
Leelamine suppresses cMyc expression in prostate cancer cells in vitro and inhibits prostate carcinogenesis in vivo

Abstract
Aim: Leelamine (LLM) inhibits the growth of human prostate cancer cells but the underlying mechanism is not fully understood. The present study was undertaken to determine the effect of LLM on cMyc, which is overexpressed in a subset of human prostate cancers.
Methods: The effect of LLM on cMyc expression and activity was determined by western blotting/confocal microscopy and luciferase reporter assay, respectively. A transgenic mouse model of prostate cancer (Hi-Myc) was used to determine the chemopreventive efficacy of LLM.
Results: Exposure of androgen-sensitive (LNCaP) and castration-resistant (22Rv1) human prostate cancer cells to LLM resulted in downregulation of protein and mRNA levels of cMyc. Overexpression of cMyc partially attenuated LLM-mediated inhibition of colony formation, cell viability, and cell migration in 22Rv1 and/or PC-3 cells. LLM treatment decreased protein levels of cMyc targets (e.g., lactate dehydrogenase), however, overexpression of cMyc did not attenuate these effects. A trend for a decrease in the expression level of cMyc protein was discernible in 22Rv1 xenografts from LLM-treated mice compared with control mice. LLM treatment (10 mg/kg body weight, 5 times/week) was well-tolerated by Hi-Myc transgenic mice. The incidence of high-grade prostatic intraepithelial neoplasia, adenocarcinoma in situ, and microinvasion were lower in LLM-treated Hi-Myc mice but the difference was not statistically significant.
Conclusion: The present study reveals that LLM inhibits cMyc expression in human prostate cancer cells in vitro but concentrations higher than 10 mg/kg may be required to achieve chemoprevention of prostate cancer.
Keywords
INTRODUCTION
Prostate cancer is a leading cause of cancer-related deaths in the United States[1]. Several oncogenic drivers of prostate cancer have been identified, including androgen receptor (AR) and cMyc transcription factors[2]. Studies using transgenic mice and human specimens indicate an oncogenic role for cMyc in prostate cancer[3-7]. For example, transgenic expression of cMyc in prostate epithelial cells resulted in transformation to prostatic intraepithelial neoplasia (PIN) at 2-4 weeks of age that progressed to adenocarcinoma by 6 months[3]. About 67% of human prostate adenocarcinoma (n = 9) exhibited high cMyc levels when compared to benign hypertrophied prostate tissue (n = 19)[4]. An androgen-responsive cell line (LNCaP) derived from lymph node metastasis of a human prostate cancer patient also showed gene amplification, rearrangement, and overexpression of cMyc[5]. The nuclear level of cMyc protein was shown to be an early event in the pathogenesis of human prostate cancer[7]. cMyc overexpression was reported in about 70% of early-stage prostate cancer and up to 37% of metastatic prostate cancer patients[8-10]. The 8q gain leading to cMyc overexpression was associated with poor survival in prostate cancer patients[11]. Moreover, cMyc overexpression is suggested to be an important mechanism in the resistance of prostate cancers to antiandrogen therapy[12]. cMyc overexpression also causes increased cell proliferation and metabolic deregulation, a hallmark of different cancers including prostate cancer, including increased glycolysis and de novo synthesis of fatty acids[13-15]. It is not surprising that considerable efforts have been devoted to the therapeutic targeting of cMyc in prostate cancer[16].
Chemoprevention using natural products or synthetic chemicals is attractive for blunting the death and suffering from prostate cancer, but a clinically effective chemopreventive intervention for this malignancy is still lacking[17-22]. Leelamine (LLM) is one such phytochemical whose effectiveness has been tested against prostate and other cancers[23-25]. The antitumor effect of LLM was initially reported in melanoma but our laboratory was the first to document its effectiveness against prostate cancer cells[24,25]. In the melanoma model, the antitumor effect of LLM was attributed to the suppression of multiple oncogenic signalling pathways, including phosphatidylinositol 3-kinase, mitogen-activated protein kinase, and signal transducer and activation of transcription 3 (STAT3)[24]. In prostate cancer cells, LLM was shown to inhibit cellular growth by suppressing the expression and activity of AR[25]. Intraperitoneal administration of about 9 mg LLM/kg body weight (5 times/week) to 22Rv1 xenograft bearing mice resulted in a significant decrease in Ki-67 expression, mitotic fraction, expression of full-length AR and AR-V7 splice variants, and prostate-specific antigen[25].
