrogate tumors for multiple different genetic abnormalities depending on the panel used
-Can easily be performed on FFPE samples (without decalcification) -High resolution: down to single digit base pairs -Highly sensitive -Sanger sequencing has a lower limit of detection as compared to NGS; however it is useful for targeted mutational testing and for confirmation of molecular alterations identified by other sequencing methods | -Longest turnaround time (1-4 weeks)
-Analysis requires complex bioinformatics pipelines and trained personnel to interpret sequencing data -Equipment and reagents currently expensive -Not available everywhere |
| Karyotype | Typically, no longer used in the routine diagnosis of primary bone tumors | Requires fresh tissue; karyotype cannot be performed on tissue that has undergone standard fixation and decalcification procedures | -Available in most academic centers
-Reasonable turnaround time (5-10 days) -May serve as a whole genome screen for structural and numerical alterations and reveal unexpected information | -Requires fresh tissue
-Technically demanding assay to set up and perform -Dependent on culture and growth of malignant cells -Low resolution: ~10mb -Provides no information on mutations |
| Array | Not commonly used for the diagnosis of primary bone tumors, although array can be used to identify amplification events in certain tumors or lesions with complex genetics and numerous copy number variations | Degradation of genomic material from decalcification interferes with proper analysis of array data | -Array available at most academic centers
-SNP array can identify CN-LOH -Can identify specific areas of gains and losses -Fast turnaround time (5-7 days) | -Intermediate resolution: 10-100 kb
-Cannot detect balanced rearrangements -Provides no information on mutations -Fresh tissue is preferable, analysis of degraded (such as FFPE) samples is difficult |
General molecular genetic landscape of bone neoplasia
Genetically bone tumors may be characterized in a similar fashion to soft tissue neoplasms, divided into lesions with simple genetics and lesions with complex genetics. These categories do not necessarily line up with the histological grade or clinical behavior of individual tumors; tumors with complex genetics are almost uniformly aggressive, although some tumors with simple genetics also behave in an aggressive fashion, and vice versa. Tumors that fall into the simple genetics category tend to have simple karyotypes and can generally be characterized by specific, recurrent molecular alterations that make them attractive targets for cytogenetic or molecular analysis. These alterations most commonly take the form of point mutations or chromosomal level abnormalities such as amplifications or translocations with various downstream effects such as the creation of oncogenic fusion proteins. Tumors from the complex category tend to display either multistep progression in their molecular profiles (such as high-grade chondrosarcoma and dedifferentiated chondrosarcoma) or are characterized by complex karyotypes with multiple molecular abnormalities present at the time of diagnosis (high-grade osteosarcoma and undifferentiated pleomorphic sarcoma). While the value of ancillary diagnostics is dependent on the aforementioned tissue processing protocols, when appropriate material is preserved, they can serve as a valuable tool in the diagnosis of difficult lesions that cannot be diagnosed on clinical, radiological, and histopathologic grounds alone.
Primary bone tumors represent a heterogenous group of tumors with a wide spectrum of differentiation. They include a variety of lesions from different categories including fibrogenic, osteogenic, chondrogenic, undifferentiated, and giant cell rich tumors. In addition, bone can be the site of various other tumors types including notochordal, vascular, myogenic and lipogenic tumors[3]. A summary of the molecular alterations found in primary bone tumors is provided in Tables 3 and 4[43-121]. While advances in molecular genetics have expanded our understanding of the molecular alterations present in many different types of tumors, the routine diagnosis of many primary bone tumors remains a clinicopathologic diagnosis rather than hinging on the identification of specific molecular alterations. As some of these lesions sometimes pose difficulty for diagnosis due to morphologic overlap with other lesions, they may benefit from ancillary molecular testing to identify specific alterations. In addition, a large variety of tumors that primarily occur within the soft tissues may rarely occur as primary osseous lesions. These tumors are mostly the subject of case reports or small series; however, they may cause problems for diagnosis when encountered as primaries outside of their usual site and many of them harbor specific genetic alterations that are helpful for diagnosis when encountered as a bone primary (see “Soft Tissue Tumors that Rarely Present as Primary Bone Tumors” section). Tables 3 and 4 provide a brief overview of selected lesions that exemplify the different categories of bone tumor genetics or in which molecular testing may be helpful.
Molecular genetic alterations in primary bone tumors (simple genetics)
| Tumor Type | Mutations | Translocations and Other Cytogenetic Aberrations | Fusion Gene or Other Effects |
|---|---|---|---|
| Osteochondroma[43-46] | EXT1 and EXT2 loss of function mutations/LOH in sporadic and hereditary osteochondromas | N/A | Disruption of EXT1/2 complex |
| Enchondroma[4,47,48] | IDH1 (R132C; R132H) or IDH2 (R172S), PTHR1 mutations | Various chromosomal level abnormalities | Altered 2-hydroxyglutarate levels, associated hypermethylation and downregulated expression of several other genes associated with IDH mutations |
| Osteoid Osteoma[16,49-51] | N/A | FOS and FOSB rearrangements | FOS/FOSB-various partners |
| Osteoblastoma[16,51-55] | N/A | FOS and FOSB rearrangements | FOS/FOSB-various partners |
| Non-ossifying Fibroma[56] | KRAS, FGFR1, and NF1 alterations | N/A | RAS-MAPK pathway activation |
| Desmoplastic Fibroma[3,57-60] | Rare cases with point mutations described (CTNNB1, APC) | Trisomy 8, Trisomy 20, 11q13 alterations reported | Dysregulation of numerous genes |
| Giant Cell Tumor of Bone[3,61-63] | H3-3A (H3F3A) p.Gly34Trp mutation, RANKL overexpression | N/A | Dysregulation of Histone H3 proteins |
| Chondroblastoma[4,61,64] | H3-3B (H3F3B) p.Lys36Met mutation
Rare cases harbor H3-3A mutations | N/A | Dysregulation of Histone H3 proteins |
| Aneurysmal Bone Cyst[65-70] | N/A | t(16;17)(q22;p13)
USP6-multiple partner genes | CDH11-USP6 Promoter swapping with multiple partner genes leads to upregulation of USP6 |
| Langerhans Cell Histiocytosis[71-75] | BRAF V600E mutations | N/A | Dysregulation of MAPK pathway |
| Fibrous Dysplasia[76-79] | GNAS activating mutations | N/A | Constitutive cAMP elevations lead to alteration in expression levels of multiple targets |
| Low-Grade Central Chondrosarcoma[3,4,80-82] | Somatic mutations in IDH1 and IDH2 | N/A | Altered 2-hydroxyglutarate levels |
| Low-Grade Peripheral Chondrosarcoma[3,4] | Somatic mutations in EXT1 and EXT2 genes | N/A | Deficiency of heparin sulfate glycotransferases |
| Low-Grade Central Osteosarcoma/Parosteal Osteosarcoma[3,83-87] | N/A | Supernumerary ring and giant chromosome markers with amplification of 12q13-15, including MDM2, FRS2, and CDK4 | Cell cycle dysregulation, overexpression of MDM2 and CDK4 |
| Ewing Sarcoma[3,20,88-92] | Mutations in STAG2, CDKN2A, and TP53 described | t(11;22)(q24;q12)
t(21;22)(q22;q12) t(7;22)(q22;q12) t(17;22)(q21;q12) t(2;22)(q36;q12) t(16;21)(p11;q22) | EWSR1-FLI1 EWSR1-ERG EWSR1-ETV1 EWSR1-ETV4 EWSR1-FEV FUS-ERG |
| Phosphaturic Mesenchymal Tumor[93-96] | N/A | Recurrent FGF gene rearrangements, some complex karyotypes have been described | FN1-FGF1 FN1-FGFR1 FGF23 overexpression |
| Mesenchymal Chondrosarcoma[97-99] | N/A | Del(8)(q13.3;q21.1)
t(1;5)(q24;q32) | HEY1-NCOA2 IRF2BP2-CDX1 |
Molecular genetic alterations in primary bone tumors (complex genetics)
| Tumor Type | Genetic Alteration | Effect |
|---|---|---|
| Chondromyxoid Fibroma[3,4,100] | GRM1 upregulation through promoter swapping and gene fusions with various fusion partners including TBL1XR1, COL12A1, BCLAF1, FRMD6, MYO1E, and MEF2A, often caused by complex rearrangement processes | Upregulated GRM1 expression transcripts across entire GRM1 coding sequence |
| High-Grade Osteosarcoma[3,101-105] | Complex karyotypes (sometimes displaying chromothripsis and kataegis) with numerous structural changes reported and multiple types of mutations across many genes (> 100) including RB1 and TP53 | Loss of multiple tumor suppressor genes |
| High-Grade[106-107] and Dedifferentiated Chondrosarcoma[108-111] | Aneuploidy and complex karyotypes, harbor IDH1 and IDH2 mutations if primary or arising from enchondroma, additional mutations in TP53, RB1, CDKN2A/2B, TERT, SUZ12, EED, and p16 | Loss of tumor suppressor function, cell cycle dysregulation, and chromatin remodeling defects |
| Adamantinoma[3,112-116] | Progressive complexity of cytogenetic alterations including increased copy number of chromosomes 7, 8, 12, 19, and 21, KMT2D alterations and rare gene fusions | Various downstream effects including altered chromatin remodeling |
| Chordoma[3,117-121] | The primary alteration in conventional chordoma is copy number gains of TBXT. In addition, chordoma displays a complex karyotype with various copy number alterations.
