
Breast-to-brain metastasis: a focus on the pre-metastatic niche

Abstract
Metastatic disease is the cause for 90% of breast cancer mortalities. For those 10%-20% of patients whose breast cancer metastasizes to the central nervous system, the one-year survival rate is just 20%. Both histology and molecular subtype have a correlation with the site of tumor metastasis, indicating an inherent preferential aspect to metastatic colony formation. The molecular differences between breast cancers may determine the site of metastasis through priming of the premetastatic niche in that site: cell surface molecules, exosomes released from the primary tumor, and soluble factors secreted from both the primary tumor and resident cells within the premetastatic niche all contribute to altering the premetastatic niche to be more favorable for the circulating tumor cells, allowing for cell invasion and growth. Here, we review breast to brain metastasis with a focus on the premetastatic niche. We discuss the secreted factors and exosomes that prime the premetastatic niche within the brain by instigating crosstalk between the resident cells of the brain microenvironment. We report on the individual roles that microglia, astrocytes, pericytes, neurons, and endothelial cells may have in the formation and maintenance of the premetastatic niche.
Keywords
BREAST CANCER AND BREAST-TO-BRAIN METASTASIS: CLINICAL AND BIOLOGICAL BASIS
It is estimated that over 42,000 women succumb to breast cancer in the United States every year[1,2]. Past data show that 90% of breast cancer mortalities are attributed to recurrent and metastatic disease[3,4]. Though some patients can develop metastases within 3 years of detection of a primary tumor, it is also common for metastases to occur more than 10 years after the initial diagnosis[3,5]. Among metastatic sites, the central nervous system (CNS) carries a grim prognosis: for breast-to-brain metastases the one-year survival rate is just 20%[6] [Table 1].
Sites of metastasis of primary breast cancer
| Study | Subtype | Location of single metastasis | % of sample | Overall survival (months) |
| Niwinska et al.[7] | Luminal/HER2- | Brain | 15 (0.2-56)* | |
| HER2+ | Brain | 9 (0.6-3.4)* | ||
| TNBC | Brain | 3.7 (0.5-52)* | ||
| Chen et al.[8] | Bone | 35.60% | 31 | |
| Brain | 1.70% | 11# | ||
| Liver | 6.40% | 19 | ||
| Lung | 9.30% | 20 | ||
| Wu et al.[9] | Luminal A | Bone | 58.52% | 14.66 ± 12.83 |
| Brain | 4.30% | |||
| Liver | 15.48% | |||
| Lung | 21.70% | |||
| Luminal B | Bone | 47.28% | 9.62 ± 10.96 | |
| Brain | 5.89% | |||
| Liver | 25.65% | |||
| Lung | 21.17% | |||
| HER2+ | Bone | 34.49% | 11.30 ± 11.71 | |
| Brain | 8.31% | |||
| Liver | 31.72% | |||
| Lung | 25.48% | |||
| TNBC | Bone | 36.39% | 12.48 ± 12.23 | |
| Brain | 9.12% | |||
| Liver | 22.40% | |||
| Lung | 32.09% | |||
| Gong et al.[10] | Luminal A | Bone | 797% | 36 |
| Brain | 1.2% | |||
| Liver | 8.1% | |||
| Lung | 11.0% | |||
| Luminal B | Bone | 61.0% | 44 | |
| Brain | 1.6% | |||
| Liver | 20.3% | |||
| Lung | 17.1% | |||
| HER2+ | Bone | 35.8% | 34 | |
| Brain | 3.4% | |||
| Liver | 32.7% | |||
| Lung | 28.1% | |||
| TNBC | Bone | 43.0% | 13 | |
| Brain | 5.1% | |||
| Liver | 18.9% | |||
| Lung | 33.1% | |||
| Gu et al.[11] | TNBC | Bone | 40% | 13 |
| Brain | 6% | 5 | ||
| Liver | 16% | 13 | ||
| Lung | 37% | 14 |
Metastasis is the process by which tumor cells invade the stromal tissue surrounding the primary tumor, enter vasculature, and disseminate throughout the body[12]. The circulating tumor cells (CTCs) then arrest in the capillaries of distant organs, and either persist as quiescent cells or proliferate and form micrometastases[13,14]. The phases of this metastatic process are Dissemination, Dormancy, and Colonization and Outgrowth at the distal site[15].