In the present study, we investigated the effect of LLM treatment on cMyc expression and activity using human prostate cancer cell lines and the Hi-Myc transgenic mouse model. The foundation for this study was based on the following observations: (1) LLM suppresses AR activity that is a ligand-independent regulator of cMyc expression in prostate cancer[25,26]; and (2) cMyc promotes fatty acid synthesis in prostate cancer that is inhibited by LLM treatment in vitro and in vivo[15,27].
METHODS
Reagents
LLM was purchased from Cayman Chemical Company (Ann Arbor, MI). The reagents for cell culture were purchased from Life Technologies-Thermo Fisher Scientific (Waltham, MA). Antibodies against cMyc, hexokinase II (HKII), pyruvate kinase M2 (PKM2), and lactate dehydrogenase A (LDH-A) were purchased from Cell Signaling Technology (Danvers, MA). The antibodies against glucose transporter 1 (Glut1) and Cyclin D1 were purchased from Abcam (Cambridge, MA) and Santa Cruz Biotechnology (Dallas, TX), respectively. An antibody against glyceraldehyde 3-phosphate dehydrogenase (GAPDH) was purchased from GeneTex (Irvine, CA). The anti-β-Actin antibody was purchased from Sigma-Aldrich (St. Louis, MO). Alexa Fluor 488-conjugated goat anti-rabbit antibody was from Life Technologies. A kit for colorimetric measurement of lactate was purchased from BioVision (Milpitas, CA).
Cell lines
The human prostate cancer cell lines, LNCaP, 22Rv1, and PC-3, were purchased from the American Type Culture Collection (ATCC; Manassas, VA) and cultured by following the supplier’s recommendations. The University of Pittsburgh has a formal material transfer agreement with ATCC. These cell lines were last authenticated by us in March of 2017. The Myc-CaP cell line derived from the prostate tumor of a Hi-Myc mouse was a kind gift from Dr. Charles L. Sawyers (Memorial Sloan Kettering Cancer Center, New York, NY, USA). The Myc-CaP cells were cultured in Dulbecco’s modified essential medium supplemented with 4.5 g/L glucose, L-glutamine, sodium pyruvate, 10% fetal bovine serum, and antibiotic mixture. The PC-3 and 22Rv1 cells were stably transfected with an empty pcDNA3 vector or the same vector encoding cMyc (Addgene, #16011), as described previously[28].
Western blotting
The prostate tumors from control and LLM-treated mice from our previously published study were used for western blotting[25]. Details of cell lysate preparation and western blotting have been described previously[29,30].
Immunocytochemistry
LNCaP (2 × 104), 22Rv1 (3 × 104), and Myc-Cap (3 × 104) cells were plated on coverslips in 24-well plates. After overnight incubation, the cells were treated with ethanol or LLM (2.5 or 5 µM) for 24 h. The cells were then fixed and permeabilized with 2% paraformaldehyde and 0.5% Triton X-100, respectively. After blocking with phosphate-buffered saline (PBS) containing 0.5% bovine serum albumin and 0.15% glycine, the cells were treated overnight at 4 °C with the cMyc primary antibody followed by Alexa Fluor 488-conjugated goat anti-rabbit antibody (1:2000 dilution) for 1 h and then counterstained with DRAQ5 (nuclear stain) for 5 min at room temperature in the dark.
Quantitative reverse-transcriptase polymerase chain reaction
Total RNA was extracted using RNeasy Mini Kit from Qiagen (Germantown, MD) and by following the manufacturer’s instructions. Complementary DNA was synthesized and reverse transcribed using oligo (dT)20 primer and SuperScript™ III reverse transcriptase. Real-time polymerase chain reaction (PCR) was done from 1:10 diluted complementary DNA using DyNAmo HS SYBR Green qPCR kits (Thermo Fisher Scientific) on ABI StepOnePlus PCR Systems (Applied Biosystems-Life Technologies). Primers for human cMyc and GAPDH were as follows: Forward (cMyc): 5’-GCCACGTCTCCACACATCAG-3’; reverse: (cMyc) 5’-TGGTGCATTTTCGGTTGTTG-3’; forward (GAPDH): 5’-GGACCTGACCTGCCGTCTAGAA-3’; reverse (GAPDH): 5’-GGTGTCGCTGTTGAAGTCAGAG-3’. The PCR conditions were as follows: 95 °C for 10 min followed by 40 cycles at 95 °C for 15 s, 60 °C for 1 min, and 72 °C for 30 s. Gene expression change was calculated using the method of Livak and Schmittgen[31].