22q (SMARCB1) loss seen in poorly differentiated chordoma | Overexpression of brachyury secondary to copy number gain of TBXT as well as various other downstream effects including altered chromatin remodeling |
Molecular genetics of selected bone tumors
Giant cell tumor of bone
Giant cell tumor of bone (GCT) is a benign primary bone tumor characterized by a mononuclear cell population of neoplastic cells with an admixed population of multinucleated osteoclast-type giant cells. These lesions fall into the simple genetics category of bone tumors. GCT represents approximately 5% of all primary bone tumors and may behave in a locally aggressive fashion[3]. Recent molecular advances have identified that nearly all of these tumors are characterized by somatic point mutations in the H3-3A (H3F3A) gene leading to a specific amino acid substitution p.Gly34Trp that is detectable with immunohistochemistry (G34W IHC). This mutation leads to epigenetic modification and abnormal function of histone protein H3[61]. In addition, studies have shown that other alterations including 20q11.1 amplification, IDH mutations (although this is disputed), and RANKL overexpression are present in GCT[62-64]. While the diagnosis of GCT is generally straight forward some cases may display morphologic overlap with other giant cell rich lesions including chondroblastoma. For this reason, immunohistochemistry or molecular testing for the H3-3A mutation may be useful in clinching the diagnosis. Achieving a correct diagnosis for these lesions is particularly useful as the tumors are amenable to targeted therapy with denosumab[64].
Chondroblastoma
Chondroblastoma is a rare benign primary bone tumor of young adulthood that displays chondrogenic differentiation and is part of the simple genetics category of lesions. It is characterized by a monotonous population of what appear to be primitive chondroblastic cells [Figure 1][4]. These tumors are driven by mutations primarily in the H3-3B (H3F3B) gene, with an extremely small percentage harboring mutations in H3-3A[4,61]. The mutations result in an amino acid change; p.Lys36Met that is detectable by immunohistochemistry (K36M IHC) [Figure 2][3,4,64]. As in GCT, this mutation interferes with histone protein H3[61]. Some cytogenetic abnormalities have been described including some rearrangements, although they do not appear to be recurrent[3]. Chondroblastoma has also been shown to harbor RANKL overexpression similar to GCT and in rare cases has shown response to denosumab therapy in refractory tumors[64]. Chondroblastoma may in some cases display morphologic overlap with both giant cell rich lesions as well as chondrogenic lesions. In some cases, prominent secondary aneurysmal bone cyst-like changes may be present[3], making the identification of the K36M mutation particularly useful.
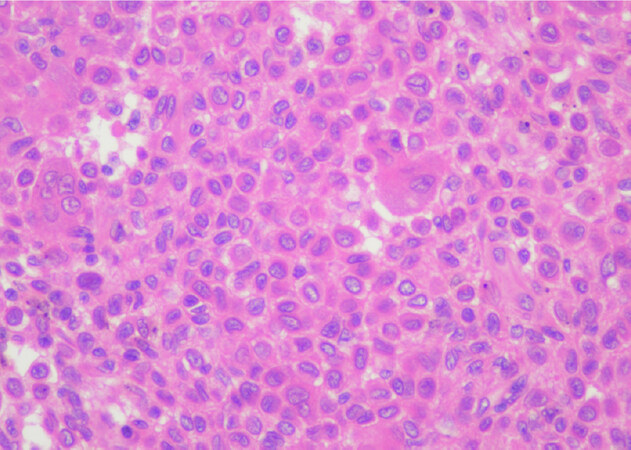
Figure 1. Chondroblastoma: high power magnification of chondroblastoma shows a monotonous appearing cell population with well-defined cell borders and ample eosinophilic cytoplasm. Some admixed multinucleated giant cells are present (Hematoxylin and Eosin, 200x magnification)
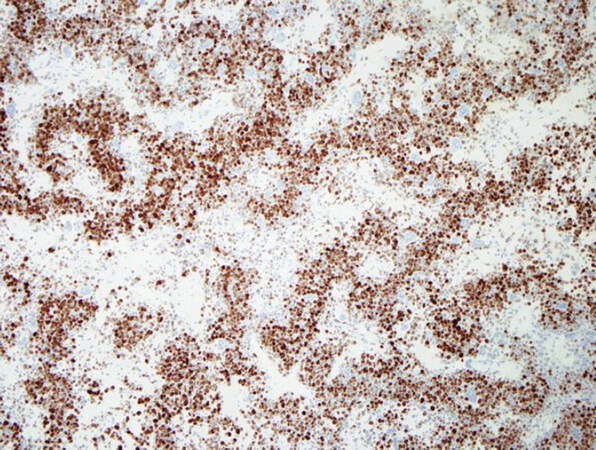
Figure 2. K36M immunohistochemistry: immunohistochemistry for the K36M antibody shows a nuclear staining pattern within the neoplastic cells of chondroblastoma consistent with H3F3B mutation (K36M immunohistochemistry, 20x magnification)
Osteoblastoma and osteoid osteoma
Osteoblastoma and osteoid osteoma are two related lesions that are both osteoid producing primary bone tumors composed of immature woven bone spicules, prominent stromal vessels, giant cells, and prominent osteoblasts [Figure 3][3]. Until recently molecular studies consisted of only a few cytogenetic studies that did not identify any recurrent alterations. More recently recurrent rearrangements of FOS and FOSB have been identified that in many cases can be identified with immunohistochemistry for FOS [Figure 4][16,50,51]. FOS is a tightly regulated transcription factor that has been known to be involved in the pathogenesis of bone tumors; FOS and FOSB rearrangements involve multiple partners and create a mutant fusion transcript lacking the normal regulatory elements[16]. A small subgroup of non-FOS-rearranged osteoblastomas have also been identified to be characterized by loss of NF2[54]. Osteoid osteoma in general i30s readily diagnosed on clinicopathologic grounds; however, osteoblastoma can present a problem for diagnosis as it may show overlap with some locally aggressive or outright malignant lesions such as osteosarcoma. While many osteoblastomas will show FOS immunoreactivity, up to 14% of osteosarcoma samples also showed immunoreactivity in one study[51]. This potential for non-specificity for osteoblastoma immunohistochemistry means that FISH or sequencing based assays are preferable as an ancillary molecular tool if fresh tissue is available; however, a positive FOS immunohistochemistry may help support a suspected diagnosis of osteoblastoma.
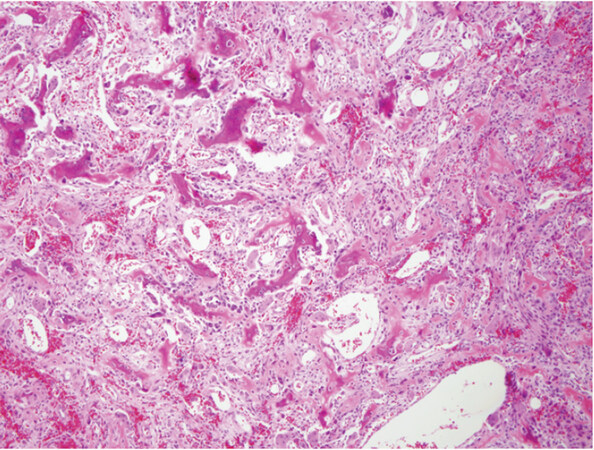
Figure 3. Osteoblastoma: low power magnification shows a highly vascular neoplasm with small spicules of immature woven production rimmed by osteoblastic cells (Hematoxylin and Eosin, 20x magnification)
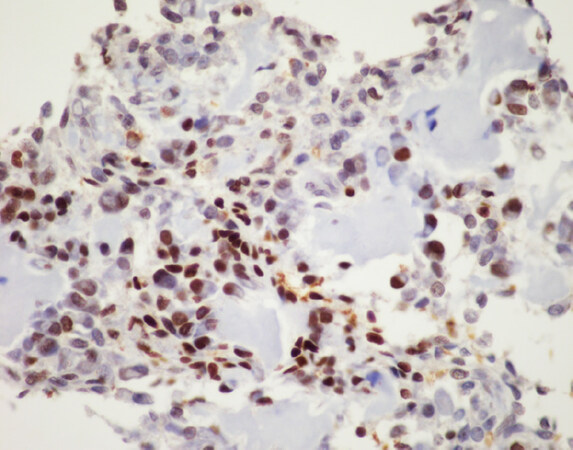
Figure 4. FOS immunohistochemistry: immunohistochemistry for FOS shows nuclear positivity for the majority of the neoplastic tumor cells in osteoblastoma, this serves as a surrogate for the presence of FOS rearrangements, although it is not entirely specific (FOS immunohistochemistry, 20x magnification)
Aneurysmal bone cyst
Aneurysmal bone cyst (ABC) is a benign, yet destructive, primary bone lesion composed of multiple cystic blood-filled spaces that has a characteristic radiological impression with fluid-fluid levels[3]. Cytogenetic characterization of these lesions has shown recurrent aberrations of chromosome 17p[65]. These aberrations are now known to be recurrent rearrangements involving the USP6 gene[66]. A CDH11-USP6 rearrangement was the first described, although numerous different translocation partners have now been identified with a common mechanism involving promoter swapping with upregulation of USP6[66-70]. The upregulation of USP6 leads to increased production of matrix metalloproteinases that lead to osteolysis, inflammation, and vascularization[66]. USP6 rearrangements are most easily identified using FISH break apart probes or next generation sequencing based fusion panels and are useful in the diagnosis of these lesions as there can be morphologic overlap between ABC and various benign and malignant lesions that can show secondary ABC-like areas.