DISSEMINATION
Dissemination can start very early during primary tumor progression and can continue until the primary tumor is removed. As cancer cells undergo epithelial-to-mesenchymal transition (EMT), a combination of expression changes of cell adhesion, migratory and invasive genes of the cancer cells occur that lead to the remodeling of the ECM, and intravasation through a leaky vasculature or lymphatics[15]. The contribution of the lymphatic system in metastasis is debated, since clinical trials have shown that breast or ovarian cancer patients who had undergone regional lymph node dissection did not exhibit any survival benefit over control patients[16]; however, it has been proposed that the lymphatic system may contribute to the CTCs from the blood stream or the lymphatic system extravasate at the distal site, and then enter the phase of Dormancy during which the cancer cells at the distal site do not yet form a clinically defined lesion because the host environment presents barriers, physical, metabolic, and immune, which hinder tumor growth. Finally at the host tissue the metastatic process enters the Colonization and then Outgrowth phase. The most common sites of metastasis can change depending on breast cancer subtypes[9].
There are numerous risk factors that increase the likelihood of metastasis in breast cancer patients, such as breast cancer molecular subtypes, patient age, number of metastatic sites, and tumor size[17-22].
The overall likelihood of metastasis depends upon molecular subtypes. Currently four major molecular subtypes of breast cancer have been characterized, depending on the expression of the estrogen receptor (ER), progesterone receptor (PR) and of human epidermal growth factor receptor 2 ERBB2/HER2: Luminal A (described as ER+, PR±, HER2- with low Ki67 expression), Luminal B (ER+, PR±, HER2± with high expression of the proliferation marker Ki67), HER2+ (ER-, PR-, HER2+) and Triple Negative (TNBC: ER-, PR-, HER2-). In breast cancers with hormone receptor expression, concerted Ki67 expression increases the chance of metastasis from 27.8% to 42.9% whereas concerted ERBB2/HER2 expression increases the chance of metastasis to 47.9%. Sole expression of ERBB2/HER2 results in a 51.4% chance of metastasis and an absence of receptor expression yields a 43.1% chance[23]. Independent of molecular subtype, breast cancer most commonly metastasizes to bone. The frequency of metastasis to distant sites, however, is dependent on subtype. Breast cancers with hormone receptor expression are the most frequent of all subtypes to form distant metastases in bone; with the additional expression of ERBB2, the second most frequent metastatic site switches from the lungs to liver. With sole ERBB2/HER2 expression, the likelihood of forming liver metastases is similar to that of bone metastases. TNBC drives both bone and lung metastases at similar rates[9]. Though each subtype has the propensity to form brain metastases, ERBB2/HER2-positive and TNBC are the most common subtypes to drive metastases to the brain. Of the two, TNBC has been reported to have a higher propensity to metastasize solely to the brain[7,24,25]. Luminal A breast cancers are more likely to metastasize to bone and less likely metastasize to brain, liver, and lung, and are less likely to present as multiple metastases[26]. Both ERBB2/HER2-positive and TNBC subtypes are also associated with the lowest median survival with brain metastases at 10 and 4 months. Patients with concurrent hormone receptor and ERBB2 expression have a median survival of thirteen months[7].
Patient age is linked to location and number of metastatic sites. Younger patients are more likely to form single metastases in the brain or liver than in other organs[25]. None the less, those who present with multiple metastatic sites also tend to be younger or middle-aged[8,25]. The increased incidence of metastases in younger patients may be in part due to the higher rate of diagnosis of TNBC in younger patients and the difficulties that surround its treatment[8]. Elderly patients (> 69 years) are more likely to have metastases in the lungs and less likely to solely have distant lymphatic metastases[8,25]. Among all patients with metastatic disease, age was linked to overall survival, with the median overall survival rates for younger and elder groups decreasing from 32 to 16 months[25].