Luciferase reporter assay
The LNCaP or 22Rv1 cells were co-transfected with 2 µg pBV-Luc wt MBS1-4 plasmid and 0.2 µg of pCMV-RL using FuGENE6. Twenty-four hours after co-transfection, the cells were treated with ethanol or the indicated doses of LLM for 12 h. Luciferase activity was determined using Dual-Luciferase® Reporter Assay kit from Promega, and by following the manufacturer’s instructions. The values were corrected for protein concentration and renilla luciferase.
Clonogenic assay
Empty vector (hereafter abbreviated as EV) transfected control cells and cMyc overexpressing 22Rv1 (1000 cells) were seeded in 6-well plates in triplicate and allowed to incubate overnight. The cells were then treated with ethanol or desired doses of LLM. The medium containing ethanol or LLM was replaced every third day. After 10 days, the cells were fixed with 100% methanol for 5 min at room temperature and stained with 0.5% crystal violet solution in 20% methanol for 30 min at room temperature. The colonies from control and LLM treatment groups were counted under an inverted microscope.
Cell viability and cell migration assay
Trypan blue dye exclusion assay was performed to determine the effect of LLM on cell viability as described previously[32]. Cell migration assay was performed as described previously[33].
Determination of lactate
The lactate level of plasma and tumors archived from our previous study[25] was measured using a colorimetric assay kit. Plasma specimens and tumor lysates were filtered using a 10 kDa filter. Assays were performed according to the manufacturer’s instructions.
The in vivo study using Hi-Myc mice
Female transgenic FVB-Tg (ARR2/Pbsn-MYC) mice were crossed with male wild-type FVB/N mice, both purchased from the NCI Mouse Repository, to establish a colony. Following transgene verification, 5-week-old male transgenic Hi-Myc mice were divided into two groups. Mice of group 1 were treated with 100 µL vehicle, while group 2 mice received 10 mg LLM/kg body weight by intraperitoneal injection 5 times/week (Monday through Friday). The vehicle consisted of 10% ethanol, 10% dimethyl sulfoxide, 30% kolliphor EL (Sigma-Aldrich, St. Louis, MO), and 50% PBS. After 5 weeks of treatment, the animals were euthanized by CO2 overdose (supplied via compressed gas cylinder), and blood and prostate tissue were collected.
Statistical tests
The Student’s t-test was used for two-group comparisons. One-way analysis of variance (ANOVA) followed by Bonferroni’s test was used for multiple comparisons. ANOVA followed by Dunnett’s test was used for dose-response comparisons. Statistical significance for the effect of LLM on the incidence of hyperplasia (HYP), low-grade PIN (LG-PIN), high-grade PIN (HG-PIN), adenocarcinoma (AC) in situ, and microinvasion (Mic-inv) was determined by Fisher’s test. GraphPad Prism (version 8.0.0) was used to perform statistical analyses.
RESULTS
Effect of LLM on the expression of cMyc
Pharmacokinetics of LLM has been investigated using male ICR mice after a single oral administration with 10 mg/kg body weight dose[33]. The peak plasma concentration of LLM was shown to be about 2.8 µM[33]. Thus, LLM concentrations of 2.5 and 5 μM were used to determine its effect on the expression of cMyc. As can be seen in Figure 1A, the expression of cMyc protein was decreased in a concentration-dependent manner. The LLM-mediated downregulation of the cMyc protein was confirmed by confocal microscopy [Figure 1B]. LLM treatment also inhibited mRNA levels of cMyc [Figure 1C] and suppressed its transcriptional activity [Figure 1D]. Collectively, these results indicated suppression of cMyc expression and activity following LLM treatment in LNCaP and 22Rv1 human prostate cancer cell lines.