Chondrosarcoma
Chondrosarcoma is defined as a malignant cartilage producing tumor and is comprised of a large family of tumors that includes multiple types such as conventional chondrosarcoma, periosteal chondrosarcoma, mesenchymal chondrosarcoma, and clear cell chondrosarcoma[3]. The most commonly encountered of these entities is conventional chondrosarcoma which can be further classified into peripheral (located in the appendicular skeleton), central (located within the axial skeleton), primary (arising in the absence of a precursor lesion), or secondary (arising from a pre-existing bone tumor, usually enchondroma or osteochondroma)[3,4]. Chondrosarcoma is graded based primarily on the degree of cytologic atypia and some other features including overall cellularity, cartilage matrix degeneration and mitotic activity. Low-grade lesions occurring in the appendicular skeleton are termed atypical cartilaginous tumors. Genetically, primary peripheral and central chondrosarcomas are characterized by somatic point mutations in the isocitrate dehydrogenase genes, IDH1 (R132C/H/G) and IDH2 (R172S) leading to altered hydroxyglutarate levels[3,4]. Secondary chondrosarcomas arising from enchondromas, a finding that often occurs as part of the genetic syndromes: Ollier disease and Maffuci syndrome, are also characterized by IDH mutations. Secondary peripheral chondrosarcomas arising in association with osteochondromas are characterized by mutations in EXT1 and EXT2, the same genes mutated in patients with multiple hereditary exostosis[4,80-82]. High-grade chondrosarcoma shows progressive molecular alterations including aneuploidy and complex karyotypes, and mutations in RB1, TP53, and COL2A1[3]. Finally, periosteal chondrosarcoma has been reported to harbor IDH gene mutations while mesenchymal chondrosarcoma shows a characteristic HEY1-NCOA2 rearrangement[3,98]. Though some genetic abnormalities have been reported in clear cell chondrosarcoma its genetic profile has not been completely elucidated[3]. Despite the presence of various molecular alterations in the different subtypes of chondrosarcoma it is worth noting that the diagnosis of conventional chondrosarcoma in general does not require ancillary molecular genetic testing and is instead established based on clinical, radiological, and histological criteria in routine clinical practice. However, associated molecular alterations may be useful in some situations, particularly in secondary lesions that may be related to germline conditions, when dealing with mesenchymal chondrosarcoma presenting as predominantly poorly differentiated small round blue cells, and in some cases separating conventional chondrosarcoma from other cartilaginous lesions when limited tissue is available for examination.
Osteosarcoma
Osteosarcoma is a malignant bone forming tumor that is the prototypical example of a primary bone sarcoma with complex genetics, but also includes some subtypes that are characterized by well-defined recurrent genetic alterations. These tumors are categorized by neoplastic osteoid production and a population of atypical osteocytes that can display a wide spectrum of cytological appearances from low-grade, bland appearing spindle cell lesions to high grade lesions with bizarre, atypical forms[3]. The molecular genetics of high-grade osteosarcoma are complex; cytogenetic and large-scale sequencing studies have identified a number of somatic mutations as well as numerous, generally non-recurrent, copy number alterations at the chromosomal level[102-104]. Somatic mutations in tumor suppressor genes and proto-oncogenes include TP53, RB1, BRCA2, BAP1, RET, CDKN2A PTEN, WRN, ATRX, and many others. Many of these genes such as TP53, RB1, and WRN are associated with hereditary cancer syndromes including Li-Fraumeni, hereditary retinoblastoma, and Werner syndrome, that predispose patients to an increased risk of osteosarcoma[102,103]. Reported mutations are numerous and number in the hundreds[102]. High-grade osteosarcoma is also characterized cytogenetically by chromothripsis (Greek origin, “thripsis” meaning shattering) and kataegis (Greek origin, “Kataegis” meaning thunderstorm) in which catastrophic chromosomal breakage occurs sometimes in combination with regional hypermutation that occurs through complex mechanisms [Figures 5 and 6][102,104].
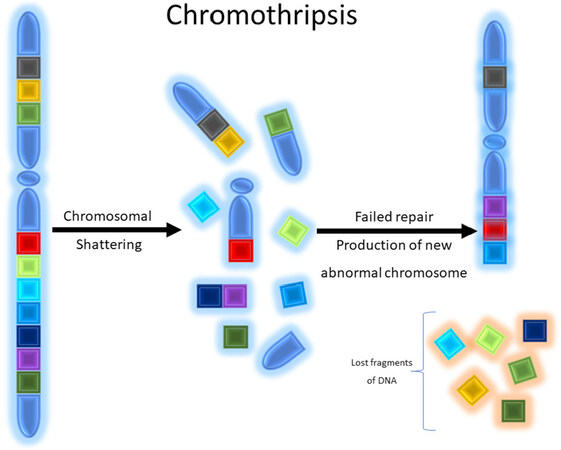
Figure 5. Graphical schematic depicting chromothripsis: chromosomal shattering occurs with subsequent failure of genomic repair mechanisms resulting in inappropriate recombination of chromosomal fragments. During this process fragments of the chromosome may be lost or remain and reassembled erroneously
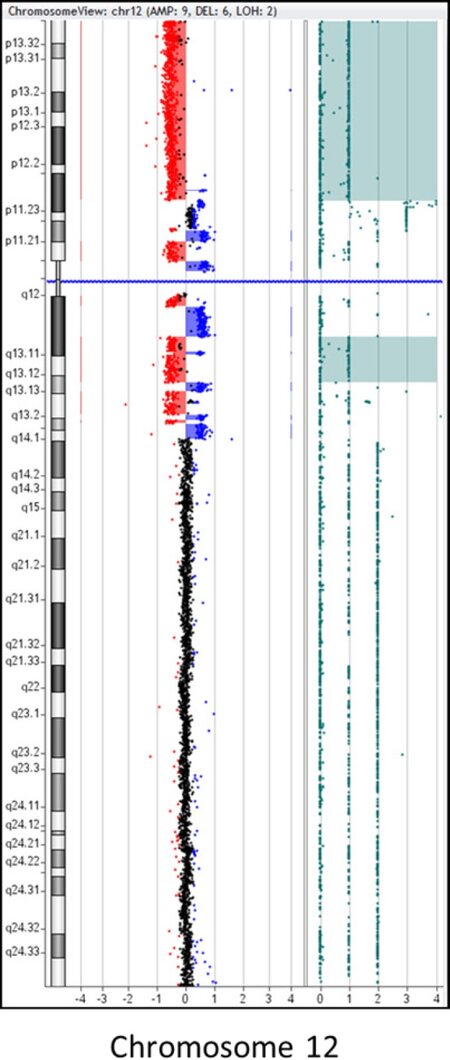
Figure 6. Representative example of chromothripsis: CMA plot of chromosome 12 demonstrating 14 alternating CNAs involving the proximal region of the short (p) and long (q) arms of chromosome 12. This pattern of alternating CNAs is suggestive of “chromothripsis”. In addition, there is a deletion of the remaining distal portion of 12p. Sample was tested using the Agilent 180k aCGH+SNP oligo array, and data analysis was performed using the Agilent CytoGenomics software v4.0 (Image courtesy of Dr. Fady M. Mikhail, MD PHD, University of Alabama at Birmingham). CMA: chromosomal microarray; CNAs: copy number abnormalities
Low-grade variants of osteosarcoma include parosteal osteosarcoma and low-grade central osteosarcoma[3]. The low-grade variants of osteosarcoma are characterized by a bland appearing spindle cell population of cells rather than the overtly malignant cells present in high-grade osteosarcoma [Figure 7]. Both of these subtypes display less complex genetics than their high-grade counterparts harboring known specific, recurrent genetic alterations in the form of supernumerary ring chromosomes containing amplified material from 12q13-15 similar to that seen in some liposarcomas [Figure 8][83-87]. In some cases, these changes have been identified in high-grade osteosarcoma as well, although it is unclear if this represents an isolated finding in a genetically complex lesion or a marker of transition from a previously low-grade lesion (dedifferentiation)[83]. Amplification of material from 12q13-15 leads to the amplification of multiple genes involved in tumorigenesis including MDM2, CDK4, and FRS2[83]. These amplifications can be detected by immunohistochemistry for MDM2 and CDK4, although testing by FISH or chromosomal microarray provides a more sensitive test[83]. These findings are quite helpful when dealing with a low-grade fibroblastic proliferation as these tumors can share morphologic overlap with other fibroblastic tumors of bone.
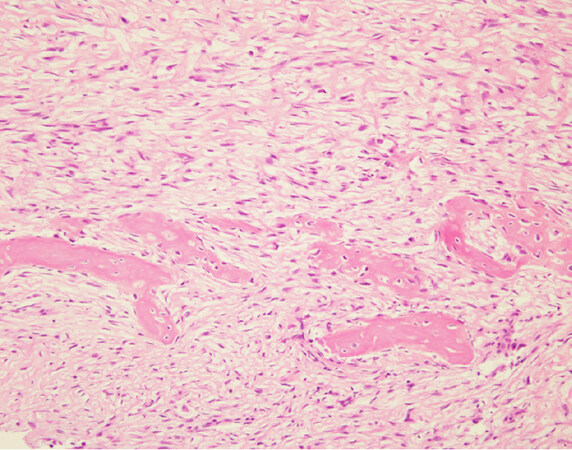
Figure 7. Parosteal osteosarcoma: high power magnification shows a bland appearing spindle cell population infiltrating around small bony fragments. Lesions such as this are difficult to distinguish from reactive processes or other fibroblastic lesions based on histomorphology alone (Hematoxylin and Eosin, 20x magnification)
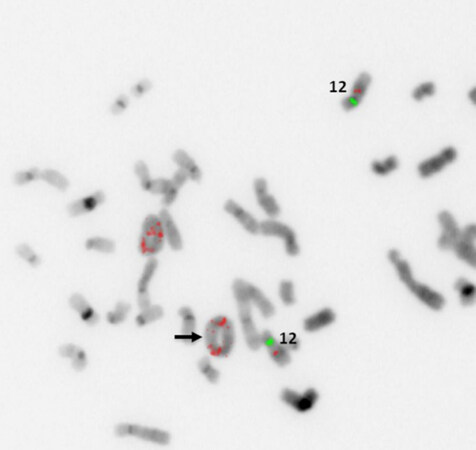
Figure 8. MDM2 amplification by FISH: representative example of supernumerary ring chromosome formation containing amplified material from 12q13-15. Increased copy numbers of MDM2 (red probe) signals (black arrow) are seen within ringed chromosomes (Image courtesy of Dr. Christine Bryke MD, Beth Israel Deaconess Medical Center). FISH: fluorescence in situ hybridization
Ewing Sarcoma/Primitive Neuroectodermal Tumor
Ewing sarcoma is the prototypical example a small round blue cell sarcoma that may occur as primary bone tumor. Ewing sarcoma is characterized by a recurrent, specific t(11;22)(q24;q12) that leads to the production of an oncogenic fusion protein: EWSR1-FLI1[88]. Various other fusion partners exist for EWSR1[Table 3], although many of these tumors are considered to fall within the Ewing sarcoma family and are treated in a similar fashion[89-92].
