
Identification of CYP 2A6 inhibitors in an effort to mitigate the harmful effects of the phytochemical nicotine

Abstract
Aim: In this study, our goal was to study the inhibition of nicotine metabolism by P450 2A6, as a means for reduction in tobacco use and consequently the prevention of smoking-related cancers. Nicotine, a phytochemical, is an addictive stimulant, responsible for the tobacco-dependence in smokers. Many of the other phytochemicals in tobacco, including polycyclic aromatic hydrocarbons, N-nitrosamines, and aromatic amines, are potent systemic carcinogens. Tobacco smoking causes about one of every five deaths in the United States annually. Nicotine plasma concentration is maintained by the smokers’ smoking behavior within a small range. Nicotine is metabolized by cytochrome P450s 2A6 and 2A13 to cotinine. This metabolism causes a decrease in nicotine plasma levels, which in turn leads to increased tobacco smoking, and increased exposure to the tobacco carcinogens.
Methods: Using the phytochemical nicotine as a lead structure, and taking its interactions with the P450 2A6 binding pocket into consideration, new pyridine derivatives were designed and synthesized as potential selective mechanism-based inhibitors for this enzyme.
Results: The design and synthesis of two series of novel pyridine-based compounds, with varying substituents and substitution locations on the pyridine ring, as well as their inhibitory activities on cytochrome P450 2A6 and their interactions with its active site are discussed here. Substitutions at position 3 of the pyridine ring with an imidazole or propargyl ether containing group showed the most optimal interactions with the P4502A6 active site.
Conclusion: The pyridine compounds with an imidazole or propargyl ether containing substituent on position 3 were found to be promising lead compounds for further development. Hydrogen-bonding interactions were determined to be crucial for effective binding of these molecules within the P450 2A6 active site.
Keywords
INTRODUCTION
Phytochemicals are plant-produced natural products, and are commonly found in human and animal diets as well as in plant-based medications and natural remedies[1-3]. While most of these chemicals have basic nutritional value, some have preventive, therapeutic or toxic effects. Phytochemicals can play a preventive role in carcinogenesis in multiple ways; as primary preventive agents that can be used to prevent the advent of cancer; as secondary agents that can prevent progression of cancer as in the case of premalignant lesions; and as tertiary agents that can prevent the recurrence of cancer[4,5]. Some examples of such phytochemicals are capsaicin from chili pepper[6,7], polyphenols from green tea, fruits, and vegetables[8,9], carotenoids such as lycopene found in fruits[10,11], cucurbitacin B from Chinese medicinal plants[12-14], isoflavones from legumes[15,16], etc. Phytochemicals have also been shown to modulate multiple mechanisms in cancer, resulting in their anti-cancer activities[5,17-21]. Phytochemicals are known to have similar preventive and/or therapeutic effects on many other diseases[10,11,16,19,22].
Phytochemicals are often toxins produced by plants as a defense mechanism against disease-causing organisms or herbivorous animals. Certain herbs and plants routinely used by humans, contain phytotoxins with carcinogenic, teratogenic and/or endocrine influencing activities[1]. While some of these phytochemicals are direct-acting toxins, others, such as procarcinogenic agents, need metabolic activation. Certain procarcinogenic alkenylbenzenes, pyrrolizidine alkaloids, ptaquiloside, aristolochic acids, and furanocoumarins are known DNA-alkylating agents[3,22-25]. Metabolic activation of phytochemical procarcinogens into their ultimate carcinogenic forms by phase I and phase II enzymes has been well established[26,27]. Cytochrome P450 enzymes, a superfamily of Phase I enzymes, metabolize endogenous and xenobiotic compounds including phytochemicals, through monooxygenation reactions[26-29].
Nicotine, a phytotoxin present in many plants and vegetables, including tobacco, and in much smaller concentrations in potatoes, tomatoes, eggplants, and green peppers, is primarily metabolized by human liver P450 2A6, and to a smaller extent by human lung P450 2A13. The metabolism of nicotine to cotinine takes place in two steps - initial oxidation to the intermediate nicotine-△1’(5’)-iminium ion by either of the two P450 enzymes[29-31], and further oxidation to cotinine by cytosolic aldehyde oxidase[29,32]. Nicotine plays a critical role in tobacco-dependence, and is the main cause of lung cancer deaths in men and women. It is estimated that more than 16 million Americans are affected by cigarette smoking, and more than 480,000 deaths per year are attributed to tobacco use[33]. Several strategies have been developed and implemented for cessation of cigarette smoking, such as nicotine replacement therapies alone or in combination with varenicline, a nicotinic acetylcholine receptor partial agonist[34]. However, only about 6% of smokers are able to overcome the addiction to nicotine[35]. The addiction to nicotine in smokers modulating their smoking behaviors has a direct correlation to the levels of nicotine in the blood and brain[36-38]. Drop of the levels of nicotine in the blood and brain due to the P450 2A6 metabolism, causes the smokers to adjust their tobacco use to maintain these levels. The impact of P450 2A6 activity on tobacco addiction is evidenced in individuals who are slow metabolizers due to P450 2A6 polymorphism, and thus smoke less[37,39,40]. Inhibition of P450 2A6, which is known to be well tolerated by humans[41], is emerging as the most promising strategy for smoking cessation and treatment of tobacco-dependence. Development of P450 2A6-specific inhibitors is currently pursued by many research groups[42-47]. Recent developments in this field include the use of bioelectrochemical platforms that use the “molecular lego” approach[48,49]. In the study reported by Castrignanò et al[50], genetically-fused P450 2A6 with Desulfovibrio vulgaris flavodoxin (FLD) module is used for investigating the inhibitory effects of coumarins and nicotine. Such an approach could improve and hasten the process of P450 enzyme inhibitor development. The development of mimicking agents with structural similarities to nicotine through the modification of its pyridine ring substituents, by our research group and the Cashman and Lazarus Research Groups[51-54], has led to potent (with low micromolar IC50 values) inhibitors of P450 2A6[42,51,54]. For this study, we have identified two series of such pyridine-based P450 2A6 inhibitors.