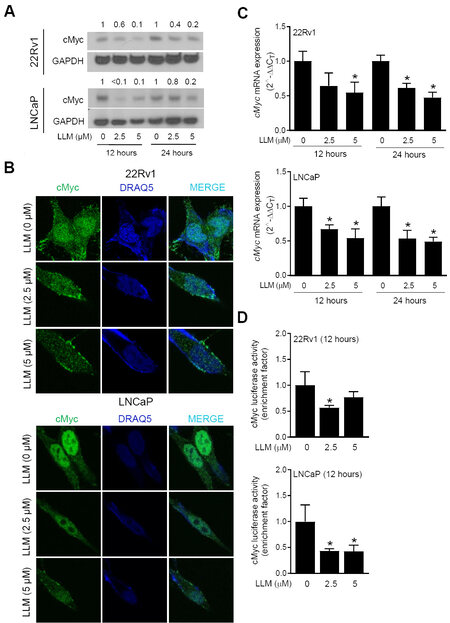
Figure 1. Effect of leelamine (LLM) treatment on expression and activity of cMyc in human prostate cancer cells. The effect of LLM treatment on the protein level of cMyc in 22Rv1 and LNCaP cells as determined by western blotting. The blots were probed with an anti-GAPDH antibody to correct for differences in protein loading. The numbers above bands represent quantitation of the cMyc protein level change relative to corresponding ethanol-treated control (A). Confocal microscopic images of cMyc protein (green fluorescence) in vehicle-treated control and LLM-treated 22Rv1 and LNCaP cells. DRAQ5 (blue fluorescence) was used for nuclear staining (B). The effect of LLM treatment on mRNA level of cMyc. The results shown are mean ± SD (n = 3). *Significantly different (P < 0.05) compared with corresponding ethanol-treated control by one-way ANOVA followed by Dunnett’s test (C). The effect of LLM treatment on the activity of cMyc as determined by luciferase reporter assay. The results shown are mean ± SD (n = 3). *Significantly different (P < 0.05) compared with ethanol-treated control by one-way ANOVA followed by Dunnett’s test (D). Each experiment was performed twice, and the results were generally consistent.
Effect of cMyc overexpression on anticancer effects of LLM
Figure 2A shows overexpression of cMyc protein in 22Rv1 and PC-3 cells stably transfected with the cMyc plasmid (hereafter abbreviated as cMyc) in comparison with EV control cells. The clonogenicity of EV cells was inhibited by LLM treatment in a dose-dependent manner [Figure 2A]. The inhibition of clonogenicity resulting from LLM exposure was partly attenuated by cMyc overexpression [Figure 2A]. Similarly, the cMyc overexpression conferred partial protection against LLM-mediated inhibition of cell viability
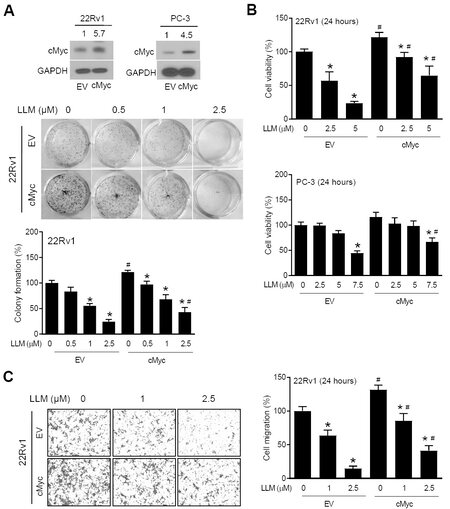
Figure 2. Effect of cMyc overexpression on the anticancer effects of leelamine (LLM). upper: Western blots showing overexpression of the cMyc protein in stable cMyc overexpressing cells (cMyc) compared to empty vector (EV) transfected control cells. The numbers above bands represent quantitation of the cMyc protein level relative to corresponding EV cells. middle and bottom: Clonogenic assay using EV and cMyc cells following treatment with vehicle or LLM. The results shown are mean ± SD (n = 3). Significantly different
Effect of LLM treatment on protein levels of cMyc regulated proteins
Next, we determined the effect of LLM treatment on the levels of cMyc target proteins. Expression of several glycolysis-related proteins was decreased by LLM treatment in 22Rv1 and LNCaP cells [Figure 3A]. There was a decrease in circulating level of lactic acid, the end product of glycolysis, in LLM-treated mice than in control mice but the difference was not significant. The 22Rv1 xenograft level of lactate was also not suppressed by LLM treatment [Figure 3B]. The expression of cMyc and some of its target proteins was also not suppressed to a significant extent by LLM administration to 22Rv1 xenograft bearing mice
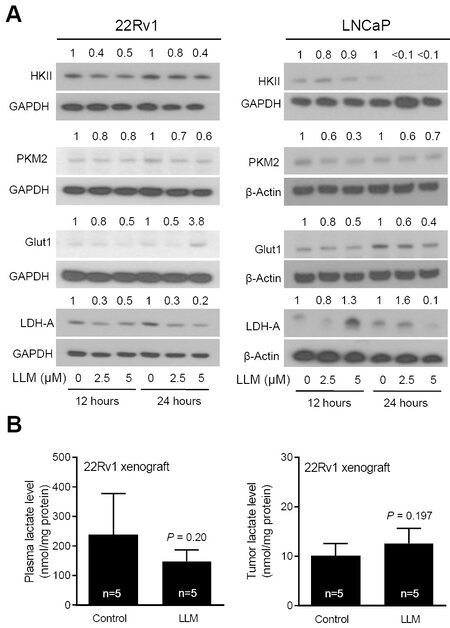
Figure 3. The effect of leelamine (LLM) treatment on glycolysis-related proteins. Western blots showing the effect of LLM treatment on expression levels of glycolysis-related proteins. The numbers above the bands represent quantitation of the protein level change relative to corresponding ethanol-treated control. The experiment was performed twice, and the results were generally consistent (A). Levels of lactate in the plasma and 22Rv1 xenograft tumors of control and LLM-treated mice (n = 5). The results shown are mean ± SD and statistical significance was determined by Student’s t-test (B).