The differential of small round blue cell sarcomas within bone has expanded considerably in the past years and now includes various non-Ewing translocation associated sarcomas, mesenchymal chondrosarcoma, myoepithelial tumors, and other poorly differentiated malignancies including high-grade osteosarcoma[90]. The wide range of lesions that can share morphologic overlap makes molecular testing for EWSR1 essential in the diagnosis of these lesions. While FISH is helpful in identifying the involvement of EWSR1 in a rearrangement, more advanced molecular techniques such as PCR or NGS may be required in order to identify fusion partners and determine the exact nature of a small round blue cell sarcoma.
Soft tissue tumors that rarely present as primary bone tumors
Apart from tumors that occur commonly within bone or primarily within bone there is a large group of mesenchymal neoplasms that may also rarely occur in bone. This list includes, but is not limited to, neural tumors (e.g., malignant peripheral nerve sheath tumor), adipocytic tumors (e.g., liposarcoma), histiocytic tumors (e.g., Rosai-Dorfman disease), tumors of uncertain histogenesis (e.g., synovial sarcoma), vascular tumors (e.g., epithelioid hemangioma and angiosarcoma), and some undifferentiated sarcomas [Table 5][122-183]. These lesions tend to share similar genetic profiles to their soft tissue counterparts when occurring as a bone primary and include lesions that fall into the simple and complex genetic category. Molecular diagnostic testing is indicated in many of these tumors as several of them are characterized by specific, recurrent alterations. An example of this is sclerosing epithelioid fibrosarcoma/low-grade fibromyxoid sarcoma - a bland appearing fibroblastic lesion that can share morphologic overlap with reactive changes, low-grade variants of fibroblastic osteosarcoma, desmoplastic fibroma, solitary fibrous tumor, and others [Figure 9]. These tumors are characterized by FUS rearrangements, usually partnering with CREB3L2 or CREB3L1 and rarely occur within the bone; however, may pose problems for diagnosis when encountered[131-133]. Although immunohistochemistry for MUC4 is helpful for the diagnosis, ancillary molecular testing through FISH, PCR, or sequencing can help solidify the diagnosis [Figure 10]. While some of these lesions can be diagnosed based on clinicopathologic features and immunohistochemistry, oftentimes they require expanded diagnostic testing for specific molecular alterations.
Molecular genetic alterations in soft tissue tumors that may also rarely occur as primary bone tumors (simple and complex genetics)
| Tumor Type | Genetic Abnormality | Recurrent Fusion or Abnormality |
|---|---|---|
| Malignant Peripheral Nerve Sheath Tumor[122-125] | Various somatic alterations in CDKN2A, NF1, EED, SUZ12, SMARCB1 (epithelioid variant) | Dysregulation of polycomb repressive complex 2 (PRC2) |
| Extraskeletal Myxoid Chondrosarcoma[126-128] | t(9;22)(q22;q12)
t(9;17)(q22;q11) t(9;15)(q22;q21) | EWSR1-NR4A3 TAF15-NR4A3 TCF12-NR4A3 |
| Epithelioid Sarcoma[129,130] | Loss of SMARCB1 (INI1) secondary to biallelic loss of function mutations or heterozygous mutations in subunits of the SWI/SNF (BAF) complex | Dysregulation of SWI/SNF (BAF) complex - involved in chromatin remodeling |
| Sclerosing Epithelioid Fibrosarcoma/Low-Grade Fibromyxoid Sarcoma[131-133] | t(7;16)(q32-33;p11)
t(11;16)(p11;p11) | FUS-CREB3L2 FUS-CREB3L1 |
| Liposarcoma[134-136] | Supernumerary ring and giant chromosome markers with amplification of 12q13-15, including MDM2 and CDK4 Pleomorphic subtype: complex genetics with various CNV | Cell cycle dysregulation, overexpression of MDM2 and CDK4 |
| Myxoid/Round Cell
Liposarcoma[137,138] | t(12;16)(q13;p11)
t(12;22)(q13;q12) | FUS-DDIT3 EWSR1-DDIT3 |
| Alveolar Soft Part Sarcoma[139,140] | t(X;17)(p11;q25) | ASPCR1-TFE3 |
| Undifferentiated Round Cell Sarcomas[4,18,91,141-144] | t(4;19)(q35;q13)
t(10;19)(q26;q13) Paracentric inv(X)(p11.4p11.22) t(20;22)(q13.2;q12.2) | CIC-DUX4 CIC-DUX4 BCOR-CCNB3 EWSR1-NFATc2^ FUS-NFATc2^ |
| Alveolar Rhabdomyosarcoma[145-147] | t(2;13)(q35;q14)
t(1;13)(p36;q14) t(2;2)(q35;p23) t(2;8)(q35;q13) | PAX3-FOXO1 PAX7-FOXO1 PAX3-NCOA1 PAX3-NCOA2 |
| Embryonal Rhabdomyosarcoma[145,148] | Loss of heterozygosity on 11p15.5 | Imprinting defects in IGF2, H19, and p57kip2 |
| Leiomyosarcoma[149,150] | Generally complex karyotypes with numerous gains and losses | Various |
| Solitary Fibrous Tumor[151-153] | Intrachromosomal inversion of 12q13 region | NAB2-STAT6 |
| Synovial Sarcoma[154-157] | t(X;18)(p11;q11)
t(X;20)(p11;q13) | SS18-SSX1 SS18-SSX2 SS18-SSX4 SS18L1-SSX1 |
| Myoepithelial Tumors[158-162] | t(6;22)(p21;q12)
t(1;22)(q23;q12) t(19;22)(q13;q12) | EWSR1-POU5F1 EWSR1-PBX1 EWSR1-ZNF444 |
| Epithelioid Hemangioma[163-165] | t(1;14)(q22;q24.3) | FOS-LMNA |
| Epithelioid Hemangioendothelioma[166,167] | t(1;3)(p36;q25)
t(X;11)(q22;p11) | WWTR1-CAMTA1 YAP1-TFE3 |
| Pseudomyogenic Hemangioendothelioma[168,170] | t(7:19)(q22;13) | SERPINE1-FOSB |
| High-Grade Angiosarcoma[4,171-174] | Gene alterations in CIC, KDR, PLCG1, FLT4, and
MYC overexpression seen in some cases | CIC fusions rare |
| Undifferentiated Pleomorphic Sarcoma[175-178] | Rare targetable fusions identified in some cases, complex karyotypes, mutations in TP53, ATRX, and RB1 and others | Various |
| Rosai-Dorfman Disease[75,179-182] | KRAS and MAP2K1 mutations | MAPK pathway alteration |
| Erdheim Chester Disease&[71,75,183] | BRAF V600E | MAPK pathway alteration |
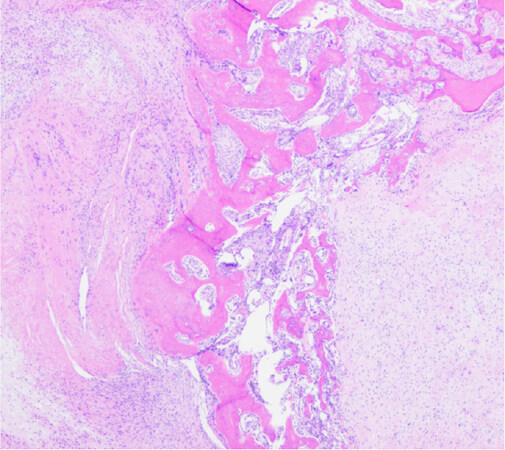
Figure 9. Sclerosing epithelioid fibrosarcoma/low-grade fibromyxoid sarcoma: Scanning magnification of FUS/LGFMS showing an infiltrative, but bland appearing fibroblastic lesion showing both intramedullary and extramedullary growth (Hematoxylin and Eosin, 20x magnification)
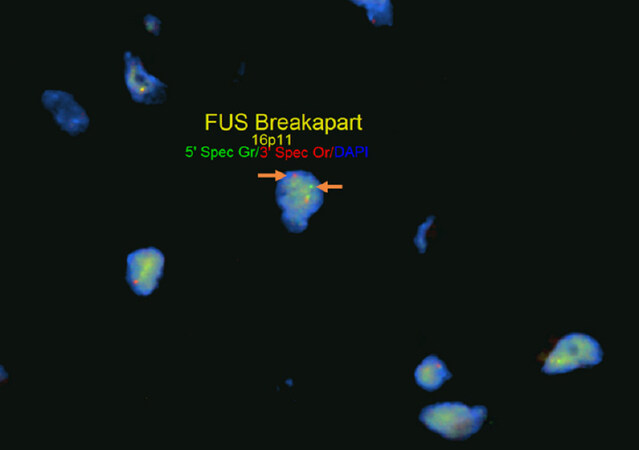
Figure 10. FUS fluorescence in situ hybridization (FISH): FISH for FUS in sclerosing epithelioid fibrosarcoma/low-grade fibromyxoid sarcoma using break apart FISH probes identifies a rearrangement of FUS indicated by separate signals (white arrows, 5’ green probe and 3’ red probe). Testing for rearrangements through FISH does not provide information on partner genes or chromosomes involved in rearrangements
Conclusion
Bone tumors represent a heterogenous category of benign and malignant lesions with a varied genomic landscape. Advances in molecular technology have vastly increased our knowledge of the molecular features of many of these lesions although the diagnosis of many bone tumors is still based entirely on histopathologic, clinical, and radiological features. Ancillary molecular diagnostics are increasingly becoming necessary for the diagnosis of bone tumors, facilitated by the development of new technologies in the past few decades. Knowledge of the molecular alterations as well as specimen handling considerations that may affect molecular testing is of utmost important as our base of knowledge continues to grow.