The likelihood of developing metastases increases with the presence of cancer cells in the lymph nodes, making it a commonly used prognostic marker[27,28]. The presence of lymph node metastases in patients with ERBB2-overexpressing breast cancer is associated with worse prognosis than those without lymph metastases[29]. However, its use as a marker is not absolute, as approximately one third of patients without lymph-node metastases will still develop distant metastases, while approximately one third of patients with nodal metastases will not form distant metastases 10 years after therapy[30,31]. Though the number of nodal metastases is often correlated with the primary tumor size, the two act as independent but additive indicators of disease state. With an increase in lymph involvement, regardless of tumor size, patient survival decreases[27].
Tumor size has long been considered a prognostic marker for metastasis, with primary tumors larger than
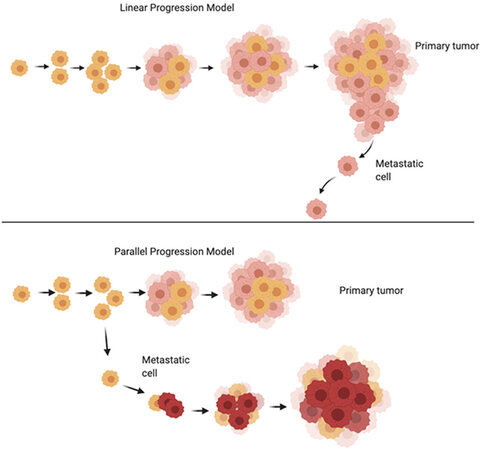
Figure 1. Linear Progression model vs. Parallel Progression model of cancer growth (adapted from[44]). Please see description of the models in the text. Image made using BioRender.
It is notable that the molecular signatures of primary tumors are not always conserved in the resulting metastases[41,42]. Though studies on the topic are limited, it has been shown that some colorectal tumors metastasizing to the liver and some breast tumors metastasizing to the brain have differing subtype expressions or molecular classifications. Priedigkeit et al.[42] performed a study with 20 cases of primary breast tumor and resulting brain metastases. Of the 20, 17 samples harbored transcriptional changes in the genes expressed in the brain metastasis samples. The most common change was reported to be increased expression of ERBB2/HER2 (n = 7), fibroblast growth factor receptor 4 (FGFR4, n = 6), Fms related receptor tyrosine kinase 1 (FLT1, n = 4), and aurora kinase A (AURKA, n = 2) as well as loss of estrogen receptor 1 expression (ESR1, n = 9). Of the seven samples with increased ERBB2, three were classified as ERBB2/HER2 negative in the original tumor, showing a shift in molecular subtype[42]. An additional study from
Here, we focus on breast cancer metastasis to the central nervous system. This affects 10%-30% of patients with metastatic disease and is most frequently not the first site of metastasis. Brain metastases are associated with a marked decrease in patients’ quality of life and have the worst prognosis in terms of patient survival with the average duration of survival after diagnosis of brain metastases ranging from 6-16 months[3,23,24,44]. Brain metastases are most common in TNBC and ERBB2 expressing breast cancers and are most likely to be lethal in patients with TNBC[23-25,44,45]. Younger patients and those diagnosed with an increased tumor stage, detection of lymph node metastases, greater than 2 metastatic sites other than the brain, or large tumor diameter are more likely to form brain metastases[17-22,46]. A study performed using patient data from the Metropolitan Detroit Cancer Surveillance System revealed that African American women were more likely to form distant metastases in the brain[47]. However, a study using patient data from the SEER database showed that patient race or ethnicity did not appear to affect the likelihood of forming brain metastases[46]. Another study that included 204,941 patients reported that as expected the TNBC subtype was associated with higher likelihood of de novo metastases occurring in the brain in both non-Hispanic-Black (n = 36) and non-Hispanic-White patients (n = 92), with higher percentages among non-Hispanic-Black patients (38.9 vs. 26.1). Surprisingly, the survival of those patients was on average 8 months for the non-Hispanic-Black patients and 6 for the non-Hispanic-White patients[48].