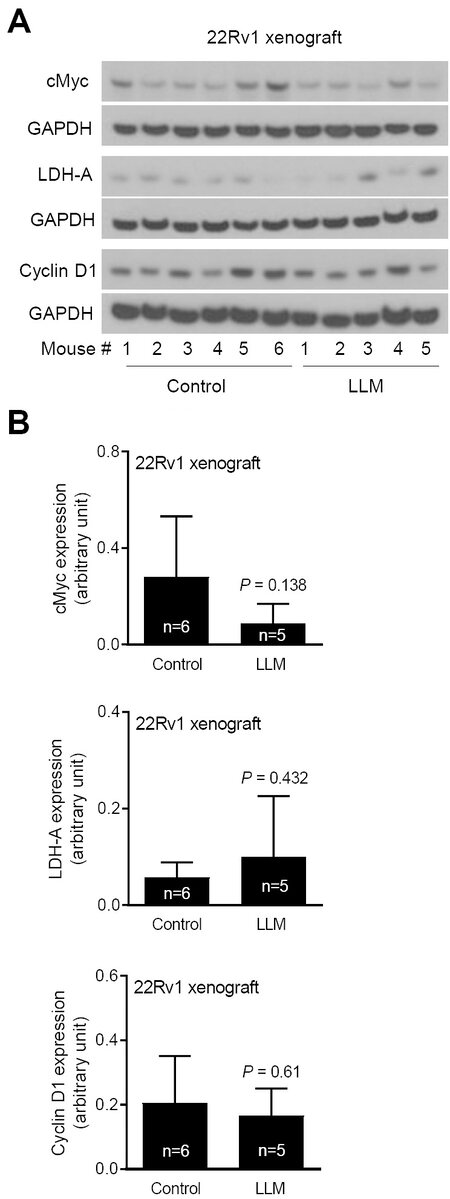
Figure 4. The effect of leelamine (LLM) treatment on cMyc and its targets using lysates from 22Rv1 xenografts. Western blots for cMyc, LDH-A, Cyclin D1, and GAPDH using tumor lysates from control and LLM-treated mice (A). Quantitation of cMyc, LDH-A, and Cyclin D1 expression. The results shown are mean ± SD (n = 5-6). Statistical significance was determined by Student’s t-test (B).
Effect of LLM treatment on cancer incidence in Hi-Myc mice
Initially, we used a cell line (Myc-CaP) derived from the prostate tumor of a Hi-Myc transgenic mouse to determine the effect of LLM treatment on cMyc protein level. Similar to human prostate cancer cells
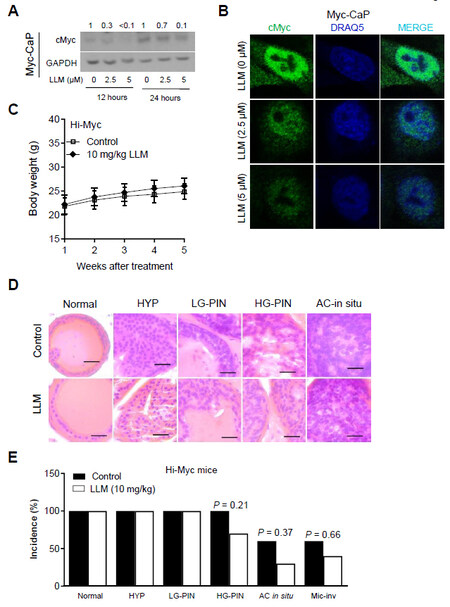
Figure 5. The effect of leelamine (LLM) treatment on cancer incidence in Hi-Myc mice. The effect of LLM treatment on the protein level of cMyc in Myc-CaP cell line as determined by western blotting. The blot was probed with an anti-GAPDH antibody to correct for differences in protein loading. The numbers above bands represent quantitation of the cMyc protein level change relative to corresponding ethanol-treated control. Similar results were obtained in repeated experiments (A). Confocal microscopic images of cMyc protein (green fluorescence) in vehicle-treated control and LLM-treated Myc-CaP cells. DRAQ5 (blue fluorescence) was used for nuclear staining. Similar results were obtained in repeated experiments (B). Body weights of control and LLM-treated Hi-Myc mice (n = 10). Statistical significance was determined by Student’s t-test. LLM treatment did not cause a significant weight loss in Hi-Myc mice (C). Representative H&E-stained sections of the prostate from control and LLM-treated Hi-Myc mice (40x objective magnification, scale bar = 50 µm) (D). The effect of LLM administration on incidence of different histopathologic stages of prostate cancer in Hi-Myc mice (n = 10). Statistical significance was determined by Fisher’s test (E).