METHODS
Synthesis of 3-((prop-2-yn-1-yloxy)methyl)pyridine (6): 3-Hydroxymethylpyridine (1.0 eq) in tetrahydrofuran (THF) was added dropwise to a cooled (0 oC) suspension of sodium hydride (NaH, 95%, 2.1 eq) in dry THF. After 20 min, propargyl bromide (80% solution in toluene, 2.0 eq) was added slowly. The reaction mixture was heated at 50 oC overnight. It was then allowed to cool to room temperature before careful quenching by the addition of water. The crude was extracted with ethyl acetate. The organic layer was washed with brine and dried over anhydrous sodium sulfate (Na2SO4). The solvent was removed under vacuum. The residue was then purified by silica gel column chromatography using hexane: ethyl acetate (1:3) as eluent to afford the desired product.
Compound 6: (96% yield; brown liquid) GC-MS showed > 99% purity. m/z: 146, 108, 92, 80, 65, 51. 1HNMR (CDCl3, 300 MHz) δ = 2.45 (t, J = 2.4 Hz, 1H), 3.42 (s, 2H), 4.51 (s, 2H), 7.32 (m, 1H), 7.62 (m, 1H), and 8.55 (m, 1H). 13C NMR (CDCl3, 75 MHz) δ = 57.7, 69.1, 75.3, 79.3, 123.5, 133.0, 135.8, 149.5, and 149.6.
Syntheses of 4-(prop-2-yn-1-yloxy)pyridine (2), 2-(prop-2-yn-1-yloxy)pyridine (3) and 4-(3-(prop-2-yn-1-yloxy)propyl)pyridine (5) were achieved using the same procedure.
Compound 2: (82% yield; white solid) GC-MS showed > 98% purity, m/z: 133.1, 104.1, 78.0, 52.0. 1HNMR (CDCl3, 300 MHz) δ = 2.63 (t, J = 2.56 Hz, 1H), 4.50 (d, J = 2.6 Hz, 2H), 6.35 (d, J = 7.3 Hz, 2H), and 7.42 (d, J = 7.6 Hz, 2H). 13C NMR (CDCl3, 75 MHz) δ = 45.5, 75.7, 77.3, 118.8, 139.6, and 179.1.
Compound 3: (76% yield; white solid) GC-MS showed > 99% purity, m/z: 133.1, 104.1, 78.0, 52.0. 1HNMR (CDCl3, 300 MHz) δ = 2.48 (t, J = 2.4 Hz, 1H), 4.72 (d, J = 2.3 Hz, 2H), 6.22 (t, J = 6.1 Hz, 1H), 6.54 (d, J = 5.2 Hz, 1H), 7.36 (d, J = 2.8 Hz, 1H), and 7.63 (d, J = 4.6 Hz, 1H). 13C NMR (CDCl3, 75 MHz) δ = 37.5, 75.3, 77.3, 106.2, 120.1, 136.5, 139.9, and 161.6.
Compound 5: (67% yield; yellow liquid) GC-MS showed > 98% purity. m/z: 174, 158, 145, 118, 110, 93. 1HNMR (CDCl3, 300 MHz) δ = 2.0 (m, 2H), 2.42(t, J = 2.6 Hz, 1H), 2.89 (t, J = 7.4 Hz, 2H), 3.56 (t, J = 6.3 Hz, 2H), 4.14 (d, J = 2.3 Hz, 2H), 7.15 (m, 2H), 7.60 (m, 1H), and 8.52 (m, 1H). 13C NMR (CDCl3, 75 MHz) δ = 30.1, 31.6, 58.2, 68.8, 74.6, 77.1, 124.1, 149.8, and 150.8.
Synthesis of 3-((1H-imidazol-1-yl)methyl)pyridine (4): The solution of 3-chloromethylpyridine hydrochloride salt (1.0 eq) and imidazole (10.0 eq) in dimethylformamide (DMF) (10 mL) was heated at 100 oC until all the salt starting material was consumed (~3 h). The light brown solution was cooled down to room temperature, and the DMF was removed under vacuum. The residue was dissolved in dichloromethane (DCM), and washed with water. The DCM layer was dried over anhydrous Na2SO4. The solvent was evaporated under vacuum to afford the desired product.
Compound 4: (14% yield; brown solid) GC-MS showed > 99% purity. m/z: 159, 132, 92. 65. 1HNMR (CDCl3, 300 MHz) δ = 5.10 (s, 2H), 6.88 (s, 1H), 7.00 (s, 1H), 7.31 (m, 2H), 7.65 (s, 1H), 8.46 (m, 1H), and 8.56 (m, 1H). 13C NMR (CDCl3, 75 MHz) δ = 48.3, 119.3, 123.9, 130.0, 135.0, 137.3, 148.7, and 149.7.
Syntheses of 3-(3-(2-methyl-1H-imidazol-1-yl))propyl]pyridine (1) and 3-[(2-methyl-1H-imidazol-1-yl)methyl] pyridine (7) were achieved using the same procedure.
