
Systemic treatment of advanced, metastatic, medullary thyroid carcinoma

 , ...
, ... Abstract
Medullary thyroid carcinoma (MTC) is a rare endocrine tumor, which arises from thyroid parafollicular C cells. Through its ability to metastasize by blood and lymphatic vessels, it can show a more aggressive clinical behavior than differentiated thyroid cancers. Mutation of RET gene is the main molecular alteration involved in MTC origin. In the case of germline RET mutation, MTC can be inherited in an autosomal dominant way and show three different phenotypes: familial medullary thyroid carcinoma and multiple endocrine neoplasia types IIA and IIB. In addition, in sporadic cases, somatic RET mutation remains the key molecular alteration in most of cases. Total thyroidectomy with prophylactic or therapeutic central compartment lymph nodes dissection is the surgical treatment of choice. Further surgical treatments and local therapies should be used in the case of single or few local or distant metastasis. However, in cases with large metastatic spread of the disease, particularly in those with significant tumor progression, additional systemic treatments are needed. In this review, we discuss the key points of systemic treatment in advanced, metastatic MTC. We provide an update on the main aspects (from biological rationale to clinical experience) of each treatment, focusing our attention on the drugs used in clinical practice in the last years. Finally, we give insights about the emerging treatments from highly selective RET inhibitors to new radionuclide therapy.
Keywords
INTRODUCTION
Medullary thyroid carcinoma (MTC) is a rare neuroendocrine tumor originating from parafollicular C cells, which represent only the 0.1% of all thyroid cells[1,2]. The peculiarity of parafollicular cells is to produce and secrete calcitonin (CT), as well as, to a lesser extent, other peptides such as chromogranin, serotonin, somatostatin, or calcitonin gene-related peptide[3]. These cells are usually located in the upper and middle thirds of the thyroid, but the hyperplastic ones are prevalently located in the middle and lower thirds of the lateral lobes and only exceptionally in the isthmus[4]. Because of its origin, MTC can be considered a separate entity from differentiated thyroid carcinoma (DTC), which originates in the epithelial follicular thyroid cells.
The prevalence of MTC is variable according to the different series, however it is generally reported as 5%-10% of all thyroid malignancies, 0.4%-1.4% of all thyroid nodules, and less than 1% in thyroids of subjects submitted to autopsy. For this reason, to date, MTC is officially considered a rare disease by the national Health Institute (NIH)[5]. Unlike DTC, MTC shows no difference in sex distribution, and the median age at diagnosis is 45-55 years[6-9].
MTC can occur in a sporadic (about 75% of cases) or hereditary (25% of cases) form. In hereditary cases, MTC can be the only clinically expressed disease [familial medullary thyroid carcinoma (FMTC)] or associated with other endocrine neoplasia, in the context of the multiple endocrine neoplasia syndromes (MEN types IIA and IIB), such as pheocromocytoma (PHEO) and/or hyperparathyroidism due to parathyroid hyperplasia or multiple adenomatosis (PTHAd)[10]. Children can only be affected by inherited MTC, and the more aggressive is the syndrome (i.e., MEN IIB), the younger is the affected child[11-14].
The pathogenesis of MTC is highly associated to the activation of the RET protooncogene, both in hereditary[15-18] and in sporadic cases[19-21]. In hereditary cases, the RET protooncogene alteration is transmitted in a dominant mendelian autosomal way and is found at germline level, while, in sporadic cases, this alteration is somatic and found only in the tumoral cells.
Through its dissemination by both lymphatic and hematic vessels, the clinical behavior of MTC is less favorable when compared with most DTC, however it is not as unfavorable as the anaplastic one (ATC)[22,23]. Five-year survival rates vary from 62% to 87% according to the different series, and the 10-year survival could decrease to 50%[24-29]. Survival is dependent on several factors, such as age at diagnosis[30]. However, the staging at diagnosis remains the most relevant clinical prognostic factor of survival of these patients. When an early diagnosis is performed, and the tumor is still intrathyroidal, 90% of patients can survive up to 35 years[29,31].
Usually, the most common clinical presentation of a sporadic MTC is a thyroid nodule, single or in a clinical picture of a multinodular goiter. At the diagnosis, lymph node metastases are frequent and distant metastases are already present in about 10% of patients[32]. In advanced metastatic cases associated with high levels of serum CT, symptoms such as diarrhea and/or flushing syndrome could be present.
Conversely, the hereditary forms can be easily suspected according to a familial history of MTC or if the same patient has already been diagnosed with PHEO and/or PTHAd. The presence of mucosal neurinomas of the tongue or conjunctivas, in particular if associated to marfanoid habitus and/or skeletal alterations, should immediately suggest the diagnosis of MEN IIB[11]. Similarly, the detection of an interscapular cutaneous itchy lesion, defined as cutaneous lichen amyloidosis, is highly suspicious of MEN IIA[33], since this lesion is almost exclusively found in this syndrome. While thyroid function, assessed by the measurement of free triiodothyronine (fT3), free thyroxine (fT4), and thyroid stimulating hormone (TSH), is commonly normal, serum CT is elevated, and sometimes this finding can represent the first suspicion of the presence of MTC, thus requiring further diagnostic procedures[34]. In advanced cases, carcinoembryonic antigen (CEA) can also be elevated. CT and CEA also represent the biochemical markers to follow-up MTC patients after surgery and during local or systemic treatments.
Initial treatment of MTC depends on its clinical presentation. Total thyroidectomy with central compartment lymph nodes dissection is considered the correct treatment for MTC in the absence of preoperative evidence of latero-cervical lymph nodes metastases. When during pre- or intra-operative evaluation latero-cervical lymph nodes metastases are detected, an oriented compartment lymph node dissection is advocated[35]. Surgical removal of the primary tumor and lymph nodes metastases of the neck is suggested also in cases in which distant metastases are already present[35]. In these latter cases, and in the case of larger tumors associated to invasion of the vital structure of the neck, in which surgical removal of the primary tumor and lymph nodes metastases is not feasible, additional therapies should be performed[36,37].
The aim of this review is to elucidate the key points about the systemic treatment of advanced, metastatic MTC. We provide an update of the main aspects of systemic treatment of MTC, focusing the attention on the drugs which have been developed in the last years, from the tyrosine kinase inhibitors (TKIs) to radionuclide therapies.
TYROSINE KINASE INHIBITORS
Rationale of treatment
In the last years, several molecular aberrations located in the cell signaling pathways of malignant cells were discovered. In particular, several tyrosine kinases (TKs), mainly TK receptors (TKRs) involved in cell growth, differentiation, and angiogenesis, were found to be mutated or overexpressed in tumor cells[38,39]. The importance of these receptors is linked to the ability of several drugs, named TKIs, to inhibit their activity[40]. To date, TKIs are firmly used in the clinical practice for the treatment of several advanced tumors, from leukemia[41] to solid tumors[42,43], including thyroid cancer[36,44-46]. TKIs can act through different mechanisms: (1) through competition with the adenosine triphosphate (ATP) at the binding site of a TKR, competition with the substrate, or both; and (2) in an allosteric modality by binding to a site located outside the active site, thus affecting its activity by determining a conformational change of the kinase[44,47,48]. Moreover, TKIs can act on tyrosine, serine, threonine, or even histidine residues, therefore are able to simultaneously inhibit the action of one or more kinases, although with different binding affinities[40,49].
Several genetic alterations, leading to dysregulation of multiple signaling pathways, have been reported in all thyroid cancers[50]. TKIs are designed to mainly interact with altered TKRs, and thus with the two main signaling pathways involved in cell growth and proliferation: the mitogen-activated protein (MAP) kinase/extracellular signal-regulated (ERK) pathway and the phosphatidylinositol-3 kinases (PI3K)/AKT/mTOR pathway [Figure 1].
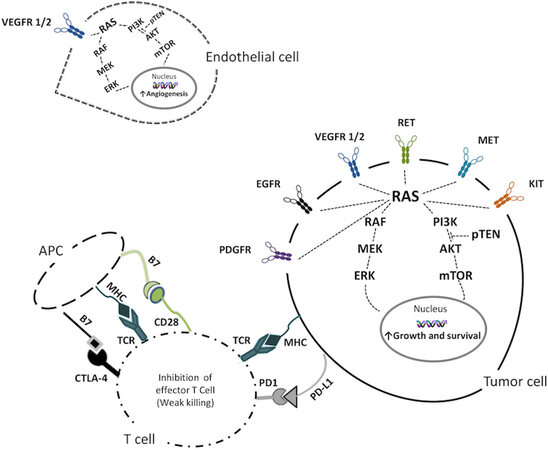
Figure 1. Schematic representation of the over activation of TKRs involved in enhancing the growth and survival in the tumor cell, promoting angiogenesis in the endothelial cell, and the inhibition of killing effect of the T cell on tumor cell. VEGFR 1/2: Vascular endothelial growth factor receptor 1/2; PDGFR: platelet-derived growth factor receptor; EGFR: epidermal growth factor receptor; RET: rearranged during transfection; MET: hepatocyte growth factor receptor; KIT: mast/stem cell growth factor receptor; RAS: rat sarcoma; RAF: v-raf murine sarcoma viral oncogene homolog; MEK: mitogen activated protein kinase; ERK: extracellular signal-regulated kinases; PI3K: phosphoinositide 3-kinase; pTEN: phosphatase and tensin homolog; AKT: protein kinase B; mTOR: mammalian target of rapamycin; MHC: major histocompatibility complex; TCR: T cell receptor; PD1: programmed cell death protein 1; PD-L1: programmed death ligand 1; CTLA-4: cytotoxic T-lymphocyte antigen 4; APC: antigen presenting cell.