DISCUSSION
The present study shows the inhibitory effect of LLM on cMyc expression and activity in prostate cancer cells. The cMyc inhibition is functionally significant because the anticancer effects of LLM, including clonogenicity, cell viability, and cell migration, are partially attenuated by cMyc overexpression. It is also clear that cMyc suppression alone is not responsible for the growth inhibitory effects of LLM on prostate cancer cells. Several other oncogenes have been shown as targets of LLM in other cancer cell types like melanoma, including phosphatidylinositol 3-kinase, mitogen-activated protein kinase, and STAT3[24]. All these oncogenic signalling pathways are known to contribute to the pathogenesis of human prostate cancer[34-36]. For example, analysis of the Oncomine database reveals upregulation of c-Jun N-terminal kinases in prostate cancer[35]. Likewise, STAT3 plays an important role in the progression of prostate cancer to incurable metastatic castration-resistant disease by integrating multiple signalling pathways involved in the reactivation of AR pathway, stem-like cells, and the epithelial to mesenchymal transition[36]. It is conceivable that LLM inhibits these signalling pathways to, in turn, inhibit the growth of prostate cancer cells. However, additional work is necessary to systematically explore this possibility.
Chemoprevention represents a worthwhile strategy for reducing the mortality and morbidity associated with prostate cancer[20-22]. The feasibility of prostate cancer chemoprevention has been explored clinically with 5α-reductase inhibitors (PCPT and REDUCE trials) as well as a novel combination of selenium and vitamin E (e.g., SELECT trial)[17-19]. Whereas administration of 5α-reductase inhibitors resulted in ~23%-25% decrease in overall relative risk, these agents were not approved by the FDA for chemoprevention of prostate cancer due to high-grade tumors in the treatment arm[17,18]. In an 18-year follow-up study, high-grade prostate cancer was still more common in the finasteride treatment group[37]. Similarly, the SELECT trial did not show any preventative benefit of vitamin E and selenium supplementation[18]. Therefore, a safe and efficacious intervention for chemoprevention of prostate cancer is still a clinically unmet need. In this study, we explored the possibility of chemoprevention by LLM using a transgenic mouse model in which prostate cancer is driven by prostate epithelial-specific overexpression of cMyc. There was a trend for a decrease in the incidence of HG-PIN, AC in situ, and Mic-inv but the difference was not significant at the 10 mg/kg body weight dose of LLM. These results are surprising because, in melanoma models, LLM doses of 5 and 7.5 mg/kg body weight were effective for in vivo inhibition of tumor growth[24].
In conclusion, the present study reveals in vitro, the inhibitory effect of LLM on cMyc expression and activity in prostate cancer cells. However, LLM doses higher than 10 mg/kg body weight might be required to achieve chemoprevention of prostate cancer in vivo. Thus, similar to phytochemicals like curcumin, resveratrol, and neem extracts, LLM may be useful for the prevention of prostate cancer[38-40]. In one such study, supercritical extract of neem leaves (SENL) was shown to inhibit integrin β1, calreticulin, and focal adhesion kinase activation in LNCaP-luc2 and PC3 prostate cancer cells[40]. The growth of LNCaP-luc2 xenograft was also reduced significantly by SENL administration that was associated with the formation of hyalinized fibrous tumor tissue, reduction in the prostate-specific antigen, and increase in AKR1C2 levels[40].
DECLARATIONS
Authors’ contributionsStudy design: Singh KB, Hahm ER, Singh SV
Performed experiments: Singh KB, Hahm ER
Study supervision: Singh SV
Data interpretation: Singh KB, Hahm ER, Singh SV
Final manuscript preparation: Singh KB, Hahm ER, Singh SV
Availability of data and materialsNot applicable.
Financial support and sponsorshipThis study was supported by the National Cancer Institute grant R01 CA225716 (to S.V.S.). The following UPMC Hillman Cancer Center core facilities partly supported by the National Cancer Institute grant P30 CA047904 were utilized in this study: Animal Facility, and Cell and Tissue Imaging Facility.