Declarations
Authors’ contributionsResponsible for conceptualization, image procurement, and content review: Suster S
Rresponsible for drafting the primary manuscript, image procurement, and figure creation: Suster D
Availability of data and materialsNot Applicable.
Financial support and sponsorshipNone.
Conflicts of interestAll authors declared that there are no conflicts of interest.
Ethical approval and consent to participateNot Applicable.
Consent for publicationNot Applicable.
Copyright© The Author(s) 2021.
REFERENCES
1. Howlader N, Noone AM, et al (eds). SEER Cancer Statistics Review, 1975-2017, National Cancer Institute. Available from: https://seer.cancer.gov/csr/1975_2017/. [Last accessed on 4 Jan 2021].
3. Bovee JVMG, Flanagan AM, Lazar AJ, Nielsen GP, Yoshida A. Bone tumours. In: WHO classification of tumours editorial board. WHO classification of tumors: soft tissue and bone tumors 5th edition. Lyon France: International Agency for Research on Cancer; 2020. pp. 338-499.
4. Suster D, Hung YP, Nielsen GP. Differential diagnosis of cartilaginous lesions of bone. Arch Pathol Lab Med 2020;144:71-82.
5. Scheitza P, Uhl M, Hauschild O, et al. Interobserver variability in the differential diagnosis of benign bone tumors and tumor-like lesions. Rofo 2016;188:479-87.
6. Gemescu IN, Thierfelder KM, Rehnitz C, Weber MA. Imaging features of bone tumors: conventional radiographs and MR imaging correlation. Magn Reson Imaging Clin N Am 2019;27:753-67.
7. Mehta K, McBee MP, Mihal DC, England EB. Radiographic analysis of bone tumors: a systematic approach. Semin Roentgenol 2017;52:194-208.
8. Larousserie F, Kreshak J, Gambarotti M, Alberghini M, Vanel D. The importance of radiographic imaging in the microscopic assessment of bone tumors. Eur J Radiol 2013;82:2100-14.
9. Andreou D, Hardes J, Gosheger G, et al. Interdisciplinary diagnostic and treatment of bone sarcomas of the extremities and trunk. Handchir Mikrochir Plast Chir 2015;47:90-9.
10. Ferguson JL, Turner SP. Bone cancer: diagnosis and treatment principles. Am Fam Physician 2018;98:205-213.
11. Mueller C, Gambarotti M, Benini S, et al. Unlocking bone for proteomic analysis and FISH. Lab Invest 2019;99:708-21.
12. Donner LR. Cytogenetics of tumors of soft tissue and bone. Cancer Genet Cytogenet 1994;78:115-26.
13. arkkanen M, Nordling S, Böhling T, et al. Comparison of cytogenetics, interphase cytogenetics, and DNA flow cytometry in bone tumors. Cytometry 1996;26:185-91.
14. Vargas AC, Selinger C, Satgunaseelan L, et al. FISH analysis of selected soft tissue tumors: diagnostic experience in a tertiary center. Asia Pac J Clin Oncol 2019;15:38-47.
15. Tanas MR, Goldblum JR. Fluorescence in situ hybridization in the diagnosis of soft tissue neoplasms: a review. Adv Anat Pathol 2009;16:383-91.
16. Fittall MW, Mifsud W, Pillay N, et al. Recurrent rearrangements of FOS and FOSB define osteoblastoma. Nat Commun 2018;9:2150.
17. Schaefer IM, Hornick JL. Diagnostic immunohistochemistry for soft tissue and bone tumors: an update. Adv Anat Pathol 2018;25:400-12.
18. Wang GY, Thomas DG, Davis JL, et al. EWSR1-NFATC2 translocation-associated sarcoma clinicopathologic findings in a rare aggressive primary bone or soft tissue tumor. Am J Surg Pathol 2019;43:1112-22.
19. Bahrami A, Weiss SW, Montgomery E, et al. RT-PCR analysis for FGF23 using paraffin sections in the diagnosis of phosphaturic mesenchymal tumors with and without known tumor induced osteomalacia. Am J Surg Pathol 2009;33:1348-54.
20. Bridge RS, Rajaram V, Dehner LP, Pfeifer JD, Perry A. Molecular diagnosis of Ewing sarcoma/primitive neuroectodermal tumor in routinely processed tissue: a comparison of two FISH strategies and RT-PCR in malignant round cell tumors. Mod Pathol 2006;19:1-8.
21. Baumhoer D, Amary F, Flanagan AM. An update of molecular pathology of bone tumors. Lessons learned from investigating samples by next generation sequencing. Genes Chromosomes Cancer 2019;58:88-99.
22. Szurian K, Kashofer K, Liegl-Atzwanger B. Role of next-generation sequencing as a diagnostic tool for the evaluation of bone and soft-tissue tumors. Pathobiology 2017;84:323-38.
23. Lam SW, Cleton-Jansen AM, Cleven AHG, et al. Molecular analysis of gene fusions in bone and soft tissue tumors by anchored multiplex PCR-based targeted next-generation sequencing. J Mol Diagn 2018;20:653-63.
24. Hogendoorn PC, Athanasou N, Bielack S, et al; ESMO/EUROBONET working group. Bone sarcomas: ESMO clinical practice guidelines for diagnosis, treatment and follow-up. Ann Oncol 2010;21:v204-13.
25. Rubin BP, Antonescu CR, Gannon FH, et al; Members of the cancer committee, college of American pathologists. Protocol for the examination of specimens from patients with tumors of bone. Arch Pathol Lab Med 2010;134:e1-7.
26. Rubin BP, Fletcher CD, Inwards C, et al. Protocol for the examination of specimens from patients with soft tissue tumors of intermediate malignant potential, malignant soft tissue tumors, and benign/locally aggressive and malignant bone tumors. Arch Pathol Lab Med 2006;130:1616-29.
27. Singh VM, Salunga RC, Huang VJ, et al. Analysis of the effect of various decalcification agents on the quantity and quality of nucleic acid (DNA and RNA) recovered from bone biopsies. Ann Diagn Pathol 2013;17:322-6.
28. Choi SE, Hong SW, Yoon SO. Proposal of an appropriate decalcification method of bone marrow biopsy specimens in the era of expanding genetic molecular study. J Pathol Transl Med 2015;49:236-42.
29. Mueller C, Harpole MG, Espina V. One-step preservation and decalcification of bony tissue for molecular profiling. Methods Mol Biol 2017;1606:85-102.
30. Babic A, Loftin IR, Stanislaw S, et al. The impact of pre-analytical processing on staining quality for H&E, dual hapten, dual color in situ hybridization and fluorescent in situ hybridization assays. Methods 2010;52:287-300.
31. Goswami RS, Luthra R, Singh RR, et al. Identification of factors affecting the success of next-generation sequencing testing in solid tumors. Am J Clin Pathol 2016;145:222-37.
32. Nagai M, Minegishi K, Komada M, Tsuchiya M, Kameda T, Yamada S. Extraction of DNA from human embryos after long-term preservation in formalin and Bouin’s solutions. Congenit Anom (Kyoto) 2016;56:112-8.
33. Alers JC, Krijtenburg PJ, Vissers KJ, van Dekken H. Effect of bone decalcification procedures on DNA in situ hybridization and comparative genomic hybridization. EDTA is highly preferable to a routinely used acid decalcifier. J Histochem Cytochem 1999;47:703-10.
34. Raj AT, Patil S, Rao RS. A Comparison of conventional and microwave decalcification and processing of tooth and mandibular bone specimens. J Clin Diagn Res 2016;10:ZC121-6.
35. Imaizumi K, Taniguchi K, Ogawa Y. An evaluation of the effect of microwave irradiation on bone decalcification aimed to DNA extraction. Leg Med (Tokyo) 2013;15:272-7.
36. Chow DH, Zheng LZ, Tian L, Ho KS, Qin L, Guo X. Application of ultrasound accelerates the decalcification process of bone matrix without affecting histological and immunohistochemical analysis. J Orthop Translat 2019;17:112-20.
37. Mueller C, Harpole MG, Espina V. One-step preservation and decalcification of bony tissue for molecular profiling. In: Espina V, editor. Molecular profiling. New York: Springer; 2017. pp. 85-102.
38. Kapila SN, Natarajan S, Boaz K, Pandya JA, Yinti SR. Driving the mineral out faster: simple modifications of the decalcification technique. J Clin Diagn Res 2015;9:ZC93-7.
39. Lam SW, van IJzendoorn DGP, Cleton-Jansen AM, Szuhai K, Bovée JVMG. Molecular pathology of bone tumors. J Mol Diagn 2019;21:171-82.
41. Bridge JA, Cushman-Vokoun AM. Molecular diagnostics of soft tissue tumors. Arch Pathol Lab Med 2011;135:588-601.
42. Gu J, Smith JL, Dowling PK. Fluorescence in situ hybridization probe validation for clinical use. In: Wan TS, editor. Cancer cytogenetics. New York: Springer; 2017. pp. 101-18.
43. Jochmann K, Bachvarova V, Vortkamp A. Reprint of: heparan sulfate as a regulator of endochondral ossification and osteochondroma development. Matrix Biol 2014;35:239-47.
44. Bovée JV, Cleton-Jansen AM, Wuyts W, et al. EXT-mutation analysis and loss of heterozygosity in sporadic and hereditary osteochondromas and secondary chondrosarcomas. Am J Hum Genet 1999;65:689-98.
45. Santos SCL, Rizzo IMPO, Takata RI, Speck-Martins CE, Brum JM, Sollaci C. Analysis of mutations in EXT1 and EXT2 in Brazilian patients with multiple osteochondromas. Mol Genet Genomic Med 2018;6:382-92.