Compound 1: (86% yield; yellow oil) GC-MS showed > 99% purity. m/z: 201, 186, 118, 96, 55. 1HNMR (CDCl3, 300 MHz) δ = 2.05 (m, 2H), 2.32 (s, 3H), 2.62 (t, J = 7.7 Hz, 2H), 3.85 (t, J = 7.4 Hz, 2H), 6.80 (d, J = 1.0 Hz, 1H), 6.90 (d, J = 1.1 Hz, 1H), 7.22 (m, 1H), 7.45 (m, 1H), and 8.45 (m, 2H). 13C NMR (CDCl3, 75 MHz) δ = 12.6, 29.3, 31.3, 44.7, 118.7, 120.9, 123.1, 126.6, 135.3, 135.6, 143.8, 147.3, and 149.3.
Compound 7: (15% yield; brown oil) GC-MS showed > 98% purity. m/z: 173, 146, 92, 65. 1HNMR (CDCl3, 300 MHz) δ = 2.35 (s, 3H), 5.0 (s, 2H), 6.84 (s, 1H), 6.97 (s, 1H), 7.31 (m, 2H), 8.46 (m, 1H), and 8.56 (m, 1H). 13C NMR (CDCl3, 75 MHz) δ = 13.1, 47.4, 119.8, 123.9, 127.8, 132.1, 132.4, 144.8, 148.4, and 149.6.
P450 2A6 inhibition assay
Cytochrome P450 2A6 (Cyp2A6) activity was determined using the Vivid CYP450 Screening Kit (Life Technologies, catalog #PV6140) according to the manufacturer’s instructions. Briefly, a master pre-mix containing baculosomes and regeneration system was prepared using 0.5× Vivid reaction buffer II. The test compounds were dissolved in dimethyl sulfoxide (DMSO) at a concentration of 100 mM. From the stock solutions, each compound was serially diluted in 0.5× Vivid reaction buffer II to make working stocks of 100 µM, 50 µM, 25 µM and so on up to 10 dilutions. It is essential to dilute the DMSO at least 1000-fold when making the working dilutions to prevent its interference with the enzyme activity. An initial high-throughput screening was performed at 10 µM concentration for each compound. For the dose-response curve determination, in a 96-well plate, 40 µL of the diluted solutions of each test compound were added to each well, followed by 50 µl of the master pre-mix, before incubation for 10 min at room temperature. A 10× mixture of Vivid substrate (reconstituted with acetonitrile) and NADP+ was then prepared. At the end of the incubation period, 10 µL of this solution was added to each well to start the reaction. After 2 hours of incubation in the dark at room temperature, the plate was read at 415 nm on a plate reader (Synergy H1, Biotek). For a positive inhibition control, tranylcypromine (Sigma-Aldrich, cat. #P8511) was used at a final concentration of 100 µM. For a negative (no inhibitor) control, a 1:1000 dilution of pure DMSO in 0.5× Vivid reaction buffer was used. Each concentration in the dose-response curve was set up in triplicates, and each data point was the average of triplicate wells. The % inhibition was calculated using the following equation.
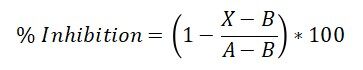
where X is the fluorescence observed in presence of the test compound; A is the fluorescence observed in the absence of an inhibitor (no inhibitor control); and B is the fluorescence observed for the positive control. For graphing purposes, percent inhibition vs. anti-log[drug concentration] was plotted. A logistic sigmoidal model was used to fit the data and obtain IC50 values using Graphpad Prism software.
Docking studies
Docking studies were performed using methods previously published[55-57]. The coordinates of the reported X-ray crystal structure of P450 2A6 bound to nicotine[58] (4EJJ.pdb) were downloaded from the Protein Data Bank website (http://www.rcsb.org/), and used for the docking studies. Heme was considered as part of the receptor for docking purposes. Standard force fields were used for the compounds. MOE energy minimization method was used using the MOE software platform from the ChemComp group.
RESULTS
Nicotine, the main addictive ingredient of tobacco products, is mainly (70%-80%) metabolized to cotinine by P450 2A6 enzyme in the liver. Cotinine has a longer half-life but is much less active in inducing dopamine release in smokers than nicotine[30]. As nicotine is metabolized, maintenance of its blood plasma levels compels the smokers to modify their smoking frequency to compensate. Tobacco usage has been linked to several debilitating diseases, with lung cancer being the most common disease among smokers. Inhibition of P450 2A6 can prove to be one of the most effective strategies for smoking cessation. Our research group has been exploring several classes of compounds as possible P450 2A6 inhibitors. The use of the pyridine molecule as a scaffold for P450 2A6 inhibitors has been explored by our research group and the Cashman and Lazarus Research Groups[51-54]. The incorporation of 5-membered heterocyclic rings has been pursued by both research groups. One of the distinctive features of the P450 2A6 inhibitors developed by the Cashman and Lazarus Research Groups has been the use of a primary amino group as the main functional group that binds to the heme-Fe of the enzyme. In contrast, our research group is focused on (1) the incorporation of a triple bond that can lead to mechanism-based inhibition of the enzyme; and (2) the incorporation of an imidazole ring that can mimic the pyrrolidine ring of nicotine, in which the -CH of the 5-membered ring faces the heme-Fe of the enzyme.