TKRs are upstream of the MAPK and PI3K pathways, and mutations or gene fusions at this level can affect the signaling transduction to the downstream, leading to oncogenic transformation and progression. Similarly, mutations occurring in the MAPK and PI3K pathways can promote tumorigenesis.
In MTCs, the most common TKR alterations are the gain of function point mutations in the RET oncogene, which are responsible for most of the hereditary and 40%-70% of the sporadic cases of MTC[51]. The RET mutations in MTC are usually detected in the cysteine-rich or tyrosine kinase domains, located within seven exons (8, 10, 11, 13, 14, 15, and 16)[19,52]. These mutations are responsible for MEN type IIA and IIB and FMTC. In MEN IIA, the most frequently detected RET point mutation is located at codon 634[53]; conversely, in MEN IIB and most sporadic cases, it is at codon 918 (M918T)[11,15]. Different RET mutations are associated with different age of onset and aggressiveness of MTC, and the presence/absence of other endocrine malignancies[35,54].
In sporadic cases, the presence of the somatic RET mutation, particularly of M918T is a prognostic factor of a bad outcome[21]. As a matter of fact, almost 85% of advanced MTC requiring a systemic therapy for the aggressiveness of the disease are carrying a somatic RET mutation[55].
The only other oncogene found to be altered at somatic level in MTC is RAS gene. Mutations in K- and HRAS genes have been identified in 20% of sporadic MTC and have been demonstrated to be mutually exclusive events with RET mutations. Moreover, RAS mutated MTC cases are apparently less aggressive that RET mutated ones. Thus far, about 20% of sporadic MTC are still orphan of a driver oncogene alteration[20,56].
TKIs in clinical practice
Several TKIs have been tested for the treatment of advanced progressive MTC, including imatinib[57], axitinib[58], motesanib[59], sorafenib[60], sunitinib[61], pazopanib[62], ponatinib[63], lenvatinib[64], and anlotinib[65].
However, only two of them, vandetanib and cabozantinib, have been approved by the Food and Drug Administration (FDA) and the European Medicine Agency (EMA), after the phase III studies, ZETA and EXAM trials[66,67].
Vandetanib
Vandetanib (also known as ZD6474) is an inhibitor of VEGFR2 and -3, RET, and EGFR kinases[36,68,69] [Figure 2].
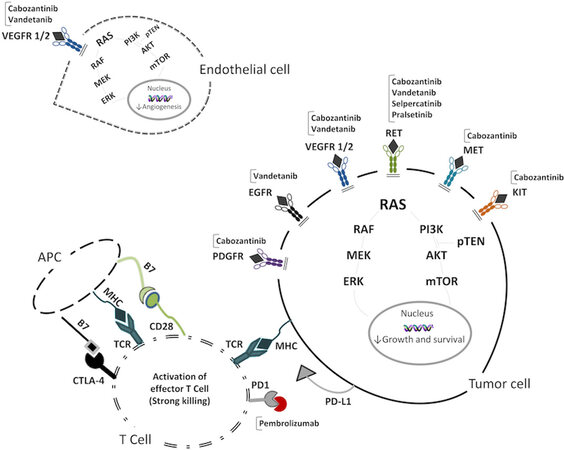
Figure 2. Molecular target of TKIs and immunotherapy used in MTC on tumor cell, endothelial cell, and T cell and the consequent effects of treatments. VEGFR 1/2: Vascular endothelial growth factor receptor 1/2; PDGFR: platelet-derived growth factor receptor; EGFR: epidermal growth factor receptor; RET: rearranged during transfection; MET: hepatocyte growth factor receptor; KIT: mast/stem cell growth factor receptor; RAS: rat sarcoma; RAF: v-raf murine sarcoma viral oncogene homolog; MEK: mitogen activated protein kinase; ERK: extracellular signal-regulated kinases; PI3K: phosphoinositide 3-kinase; pTEN: phosphatase and tensin homolog; AKT: protein kinase B; mTOR: mammalian target of rapamycin; MHC: major histocompatibility complex; TCR: T cell receptor; PD1: programmed cell death protein 1; PD-L1: programmed death ligand 1; CTLA-4: cytotoxic T-lymphocyte antigen 4; APC: antigen presenting cell.
In 2012, the results of the international randomized phase III ZETA trial (ClinicalTrials.gov, number NCT00410761) demonstrated an efficacy of vandetanib, at dosage of 300 mg/daily, in prolonging progression free survival (PFS) in 331 patients with advanced progressive MTC, compared to placebo (30.5 months vs. 19.3 months; HR = 0.46; 95%CI: 0.31-0.69). However, no differences in the overall survival (OS) between the two groups was shown[66]. The clinical benefit of vandetanib treatment was also confirmed in a recent post-hoc analysis, when patients were divided into four disease severity subgroups: patients with both progression and symptoms at baseline, those with symptoms only, those with progression only, and those with no progression or symptoms at baseline[70]. In 2014, Massicotte et al.[71], in a retrospective study on a small subgroup of advanced MTC (n = 11), confirmed that a partial response was obtained in 36% of the study group.
In addition, outside of clinical trial, vandetanib treatment showed its efficacy. In a multicentric French study, 60 patients with advanced MTC and diffuse metastatic involvement were treated with this drug and the data were analyzed. After a median follow-up of 20 months and a median duration of treatment of 9.7 months, median PFS was 16.1 months. Moreover, a clinical benefit defined as best tumor response was observed in most of the patients (75%)[72]. Interestingly, in this study, one patient showed a complete response, while, conversely, one patient died because of vandetanib-induced cardiac toxicity.
Recently, clinical experiences in real-life settings have been reported[73,74]. Valerio et al.[74] evaluated 79 MTC patients, treated with vandetanib and followed at a tertiary care center. Patients were divided according to the time of treatment into short- (< 1 year) and long-term (> 1 year) responders. Median PFS of the entire study group was 47 months, longer than in ZETA trial (30.5 months), and, when considering only the long-term responders, PFS was even longer (54.5 months). Similar results were reported in a following study by Ramos et al[73]. showing that that PFS of those patients who continued vandetanib treatment for more than 4 years was significantly longer (73.2 months) than that of ZETA trial. Interestingly, in both studies, a better and durable clinical response was experienced in younger patients and in those in whom vandetanib treatment was started without evidence of tumor progression but just for severe symptoms.
In several patients during the clinical trials as well as in the real-life studies, vandetanib treatment was withdrawn during the follow-up either for progression of the disease or for the onset of adverse events (AEs). Indeed, several AEs are commonly experienced during vandetanib treatment[66]. According to the Common Terminology Criteria for Adverse Events (CTCAE)[75], AEs are classified into grades of severity. The most common mild and severe adverse events experienced in clinical trials and real-life studies are reported in Table 1.
Safety and efficacy data of clinical trials evaluating vandetanib, cabozantinib, selpercatinib, and pralsetinib treatments in MTC patients
| Vandetanib[66] | Cabozantinib[67] | Selpercatinib[76] | Pralsetinib[77] | ||
| Trial design | Double-blind, randomized, placebo-controlled | Double-blind, randomized, placebo-controlled | Open label | Open label | |
| Clinical phase | III | III | I (dose escalation) II (dose expansion) | I (dose escalation) II (dose expansion) | |
| Number of patients | Treatment | 231 | 219 | 143 | 92 |
| Placebo | 100 | 111 | -- | -- | |
| Initial drug dose | 300 mg | 140 mg | Phase I: from 20 mg once/day to 240 mg twice/day Phase II: 160 mg twice/day | Phase I: from 30 mg once/day to 600 mg twice/day Phase II: 400 mg once/day | |
| TKIs naïve patients | 90/231 (39%) | 44/219 (20.1%) | 55/143 (38.5%) | 53/72* (73.6%) | |
| ECOG status | 0 | 154 (66.7%) | 123 (56.2%) | 54 (37.7%) | --** |
| 1 | 67 (29.0%) | 95 (43.4%) | 83 (58.0%) | --** | |
| 2 | 10 (4.3%) | 6 (4.2%) | --** | ||
| Primary outcome | PFS | PFS | ORR | MTD and RP2D Safety | |
| Secondary outcome | ORR, DCR, OS, biochemical response, time to worsening pain | OS and ORR | DOR, PFS, and safety | ORR, CBR, DOR, DCR, PFS, OS, pharmacokinetic features | |
| PFS | 30.5 months (treatment) 19.3 months (placebo) | 11.2 months (treatment) 4.0 months (placebo) | 23.6 months (TKIs naïve) 27.4 months (Non-naïve) | Not reached | |
| ORR | 45% | 28% | 70% (TKIs naïve) 61% (Non-naïve) | 74% (TKIs naïve) 60% (Non-naïve) | |
| DCR | 87% | 76% | 87% (TKIs naïve) 84% (Non-naïve) | 100% (TKIs naïve) 96% (Non-naïve) | |
| Most common AE | Diarrhea, nausea, rash and hypertension | Diarrhea, palmar-plantar erythrodysesthesia, decreased weight or appetite | Dry mouth, hypertension, diarrhea, fatigue | Increase of liver enzymes, anemia, constipation, hypertension | |
| Most common AE ≥ CTCAE Gr. 3 | Diarrhea, hypertension, QT prolongation | Diarrhea, palmar-plantar erythrodysesthesia, fatigue, hypertension | Hypertension, increase of liver enzymes | --* | |
Since the good clinical response was demonstrated and AEs were common findings, a clinical trial (Nbib1496313) evaluating the risk-benefit of two different starting doses (150 mg/daily vs. 300 mg/daily) of vandetanib was performed[78]. Patients were randomized 1:1 to receive vandetanib 150 or 300 mg/daily for a maximum time of 14 months (Part A). At the end of Part A, the possibility to enter in the open-label phase (Part B), investigating vandetanib at 100, 150, 200, and 300 mg/daily doses, was given to all patients. Eighty-one patients were randomized in Part A and 61 patients accepted to continue in Part B, 37 (60.7%) of whom received 2 years of treatment. One quarter of the patients experienced an objective response (OR) after 14 months, statistically significant for both doses of 300 mg (HR = 0.29; 95%CI: 0.176-0.445) and 150 mg (HR = 0.20; 95%CI: 0.105-0.348). In addition, in Part B, safety and tolerability were similar to those in Part A. However, AEs such as diarrhea, hypocalcemia, asthenia, QTc prolongation, hypokalemia, and keratopathy were more common in patients who were treated with 300 mg.