Conflicts of interestAll authors declared that there are no conflicts of interest.
Ethical approval and consent to participateThe care of Hi-Myc mice was consistent with the University of Pittsburgh Animal Care and Use Committee guidelines.
Consent for publicationNot applicable.
Copyright© The Author(s) 2021.
REFERENCES
2. Labbé DP, Brown M. Transcriptional regulation in prostate cancer. Cold Spring Harb Perspect Med 2018;8:a030437.
3. Ellwood-yen K, Graeber TG, Wongvipat J, et al. Myc-driven murine prostate cancer shares molecular features with human prostate tumors. Cancer Cell 2003;4:223-38.
4. Buttyan R, Sawczuk IS, Benson MC, Siegal JD, Olsson CA. Enhanced expression of the c-myc protooncogene in high-grade human prostate cancers. Prostate 1987;11:327-37.
5. Nag A, Smith RG. Amplification, rearrangement, and elevated expression of c-myc in the human prostatic carcinoma cell line LNCaP. Prostate 1989;15:115-22.
6. Edwards J, Krishna NS, Witton CJ, Bartlett JM. Gene amplifications associated with the development of hormone-resistant prostate cancer. Clin Cancer Res 2003;9:5271-81.
7. Gurel B, Iwata T, Koh CM, et al. Nuclear MYC protein overexpression is an early alteration in human prostate carcinogenesis. Mod Pathol 2008;21:1156-67.
8. Fox SB, Persad RA, Royds J, Kore RN, Silcocks PB, Collins CC. p53 and c-myc expression in stage A1 prostatic adenocarcinoma: Useful prognostic determinants? Journal of Urology 1993;150:490-4.
9. Zeng W, Sun H, Meng F, et al. Nuclear C-MYC expression level is associated with disease progression and potentially predictive of two year overall survival in prostate cancer. Int J Clin Exp Pathol 2015;8:1878-88.
10. Kumar A, Coleman I, Morrissey C, et al. Substantial interindividual and limited intraindividual genomic diversity among tumors from men with metastatic prostate cancer. Nat Med 2016;22:369-78.
11. Ribeiro FR, Jerónimo C, Henrique R, et al. 8q gain is an independent predictor of poor survival in diagnostic needle biopsies from prostate cancer suspects. Clin Cancer Res 2006;12:3961-70.
12. Asangani IA, Dommeti VL, Wang X, et al. Therapeutic targeting of BET bromodomain proteins in castration-resistant prostate cancer. Nature 2014;510:278-82.
14. Stine ZE, Walton ZE, Altman BJ, Hsieh AL, Dang CV. MYC, metabolism, and cancer. Cancer Discov 2015;5:1024-39.
15. Singh KB, Hahm ER, Kim SH, Wendell SG, Singh SV. A novel metabolic function of Myc in regulation of fatty acid synthesis in prostate cancer. Oncogene 2021;40:592-602.
16. Rebello RJ, Pearson RB, Hannan RD, Furic L. Therapeutic approaches targeting MYC-driven prostate cancer. Genes (Basel) 2017;8:71.
17. Thompson IM, Goodman PJ, Tangen CM, et al. The influence of finasteride on the development of prostate cancer. N Engl J Med 2003;349:215-24.
18. Lippman SM, Klein EA, Goodman PJ, et al. Effect of selenium and vitamin E on risk of prostate cancer and other cancers: the Selenium and Vitamin E Cancer Prevention Trial (SELECT). JAMA 2009;301:39-51.
19. Andriole GL, Bostwick DG, Brawley OW, et al. REDUCE Study Group. Effect of dutasteride on the risk of prostate cancer. N Engl J Med 2010;362:1192-202.
20. Ju J, Picinich SC, Yang Z, et al. Cancer-preventive activities of tocopherols and tocotrienols. Carcinogenesis 2010;31:533-42.
21. Ting H, Deep G, Agarwal C, Agarwal R. The strategies to control prostate cancer by chemoprevention approaches. Mutat Res 2014;760:1-15.
22. Mokbel K, Wazir U, Mokbel K. Chemoprevention of prostate cancer by natural agents: Evidence from molecular and epidemiological studies. Anticancer Res 2019;39:5231-59.
23. Merarchi M, Jung YY, Fan L, Sethi G, Ahn KS. A brief overview of the antitumoral actions of leelamine. Biomedicines 2019;7:53.
24. Gowda R, Madhunapantula SV, Kuzu OF, Sharma A, Robertson GP. Targeting multiple key signaling pathways in melanoma using leelamine. Mol Cancer Ther 2014;13:1679-89.