DORMANCY
The phase of dormancy is very difficult to model in an experimental setting, as the majority of carcinoma cells selected are metastatically aggressive. The theory that underlies exit from dormancy implicates a balance between active metastasis initiating cells and antagonistic immune surveillance. The dormant cancer cells receive growth-inhibitory signals in the host microenvironment, bypassing TGFβ1 brain-specific signaling, secreting DKK1[49] to inhibit Wnt, and exhibit resistance to antimitotic therapy[50]. Exit from dormancy is not well understood, although changes in cell adhesion molecules, in neutrophil extracellular traps, and autophagy genes have been documented to affect the process[51].
Using gene analysis approaches, seventeen genes have been described that promote metastatic tropism to the breast cancer cells. Among them there are genes that may modify the cancer cell surface protein expression, like α2,6-sialyltransferase ST6GALNAC5[52], MMPs and chemokines[53] or affect the ECM protein expression[54], and these are reported to specifically drive metastasis to the brain. Once the cancer cells enter the distant site of metastasis, two processes are initiated in an effort to overcome the hostility of and co-opt the local environment and adapt metabolically: colonization and outgrowth of the cancer cells.
COLONIZATION
The development of BCBM depends on effectiveness of colonization of the perivascular space in the brain and the local initial growth of the cancer cells. During the phase of colonization the cancer cells exhibit high levels of oxidative stress and the formation of reactive oxygen species. Within the circulation the cancer cells decrease their aerobic metabolism and oxidative phosphorylation, which is re-activated as the cells reach their target organ location[55,56]. In that space (brain) the metastatic cancer cells interact with the brain parenchymal microenvironment[57] and eventually establish a metabolically-favorable tumor microenvironment for metastasis (TME)[58]. One of the events that lead to changes in the metabolism of the proximal parenchyma is the restriction of blood flow and limited nutrient availability. Levels of glutamine and glutamate are affected by these restrictions and the local metabolism is altered[59]. A factor involved in this metabolic change is the lymphoid enhancer-binding factor 1 (LEF1), which is frequently overexpressed in cancer cells as they colonize the brain[60]. LEF1 has recently been described to regulate glutathione metabolism, thus protecting the cancer cells from undergoing apoptosis. Other reports have indicated that cancer cells colonizing the CNS upregulate the expression of GABA receptors or GABA transporters as an effort to be able to metabolize GABA, which is abundantly available in the CNS, as a source for protein synthesis[61].
OUTGROWTH
The metabolically-flexible breast cancer cells that colonize the brain have the ability to utilize glutamine as energy source, upregulate factors like the glucose regulated protein 94 (GRP94), and regulate the process of autophagy. The events allow the cells to grow and form the metastatic lesion at target site of the brain. This regulation of autophagy has been recognized as an attractive strategy to target BCBM[53]. Amplification of epidermal growth factor receptor (EGFR) in the cancer cells is associated with loss of PTEN, which both promotes survival and regulates c-Myc to facilitate metabolic reprogramming[62]. A critical factor for the colonizing cancer cells is their ability to modify their exposure to immune attack by changing the expression of MHC I, of the ligands of NK cells and effector T cells, as well as the cGAS-Sting and cytosolic DNA-sensing pathways, and damage-associated RNAs as ligands for pattern-recognition receptors. Modifying the presumptive tumor microenvironment to support the infiltrating tumor cells becomes a necessary step for the outgrowth of the breast cancer cells in the brain[63].
DEFINING THE PRE-METASTATIC NICHE
To successfully form secondary tumors in organs distant from the primary neoplasm, tumor cells require a permissive environment to seed and grow. The pre-metastatic niche is defined as a supportive environment in a tissue prior to tumor spread. The formation of a pre-metastatic niche was first described when hematopoietic progenitor cells expressing vascular endothelial growth factor receptor 1 (VEGFR1) localized and clustered at pre-metastatic sites prior to the arrival or detection of tumor cells[64-66]. Appropriate expression of matrix metalloproteases and deposition of fibronectin are thought to contribute to the permissive environment of the pre-metastatic niche[66].