At variance with other TKIs, vandetanib treatment has also been tested in children, and it has been demonstrated to be safe and effective in controlling childhood MTC[79]. Moreover, evidence show the usefulness of vandetanib in reverting ectopic ACTH secretion and the consequent paraneoplastic Cushing’s syndrome which could develop in patients with very advanced disease[80-83].
Cabozantinib
Cabozantinib (also known as XL184) is an inhibitor of VEGFR1 and -2, RET, c-KIT, MET, and KIF 5B rearrangements[84] [Figure 2]. The efficacy of cabozantinib was demonstrated in a phase I study[85]. In 2013, after the results of the double-blinded, phase III trial EXAM study (ClinicalTrials.gov, number NCT00704730), cabozantinib was approved by FDA and EMA. In this study, a significant improvement in PFS was demonstrated in patients with progressive MTC, compared to placebo[67]. Patients (n = 330) with progressive MTC were enrolled and treated with cabozantinib (140 mg/daily), at 2:1 compared to placebo. The estimated median PFS was 11.2 months for cabozantinib group vs. 4.0 months for placebo (HR = 0.28; 95%CI: 0.19-0.40; P < 0.001). The analysis of specific subgroups of patients according to age, previous TKIs treatment, and hereditary or sporadic cases showed an improved PFS in treated patients compared to placebo. In addition, response rate (28% vs. 0%) and estimation of progression-free at 1 year (47.3% vs. 7.2%) were higher in patients treated with cabozantinib.
AEs were commonly described and included diarrhea, palmar-plantar erythrodysesthesia, decreased weight and appetite, nausea, and fatigue. To manage these AEs, most patients (79%) experienced a dose reduction and some of them (16%) a treatment discontinuation. Some severe AEs, e.g., hemorrhages and intestinal perforation, related to the high activity of cabozantinib against VEGFR, were reported during the EXAM clinical trial.
In 2016, an exploratory analysis in patients of a phase III study was performed to evaluate the clinical response to cabozantinib, according to RET or RAS (HRAS, KRAS, and NRAS) mutations[86]. Half of the patients (51.2%) harbored RET mutation, prevalently M918T (38.2%), while the remaining were RET negative (13.9%) or RET unknown (34.8%). Only 16 patients (4.8%) showed RAS mutation. PFS was longer in cabozantinib group compared to placebo in three out of four subgroups of patients: those who harbored RET mutations (60 weeks vs. 20 weeks; HR = 0.23; 95%CI: 0.14-0.38; P < 0.0001), those with RET unknown (HR = 0.30; 95%CI: 0.16-0.57; P = 0.0001), and those with RAS mutations (HR = 0.15; 95%CI: 0.02-1.10; P = 0.0317). Interestingly, in the RET-mutated patients, those harboring the M918T mutation showed the greatest benefit in PFS from cabozantinib therapy compared to placebo (61 weeks vs. 17 weeks; HR = 0.15; 95%CI: 0.08-0.28; P < 0.0001). In patients without RET or RAS mutation, no benefit in PFS was observed. All subgroups showed similar safety profile.
Overall survival was evaluated in another exploratory analysis assessing the data of patients included in a phase III study, after long-term follow up[87]. After a minimum follow-up of 42 months, a 5.5-month increase in median OS was observed in cabozantinib vs. placebo group (26.6 months vs. 21.1 months; HR = 0.85; 95%CI: 0.64-1.12; P = 0.24), although not statistically significant. However, in this analysis, cabozantinib treated patients also experienced longer OS if harboring RET M918T mutation, compared to placebo (44.3 months vs. 18.9 months; HR = 0.60; 95%CI: 0.38-0.94; P = 0.03). The safety profile for cabozantinib patients remained similar to the data of the primary analysis. The authors concluded that, although the OS was not significantly longer in the cabozantinib group, patients with RET M918T mutation could greatly benefit from cabozantinib treatment.
Clinical point of view
At present, vandetanib and cabozantinib are both valid options in the treatment of advanced metastatic MTC. Both should be started when a progression of the structural disease according to the RECIST[88] is documented or according to clinical judgement in peculiar cases. The choice of the drug is dependent on the drug availability in different countries, because not in all countries are approved and reimbursed.
However, if both were available, the choice should consider both drug characteristics and patient features. According to the results of the studies, cabozantinib seems to be more rapid in inducing the shrinkage of the tumor tissue and could be useful when there is the need to rapidly control the tumor burden (e.g., vertebral lesion compressing spinal cord). Conversely, vandetanib showed a better safety profile and an easier manageability of its specific AE.
When choosing the drug, it should also be considered that cabozantinib has been tested as second line treatment after other TKIs and showed advantages in prolongation of PFS, while this information is unavailable for vandetanib. Conversely, vandetanib is contraindicated in patients who have a prolonged QTc (> 450 ms in men and > 470 ms in females)[89-91], while cabozantinib should not be used in patients with a history of diverticulitis[67], and renal function should be carefully monitored during treatment because of the potential onset of renal damage with proteinuria[92]. Moreover, both treatments could modify thyroid function, in patients with thyroid gland in situ and thyroid hormone metabolism; thus, the evaluation of thyroid function tests is mandatory for all patients undergoing TKI treatment[93]. Furthermore, for both treatments, particular caution should be taken in evaluating the medical history of hemoptysis and hemorrhages. If the tumor invades the vital structures of the neck and if patients experienced radiation treatment of the neck or mediastinum, they carry a higher risk for hemorrhages and fistula formation, which is a life-threatening AE[94]. Moreover, as mentioned above, vandetanib has been approved for the treatment of children with MTC, mainly MENII patients, while this is not the case for cabozantinib. Similarly, no data have been reported on the benefit that cabozantinib has in the treatment of ACTH ectopic syndrome. Thus, it is intuitive that, in either children or patients with ectopic ACTH, vandetanib should be preferred if no other elements contraindicate its use.
Furthermore, in vitro studies showed the V804 RET mutation confers resistance to vandetanib[95], but this seems to not be a limitation for cabozantinib. Therefore, in patients with V804M, cabozantinib should be preferred.
Lastly, AEs are common and similar for both drugs, particularly those of mild intensity (CTCAE Grades 1 and 2), but their prevalence is different according to the drug used [Table 1].
Vandetanib and cabozantinib are two cytostatic drugs, therefore they inhibit cellular growth but do not kill the neoplastic cells. For this reason, the treatment should be continued as long as there is evidence of clinical benefit.
To date, no advantages in OS were shown in the exploratory analysis of both phase III studies[70,87] when comparing vandetanib or cabozantinib to placebo. Moreover, several clinical data indicate that patients on TKIs treatment will eventually develop a mechanism of resistance to the drug, associated with progression of the disease. This mechanism, highlighted in all TKIs treatment, seems to be independent of the TKI used[96] and could be related to secondary mutations in the kinase domains that block the TKIs binding in the target genes[97]. For these reasons, in the last years, further studies have been performed to evaluate new drugs able to implement the therapeutic landscape of systemic treatment of advanced MTC.
RET SELECTIVE INHIBITORS
Rationale of treatment
RET mutations are found in more than 40% of sporadic and virtually 100% of hereditary cases[15,20,98]. Moreover, about 85% of advanced metastatic MTC cases carried RET mutations, which are able to induce higher risk of lymph nodes metastases, disease recurrence/persistence after surgery, and worst survival compared to other mutations[20,55,99].
In addition to MTC, RET is a driver gene in other tumors such as papillary thyroid, lung, breast, and colon cancers. The group of cancers characterized by these driver mutations can be defined as REToma[100]. According to its role in REToma carcinogenesis, RET protein has been considered an ideal target for highly selective drugs.
Thus, a second generation of highly selective RET inhibitors have been developed both to maintain a significant anti-tumor efficacy, similar to vandetanib and cabozantinib, and to improve the safety profile. In 2018, two molecules, selpercatinib and pralsetinib[101,102], were proposed. These drugs showed a stronger inhibition of RET and a weaker activity against other targets, such as VEGFR, compared to vandetanib and cabozantinib[103] [Figure 2]. Moreover, selpercatinib and pralsetinib inhibited the proliferation of cells harboring many kinds of RET mutations, both gene fusions and point mutations, including the V804M mutation which, as mentioned above, showed in vitro resistance to vandetanib[95]. These results were further confirmed in animal models and clinical studies[101,102], in which a relevant tumor shrinkage was shown. Interestingly, in mouse models, selpercatinib showed anti-neoplastic activity against brain metastasis, too[101].