46. Hameetman L, David G, Yavas A, et al. Decreased EXT expression and intracellular accumulation of heparan sulphate proteoglycan in osteochondromas and peripheral chondrosarcomas. J Pathol 2007;211:399-409.
47. Bell WC, Klein MJ, Pitt MJ, Siegal GP. Molecular pathology of chondroid neoplasms: part 1, benign lesions. Skeletal Radiol 2006;35:805-13.
48. Kumar A, Jain VK, Bharadwaj M, Arya RK. Ollier disease: pathogenesis, diagnosis, and management. Orthopedics 2015;38:e497-506.
49. Franceschini N, Lam SW, Cleton-Jansen AM, Bovée JVMG. What’s new in bone forming tumours of the skeleton? Virchows Arch 2020;476:147-57.
50. Amary F, Markert E, Berisha F, et al. FOS Expression in osteoid osteoma and osteoblastoma: a valuable ancillary diagnostic tool. Am J Surg Pathol 2019;43:1661-7.
51. Lam SW, Cleven AHG, Kroon HM, Briaire-de Bruijn IH, Szuhai K, Bovée JVMG. Utility of FOS as diagnostic marker for osteoid osteoma and osteoblastoma. Virchows Arch 2020;476:455-63.
52. Panagopoulos I, Gorunova L, Lobmaier I, et al.
53. Giannico G, Holt GE, Homlar KC, Johnson J, Pinnt J, Bridge JA. Osteoblastoma characterized by a three-way translocation: report of a case and review of the literature. Cancer Genet Cytogenet 2009;195:168-71.
54. Saba KH, Cornmark L, Hofvander J, et al. Loss of NF2 defines a genetic subgroup of non-FOS-rearranged osteoblastoma. J Pathol Clin Res 2020;6:231-7.
55. Nord KH, Nilsson J, Arbajian E, et al. Recurrent chromosome 22 deletions in osteoblastoma affect inhibitors of the Wnt/beta-catenin signaling pathway. PLoS One 2013;8:e80725.
56. Bovée JV, Hogendoorn PC. Non-ossifying fibroma: A RAS-MAPK driven benign bone neoplasm. J Pathol 2019;248:127-30.
57. Bridge JA, Swarts SJ, Buresh C, et al. Trisomies 8 and 20 characterize a subgroup of benign fibrous lesions arising in both soft tissue and bone. Am J Pathol 1999;154:729-33.
58. E Horvai A, C Jordan R. Fibro-osseous lesions of the craniofacial bones: β-catenin immunohistochemical analysis and CTNNB1 and APC mutation analysis. Head Neck Pathol 2014;8:291-7.
59. Trombetta D, Macchia G, Mandahl N, Nord KH, Mertens F. Molecular genetic characterization of the 11q13 breakpoint in a desmoplastic fibroma of bone. Cancer Genet 2012;205:410-3.
60. Song W, van den Berg E, Kwee TC, et al. Low-grade central fibroblastic osteosarcoma may be differentiated from its mimicker desmoplastic fibroma by genetic analysis. Clin Sarcoma Res 2018;8:16.
61. Behjati S, Tarpey PS, Presneau N, et al. Distinct H3F3A and H3F3B driver mutations define chondroblastoma and giant cell tumor of bone. Nat Genet 2013;45:1479-82.
62. Smith LT, Mayerson J, Nowak NJ, et al. 20q11.1 amplification in giant-cell tumor of bone: Array CGH, FISH, and association with outcome. Genes Chromosomes Cancer 2006;45:957-66.
63. Kato Kaneko M, Liu X, Oki H, et al. Isocitrate dehydrogenase mutation is frequently observed in giant cell tumor of bone. Cancer Sci 2014;105:744-8.
64. Suster DI, Kurzawa P, Neyaz A, et al. Chondroblastoma expresses RANKL by RNA in situ hybridization and may respond to denosumab therapy. Am J Surg Pathol 2020;44:1581-90.
65. Althof PA, Ohmori K, Zhou M, et al. Cytogenetic and molecular cytogenetic findings in 43 aneurysmal bone cysts: aberrations of 17p mapped to 17p13.2 by fluorescence in situ hybridization. Mod Pathol 2004;17:518-25.
66. Oliveira AM, Chou MM. USP6-induced neoplasms: the biologic spectrum of aneurysmal bone cyst and nodular fasciitis. Hum Pathol 2014;45:1-11.
67. Blackburn PR, Davila JI, Jackson RA, et al. RNA sequencing identifies a novel USP9X-USP6 promoter swap gene fusion in a primary aneurysmal bone cyst. Genes Chromosomes Cancer 2019;58:589-94.
68. Guseva NV, Jaber O, Tanas MR, et al. Anchored multiplex PCR for targeted next-generation sequencing reveals recurrent and novel USP6 fusions and upregulation of USP6 expression in aneurysmal bone cyst. Genes Chromosomes Cancer 2017;56:266-77.
69. Šekoranja D, Boštjančič E, Salapura V, Mavčič B, Pižem J. Primary aneurysmal bone cyst with a novel SPARC-USP6 translocation identified by next-generation sequencing. Cancer Genet 2018;228-229:12-6.
70. Šekoranja D, Zupan A, Mavčič B, et al. Novel ASAP1-USP6, FAT1-USP6, SAR1A-USP6, and TNC-USP6 fusions in primary aneurysmal bone cyst. Genes Chromosomes Cancer 2020;59:357-65.
71. Milne P, Bigley V, Bacon CM, et al. Hematopoietic origin of Langerhans cell histiocytosis and Erdheim-Chester disease in adults. Blood 2017;130:167-75.
72. Postini AM, Andreacchio A, Boffano M, Pagano M, Brach Del Prever A, Fagioli F. Langerhans cell histiocytosis of bone in children: a long-term retrospective study. J Pediatr Orthop B 2012;21:457-62.
73. Maria Postini A, del Prever AB, Pagano M, et al. Langerhans cell histiocytosis: 40 years’ experience. J Pediatr Hematol Oncol 2012;34:353-8.
74. Badalian-Very G, Vergilio JA, Degar BA, et al. Recurrent BRAF mutations in Langerhans cell histiocytosis. Blood 2010;116:1919-23.
75. Papo M, Cohen-Aubart F, Trefond L, et al. Systemic histiocytosis (langerhans cell histiocytosis, erdheim-chester disease, destombes-rosai-dorfman disease): from oncogenic mutations to inflammatory disorders. Curr Oncol Rep 2019;21:62.
76. Jour G, Oultache A, Sadowska J, et al. GNAS mutations in fibrous dysplasia: a comparative study of standard sequencing and locked nucleic acid PCR sequencing on decalcified and nondecalcified formalin-fixed paraffin-embedded tissues. Appl Immunohistochem Mol Morphol 2016;24:660-7.
78. Pereira TDSF, Gomes CC, Brennan PA, Fonseca FP, Gomez RS. Fibrous dysplasia of the jaws: integrating molecular pathogenesis with clinical, radiological, and histopathological features. J Oral Pathol Med 2019;48:3-9.
80. Amary MF, Bacsi K, Maggiani F, et al. IDH1 and IDH2 mutations are frequent events in central chondrosarcoma and central and periosteal chondromas but not in other mesenchymal tumours. J Pathol 2011;224:334-43.
81. Nicolle R, Ayadi M, Gomez-Brouchet A, et al. Integrated molecular characterization of chondrosarcoma reveals critical determinants of disease progression. Nat Commun 2019;10:4622.
82. Parker SJ, Metallo CM. Metabolic consequences of oncogenic IDH mutations. Pharmacol Ther 2015;152:54-62.
83. He X, Pang ZG, Zhang XL, et al. Consistent amplification of FRS2 and MDM2 in low-grade osteosarcoma: a genetic study of 22 cases with clinicopathologic analysis. Am J Surg Pathol 2018;42:1143-55.
84. Limbach AL, Lingen MW, McElherne J, et al. The utility of MDM2 and CDK4 immunohistochemistry and MDM2 FISH in craniofacial osteosarcoma. Head Neck Pathol 2020;14:889-98.
85. Righi A, Gambarotti M, Benini S, et al. MDM2 and CDK4 expression in periosteal osteosarcoma. Hum Pathol 2015;46:549-53.
86. Chen PC, Yen CC, Hung GY, Pan CC, Chen WM. Gene amplification and tumor grading in parosteal osteosarcoma. J Chin Med Assoc 2019;82:889-94.
87. Olivos DJ 3rd, Perrien DS, Hooker A, et al. The proto-oncogene function of Mdm2 in bone. J Cell Biochem 2018;119:8830-40.
88. Shing DC, McMullan DJ, Roberts P, et al. FUS/ERG gene fusions in Ewing’s tumors. Cancer Res 2003;63:4568-4576.
89. Delattre O, Zucman J, Plougastel B, et al. Gene fusion with an ETS DNA-binding domain caused by chromosome translocation in human tumours. Nature 1992;359:162-5.
90. Delattre O, Zucman J, Melot T, et al. The Ewing family of tumors - a subgroup of small-round-cell tumors defined by specific chimeric transcripts. N Engl J Med 1994;331:294-9.
91. Watson S, Perrin V, Guillemot D, et al. Transcriptomic definition of molecular subgroups of small round cell sarcomas. J Pathol 2018;245:29-40.
93. Folpe AL. Phosphaturic mesenchymal tumors: a review and update. Semin Diagn Pathol 2019;36:260-8.
94. Lee JC, Su SY, Changou CA, et al. Characterization of FN1-FGFR1 and novel FN1-FGF1 fusion genes in a large series of phosphaturic mesenchymal tumors. Mod Pathol 2016;29:1335-46.