In addition to the bone marrow-derived progenitor cells, tumor cell-derived factors also play a role in the preparation of the pre-metastatic niche in a distant organ. These factors secreted by the tumor cells include both soluble factors, such as cytokines, and extracellular vesicles (EV) that interact with and influence cells within the pre-metastatic niche. Through these interactions, the secreted factors are involved in the processes of cell activation, extracellular matrix remodeling, and overall priming of the pre-metastatic niche[5,67-70]. Analysis of the secretome of a tumor can be useful to define these interactions and for therapeutic purposes as it can provide biomarkers for tumor progression and potential targets to inhibit the interactions between the secreted macromolecules and resident cells of the pre-metastatic niche[71].
RESIDENT CELL CONTRIBUTIONS TO THE ESTABLISHMENT OF THE BRAIN PREMETASTATIC NICHE
Within the brain microenvironment microglia, astrocytes, pericytes, neurons, and endothelial cells all interact through a network of cell signaling that results in changes in both gene expression and secretome [Figure 2]. Crosstalk between these CNS-residing cells and circulating tumor cells can result in the establishment of a more favorable environment for metastatic colonization [Figure 3][72]. All cells within the microenvironment collaborate to prime the premetastatic niche. Recent analyses of the eventual microenvironment around brain tumors have indicated that the resident CNS cells respond differently to primary brain tumors vs secondary, metastatic tumors, with the latter, and specifically the breast metastases, characterized by significant accumulation of neutrophils[73].
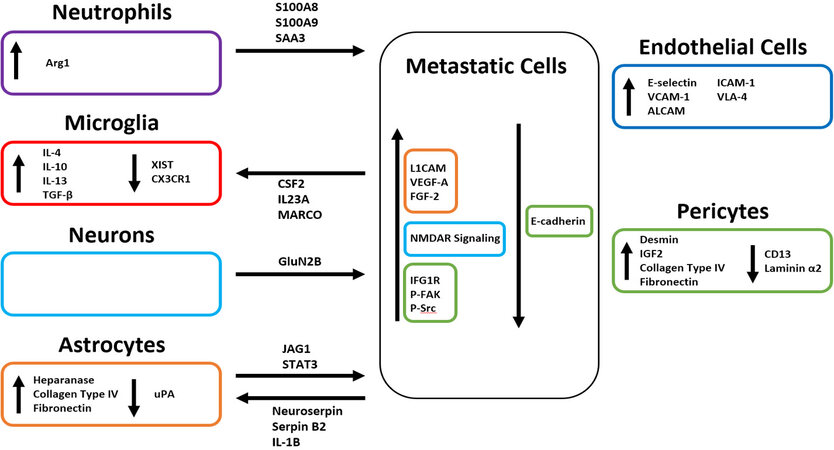
Figure 2. Changes in gene expression and secretome in the brain parenchyma.
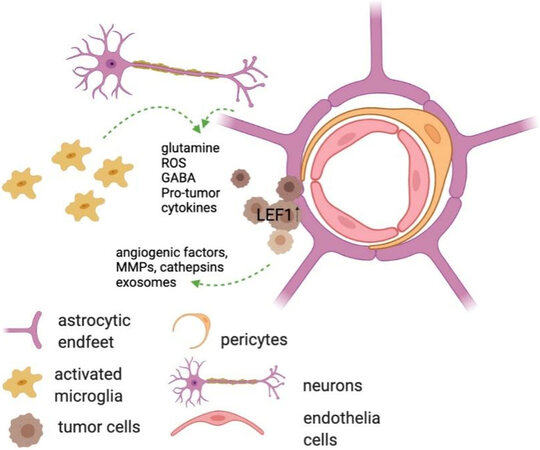
Figure 3. Tumor cell interactions with the cells and cell processes in the brain parenchyma. Image made using BioRender.