According to their highly selective activity against RET protein, regardless of tumor type, both drugs were involved in basket clinical trials with many cancers of the REToma network[76,77].
Selpercatinib
Between May 2017 and June 2019, 162 patients were enrolled in the phase I/II LIBRETTO-001 trial (ClinicalTrials.gov, number NCT03157128) to evaluate the OR rate in different histological types of thyroid cancers harboring RET mutations. Patients enrolled in phase I trial received selpercatinib with different starting doses (from 20 mg once daily to 240 mg twice daily) and progressive decreases to the highest safe dose were performed. Conversely, all patients in phase II study started with the same dose of 160 mg twice daily.
Most patients (143/162, 88.3%) had MTC, and 55 (38.5%) of them were already treated with vandetanib and/or cabozantinb while 88 (61.5%) were treatment naïve. The remaining patients (19/162, 11.7%) showed a RET fusion-positive thyroid cancer (PTC, poorly differentiated thyroid carcinoma, Huerthle cell carcinoma, and anaplastic carcinoma). OR rate was 69% in patients already treated with vandetanib and/or cabozantinib and 73% in treatment naïve patients. In the remaining patients with RET mutated thyroid cancers, OR rate was similar (79%). This result was confirmed for all RET mutation profiles, including V804M. After 1 year, 82% of MTC patients previously treated with vandetanib and/or cabozantinib, 92% of treatment naïve patients, and 64% of other thyroid cancer patients continued to be free of progression. However, longer follow-up data are needed to evaluate the duration of the clinical response.
Several AEs were experienced by most of the patients; however, the prevalence of AEs for single patient was lower and most of them were of lesser intensity (CTCAE Grade 1 or 2), compared with vandetanib[66] and cabozantinib[67] [Table 1]. Moreover, the safety profile was the same across all cancers treated in LIBRETTO-001[76].
Pralsetinib
ARROW is a phase I dose escalation trial to establish the recommended phase II dose (400 mg once daily) and phase II expansion cohorts defined by tumor type and/or RET alteration (ClinicalTrials.gov, number NCT03037385)[77].
Although the ARROW trial is not completed yet, recently, the ad interim results were presented, and efficacy and safety were assessed at the data cut-off of 13 February 2020[77]. Since March 2017, 79 patients with RET positive MTC were enrolled, either previously treated with vandetanib and/or cabozantinib or treatment naïve. Overall OR rate was evaluated on 51 patients and was 65%. In patients previously treated with vandetanib and/or cabozantinib, OR was 60%, while, in naive patients, it was 74% (14/19). PFS after 18 months was 71% in patients previously treated with other TKIs and 85% in treatment naïve patients. The authors did not find any influence due to different RET genotype on clinical response. Although the data about safety are not completed, the AEs were mainly of lower intensity (CTCAE Grade 1 or 2), and only 4% of patients discontinued the drug because of AEs.
Mechanisms of resistance
Despite the very interesting profile of these new drugs, several emerging reports also observed a potential resistance to these RET inhibitors. Recently, a patient with KIF5B-RET non-small cells lung carcinoma (NSCLC) developed progression after an initial partial response on selpercatinib treatment[104]. Circulating tumor DNA and post-mortem biopsy analysis showed the appearance of a wide spectrum of RET mutations on G810 residue, which could be considered a key spot for drug resistance. The appearance of these mutations was further confirmed in other progressing NSCLC and MTC cases and in vivo and in vitro models[104]. Structural analysis showed that mutations occurring at 810 residue can sterically alter selpercatinib binding. Subbiah et al.[105], using X-ray crystal analysis, revealed that selpercatinib and pralsetinib showed a peculiar binding with RET protein, compared to previous TKIs. This binding permits avoiding the resistance to 804 mutation, but it can be susceptible to mutations occurring at 738, 806, and 810 residues. In addition, RET-dependent resistance mechanisms, other RET-independent mechanisms have been described such as KRAS and MET amplification, which have recently been showed in progressing NSCLC patients[106,107].
Clinical point of view
Selpercatinib and pralsetinib are two promising drugs which showed a relevant clinical response in MTC patients, regardless of both previous TKI treatment experienced and different types of RET mutations. Moreover, their safety profile seems to be very encouraging, compared to previous TKIs. However, they can only be used in MTC patients harboring RET mutation, which represent the majority, but not all cases[20]. Furthermore, these drugs are susceptible to induce peculiar resistance mechanisms, and further studies are needed to completely understand the ideal clinical and molecular scenario to use these drugs.
At the same time, a third generation of TKIs, RET mutation selective ones, is under evaluation and could represent the future of the treatment of advanced MTC[108].
IMMUNOTHERAPY
Rationale of treatment
In the last decades, many promising results in the treatment of several human cancers derived from the development of immunotherapy, and in particular of drugs called immune checkpoints inhibitors (ICIs). The rationale for the use of immunotherapy is the close linkage among immune system, tumors, and tumor microenvironment (fibroblasts, pericytes, adipocytes, and other stromal cells)[109-111]. The immune system acts as a mediator of “immune surveillance”, recognizing and removing cancer cells, which try to escape the immune control through several mechanisms[112].
Several molecules are able to regulate the immune response, mainly through its inhibition. Immunotherapy aims to enhance the immune response against cancer cells, and, particularly, ICIs block the inhibitory activity of immune checkpoints. ICIs are monoclonal antibodies which target several immune checkpoints such as the cytotoxic T-lymphocyte antigen (CTLA-4), the programmed cell death 1 receptor (PD-1), and its ligand (PD-L1) [Figures 1 and 2]. To our knowledge, there have been no studies or clinical trial designed against thyroid cancer for ICIs targeting CTLA-4. For this reason, we focus our attention on the PD-1/PD-L1 pathway.
PD-1/PD-L1 pathway
Most studies evaluating the expression of the PD-1/PD-L1 pathway in thyroid cancer have been focused on follicular cell-derived tumors, including DTC, PDTC, and ATC. The PD1/PD-L1 pathway is highly expressed in DTC compared to benign tissues[113]. It is more diffuse in ATC patients[114], and its level is higher in BRAFV600E PTC compared to BRAF wild-type tumors[115]. The prognostic role of PD-L1 is still controversial since it is associated with higher risk of recurrence and a shorter disease-free survival[116]. Conversely, in a large cohort of follicular-derived thyroid cancer patients, it is not associated with disease progression[117]. According to these findings, clinical trials were performed to evaluate the efficacy of immunotherapy. The antitumor activity of pembrolizumab (anti-PD-1 antibody) in patients with advanced PTC and FTC was limited (9.1% of patients showed a partial response)[118]. Other trials are ongoing, but the results are still not available (ClinicalTrials.gov, numbers NCT03012620 and NCT02973997).
Concerning MTC, the potential role of immunotherapy could be associated to the immune reactivity of tumor cell lysates, which usually allowed the exposure of a wide variety of antigens[119,120], and the subsequent use of dendritic cell-based vaccinations using CT or CEA antigen[121-123].
The knowledge about the expression of immune checkpoint in MTC is increasing in the last years. The expression of PD-1/PD-L1 in MTC seems to be lower than in DTC. Bongiovanni et al.[124] showed low expression of PD-L1 in 16 MTC: only in one case was the PD-L1 percentage of expression higher than 1% in malignant cells, considered as positive cut-off, and two cases showed positivity for PD-L1 in immune cells.
Conversely, Bi et al.[125] showed higher level of expression of PD-1/PD-L1 in MTC patients: PD-1 was present on 25.3% (22/87) of the tumor-infiltrating immune cells, and 21.8% (19/87) had positive PD-L1 staining on tumor and immune cells. Moreover, in this study, the expression of PD-L1 in tumor cells and immune cells correlated with distant metastases at the time of surgery. Pozdeyev et al.[126] showed PD-L1 expression in 46 MTC patients: it was detected in 32% and 26% of primary tumors and metastases, respectively. Shi et al.[127] reported a lower positivity of the PD-L1 expression (14%) in 201 MTC patients at immunohistochemical staining. PD‐L1 positivity was associated with larger tumor size, lymph node metastases, higher TNM staging, structural recurrence, biochemical recurrence/persistent disease and a lower 5-year structural recurrence‐free survival rate (57.9% vs. 85.4%). In a subsequent study[128], the same authors explored the expression of multiple immune checkpoints in MTC. They found that TIM-3 (T-cell immunoglobulin and mucin-domain containing-3) was the most expressed (48%), and, in most of the cases, it was expressed exclusively on tumor cells and correlated with older age at diagnosis and advanced staging. PD-1 positivity was observed in 13.5% of cases, and about half (44.4%) of them had concurrent PD-L1 expression; CTLA-4 was present in 12.5%, while LAG-3 (lymphocyte activation gene-3) and TIGIT (T cell immunoglobulin and ITIM domain) were positive in 3% of patients. Moreover, the expression of these molecules correlated with clinical outcome: the rate of structural recurrence was significantly higher in positive patients for TIM-3, CTLA-4, and PD-1/PD-L1, and 80% of 20 patients who developed advanced disease during the follow-up period had single or multiple immune checkpoint expressions.
Recent evidence shows that MTC harbored a greater mutational load, with a potentially relevant immunogenicity[125,127,128]. In addition, the presence of RET-mutated cells could induce changes in the tumor microenvironment, influencing the surrounding stroma, activating cancer-associated fibroblasts, promoting cancer-associated inflammation, and suppressing anti-cancer immune response[129]. These findings, showing the expression of single or multiple immune checkpoints in MTC tumor and immune cells, could be hypothesized as a potential target for ICI.