95. Lee JC, Jeng YM, Su SY, et al. Identification of a novel FN1-FGFR1 genetic fusion as a frequent event in phosphaturic mesenchymal tumour. J Pathol 2015;235:539-45.
96. Graham RP, Hodge JC, Folpe AL, et al. A cytogenetic analysis of 2 cases of phosphaturic mesenchymal tumor of mixed connective tissue type. Hum Pathol 2012;43:1334-8.
97. Xu J, Li D, Xie L, Tang S, Guo W. Mesenchymal chondrosarcoma of bone and soft tissue: a systematic review of 107 patients in the past 20 years. PLoS One 2015;10:e0122216.
98. Wang L, Motoi T, Khanin R, et al. Identification of a novel, recurrent HEY1-NCOA2 fusion in mesenchymal chondrosarcoma based on a genome-wide screen of exon-level expression data. Genes Chromosomes Cancer 2012;51:127-39.
99. Naumann S, Krallman PA, Unni KK, Fidler ME, Neff JR, Bridge JA. Translocation der(13;21)(q10;q10) in skeletal and extraskeletal mesenchymal chondrosarcoma. Mod Pathol 2002;15:572-6.
100. Nord KH, Lilljebjörn H, Vezzi F, et al. GRM1 is upregulated through gene fusion and promoter swapping in chondromyxoid fibroma. Nat Genet 2014;46:474-7.
101. Sandberg AA, Bridge JA. Updates on the cytogenetics and molecular genetics of bone and soft tissue tumors: osteosarcoma and related tumors. Cancer Genet Cytogenet 2003;145:1-30.
103. Lin YH, Jewell BE, Gingold J, et al. Osteosarcoma: molecular pathogenesis and iPSC modeling. Trends Mol Med 2017;23:737-55.
104. Li CJ, Cong Y, Liu XZ, et al. The progress of molecular diagnostics of osteosarcoma. Front Biosci (Landmark Ed) 2016;21:20-30.
105. Negri GL, Grande BM, Delaidelli A, et al. Integrative genomic analysis of matched primary and metastatic pediatric osteosarcoma. J Pathol 2019;249:319-31.
106. Lin Y, Seger N, Chen Y, et al. hTERT promoter mutations in chondrosarcomas associate with progression and disease-related mortality. Mod Pathol 2018;31:1834-41.
107. van Oosterwijk JG, de Jong D, van Ruler MA, et al. Three new chondrosarcoma cell lines: one grade III conventional central chondrosarcoma and two dedifferentiated chondrosarcomas of bone. BMC Cancer 2012;12:375.
108. Zhu GG, Nafa K, Agaram N, et al. Genomic profiling identifies association of
109. Sakamoto A. The molecular pathogenesis of dedifferentiated chondrosarcoma. Indian J Orthop 2014;48:262-5.
110. Swarts SJ, Neff JR, Johansson SL, Bridge JA. Cytogenetic analysis of dedifferentiated chondrosarcoma. Cancer Genet Cytogenet 1996;89:49-51.
111. Makise N, Sekimizu M, Konishi E, et al. H3K27me3 deficiency defines a subset of dedifferentiated chondrosarcomas with characteristic clinicopathological features. Mod Pathol 2019;32:435-45.
112. Ali NM, Niada S, Morris MR, et al. Comprehensive molecular characterization of adamantinoma and OFD-like adamantinoma bone tumors. Am J Surg Pathol 2019;43:965-74.
113. Gleason BC, Liegl-Atzwanger B, Kozakewich HP, et al. Osteofibrous dysplasia and adamantinoma in children and adolescents: a clinicopathologic reappraisal. Am J Surg Pathol 2008;32:363-76.
114. Taylor RM, Kashima TG, Ferguson DJ, Szuhai K, Hogendoorn PC, Athanasou NA. Analysis of stromal cells in osteofibrous dysplasia and adamantinoma of long bones. Mod Pathol 2012;25:56-64.
115. Kanamori M, Antonescu CR, Scott M, et al. Extra copies of chromosomes 7, 8, 12, 19, and 21 are recurrent in adamantinoma. J Mol Diagn 2001;3:16-21.
116. Hazelbag HM, Wessels JW, Mollevangers P, van den Berg E, Molenaar WM, Hogendoorn PC. Cytogenetic analysis of adamantinoma of long bones: further indications for a common histogenesis with osteofibrous dysplasia. Cancer Genet Cytogenet 1997;97:5-11.
117. Shih AR, Chebib I, Deshpande V, Dickson BC, Iafrate AJ, Nielsen GP. Molecular characteristics of poorly differentiated chordoma. Genes Chromosomes Cancer 2019;58:804-8.
118. Scheil-Bertram S, Kappler R, von Baer A, et al. Molecular profiling of chordoma. Int J Oncol 2014;44:1041-55.
119. Gil Z, Fliss DM. Cytogenetic analysis of skull base tumors: where do we stand? Curr Opin Otolaryngol Head Neck Surg 2012;20:130-6.
120. Gil Z, Fliss DM, Voskoboinik N, et al. Cytogenetic analysis of three variants of clival chordoma. Cancer Genet Cytogenet 2004;154:124-30.
121. Scheil S, Brüderlein S, Liehr T, et al. Genome-wide analysis of sixteen chordomas by comparative genomic hybridization and cytogenetics of the first human chordoma cell line, U-CH1. Genes Chromosomes Cancer 2001;32:203-11.
122. Brohl AS, Kahen E, Yoder SJ, Teer JK, Reed DR. The genomic landscape of malignant peripheral nerve sheath tumors: diverse drivers of Ras pathway activation. Sci Rep 2017;7:14992.
123. Wesche WA, Khare V, Rao BN, Bowman LC, Parham DM. Malignant peripheral nerve sheath tumor of bone in children and adolescents. Pediatr Dev Pathol 1999;2:159-67.
124. Liu WJ, Zhang S, Liu JX, Shao ZW. Intraosseous malignant peripheral nerve sheath tumor of 2 consecutive lumbar vertebrae: a case report and literature review. World Neurosurg 2019;130:459-66.
125. Davila A, Conrad EU, Ayala GE, Beckmann NM. Intraosseous malignant peripheral nerve sheath tumor of the sacrum in a patient with neurofibromatosis type I. Radiol Case Rep 2019;14:880-4.
126. Demicco EG, Wang WL, Madewell JE, et al. Osseous myxochondroid sarcoma: a detailed study of 5 cases of extraskeletal myxoid chondrosarcoma of the bone. Am J Surg Pathol 2013;37:752-62.
127. Finos L, Righi A, Frisoni T, et al. Primary extraskeletal myxoid chondrosarcoma of bone: Report of three cases and review of the literature. Pathol Res Pract 2017;213:461-6.
128. Hisaoka M, Hashimoto H. Extraskeletal myxoid chondrosarcoma: updated clinicopathological and molecular genetic characteristics. Pathol Int 2005;55:453-63.
129. Tran H, Shillingford N, Thomas S, Hammoudeh J, Zhou S. Primary epithelioid sarcoma of the zygomatic bone. Pediatr Dev Pathol 2019;22:252-7.
130. Raoux D, Péoc’h M, Pedeutour F, Vaunois B, Decouvelaere AV, Folpe AL. Primary epithelioid sarcoma of bone: report of a unique case, with immunohistochemical and fluorescent in situ hybridization confirmation of INI1 deletion. Am J Surg Pathol 2009;33:954-8.
131. Wojcik JB, Bellizzi AM, Dal Cin P, et al. Primary sclerosing epithelioid fibrosarcoma of bone: analysis of a series. Am J Surg Pathol 2014;38:1538-44.
132. Tsuda Y, Dickson BC, Dry SM, et al. Clinical and molecular characterization of primary sclerosing epithelioid fibrosarcoma of bone and review of the literature. Genes Chromosomes Cancer 2020;59:217-24.
133. Kosemehmetoglu K, Ardic F, Kilpatrick SE, Aydingoz U, Sumathi VP, Michal M. Sclerosing epithelioid fibrosarcoma of bone: morphological, immunophenotypical, and molecular findings of 9 cases. Virchows Arch 2020; doi: 10.1007/s00428-020-02953-y.
134. Kalil RK. Liposarcoma of bone. In: Santini-Araujo E, Kalil R, Bertoni F, Park YK, editors. Tumors and tumor-like lesions of bone. London: Springer; 2015. pp. 569-573.
135. Tiemeier GL, Brown JM, Pratap SE, et al. Pleomorphic liposarcoma of bone: a rare primary malignant bone tumour. Clin Sarcoma Res 2018;8:2.
136. Zhang S, Wang XQ. Primary dedifferentiated liposarcoma of the femur presenting with malignant fibrous histiocytoma: a case report and review of the literature. Oncol Lett 2014;8:663-6.
137. Rovlias A, Balanika A, Nomikos A, Melissaris S. Primary myxoid liposarcoma of the upper thoracic spine in an elderly patient. J Neurosci Rural Pract 2017;8:S120-2.
138. Sandberg AA. Updates on the cytogenetics and molecular genetics of bone and soft tissue tumors: liposarcoma. Cancer Genet Cytogenet 2004;155:1-24.
139. Park YK, Unni KK, Kim YW, et al. Primary alveolar soft part sarcoma of bone. Histopathology 1999;35:411-7.
141. Kawamura-Saito M, Yamazaki Y, Kaneko K, et al. Fusion between CIC and DUX4 up-regulates PEA3 family genes in Ewing-like sarcomas with t(4;19)(q35;q13) translocation. Hum Mol Genet 2006;15:2125-37.
142. Pižem J, Šekoranja D, Zupan A, et al. FUS-NFATC2 or EWSR1-NFATC2 fusions are present in a large proportion of simple bone cysts. Am J Surg Pathol 2020;44:1623-34.
143. Pierron G, Tirode F, Lucchesi C, et al. A new subtype of bone sarcoma defined by BCOR-CCNB3 gene fusion. Nat Genet 2012;44:461-6.