The syntheses of compounds 1 to 7 [Figure 1] were accomplished using the synthetic Schemes 1 and 2. The binding mode of nicotine to the P450 enzyme in the X-ray crystal structure (4EJJ.pdb) indicates that substituents at position 3 of the pyridine ring would be ideal. Nicotine-mimicking compounds containing imidazole or methyl substituted imidazole side chains with varied sizes of 1-3 carbon alkyl spacers at position 3 of the pyridine ring were synthesized using Scheme 1 (compounds 1, 4 and 7). Based on our previous findings that acetylenic substituents can interact with the P450 active site amino acids and lead to mechanism-based inhibition, triple bond in the form of propargyl ether was incorporated in a second series of compounds at positions 2, 3 or 4 of the pyridine ring as an ether linkage (compounds 3, 6 and 2, respectively). Positions 2 and 4 were used to investigate whether substituted compounds at those positions would show a difference in their inhibition activities compared to the 3-position substituted compound. A longer spacer in between the propargyl ether and the 4-position of the pyridine ring was introduced to investigate whether a greater flexibility in the positioning of the key functional groups (pyridine ring and the alkyne moiety) in the binding site would be achieved. The Vivid CYP450 Screening Kit (Life Technologies, catalog #PV6140) was used for the P450 2A6 inhibition assays. An initial high-throughput screening was performed at a 10 µM concentration followed by a dose-response curve determination. Compounds 1, 4 and 6, all three containing a substituent on position 3 of the pyridine ring, were found to have single digit micromolar IC50 values [Figure 2, Table 1]. Compounds 5 and 7 were found to have moderate inhibition activity, with compound 7, a 3-position substituted compound, showing better inhibition than the 4-substituted compound 5. Compound 2, a 4-position propargyl ether substituted compound, exhibited much lower inhibition, while compound 3, a 2-postion propargyl ether substituted compound, did not inhibit P450 2A6 enzyme activity.
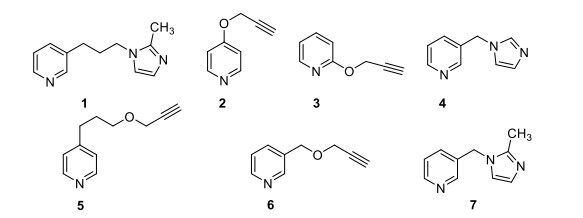
Figure 1. Structures of Compounds 1 to 7.
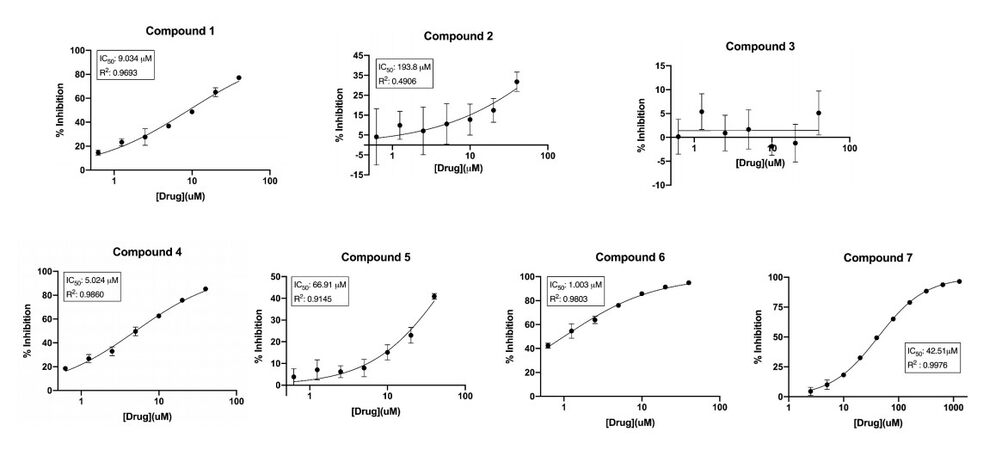
Figure 2. Dose-response curves for the inhibition of P450 2A6 by compounds 1 to 7. The concentrations of the compounds are represented on the X-axis as an antilog scale. The highest compound concentrations used for dose-response curves is 40 µM.
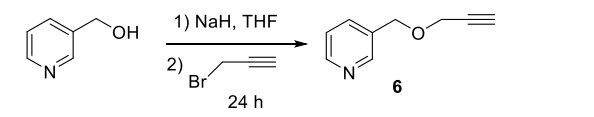
Scheme 1. Synthesis of compounds 2, 3, 5 and 6 were accomplished using the following scheme.
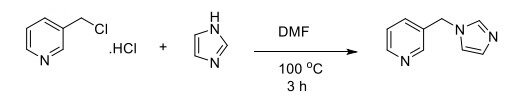
Scheme 2. Synthesis of compounds 1, 4 and 7 were accomplished using the following scheme.
IC50 values of P450 2A6 inhibition by compounds 1 to 7
| Compound | IC50 (µM) |
| 1 | 9.034 |
| 2 | 193.8 |
| 3 | 0 |
| 4 | 5.024 |
| 5 | 66.91 |
| 6 | 1.003 |
| 7 | 42.51 |
Docking studies of compounds 1 through 7 with the reported X-ray crystal structure of P450 2A6 with nicotine in its active site (4EJJ.pdb) were conducted using the MOE software platform from the ChemComp group. The nicotine molecule makes two H-bonds with Thr305 and Asn297 [Figure 3A and E], and the -CH2 group of the pyrrolidine ring is in close proximity to the heme-Fe atom. The binding poses of the imidazole substituted compounds 1, 4 and 7 showed that all three molecules made H-bonds with the active site residues. Similar to the substitution pattern of nicotine, the imidazole substituents of these compounds are connected with varying alkyl chain lengths (1 or 3 carbons) at position 3 of the pyridine ring. Compound 1 made two H-bonds with Thr305 and Asn297, and had the -CH of the imidazole ring in close proximity to the heme-Fe atom [Figure 3B and F]. Compound 4 made one H-bond with Thr305, and aromatic π-H interaction with Phe107 [Figure 3C and G]. The -CH group at position 2 of the imidazole ring was facing the heme-Fe atom. Compound 7 made one H-bond with Asn297, and the sp2 nitrogen atom of the imidazole ring was in close proximity to the heme-Fe atom [Figure 3D and H]. The presence of the methyl substituent at the 2-position of the imidazole ring in this compound caused a change in the orientation of the methyl group, away from the heme-Fe atom, causing a flip in the positioning of this ring in the active site. The propargyl ether pyridine compounds 2, 3, 5 and 6 showed variations in binding poses, based on the position of the substituent [Figure 4]. Compounds 2 and 5 had substituents at position 4 of the pyridine ring. Compound 2 did not have a linker alkyl chain connecting the propargyl ether group to the pyridine ring [Figure 4A]. Compound 2 did not make any H-bond interactions with the active site residues, and the alkyne carbons were in close proximity to the heme-Fe atom. Compound 5 with a 3-carbon alkyl chain linker between the propargyl ether group and the pyridine ring depicted a flipped binding pose with the pyridine ring oriented towards the heme-Fe [Figure 4C]. Compound 3 with the propargyl ether group at position 2 of the pyridine ring depicted a binding pose identical to that of compound 2, with the alkyne carbons facing the heme-Fe atom [Figure 4B]. Compound 6 with the propargyl ether group connected by a methylene linker to position 2 of the pyridine ring had a binding pose similar to that of compound 5, with the pyridine ring facing the heme-Fe atom [Figure 4D and E]. Compound 6 was the only propargyl ether derivative to make a H-bond with the active site residue Thr305. The P450 2A6 inhibition studies clearly confirmed these observations and indicated that substitution at position 3 of the pyridine ring was the most ideal. The docking studies showed that the close proximity of the heterocyclic rings to the heme-Fe atom was most favored, and increases in H-bond interactions with the active site residues increased the inhibition potency.