Thus far, a phase II clinical trial (ClinicalTrials.gov, number NCT03072160) to evaluate the efficacy of pembrolizumab in advanced MTC has been planned. Two arms were designed, one with a previous immune stimulatory vaccine and the other without. The enrollment was completed, but to date no results are available.
Clinical point of view
To date, no clinical results on the effects of pembrolizumab on MTC patients are available. However, immunotherapy is at the beginning of its clinical application in several tumors and the promising results should be a stimulus to continue the research also for thyroid cancers. However, at the moment, no clinical application, either in clinical trials or as a compassionate or off-label use, is possible.
PEPTIDE RECEPTOR RADIONUCLIDE THERAPY
Rationale of treatment
MTC cells, similar to other neuroendocrine tumors, can express somatostatin receptors (SSTRs)[130-132]. For this reason, diagnostic tools based on radionuclides with high affinity for SSTRs, such as octreoscan[133] and 68Ga-DOTATATE or DOTATOC PET/CT[134], could be performed. The presence of SSTRs is the rationale for peptide receptor radionuclide therapy (PRRT) with radiolabeled somatostatin analogs (SSAs). Several radiolabeled SSAs have been explored as potential therapeutic agents. Most of the studies about PRRT in MTC investigated the use of SSAs labeled with the radionuclide(s) 90 yttrium (90Y) and/or 177 lutetium (177Lu)[135-143]. In addition, the use of 111 indium (111In) has been evaluated[144-146].
Several of these studies showed a decrease ≥ 50% in post treatment CT values, compared to baseline, in 24%-30% of patients treated with 90Y[137] and 177Lu[135]. Another study showed a prolongation of CT doubling time (≥ 100%) in 18/31 (58%) patients[147], indicating a relevant reduction of the tumor growth rate. However, most of these studies, although with a limited number of patients, showed that a stabilization of the disease (43%-100%) was the main clinical response obtained after PRRT treatments with Y90 or 177Lu[136,140,142,148]. These results were confirmed in studies with greater numbers of patients in which stable disease accounted for 57%-58% of cases[135,137]. Few and scattered data are available on the outcome of these MTC patients, and Parghane et al.[137] reported a median OS of 26 months in 43 patients treated with 177Lu‐DOTATATE PRRT.
Since, in MTC cells, cholecystokinin 2 receptor (CCK2R) could be expressed, this receptor may represent a potential target for PRRT[149], but this possibility has to be better explored. The possibility to use PRRT in MTC seems to be another possible therapeutic way, however current experiences are very limited. More reliable data could derive from several ongoing clinical trials (ClinicalTrials.gov, numbers NCT00002947, NCT03647657, NCT02088645, and NCT04106843).
Clinical point of view
The knowledge about the efficacy and safety of the PRRT on MTC is still under construction. However, reported data are in favor of using PRRT in specific cases such as those patients with metastatic advanced MTC who experienced no benefit with other therapies and express SSTr as assessed with a diagnostic tool exploring the presence of these receptors.
CONCLUSION
MTC is a rare disease with a high risk of not being cured by the initial treatment. Nowadays, advanced and progressive MTC can be treated with at least two different types of TKI, vandetanib and cabozantinib. The choice is highly dependent on the availability of the drug; the location of the metastases; the need to have a more or less rapid response; the patient clinical features, such as the presence of diverticulitis or congenital QTc prolongation; and the experience of the medical team. In the near future, a second generation of TKI, which are much more selective for RET mutations and almost harmless, will be available in clinical practice, while other even more selective and mutation specific drugs are under development. The limit of these last generation drugs is that they cannot be used in RET-negative cases, which represent almost 40% of cases.
Immunotherapy, although promising, is still unavailable for MTC patients, while some therapeutic space is possible for radiometabolic therapy with radiolabeled analogs of somatostatin in those cases that are positive for the corresponding receptor, especially if no other therapeutic options are possible.
DECLARATIONS
Authors’ contributionsConceptualization: Matrone A, Elisei R
Methodology: Matrone A, Gambale C, Prete A, Elisei R
Original draft preparation: Matrone A, Gambale C, Prete A
Review and editing: Cappagli V, Lorusso L, Bottici V
Final editing: Matrone A, Elisei R
Supervision: Elisei R
All authors have read and agreed to the published version of the manuscript.
Availability of data and materialsNot applicable.
Financial support and sponsorshipThis study has been supported by grants to R.E. from Associazione Italiana per la Ricerca sul Cancro (AIRC, Investigator grant 2018, project code 21790. Title: NEW INSIGHTS IN THE GENETIC PROFILE OF MEDULLARY THYROID CARCINOMA), Agenzia Italiana del Farmaco (AIFA, project code AIFA-2016- 02365049. Title: Circulating microRNAs and DNA (cfDNA) as novel biomarkers for diagnostic, prognostic and therapeutic use in Medullary Thyroid Carcinoma).
Conflict of interestElisei R is consultant for Eisai, Loxo, Ipsen, Lilly, but the content of this paper was not influenced by this activity.
All the other authors declared that there have no conflicts of interest.
Ethical approval and consent to participateNot applicable.
Consent for publicationNot applicable.
Copyright©The Author(s) 2021.
REFERENCES
1. Melvin KE, Tashjian AH Jr. The syndrome of excessive thyrocalcitonin produced by medullary carcinoma of the thyroid. Proc Natl Acad Sci U S A 1968;59:1216-22.
2. Hazard JB, Hawk WA, Crile G Jr. Medullary (solid) carcinoma of the thyroid; a clinicopathologic entity. J Clin Endocrinol Metab 1959;19:152-61.
3. Pacini F, Basolo F, Elisei R, Fugazzola L, Cola A, Pinchera A. Medullary thyroid cancer. An immunohistochemical and humoral study using six separate antigens. Am J Clin Pathol 1991;95:300-8.
4. Gmunder-Lehner RB, Okamoto E, Hedinger C. Distribution of C cells in the human thyroid gland. Schweiz Med Wochenschr 1983;113:1385-94.
5. Available from: https://rarediseases.info.nih.gov/diseases/7004/thyroid-cancer-medullary. [Last accessed on 20 Apr 2021].
6. Bergholm U, Adami HO, Telenius-Berg M, Johansson H, Wilander E. Incidence of sporadic and familial medullary thyroid carcinoma in Sweden 1959 through 1981. A nationwide study in 126 patients. Swedish MCT Study Group. Acta Oncol 1990;29:9-15.
7. Bhattacharyya N. A population-based analysis of survival factors in differentiated and medullary thyroid carcinoma. Otolaryngol Head Neck Surg 2003;128:115-23.
8. Bondeson L, Ljungberg O. Occult thyroid carcinoma at autopsy in Malmö, Sweden. Cancer 1981;47:319-23.
9. Christensen SB, Ljungberg O, Tibblin S. A clinical epidemiologic study of thyroid carcinoma in Malmö, Sweden. Curr Probl Cancer 1984;8:1-49.
10. Romei C, Pardi E, Cetani F, Elisei R. Genetic and clinical features of multiple endocrine neoplasia types 1 and 2. J Oncol 2012;2012:705036.
11. Elisei R, Matrone A, Valerio L, et al. Fifty years after the first description, MEN 2B syndrome diagnosis is still late: descriptions of two recent cases. J Clin Endocrinol Metab 2019;104:2520-6.
12. Sanso GE, Domene HM, Garcia R, et al. Very early detection of RET proto-oncogene mutation is crucial for preventive thyroidectomy in multiple endocrine neoplasia type 2 children: presence of C-cell malignant disease in asymptomatic carriers. Cancer 2002;94:323-30.
13. Niccoli-Sire P, Murat A, Baudin E, et al. Early or prophylactic thyroidectomy in MEN 2/FMTC gene carriers: results in 71 thyroidectomized patients. The French Calcitonin Tumours Study Group (GETC). Eur J Endocrinol 1999;141:468-74.
14. Machens A, Niccoli-Sire P, Hoegel J, et al. Early malignant progression of hereditary medullary thyroid cancer. N Engl J Med 2003;349:1517-25.
15. Elisei R, Tacito A, Ramone T, et al. Twenty-five years experience on RET genetic screening on hereditary MTC: an update on the prevalence of germline RET mutations. Genes (Basel) 2019;10:698.
16. Donis-Keller H, Dou S, Chi D, et al. Mutations in the RET proto-oncogene are associated with MEN 2A and FMTC. Hum Mol Genet 1993;2:851-6.
17. Mulligan LM, Marsh DJ, Robinson BG, et al. Genotype-phenotype correlation in multiple endocrine neoplasia type 2: report of the International RET Mutation Consortium. J Intern Med 1995;238:343-6.
18. Mulligan LM, Kwok JB, Healey CS, et al. Germ-line mutations of the RET proto-oncogene in multiple endocrine neoplasia type 2A. Nature 1993;363:458-60.
19. Romei C, Ciampi R, Elisei R. A comprehensive overview of the role of the RET proto-oncogene in thyroid carcinoma. Nat Rev Endocrinol 2016;12:192-202.
20. Ciampi R, Romei C, Ramone T, et al. Genetic landscape of somatic mutations in a large cohort of sporadic medullary thyroid carcinomas studied by next-generation targeted sequencing. iScience 2019;20:324-36.
21. Elisei R, Cosci B, Romei C, et al. Prognostic significance of somatic RET oncogene mutations in sporadic medullary thyroid cancer: a 10-year follow-up study. J Clin Endocrinol Metab 2008;93:682-7.