Microglia are the resident innate immune cells located in the brain. Although they are known to become activated by exaggerated CNS stimulation and pathological conditions, but until recently little was known about their involvement in the formation of brain metastases[74,75]. Microglia remain in a resting state constantly surveying the CNS parenchyma. Upon pathological stimulation they are activated and shift into phenotypes that have been termed either M1 or M2[76,77]. Similar to tumor associated macrophages, microglia expressing an M1-like phenotype have pro-inflammatory and tumor suppressive properties through the secretion of cytokines such as tumor necrosis factor-α (TNF-α), interleukin-1β (IL-1β), superoxide, nitric oxide (NO), reactive oxygen species (ROS), and specific proteases[78-80]. Microglia characterized as presenting an M2-like phenotype are anti-inflammatory and pro-tumoral through the upregulation of IL-4, IL-10, IL-13, and transforming growth factor-β (TGF-β) which antagonize the functions of the previously mentioned pro-inflammatory cytokines[77,81,82]. Activated microglia do not present solely M1 or M2 markers and are not exclusively in M1-like or M2-like states, but rather express markers that form a continuum between the two states. In the presence of M1-like activated microglia, breast tumor cells are phagocytosed soon after infiltrating the brain[83]. M2-like differentiated microglia are thought to play a crucial role in the formation of the pre-metastatic niche by suppressing their innate immune function and thus allowing circulating tumor cells to enter the brain unchallenged and unharmed. The activation of M2-like microglia has been reported to promote breast cancer cell invasion in a Wnt-dependent manner[84]. In breast-to-brain metastasis, M2 polarization is also triggered by the loss of expression of the long noncoding RNA X-inactive specific transcript (XIST) which is a major regulator of X-chromosome inactivation[85,86]. The effect that the breast tumor secretome may have on the activation of microglia is not extensively studied, nor is it known whether it is a stable or dynamic secretome, changing over time and during the different phases of metastasis. In a series of publications in the last year, bioinformatic approaches using both RNASeq and CYTOF approaches have started analyzing the immune microenvironment of BCBM. The majority of these studies have not yet provided information on the early changes in the CNS during the formation of the pre-metastatic/metastatic niche but do give new views on the distinct tumor microenvironment around these metastases once established[73,87,88]. Overall samples from brain metastases present an abundance of molecules regulating myeloid and lymphocytic cell signaling[73], including CSF2, IL23A and the pattern recognition receptor MARCO. Among microglia specific markers, prominently genes of the IL-6 signaling pathway were upregulated, normally known for suppressing systemic immune responses, as well as TREM1 and CXCL5. Interestingly, CXCL8 was also upregulated in microglia around brain metastases, which functions to chemoattract neutrophils[73]. Another chemokine receptor, CX3CR1, was downregulated in the CNS around metastatic lesions[89]. In an effort to look at newly-formed metastases in the brain, Schulz et al.[90] examined immune cell changes around smaller vs. bigger tumors and reported that the infiltrating tumor cells “educate” the local immune cell environment, with microglia displaying only changes in few genes when in proximity with small or large metastatic lesions in the H2030-BrM model. A limitation of this analysis, however, was the use of non-immune-competent mice.
The function of astrocytes in the normal, physiological brain is to provide structural and functional support to the neurons, to regulate extrasynaptic neurotransmitter levels and to modulate specific steps in synapse formation and plasticity[91]. It has been speculated that astrocytes affect brain metastasis progression through crosstalk with cancer cells via paracrine pathways, since they are shown to surround metastatic lesions in the brain and their endfeet are prominent at the blood-brain barrier[52,74,92,93]. Because of this physical proximity, astrocytes interact with cells upon extravasation and have been reported to induce Fas-dependent death in both lung- and breast-derived cells that have infiltrated the brain[94-96]. Astrocytes can also release plasminogen activators (PAs), primarily urokinase PA (uPA). PA-mediated plasmin generation leads to the proteolysis of L1 cell adhesion molecule (L1CAM) which is expressed by metastatic cells to promote spreading along brain capillaries and vascular co-option[97,98]. Both breast- and lung-derived brain metastatic cells express high levels of serpins that target PA, primarily Neuroserpin and Serpin B2, effectively neutralizing the plasmin-mediated antit-metastatic effects of astrocytes and thus promoting the formation of the pre-metastatic niche[96]. Activation of Notch signaling also becomes possible as a