144. Szuhai K, Ijszenga M, de Jong D, Karseladze A, Tanke HJ, Hogendoorn PC. The NFATc2 gene is involved in a novel cloned translocation in a Ewing sarcoma variant that couples its function in immunology to oncology. Clin Cancer Res 2009;15:2259-68.
145. Davicioni E, Anderson MJ, Finckenstein FG, et al. Molecular classification of rhabdomyosarcoma - genotypic and phenotypic determinants of diagnosis: a report from the Children’s Oncology Group. Am J Pathol 2009;174:550-64.
146. Balogh P, Bánusz R, Csóka M, Váradi Z, Varga E, Sápi Z. Primary alveolar rhabdomyosarcoma of the bone: two cases and review of the literature. Diagn Pathol 2016;11:99.
147. Agaram NP, Zhang L, Sung YS, et al. Expanding the spectrum of intraosseous rhabdomyosarcoma: correlation between 2 distinct gene fusions and phenotype. Am J Surg Pathol 2019;43:695-702.
148. Anderson J, Gordon A, McManus A, Shipley J, Pritchard-Jones K. Disruption of imprinted genes at chromosome region 11p15.5 in paediatric rhabdomyosarcoma. Neoplasia 1999;1:340-8.
149. Wang GY, Lucas DR. Primary leiomyosarcoma of bone: review and update. Arch Pathol Lab Med 2019;143:1332-7.
150. Wirbel RJ, Verelst S, Hanselmann R, Remberger K, Kubale R, Mutschler WE. Primary leiomyosarcoma of bone: clinicopathologic, immunohistochemical, and molecular biologic aspects. Ann Surg Oncol 1998;5:635-41.
151. Verbeke SL, Fletcher CD, Alberghini M, et al. A reappraisal of hemangiopericytoma of bone; analysis of cases reclassified as synovial sarcoma and solitary fibrous tumor of bone. Am J Surg Pathol 2010;34:777-83.
152. Robinson DR, Wu YM, Kalyana-Sundaram S, et al. Identification of recurrent NAB2-STAT6 gene fusions in solitary fibrous tumor by integrative sequencing. Nat Genet 2013;45:180-5.
153. Ge XH, Liao JS, Choo RJ, Yan JC, Zhang JF. Solitary fibrous tumor of the ilium: a case report. Medicine (Baltimore) 2017;96:e9355.
154. Horvai A, Dashti NK, Rubin BP, et al. Genetic and molecular reappraisal of spindle cell adamantinoma of bone reveals a small subset of misclassified intraosseous synovial sarcoma. Mod Pathol 2019;32:231-41.
155. Fujibuchi T, Miyawaki J, Kidani T, et al. Intraosseous synovial sarcoma of the distal ulna: a case report and review of the literature. BMC Cancer 2019;19:116.
156. Shah WU, Shujaat SD, Ullah N, Mansoor S. Synovial sarcoma of cervicodorsal spine: a case report. J Pak Med Assoc 2018;68:1100-1104.
157. Kim J, Lee SH, Choi YL, Bae GE, Kim ES, Eoh W. Synovial sarcoma of the spine: a case involving paraspinal muscle with extensive calcification and the surgical consideration in treatment. Eur Spine J 2014;23:27-31.
158. Verma A, Rekhi B. Myoepithelial tumor of soft tissue and bone: a current perspective. Histol Histopathol 2017;32:861-77.
159. Rekhi B, Amare P, Gulia A, et al. Primary intraosseous myoepithelioma arising in the iliac bone and displaying trisomies of 11, 15, 17 with del (16q) and del (22q11)--A rare case report with review of literature. Pathol Res Pract 2011;207:780-5.
160. Rekhi B, Joshi S, Panchwagh Y, et al. Clinicopathological features of five unusual cases of intraosseous myoepithelial carcinomas, mimicking conventional primary bone tumours, including EWSR1 rearrangement in one case. APMIS 2016;124:278-90.
161. Suurmeijer AJH, Dickson BC, Swanson D, et al. A morphologic and molecular reappraisal of myoepithelial tumors of soft tissue, bone, and viscera with EWSR1 and FUS gene rearrangements. Genes Chromosomes Cancer 2020;59:348-56.
162. Yamaguchi S, Yamazaki Y, Ishikawa Y, Kawaguchi N, Mukai H, Nakamura T. EWSR1 is fused to POU5F1 in a bone tumor with translocation t(6;22)(p21;q12). Genes Chromosomes Cancer 2005;43:217-22.
163. Xian J, Righi A, Vanel D, Baldini N, Errani C. Epithelioid hemangioma of bone: a unique case with multifocal metachronous bone lesions. J Clin Orthop Trauma 2019;10:1068-72.
164. van IJzendoorn DG, de Jong D, Romagosa C, et al. Fusion events lead to truncation of FOS in epithelioid hemangioma of bone. Genes Chromosomes Cancer 2015;54:565-74.
165. Huang SC, Zhang L, Sung YS, et al. Frequent FOS gene rearrangements in epithelioid hemangioma: a molecular study of 58 cases with morphologic reappraisal. Am J Surg Pathol 2015;39:1313-21.
166. Verbeke SL, Bovée JV. Primary vascular tumors of bone: a spectrum of entities? Int J Clin Exp Pathol 2011;4:541-51.
167. Xu YJ, Chen WX, Cheng H, Lin ZY. Epithelioid hemangioendothelioma of the bone: a case report with findings of bone scintigraphy. Medicine (Baltimore) 2019;98:e15546.
168. Otani S, Nakayama R, Sekita T, et al. Pseudomyogenic hemangioendothelioma of bone treated with denosumab: a case report. BMC Cancer 2019;19:872.
169. Inyang A, Mertens F, Puls F, et al. Primary pseudomyogenic hemangioendothelioma of bone. Am J Surg Pathol 2016;40:587-98.
170. Walther C, Tayebwa J, Lilljebjörn H, et al. A novel SERPINE1-FOSB fusion gene results in transcriptional up-regulation of FOSB in pseudomyogenic haemangioendothelioma. J Pathol 2014;232:534-40.
171. Baliaka A, Balis G, Michalopoulou-Manoloutsiou E, Papanikolaou A, Nikolaidou A. Primary angiosarcoma of bone. A case report. Hippokratia 2013;17:180-2.
172. Palmerini E, Leithner A, Windhager R, et al. Angiosarcoma of bone: a retrospective study of the European Musculoskeletal Oncology Society (EMSOS). Sci Rep 2020;10:10853.
173. Deshpande V, Rosenberg AE, O’Connell JX, Nielsen GP. Epithelioid angiosarcoma of the bone: a series of 10 cases. Am J Surg Pathol 2003;27:709-16.
174. Huang SC, Zhang L, Sung YS, et al. Recurrent CIC gene abnormalities in Angiosarcomas: a molecular study of 120 cases with concurrent investigation of PLCG1, KDR, MYC, and FLT4 gene alterations. Am J Surg Pathol 2016;40:645-55.
175. Ali NM, Niada S, Brini AT, et al. Genomic and transcriptomic characterisation of undifferentiated pleomorphic sarcoma of bone. J Pathol 2019;247:166-76.
176. Li XN, Zhang ZK, Latif M, Chen W, Cui JL, Peng ZG. Synovium as a widespread pathway to the adjacent joint in undifferentiated high-grade pleomorphic sarcoma of the tibia: a case report. Medicine (Baltimore) 2018;97:e9870.
177. Zhou N, Schäfer R, Li T, Fang MY, Liu LY. A primary undifferentiated pleomorphic sarcoma of the lumbosacral region harboring a LMNA-NTRK1 gene fusion with durable clinical response to crizotinib: a case report. BMC Cancer 2018;18:842.
178. Cerejeira A, Gomes N, Costa-Silva M, Moreira E, Azevedo F. Undifferentiated pleomorphic sarcoma presenting as an exophytic pedunculated tumor on the left scapula. Dermatol Online J 2019;25:13030/qt738094fs.
179. Garces S, Medeiros LJ, Patel KP, et al. Mutually exclusive recurrent KRAS and MAP2K1 mutations in Rosai-Dorfman disease. Mod Pathol 2017;30:1367-77.
180. Mosheimer BA, Oppl B, Zandieh S, et al. Bone involvement in rosai-dorfman disease (RDD): a case report and systematic literature review. Curr Rheumatol Rep 2017;19:29.
181. Mavrogenis AF, Igoumenou VG, Antoniadou T, et al. Rare diseases of bone: Erdheim-Chester and Rosai-Dorfman non-Langerhans cell histiocytoses. EFORT Open Rev 2018;3:381-90.
182. Ross AB, Davis KW, Buehler D, Chan BY. Primary rosai-dorfman disease of bone: a report of two cases. Case Rep Radiol 2019;2019:1720131.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Suster D, Suster S. Genetic characteristics and molecular diagnostics of bone tumors. J Cancer Metastasis Treat 2021;7:8. http://dx.doi.org/10.20517/2394-4722.2020.119
AMA Style
Suster D, Suster S. Genetic characteristics and molecular diagnostics of bone tumors. Journal of Cancer Metastasis and Treatment. 2021; 7: 8. http://dx.doi.org/10.20517/2394-4722.2020.119
Chicago/Turabian Style
Suster, David, Saul Suster. 2021. "Genetic characteristics and molecular diagnostics of bone tumors" Journal of Cancer Metastasis and Treatment. 7: 8. http://dx.doi.org/10.20517/2394-4722.2020.119
ACS Style
Suster, D.; Suster S. Genetic characteristics and molecular diagnostics of bone tumors. J. Cancer. Metastasis. Treat. 2021, 7, 8. http://dx.doi.org/10.20517/2394-4722.2020.119
About This Article
Copyright

Data & Comments
Data



 Cite This Article 35 clicks
Cite This Article 35 clicks

 Like This Article 21
likes
Like This Article 21
likes






Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.