22. Molinaro E, Romei C, Biagini A, et al. Anaplastic thyroid carcinoma: from clinicopathology to genetics and advanced therapies. Nat Rev Endocrinol 2017;13:644-60.
23. Christensen SB, Ljungberg O. Mortality from thyroid carcinoma in malmö, Sweden 1960–1977. A clinical and pathologic study of 38 fatal cases. Cancer 1984;54:1629-34.
24. Kwon H, Kim WG, Sung TY, et al. Changing trends in the clinicopathological features and clinical outcomes of medullary thyroid carcinoma. J Surg Oncol 2016;113:152-8.
25. Torresan F, Mian C, Cavedon E, Iacobone M. Cure and survival of sporadic medullary thyroid carcinoma following systematic preoperative calcitonin screening. Langenbecks Arch Surg 2019;404:411-9.
26. Torresan F, Cavedon E, Mian C, Iacobone M. Long-term outcome after surgery for medullary thyroid carcinoma: a single-center experience. World J Surg 2018;42:367-75.
27. Brierley J, Tsang R, Simpson WJ, Gospodarowicz M, Sutcliffe S, Panzarella T. Medullary thyroid cancer: analyses of survival and prognostic factors and the role of radiation therapy in local control. Thyroid 1996;6:305-10.
28. Mathiesen JS, Kroustrup JP, Vestergaard P, et al. Survival and long-term biochemical cure in medullary thyroid carcinoma in denmark 1997-2014: a nationwide study. Thyroid 2019;29:368-77.
29. Gharib H, Mcconahey WM, Tiegs RD, et al. Medullary thyroid carcinoma: clinicopathologic features and long-term follow-up of 65 patients treated during 1946 through 1970. Mayo Clinic Proceedings 1992;67:934-40.
30. Matrone A, Gambale C, Prete A, et al. Impact of advanced age on the clinical presentation and outcome of sporadic medullary thyroid carcinoma. Cancers (Basel) 2020;13:94.
31. Kebebew E, Ituarte PHG, Siperstein AE, Duh Q, Clark OH. Medullary thyroid carcinoma: clinical characteristics, treatment, prognostic factors, and a comparison of staging systems. Cancer 2000;88:1139-48.
32. Moley JF. Medullary thyroid carcinoma: management of lymph node metastases. J Natl Compr Canc Netw 2010;8:549-56.
33. Romei C, Mariotti S, Fugazzola L, et al. Multiple endocrine neoplasia type 2 syndromes (MEN 2): results from the ItaMEN network analysis on the prevalence of different genotypes and phenotypes. Eur J Endocrinol 2010;163:301-8.
34. Elisei R. Routine serum calcitonin measurement in the evaluation of thyroid nodules. Best Pract Res Clin Endocrinol Metab 2008;22:941-53.
35. Wells SA Jr, Asa SL, Dralle H, et al. Revised american thyroid association guidelines for the management of medullary thyroid carcinoma. Thyroid 2015;25:567-610.
36. Valerio L, Pieruzzi L, Giani C, et al. Targeted therapy in thyroid cancer: state of the art. Clin Oncol (R Coll Radiol) 2017;29:316-24.
37. Viola D, Elisei R. Management of medullary thyroid cancer. Endocrinol Metab Clin North Am 2019;48:285-301.
40. Bible KC, Ryder M. Evolving molecularly targeted therapies for advanced-stage thyroid cancers. Nat Rev Clin Oncol 2016;13:403-16.
41. Druker BJ, Talpaz M, Resta DJ, et al. Efficacy and safety of a specific inhibitor of the BCR-ABL tyrosine kinase in chronic myeloid leukemia. N Engl J Med 2001;344:1031-7.
42. Tacconi EMC, Tuthill M, Protheroe A. Review of adjuvant therapies in renal cell carcinoma: evidence to date. Onco Targets Ther 2020;13:12301-16.
43. Llovet JM, Kelley RK, Villanueva A, et al. Hepatocellular carcinoma. Nat Rev Dis Primers 2021;7:6.
44. Matrone A, Valerio L, Pieruzzi L, et al. Protein kinase inhibitors for the treatment of advanced and progressive radiorefractory thyroid tumors: from the clinical trials to the real life. Best Pract Res Clin Endocrinol Metab 2017;31:319-34.
45. Brose MS, Nutting CM, Jarzab B, et al. Sorafenib in radioactive iodine-refractory, locally advanced or metastatic differentiated thyroid cancer: a randomised, double-blind, phase 3 trial. Lancet 2014;384:319-28.
46. Schlumberger M, Tahara M, Wirth LJ, et al. Lenvatinib versus placebo in radioiodine-refractory thyroid cancer. N Engl J Med 2015;372:621-30.
47. Gotink KJ, Verheul HM. Anti-angiogenic tyrosine kinase inhibitors: what is their mechanism of action? Angiogenesis 2010;13:1-14.
48. Posner I, Engel M, Gazit A, Levitzki A. Kinetics of inhibition by tyrphostins of the tyrosine kinase activity of the epidermal growth factor receptor and analysis by a new computer program. Mol Pharmacol 1994;45:673-83.
49. Viola D, Valerio L, Molinaro E, et al. Treatment of advanced thyroid cancer with targeted therapies: ten years of experience. Endocr Relat Cancer 2016;23:R185-205.
50. Fagin JA, Wells SA Jr. Biologic and clinical perspectives on thyroid cancer. N Engl J Med 2016;375:1054-67.
51. Eng C, Smith DP, Mulligan LM, et al. Point mutation within the tyrosine kinase domain of the RET proto-oncogene in multiple endocrine neoplasia type 2B and related sporadic tumours. Hum Mol Genet 1994;3:237-41.
52. Krampitz GW, Norton JA. RET gene mutations (genotype and phenotype) of multiple endocrine neoplasia type 2 and familial medullary thyroid carcinoma. Cancer 2014;120:1920-31.
53. Wells SA Jr, Pacini F, Robinson BG, Santoro M. Multiple endocrine neoplasia type 2 and familial medullary thyroid carcinoma: an update. J Clin Endocrinol Metab 2013;98:3149-64.
54. Raue F, Bruckner T, Frank-Raue K. Long-term outcomes and aggressiveness of hereditary medullary thyroid carcinoma: 40 years of experience at one center. J Clin Endocrinol Metab 2019;104:4264-72.
55. Romei C, Casella F, Tacito A, et al. New insights in the molecular signature of advanced medullary thyroid cancer: evidence of a bad outcome of cases with double RET mutations. J Med Genet 2016;53:729-34.
56. Agrawal N, Jiao Y, Sausen M, et al. Exomic sequencing of medullary thyroid cancer reveals dominant and mutually exclusive oncogenic mutations in RET and RAS. J Clin Endocrinol Metab 2013;98:E364-9.
57. de Groot JW, Zonnenberg BA, van Ufford-Mannesse PQ, et al. A phase II trial of imatinib therapy for metastatic medullary thyroid carcinoma. J Clin Endocrinol Metab 2007;92:3466-9.
58. Cohen EE, Rosen LS, Vokes EE, et al. Axitinib is an active treatment for all histologic subtypes of advanced thyroid cancer: results from a phase II study. J Clin Oncol 2008;26:4708-13.
59. Schlumberger MJ, Elisei R, Bastholt L, et al. Phase II study of safety and efficacy of motesanib in patients with progressive or symptomatic, advanced or metastatic medullary thyroid cancer. J Clin Oncol 2009;27:3794-801.
60. Lam ET, Ringel MD, Kloos RT, et al. Phase II clinical trial of sorafenib in metastatic medullary thyroid cancer. J Clin Oncol 2010;28:2323-30.
61. Carr LL, Mankoff DA, Goulart BH, et al. Phase II study of daily sunitinib in FDG-PET-positive, iodine-refractory differentiated thyroid cancer and metastatic medullary carcinoma of the thyroid with functional imaging correlation. Clin Cancer Res 2010;16:5260-8.
62. Bible KC, Suman VJ, Molina JR, et al. A multicenter phase 2 trial of pazopanib in metastatic and progressive medullary thyroid carcinoma: MC057H. J Clin Endocrinol Metab 2014;99:1687-93.
63. De Falco V, Buonocore P, Muthu M, et al. Ponatinib (AP24534) is a novel potent inhibitor of oncogenic RET mutants associated with thyroid cancer. J Clin Endocrinol Metab 2013;98:E811-9.
64. Schlumberger M, Jarzab B, Cabanillas ME, et al. A phase II trial of the multitargeted tyrosine kinase inhibitor lenvatinib (E7080) in advanced medullary thyroid cancer. Clin Cancer Res 2016;22:44-53.
65. Sun Y, Du F, Gao M, et al. Anlotinib for the treatment of patients with locally advanced or metastatic medullary thyroid cancer. Thyroid 2018;28:1455-61.
66. Wells SA Jr, Robinson BG, Gagel RF, et al. Vandetanib in patients with locally advanced or metastatic medullary thyroid cancer: a randomized, double-blind phase III trial. J Clin Oncol 2012;30:134-41.
67. Elisei R, Schlumberger MJ, Müller SP, et al. Cabozantinib in progressive medullary thyroid cancer. J Clin Oncol 2013;31:3639-46.
68. Wedge SR, Ogilvie DJ, Dukes M, et al. ZD6474 inhibits vascular endothelial growth factor signaling, angiogenesis, and tumor growth following oral administration. Cancer Res 2002;62:4645-55.