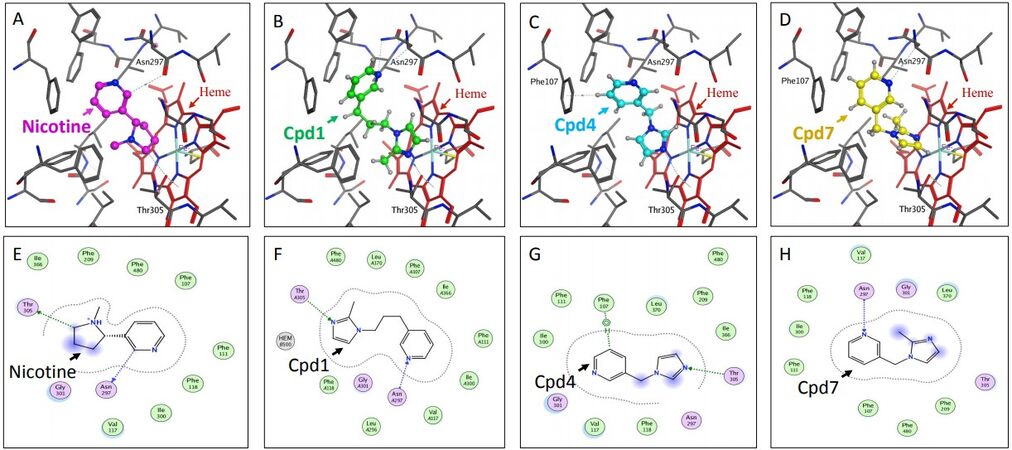
Figure 3. Docking studies on compounds (Cpd) that have pyridine with imidazole substituents (Cpd1, Cpd4 and Cpd7). Figures (A), (B), (C) and (D) show the binding modes and figures (E), (F), (G) and (H) show the ligand interactions with the active site residues for nicotine and compounds 1, 4 and 7, respectively in the active site of P450 2A6 enzyme.
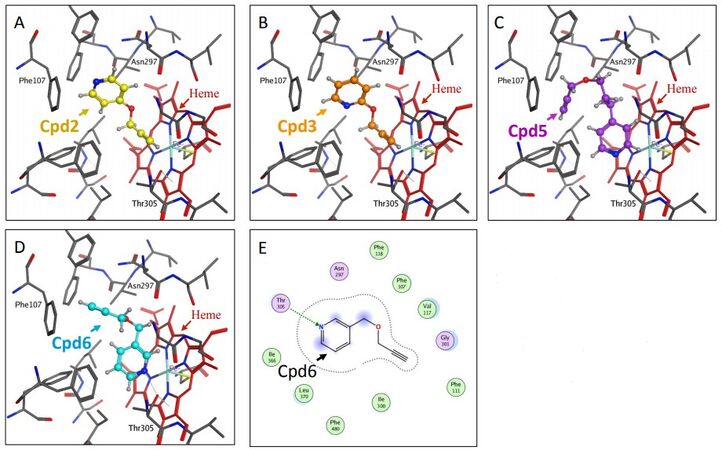
Figure 4. Docking studies on compounds (Cpd) that have pyridine with propargyl ether substituents (Cpd2, Cpd3, Cpd5 and Cpd6). Figures (A), (B), (C) and (D) show the binding modes of compounds 2, 3, 5 and 6, respectively with the active site of P450 2A6. Figure E depicts the ligand interactions of compound 6 with the active site residues.
The inhibition studies on our compounds were performed using a Vivid CYP2A6 Kit (Life Technologies, catalog #PV6140). This kit uses microsomes from insect cells stably expressing human CYP2A6 enzyme. As such all the enzyme from these microsomes is essentially CYP2A6, eliminating the possibility of the presence of other polymorphic variants of CYP2A6 that could be naturally found in liver microsomes. Therefore, the Vivid CYP2A6 Kit used in this study does not account for any polymorphism of CYP2A6. However, it is very important to understand the inhibitory effect of new compounds on CYP2A6 polymorphism as people with other allelic variants of CYP2A6 exhibit varied metabolic response. Our future studies would aim to study this phenomenon, either by developing the cell systems expressing various CYP2A6 allelic variants using baculovirus and insect cell lines by genetic engineering tools[59-61], or by deploying the “molecular lego” approach developed by Castrignano et al.[50], using a chimeric CYP2A6-flavodoxin/CYP2A6 and determining the kinetic parameters of coumarin electrocatalysis by electrochemical detection.