25. Singh KB, Ji X, Singh SV. Therapeutic potential of leelamine, a novel inhibitor of androgen receptor and castration-resistant prostate cancer. Mol Cancer Ther 2018;17:2079-90.
26. Gao L, Schwartzman J, Gibbs A, et al. Androgen receptor promotes ligand-independent prostate cancer progression through c-Myc upregulation. PLoS One 2013;8:e63563.
27. Singh KB, Hahm ER, Pore SK, Singh SV. Leelamine is a novel lipogenesis inhibitor in prostate cancer cells in vitro and in vivo. Mol Cancer Ther 2019;18:1800-10.
28. Hahm ER, Singh KB, Singh SV. c-Myc is a novel target of cell cycle arrest by honokiol in prostate cancer cells. Cell Cycle 2016;15:2309-20.
29. Xiao D, Srivastava SK, Lew KL, et al. Allyl isothiocyanate, a constituent of cruciferous vegetables, inhibits proliferation of human prostate cancer cells by causing G2/M arrest and inducing apoptosis. Carcinogenesis 2003;24:891-7.
30. Hahm ER, Lee J, Kim SH, et al. Metabolic alterations in mammary cancer prevention by withaferin A in a clinically relevant mouse model. J Natl Cancer Inst 2013;105:1111-22.
31. Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001;25:402-8.
32. Xiao D, Choi S, Johnson DE, et al. Diallyl trisulfide-induced apoptosis in human prostate cancer cells involves c-Jun N-terminal kinase and extracellular-signal regulated kinase-mediated phosphorylation of Bcl-2. Oncogene 2004;23:5594-606.
33. Song M, Lee D, Lee T, Lee S. Determination of leelamine in mouse plasma by LC-MS/MS and its pharmacokinetics. J Chromatogr B Analyt Technol Biomed Life Sci 2013;931:170-3.
34. Chen H, Zhou L, Wu X, et al. The PI3K/AKT pathway in the pathogenesis of prostate cancer. Front Biosci (Landmark Ed) 2016;21:1084-91.
35. Xu R, Hu J. The role of JNK in prostate cancer progression and therapeutic strategies. Biomed Pharmacother 2020;121:109679.
36. Bishop JL, Thaper D, Zoubeidi A. The multifaceted roles of STAT3 signaling in the progression of prostate cancer. Cancers (Basel) 2014;6:829-59.
37. Thompson IM Jr, Goodman PJ, Tangen CM, et al. Long-term survival of participants in the prostate cancer prevention trial. N Engl J Med 2013;369:603-10.
38. Dorai T, Cao YC, Dorai B, Buttyan R, Katz AE. Therapeutic potential of curcumin in human prostate cancer. III. Curcumin inhibits proliferation, induces apoptosis, and inhibits angiogenesis of LNCaP prostate cancer cells in vivo. Prostate 2001;47:293-303.
39. Wang TT, Hudson TS, Wang TC, et al. Differential effects of resveratrol on androgen-responsive LNCaP human prostate cancer cells in vitro and in vivo. Carcinogenesis 2008;29:2001-10.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Singh KB, Hahm ER, Singh SV. Leelamine suppresses cMyc expression in prostate cancer cells in vitro and inhibits prostate carcinogenesis in vivo. J Cancer Metastasis Treat 2021;7:16. http://dx.doi.org/10.20517/2394-4722.2021.08
AMA Style
Singh KB, Hahm ER, Singh SV. Leelamine suppresses cMyc expression in prostate cancer cells in vitro and inhibits prostate carcinogenesis in vivo. Journal of Cancer Metastasis and Treatment. 2021; 7: 16. http://dx.doi.org/10.20517/2394-4722.2021.08
Chicago/Turabian Style
Singh, Krishna B., Eun-Ryeong Hahm, Shivendra V. Singh. 2021. "Leelamine suppresses cMyc expression in prostate cancer cells in vitro and inhibits prostate carcinogenesis in vivo" Journal of Cancer Metastasis and Treatment. 7: 16. http://dx.doi.org/10.20517/2394-4722.2021.08
ACS Style
Singh, KB.; Hahm E.R.; Singh SV. Leelamine suppresses cMyc expression in prostate cancer cells in vitro and inhibits prostate carcinogenesis in vivo. J. Cancer. Metastasis. Treat. 2021, 7, 16. http://dx.doi.org/10.20517/2394-4722.2021.08
About This Article
Copyright

Data & Comments
Data


 Cite This Article 11 clicks
Cite This Article 11 clicks

 Like This Article 18
likes
Like This Article 18
likes






Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.