69. Carlomagno F, Vitagliano D, Guida T, et al. ZD6474, an orally available inhibitor of KDR tyrosine kinase activity, efficiently blocks oncogenic RET kinases. Cancer Res 2002;62:7284-90.
70. Kreissl MC, Bastholt L, Elisei R, et al. Efficacy and safety of vandetanib in progressive and symptomatic medullary thyroid cancer: post hoc analysis from the ZETA trial. J Clin Oncol 2020;38:2773-81.
71. Massicotte MH, Brassard M, Claude-Desroches M, et al. Tyrosine kinase inhibitor treatments in patients with metastatic thyroid carcinomas: a retrospective study of the TUTHYREF network. Eur J Endocrinol 2014;170:575-82.
72. Chougnet CN, Borget I, Leboulleux S, et al. Vandetanib for the treatment of advanced medullary thyroid cancer outside a clinical trial: results from a French cohort. Thyroid 2015;25:386-91.
73. Ramos HE, Hecht F, Berdelou A, et al. Long-term follow-up and safety of vandetanib for advanced medullary thyroid cancer. Endocrine 2021;71:434-42.
74. Valerio L, Bottici V, Matrone A, et al. Medullary thyroid cancer treated with vandetanib: predictors of a longer and durable response. Endocr Relat Cancer 2020;27:97-110.
75. Trotti A, Colevas AD, Setser A, Basch E. Patient-reported outcomes and the evolution of adverse event reporting in oncology. J Clin Oncol 2007;25:5121-7.
76. Wirth LJ, Sherman E, Robinson B, et al. Efficacy of Selpercatinib in RET-altered thyroid cancers. N Engl J Med 2020;383:825-35.
77. Hu M, Subbiah M, Wirth LJ, et al. Results from the registrational phase I/II ARROW trial of pralsetinib (BLU-667) in patients (pts) with advanced RET mutation-positive medullary thyroid cancer (RET+ MTC). Ann Oncol 2020;31:S1084.
78. Hu MI, Elisei R, Dedecjus M, et al. Safety and efficacy of two starting doses of vandetanib in advanced medullary thyroid cancer. Endocr Relat Cancer 2019;26:241-50.
79. Fox E, Widemann BC, Chuk MK, et al. Vandetanib in children and adolescents with multiple endocrine neoplasia type 2B associated medullary thyroid carcinoma. Clin Cancer Res 2013;19:4239-48.
80. Paepegaey AC, Cochand-Priollet B, Louiset E, et al. Long-term control of hypercortisolism by vandetanib in a case of medullary thyroid carcinoma with a somatic RET mutation. Thyroid 2017;27:587-90.
81. Pitoia F, Bueno F, Schmidt A, Lucas S, Cross G. Rapid response of hypercortisolism to vandetanib treatment in a patient with advanced medullary thyroid cancer and ectopic Cushing syndrome. Arch Endocrinol Metab 2015;59:343-6.
82. Nella AA, Lodish MB, Fox E, et al. Vandetanib successfully controls medullary thyroid cancer-related Cushing syndrome in an adolescent patient. J Clin Endocrinol Metab 2014;99:3055-9.
83. Baudry C, Paepegaey AC, Groussin L. Reversal of Cushing's syndrome by vandetanib in medullary thyroid carcinoma. N Engl J Med 2013;369:584-6.
84. Cabanillas ME, Ryder M, Jimenez C. Targeted therapy for advanced thyroid cancer: kinase inhibitors and beyond. Endocr Rev 2019;40:1573-604.
85. Kurzrock R, Sherman SI, Ball DW, et al. Activity of XL184 (Cabozantinib), an oral tyrosine kinase inhibitor, in patients with medullary thyroid cancer. J Clin Oncol 2011;29:2660-6.
86. Sherman SI, Clary DO, Elisei R, et al. Correlative analyses of RET and RAS mutations in a phase 3 trial of cabozantinib in patients with progressive, metastatic medullary thyroid cancer. Cancer 2016;122:3856-64.
87. Schlumberger M, Elisei R, Müller S, et al. Overall survival analysis of EXAM, a phase III trial of cabozantinib in patients with radiographically progressive medullary thyroid carcinoma. Ann Oncol 2017;28:2813-9.
88. Eisenhauer EA, Therasse P, Bogaerts J, et al. New response evaluation criteria in solid tumours: revised RECIST guideline (version 1.1). Eur J Cancer 2009;45:228-47.
89. Massicotte MH, Borget I, Broutin S, et al. Body composition variation and impact of low skeletal muscle mass in patients with advanced medullary thyroid carcinoma treated with vandetanib: results from a placebo-controlled study. J Clin Endocrinol Metab 2013;98:2401-8.
90. Cabanillas ME, Hu MI, Jimenez C. Medullary thyroid cancer in the era of tyrosine kinase inhibitors: to treat or not to treat--and with which drug--those are the questions. J Clin Endocrinol Metab 2014;99:4390-6.
91. Brose MS, Bible KC, Chow LQM, et al. Management of treatment-related toxicities in advanced medullary thyroid cancer. Cancer Treat Rev 2018;66:64-73.
92. Cappagli V, Moriconi D, Bonadio AG, et al. Proteinuria is a late-onset adverse event in patients treated with cabozantinib. J Endocrinol Invest 2021;44:95-103.
93. Basolo A, Matrone A, Elisei R, Santini F. Effects of tyrosine kinase inhibitors on thyroid function and thyroid hormone metabolism. Semin Cancer Biol 2021:S1044-579X(20)30268.
94. Blevins DP, Dadu R, Hu M, et al. Aerodigestive fistula formation as a rare side effect of antiangiogenic tyrosine kinase inhibitor therapy for thyroid cancer. Thyroid 2014;24:918-22.
95. Carlomagno F, Guida T, Anaganti S, et al. Disease associated mutations at valine 804 in the RET receptor tyrosine kinase confer resistance to selective kinase inhibitors. Oncogene 2004;23:6056-63.
96. Arao T, Matsumoto K, Furuta K, et al. Acquired drug resistance to vascular endothelial growth factor receptor 2 tyrosine kinase inhibitor in human vascular endothelial cells. Anticancer Res 2011;31:2787-96.
97. Liu X, Shen T, Mooers BHM, Hilberg F, Wu J. Drug resistance profiles of mutations in the RET kinase domain. Br J Pharmacol 2018;175:3504-15.
98. Tate JG, Bamford S, Jubb HC, et al. COSMIC: the catalogue of somatic mutations in cancer. Nucleic Acids Res 2019;47:D941-7.
99. Moura MM, Cavaco BM, Pinto AE, et al. Correlation of RET somatic mutations with clinicopathological features in sporadic medullary thyroid carcinomas. Br J Cancer 2009;100:1777-83.
100. Kohno T, Tabata J, Nakaoku T. REToma: a cancer subtype with a shared driver oncogene. Carcinogenesis 2020;41:123-9.
101. Subbiah V, Velcheti V, Tuch BB, et al. Selective RET kinase inhibition for patients with RET-altered cancers. Ann Oncol 2018;29:1869-76.
102. Subbiah V, Gainor JF, Rahal R, et al. Precision targeted therapy with BLU-667 for RET-driven cancers. Cancer Discov 2018;8:836-49.
103. Seoane J, Capdevila J. The right compound for the right target: tackling RET. Ann Oncol 2018;29:1623-5.
104. Solomon BJ, Tan L, Lin JJ, et al. RET solvent front mutations mediate acquired resistance to selective RET inhibition in RET-driven malignancies. J Thorac Oncol 2020;15:541-9.
105. Subbiah V, Shen T, Terzyan SS, et al. Structural basis of acquired resistance to selpercatinib and pralsetinib mediated by non-gatekeeper RET mutations. Ann Oncol 2021;32:261-8.
106. Lin JJ, Liu SV, McCoach CE, et al. Mechanisms of resistance to selective RET tyrosine kinase inhibitors in RET fusion-positive non-small-cell lung cancer. Ann Oncol 2020;31:1725-33.
107. Rosen EY, Johnson ML, Clifford SE, et al. Overcoming MET-dependent resistance to selective RET inhibition in patients with RET fusion-positive lung cancer by combining selpercatinib with crizotinib. Clin Cancer Res 2021;27:34-42.
108. Drilon AE, Zhai D, Rogers E, et al. The next-generation RET inhibitor TPX-0046 is active in drug-resistant and naïve RET-driven cancer models. J Clin Oncol 2020;38:3616.
109. Egeblad M, Rasch MG, Weaver VM. Dynamic interplay between the collagen scaffold and tumor evolution. Curr Opin Cell Biol 2010;22:697-706.
110. McAllister SS, Weinberg RA. The tumour-induced systemic environment as a critical regulator of cancer progression and metastasis. Nat Cell Biol 2014;16:717-27.
111. Hanahan D, Coussens LM. Accessories to the crime: functions of cells recruited to the tumor microenvironment. Cancer Cell 2012;21:309-22.
112. Velcheti V, Schalper K. Basic overview of current immunotherapy approaches in cancer. Am Soc Clin Oncol Educ Book 2016;35:298-308.
113. Cunha LL, Marcello MA, Morari EC, et al. Differentiated thyroid carcinomas may elude the immune system by B7H1 upregulation. Endocr Relat Cancer 2013;20:103-10.
114. Bastman JJ, Serracino HS, Zhu Y, et al. Tumor-infiltrating T cells and the PD-1 checkpoint pathway in advanced differentiated and anaplastic thyroid cancer. J Clin Endocrinol Metab 2016;101:2863-73.