DISCUSSION
Based on the pyridine scaffold of nicotine, two series of compounds were designed and synthesized for this study. The compounds containing an imidazole or propargyl ether substituents at position 3 of the pyridine ring were found to be promising lead compounds for further development. Our studies clearly illustrated that H-bonding interactions were very important for effective binding of these molecules within the P450 2A6 active site. Many of the pyridine derivatives developed by the Cashman and Lazarus Research Groups have a primary or secondary amine functional group that interacts with the heme iron[51-54]. The compounds developed by our group do not contain any amine functional groups, resulting in the aromatic heterocyclic rings facing the heme-Fe and also showing low P450 2A6 inhibition IC50 values (micromolar). Using these lead compounds, additional potential inhibitors with increased number of interactions with the amino acid residues of the P450 2A6 active site will be designed and synthesized.
DECLARATIONS
AcknowledgementResearch reported in this publication was supported by the Louisiana Cancer Research Center, its Tobacco Free Living Program, the National Institute of General Medical Sciences of the National Institutes of Health under Award Number 5RL5GM118966, and the NIMHD-RCMI grant number 5G12MD007595. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health or the Louisiana Cancer Research Center.
Authors’ contributionsDesign, synthesis, purification, and structural analysis of the compounds: Goyal N
Design and performance of the docking studies: Sridhar J
Synthesis and purification of the compounds: Do C
Design and performance of the inhibition assays: Bratton M, Shaik S, Jiang Q
Overall project conception and direction: Foroozesh M
Contributed significantly and equally to the manuscript: Goyal N, Sridhar J, Foroozesh M
Availability of data and materialsNot applicable.
Financial support and sponsorshipNone.
Conflict of interestAll authors declared that there are no conflicts of interest.
Ethical approval and consent to participateNot applicable.
Consent for publicationNot applicable.
Copyright© The Author(s) 2021.
REFERENCES
1. Boada LD, Henríquez-Hernández LA, Luzardo OP. The impact of red and processed meat consumption on cancer and other health outcomes: Epidemiological evidences. Food Chem Toxicol 2016;92:236-44.
2. Lin DX, Lang NP, Kadlubar FF. . Species differences in the biotransformation of the food-borne carcinogen 2-amino-1-methyl-6-phenylimidazo[4,5-b]pyridine by hepatic microsomes and cytosols from humans, rats, and mice. Drug Metab Dispos 1995;23:518-24.
3. Wink M. Plant secondary metabolism: Diversity, function and its evolution. Natural Product Communications 2008;3:1934578X0800300.
4. Ranjan A, Ramachandran S, Gupta N, et al. Role of phytochemicals in cancer prevention. Int J Mol Sci 2019;20:4981.
5. Dutta S, Mahalanobish S, Saha S, Ghosh S, Sil PC. Natural products: An upcoming therapeutic approach to cancer. Food Chem Toxicol 2019;128:240-55.
6. Chapa-Oliver AM, Mejía-Teniente L. Capsaicin: From plants to a cancer-suppressing agent. Molecules 2016;21:931.
7. Venier NA, Yamamoto T, Sugar LM, et al. Capsaicin reduces the metastatic burden in the transgenic adenocarcinoma of the mouse prostate model. Prostate 2015;75:1300-11.
8. Tsai YJ, Chen BH. Preparation of catechin extracts and nanoemulsions from green tea leaf waste and their inhibition effect on prostate cancer cell PC-3. Int J Nanomedicine 2016;11:1907-26.
9. Tu Y, Kim E, Gao Y, Rankin GO, Li B, Chen YC. Theaflavin-3, 3'-digallate induces apoptosis and G2 cell cycle arrest through the Akt/MDM2/p53 pathway in cisplatin-resistant ovarian cancer A2780/CP70 cells. Int J Oncol 2016;48:2657-65.
11. Preet R, Mohapatra P, Das D, et al. Lycopene synergistically enhances quinacrine action to inhibit Wnt-TCF signaling in breast cancer cells through APC. Carcinogenesis 2013;34:277-86.
12. Gupta P, Srivastava SK. Inhibition of Integrin-HER2 signaling by Cucurbitacin B leads to in vitro and in vivo breast tumor growth suppression. Oncotarget 2014;5:1812-28.
13. Saglam AS, Alp E, Elmazoglu Z, Menevse S. Treatment with cucurbitacin B alone and in combination with gefitinib induces cell cycle inhibition and apoptosis via EGFR and JAK/STAT pathway in human colorectal cancer cell lines. Hum Exp Toxicol 2016;35:526-43.
14. Zheng Q, Liu Y, Liu W, et al. Cucurbitacin B inhibits growth and induces apoptosis through the JAK2/STAT3 and MAPK pathways in SHSY5Y human neuroblastoma cells. Mol Med Rep 2014;10:89-94.
15. Chen J, Duan Y, Zhang X, Ye Y, Ge B, Chen J. Genistein induces apoptosis by the inactivation of the IGF-1R/p-Akt signaling pathway in MCF-7 human breast cancer cells. Food Funct 2015;6:995-1000.
16. Sarkar FH, Li Y. Mechanisms of cancer chemoprevention by soy isoflavone genistein. Cancer Metastasis Rev 2002;21:265-80.
17. Bahrami A, Fereidouni M, Pirro M, Bianconi V, Sahebkar A. Modulation of regulatory T cells by natural products in cancer. Cancer Lett 2019;459:72-85.
18. Kim C, Kim B. Anti-cancer natural products and their bioactive compounds inducing ER stress-mediated apoptosis: A Review. Nutrients 2018;10:1021.
19. Kumar A, Jaitak V. Natural products as multidrug resistance modulators in cancer. Eur J Med Chem 2019;176:268-91.
20. Bonofiglio D, Giordano C, De Amicis F, Lanzino M, Andò S. Natural products as promising antitumoral agents in breast cancer: Mechanisms of action and molecular targets. Mini Rev Med Chem 2016;16:596-604.
21. Yin B, Fang DM, Zhou XL, Gao F. Natural products as important tyrosine kinase inhibitors. Eur J Med Chem 2019;182:111664.
22. Wink M. Evolution of secondary metabolites from an ecological and molecular phylogenetic perspective. Phytochemistry 2003;64:3-19.
23. Schmeller T, Latz-Brüning B, Wink M. Biochemical activities of berberine, palmatine and sanguinarine mediating chemical defense against microorganisms and herbivores. Phytochemistry 1997;44:257-66.
24. Lu JJ, Bao JL, Chen XP, Huang M, Wang YT. Alkaloids isolated from natural herbs as the anticancer agents. Evid Based Complement Alternat Med 2012;2012:485042.
26. He X, Feng S. Role of metabolic enzymes P450 (CYP) on activating procarcinogen and their polymorphisms on the risk of cancers. Curr Drug Metab 2015;16:850-63.
27. Hrycay EG, Bandiera SM. . Involvement of cytochrome P450 in reactive oxygen species formation and cancer. Cytochrome P450 Function and Pharmacological Roles in Inflammation and Cancer. Elsevier; 2015. pp. 35-84.
28. Shimada T, Oda Y, Gillam EM, Guengerich FP, Inoue K. Metabolic activation of polycyclic aromatic hydrocarbons and other procarcinogens by cytochromes P450 1A1 and P450 1B1 allelic variants and other human cytochromes P450 in Salmonella typhimurium NM2009. Drug Metab Dispos 2001;29:1176-82.
29. Williams DE, Shigenaga MK, Castagnoli N Jr. The role of cytochromes P-450 and flavin-containing monooxygenase in the metabolism of (S)-nicotine by rabbit lung. Drug Metab Dispos 1990;18:418-28.
30. Bao Z, He XY, Ding X, Prabhu S, Hong JY. Metabolism of nicotine and cotinine by human cytochrome P450 2A13. Drug Metab Dispos 2005;33:258-61.
31. Peterson LA, Trevor A, Castagnoli N Jr. Stereochemical studies on the cytochrome P-450 catalyzed oxidation of (S)-nicotine to the (S)-nicotine delta 1'(5')-iminium species. J Med Chem 1987;30:249-54.
32. Gorrod JW, Hibberd AR. The metabolism of nicotine-delta 1'(5')-iminium ion, in vivo and in vitro. Eur J Drug Metab Pharmacokinet 1982;7:293-8.
33. Centers for Disease Control and Prevention - Data and Statistics, F. F. a. F. S., Smoking & Tobacco Use. https://www.cdc.gov/tobacco/data_statistics/fact_sheets/fast_facts/index.htm.
34. Baker TB, Piper ME, Stein JH, et al. Effects of nicotine patch vs Varenicline vs combination nicotine replacement therapy on smoking cessation at 26 weeks: A randomized clinical trial. JAMA 2016;315:371-9.
35. National Institute on Drug Abuse. Tobacco, N., and ECigarettes, Is Nicotine Addictive? https://www.drugabuse.gov/publications/research-reports/tobacco/nicotine-addictive. [Last accessed on 1 Dec 2020].
36. Mcmorrow MJ, Foxx RM. Nicotine's role in smoking: An analysis of nicotine regulation. Psychological Bulletin 1983;93:302-27.
37. Sellers EM, Kaplan HL, Tyndale RF. Inhibition of cytochrome P450 2A6 increases nicotine's oral bioavailability and decreases smoking. Clin Pharmacol Ther 2000;68:35-43.
38. Sellers EM, Ramamoorthy Y, Zeman MV, Djordjevic MV, Tyndale RF. The effect of methoxsalen on nicotine and 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone (NNK) metabolism in vivo. Nicotine Tob Res 2003;5:891-9.
39. Pianezza ML, Sellers EM, Tyndale RF. Nicotine metabolism defect reduces smoking. Nature 1998;393:750.
40. Rao Y, Hoffmann E, Zia M, et al. Duplications and defects in the CYP2A6 gene: identification, genotyping, and in vivo effects on smoking. Mol Pharmacol 2000;58:747-55.
41. Ezzeldin N, El-Lebedy D, Darwish A, et al. Association of genetic polymorphisms CYP2A6*2 rs1801272 and CYP2A6*9 rs28399433 with tobacco-induced lung Cancer: case-control study in an Egyptian population. BMC Cancer 2018;18:525.
42. Foroozesh M, Jiang Q, Sridhar J, et al. Design, synthesis, and evaluation of a family of propargyl pyridinyl ethers as potential cytochrome P450 inhibitors. J Undergrad Chem Res 2013;12:91-4.
43. Shimada T, Takenaka S, Kakimoto K, et al. Structure-function studies of naphthalene, phenanthrene, biphenyl, and their derivatives in interaction with and oxidation by cytochromes P450 2A13 and 2A6. Chem Res Toxicol 2016;29:1029-40.
44. Shimada T, Takenaka S, Murayama N, et al. Oxidation of pyrene, 1-hydroxypyrene, 1-nitropyrene and 1-acetylpyrene by human cytochrome P450 2A13. Xenobiotica 2016;46:211-24.
45. Kramlinger VM, von Weymarn LB, Murphy SE. Inhibition and inactivation of cytochrome P450 2A6 and cytochrome P450 2A13 by menthofuran, β-nicotyrine and menthol. Chem Biol Interact 2012;197:87-92.