115. Angell TE, Lechner MG, Jang JK, Correa AJ, LoPresti JS, Epstein AL. BRAF V600E in papillary thyroid carcinoma is associated with increased programmed death ligand 1 expression and suppressive immune cell infiltration. Thyroid 2014;24:1385-93.
116. Chowdhury S, Veyhl J, Jessa F, et al. Programmed death-ligand 1 overexpression is a prognostic marker for aggressive papillary thyroid cancer and its variants. Oncotarget 2016;7:32318-28.
117. Ahn S, Kim TH, Kim SW, et al. Comprehensive screening for PD-L1 expression in thyroid cancer. Endocr Relat Cancer 2017;24:97-106.
118. Mehnert JM, Varga A, Brose MS, et al. Safety and antitumor activity of the anti-PD-1 antibody pembrolizumab in patients with advanced, PD-L1-positive papillary or follicular thyroid cancer. BMC Cancer 2019;19:196.
119. Rocklin RE, Gagel R, Feldman Z, Tashjian AH Jr. Cellular immune responses in familial medullary thyroid carcinoma. N Engl J Med 1977;296:835-8.
120. George JM, Williams MA, Almoney R, Sizemore G. Medullary carcinoma of the thyroid.cellular immune response to tumor antigen in a heritable human cancer. Cancer 1975;36:1658-61.
121. Bilusic M, Heery CR, Arlen PM, et al. Phase I trial of a recombinant yeast-CEA vaccine (GI-6207) in adults with metastatic CEA-expressing carcinoma. Cancer Immunol Immunother 2014;63:225-34.
122. Schott M, Feldkamp J, Klucken M, Kobbe G, Scherbaum WA, Seissler J. Calcitonin-specific antitumor immunity in medullary thyroid carcinoma following dendritic cell vaccination. Cancer Immunol Immunother 2002;51:663-8.
123. Bachleitner-Hofmann T, Friedl J, Hassler M, et al. Pilot trial of autologous dendritic cells loaded with tumor lysate(s) from allogeneic tumor cell lines in patients with metastatic medullary thyroid carcinoma. Oncol Rep 2009;21:1585-92.
124. Bongiovanni M, Rebecchini C, Saglietti C, et al. Very low expression of PD-L1 in medullary thyroid carcinoma. Endocr Relat Cancer 2017;24:L35-8.
125. Bi Y, Ren X, Bai X, et al. PD-1/PD-L1 expressions in medullary thyroid carcinoma: clinicopathologic and prognostic analysis of Chinese population. Eur J Surg Oncol 2019;45:353-8.
126. Pozdeyev N, Erickson TA, Zhang L, et al. Comprehensive immune profiling of medullary thyroid cancer. Thyroid 2020;30:1263-79.
127. Shi X, Li CW, Tan LC, et al. Immune co-inhibitory receptors PD-1, CTLA-4, TIM-3, LAG-3, and TIGIT in medullary thyroid cancers: a large cohort study. J Clin Endocrinol Metab 2021;106:120-32.
128. Shi X, Yu PC, Lei BW, et al. Association between programmed death-ligand 1 expression and clinicopathological characteristics, structural recurrence, and biochemical recurrence/persistent disease in medullary thyroid carcinoma. Thyroid 2019;29:1269-78.
129. Castellone MD, Melillo RM. RET-mediated modulation of tumor microenvironment and immune response in multiple endocrine neoplasia type 2 (MEN2). Endocr Relat Cancer 2018;25:T105-19.
130. Faggiano A, Grimaldi F, Pezzullo L, et al. Secretive and proliferative tumor profile helps to select the best imaging technique to identify postoperative persistent or relapsing medullary thyroid cancer. Endocr Relat Cancer 2009;16:225-31.
131. De Luca S, Fonti R, Camera L, et al. Multimodal imaging with (18)F-FDG-PET/CT and (111)In-Octreotide SPECT in patients with metastatic medullary thyroid carcinoma. Ann Nucl Med 2016;30:234-41.
132. Mato E, Matías-Guiu X, Chico A, et al. Somatostatin and somatostatin receptor subtype gene expression in medullary thyroid carcinoma. J Clin Endocrinol Metab 1998;83:2417-20.
133. Baudin E, Lumbroso J, Schlumberger M, et al. Comparison of octreotide scintigraphy and conventional imaging in medullary thyroid carcinoma. J Nucl Med 1996;37:912-6.
134. Yamaga LYI, Cunha ML, Campos Neto GC, et al. 68Ga-DOTATATE PET/CT in recurrent medullary thyroid carcinoma: a lesion-by-lesion comparison with 111In-octreotide SPECT/CT and conventional imaging. Eur J Nucl Med Mol Imaging 2017;44:1695-701.
135. Bodei L, Handkiewicz-Junak D, Grana C, et al. Receptor radionuclide therapy with 90Y-DOTATOC in patients with medullary thyroid carcinomas. Cancer Biother Radiopharm 2004;19:65-71.
136. Satapathy S, Mittal BR, Sood A, Verma R, Panda N. Efficacy and safety of concomitant 177Lu-DOTATATE and low-dose capecitabine in advanced medullary thyroid carcinoma: a single-centre experience. Nucl Med Commun 2020;41:629-35.
137. Parghane RV, Naik C, Talole S, et al. Clinical utility of 177Lu-DOTATATE PRRT in somatostatin receptor-positive metastatic medullary carcinoma of thyroid patients with assessment of efficacy, survival analysis, prognostic variables, and toxicity. Head Neck 2020;42:401-16.
138. DeGroot LJ. Management of thyroid nodules: how far have we come? Hosp Pract (Off Ed) 1986;21:9-10.
139. Lapa C, Werner RA, Schmid JS, et al. Prognostic value of positron emission tomography-assessed tumor heterogeneity in patients with thyroid cancer undergoing treatment with radiopeptide therapy. Nucl Med Biol 2015;42:349-54.
140. Vaisman F, Rosado de Castro PH, Lopes FP, et al. Is there a role for peptide receptor radionuclide therapy in medullary thyroid cancer? Clin Nucl Med 2015;40:123-7.
141. Gao Z, Biersack HJ, Ezziddin S, Logvinski T, An R. The role of combined imaging in metastatic medullary thyroid carcinoma: 111In-DTPA-octreotide and 131I/123I-MIBG as predictors for radionuclide therapy. J Cancer Res Clin Oncol 2004;130:649-56.
142. Bodei L, Cremonesi M, Zoboli S, et al. Receptor-mediated radionuclide therapy with 90Y-DOTATOC in association with amino acid infusion: a phase I study. Eur J Nucl Med Mol Imaging 2003;30:207-16.
143. Paganelli G, Zoboli S, Cremonesi M, et al. Receptor-mediated radiotherapy with 90Y-DOTA-D-Phe1-Tyr3-octreotide. Eur J Nucl Med 2001;28:426-34.
144. Pasieka JL, McEwan AJ, Rorstad O. The palliative role of 131I-MIBG and 111In-octreotide therapy in patients with metastatic progressive neuroendocrine neoplasms. Surgery 2004;136:1218-26.
145. Buscombe JR, Caplin ME, Hilson AJ. Long-term efficacy of high-activity 111in-pentetreotide therapy in patients with disseminated neuroendocrine tumors. J Nucl Med 2003;44:1-6.
146. Valkema R, De Jong M, Bakker WH, et al. Phase I study of peptide receptor radionuclide therapy with [In-DTPA] octreotide: the Rotterdam experience. Semin Nucl Med 2002;32:110-22.
147. Iten F, Müller B, Schindler C, et al. Response to [90Yttrium-DOTA]-TOC treatment is associated with long-term survival benefit in metastasized medullary thyroid cancer: a phase II clinical trial. Clin Cancer Res 2007;13:6696-702.
148. Beukhof CM, Brabander T, van Nederveen FH, et al. Peptide receptor radionuclide therapy in patients with medullary thyroid carcinoma: predictors and pitfalls. BMC Cancer 2019;19:325.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Matrone A, Gambale C, Prete A, Cappagli V, Lorusso L, Bottici V, Elisei R. Systemic treatment of advanced, metastatic, medullary thyroid carcinoma. J Cancer Metastasis Treat 2021;7:23. http://dx.doi.org/10.20517/2394-4722.2021.47
AMA Style
Matrone A, Gambale C, Prete A, Cappagli V, Lorusso L, Bottici V, Elisei R. Systemic treatment of advanced, metastatic, medullary thyroid carcinoma. Journal of Cancer Metastasis and Treatment. 2021; 7: 23. http://dx.doi.org/10.20517/2394-4722.2021.47
Chicago/Turabian Style
Matrone, Antonio, Carla Gambale, Alessandro Prete, Virginia Cappagli, Loredana Lorusso, Valeria Bottici, Rossella Elisei. 2021. "Systemic treatment of advanced, metastatic, medullary thyroid carcinoma" Journal of Cancer Metastasis and Treatment. 7: 23. http://dx.doi.org/10.20517/2394-4722.2021.47
ACS Style
Matrone, A.; Gambale C.; Prete A.; Cappagli V.; Lorusso L.; Bottici V.; Elisei R. Systemic treatment of advanced, metastatic, medullary thyroid carcinoma. J. Cancer. Metastasis. Treat. 2021, 7, 23. http://dx.doi.org/10.20517/2394-4722.2021.47
About This Article
Special Issue
Copyright

Data & Comments
Data


 Cite This Article 24 clicks
Cite This Article 24 clicks

 Like This Article 19
likes
Like This Article 19
likes






Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.