46. Prasopthum A, Pouyfung P, Sarapusit S, Srisook E, Rongnoparut P. Inhibition effects of Vernonia cinerea active compounds against cytochrome P450 2A6 and human monoamine oxidases, possible targets for reduction of tobacco dependence. Drug Metab Pharmacokinet 2015;30:174-81.
47. Nakajima M, Itoh M, Yamanaka H, et al. Isoflavones inhibit nicotine C-oxidation catalyzed by human CYP2A6. J Clin Pharmacol 2006;46:337-44.
48. Gilardi G, Meharenna YT, Tsotsou GE, Sadeghi SJ, Fairhead M, Giannini S. Molecular Lego: design of molecular assemblies of P450 enzymes for nanobiotechnology. Biosensors and Bioelectronics 2002;17:133-45.
49. Dodhia VR, Fantuzzi A, Gilardi G. Engineering human cytochrome P450 enzymes into catalytically self-sufficient chimeras using molecular Lego. J Biol Inorg Chem 2006;11:903-16.
50. Castrignanò S, Ortolani A, Sadeghi SJ, Di Nardo G, Allegra P, Gilardi G. Electrochemical detection of human cytochrome P450 2A6 inhibition: a step toward reducing dependence on smoking. Anal Chem 2014;86:2760-6.
51. Denton TT, Srivastava P, Xia Z, et al. Identification of the 4-position of 3-alkynyl and 3-heteroaromatic substituted pyridine methanamines as a key modification site eliciting increased potency and enhanced selectivity for cytochrome P-450 2A6 inhibition. J Med Chem 2018;61:7065-86.
52. Denton TT, Zhang X, Cashman JR. Nicotine-related alkaloids and metabolites as inhibitors of human cytochrome P-450 2A6. Biochem Pharmacol 2004;67:751-6.
53. Denton TT, Zhang X, Cashman JR. 5-substituted, 6-substituted, and unsubstituted 3-heteroaromatic pyridine analogues of nicotine as selective inhibitors of cytochrome P-450 2A6. J Med Chem 2005;48:224-39.
54. Yano JK, Denton TT, Cerny MA, Zhang X, Johnson EF, Cashman JR. Synthetic inhibitors of cytochrome P-450 2A6: inhibitory activity, difference spectra, mechanism of inhibition, and protein cocrystallization. J Med Chem 2006;49:6987-7001.
55. Liu J, Taylor SF, Dupart PS, et al. Pyranoflavones: a group of small-molecule probes for exploring the active site cavities of cytochrome P450 enzymes 1A1, 1A2, and 1B1. J Med Chem 2013;56:4082-92.
56. Sridhar J, Jin P, Liu J, Foroozesh M, Stevens CL. In silico studies of polyaromatic hydrocarbon inhibitors of cytochrome P450 enzymes 1A1, 1A2, 2A6, and 2B1. Chem Res Toxicol 2010;23:600-7.
57. Sridhar J, Liu J, Komati R, et al. Ortho-methylarylamines as time-dependent inhibitors of cytochrome P450 1A1 enzyme. Drug Metab Lett 2017;10:270-7.
58. DeVore NM, Scott EE. Nicotine and 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone binding and access channel in human cytochrome P450 2A6 and 2A13 enzymes. J Biol Chem 2012;287:26576-85.
59. Mao W, Berenbaum MR, Schuler MA. Modifications in the N-terminus of an insect cytochrome P450 enhance production of catalytically active protein in baculovirus-Sf9 cell expression systems. Insect Biochem Mol Biol 2008;38:66-75.
60. Schwarz D, Kisselev P, Honeck H, Cascorbi I, Schunck WH, Roots I. Co-expression of human cytochrome P4501A1 (CYP1A1) variants and human NADPH-cytochrome P450 reductase in the baculovirus/insect cell system. Xenobiotica 2001;31:345-56.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Goyal N, Sridhar J, Do C, Bratton M, Shaik S, Jiang Q, Foroozesh M. Identification of CYP 2A6 inhibitors in an effort to mitigate the harmful effects of the phytochemical nicotine. J Cancer Metastasis Treat 2021;7:18. http://dx.doi.org/10.20517/2394-4722.2020.143
AMA Style
Goyal N, Sridhar J, Do C, Bratton M, Shaik S, Jiang Q, Foroozesh M. Identification of CYP 2A6 inhibitors in an effort to mitigate the harmful effects of the phytochemical nicotine. Journal of Cancer Metastasis and Treatment. 2021; 7: 18. http://dx.doi.org/10.20517/2394-4722.2020.143
Chicago/Turabian Style
Goyal, Navneet, Jayalakshmi Sridhar, Camilla Do, Melyssa Bratton, Shahensha Shaik, Quan Jiang, Maryam Foroozesh. 2021. "Identification of CYP 2A6 inhibitors in an effort to mitigate the harmful effects of the phytochemical nicotine" Journal of Cancer Metastasis and Treatment. 7: 18. http://dx.doi.org/10.20517/2394-4722.2020.143
ACS Style
Goyal, N.; Sridhar J.; Do C.; Bratton M.; Shaik S.; Jiang Q.; Foroozesh M. Identification of CYP 2A6 inhibitors in an effort to mitigate the harmful effects of the phytochemical nicotine. J. Cancer. Metastasis. Treat. 2021, 7, 18. http://dx.doi.org/10.20517/2394-4722.2020.143
About This Article
Special Issue
Copyright

Data & Comments
Data


 Cite This Article 7 clicks
Cite This Article 7 clicks

 Like This Article 25
likes
Like This Article 25
likes






Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.