result of the proximity of astrocytes[99,100]. IL-1β, secreted from tumor cells, activates Jagged-1 (JAG1) expression in astrocytes; JAG1 then interacts with tumor cells to significantly stimulate Notch signaling[99,101,102]. The bi-directional signaling mediated by the Notch pathway drives the formation of a permissive metastatic niche and eventually a tumor microenvironment. The secretion of heparanase and neurotrophins by activated astrocytes near sites of tumor cell arrest, extravasation, and invasion into the brain are also thought to initiate and promote metastatic growth through conditioning of the premetastatic niche[93,103,104]. Heparanase is an enzyme that degrades heparan sulfate proteoglycan, a major constituent of endothelial basement membranes. It is linked to increased metastasis in multiple cancer types, as degradation of the proteoglycans is thought to “loosen” the extracellular matrix and facilitate angiogenic responses through the release of heparan sulfate-associated growth factors, such as vascular endothelial growth factor A (VEGF-A) and
Pericytes surround the capillaries of the brain and have been described to have both signaling and barrier roles. Hemodynamic modulation and blood brain barrier maintenance are among the most important[112]. The role of pericytes on primary brain tumors has been studied extensively and, in this context, elimination of pericytes resulted in increase in vessel leakiness and blood supply[113]. Previously, studies have shown an inverse relationship between the number of pericytes and the likelihood of breast cancer metastasis to the lungs[114,115]. A recent study conducted by Molnár et al.[116] has reported that pericytes are crucial to the development of TNBC-derived brain metastases. Pericytes directly interact with the metastatic cells by secreting proteins of the extracellular matrix, insulin-like growth factors (IGFs), and other soluble factors which have chemoattractant, adhesion-, and proliferation-enhancing effects. Insulin-like growth factor 2 (IGF2) is highly upregulated in pericytes and interacts with insulin-like growth factor 1 receptor (IGF1R) on TNBC cells to promote proliferation. TNBC cells respond to the increased collagen type IV and fibronectin secretion from pericytes by increasing intracellular focal adhesion kinase (FAK) and Src phosphorylation to promote focal adhesion formation with the newly formed extracellular matrix. Intercellular adhesion is also disrupted by pericytes which confers a migratory and invasive phenotype onto the metastatic TNBC cells[116]. Increased permeability of the blood brain barrier and experimental TNBC metastasis to the brain was found to involve changes in pericytes, namely an increase in desmin+ pericytes, and decrease in CD13+ pericytes, as well as in extracellular laminin α2[117]. The impact that pericytes in the brain have on the formation of local metastases derived from other breast cancer subtypes remains unknown as does their effect on priming the pre-metastatic niche.
Until recently, the role that neurons had on the formation of breast-derived brain metastases had remained largely unexplored. Zeng et al.[118] revealed that neurons interact with metastatic cells to activate the glutamine-stimulated N-methyl-D-aspartate receptor (NMDAR) signaling (GluN2B-mediated) in the invading cancer cells within the brain. They demonstrated a dependence on NMDAR activation for successful colony formation in the pre-metastatic niche, thus indicating that this signaling axis may yield potential therapeutic targets for the prevention of metastasis[118].
Endothelial cells are critical for the process of extravasation of tumor cells from the CNS vasculature[119]. Soto et al.[120] have demonstrated in vitro that the adhesion molecules E-selectin, vascular cell adhesion molecule-1 (VCAM-1), activated leukocyte cellular adhesion molecule (ALCAM), intercellular adhesion molecule-1 (ICAM-1), and very late antigen 4 (VLA-4) are upregulated on the surface of CNS endothelial cells in the presence of TNBC cells. In parallel, expression of ligands to these adhesion molecules is increased on the surface of TNBC cells. Inhibition of ALCAM and VLA-4 significantly decreased the number of brain metastases, further demonstrating the importance of these adhesion molecules in the formation of the pre-metastatic niche to allow metastatic colony formation in the CNS[120]. Activation of endothelial cells has also been shown to allow for the extravasation of T cells from the periphery into the brain[119], although the process is not fully understood in the context of breast to brain metastasis. Decreases in zonula occludens (ZO)-expression and interactions of VLA1 and LFA1 with VCAM and ICAM have been described in other types of tissue injury or inflammation[119].
The role of neutrophils in the pre-metastatic niche has not been fully explored. Neutrophils are innate immune cells that maintain tissue homeostasis by adjusting their function in response to various stimuli[121,122]. Although initially thought to not play an active role in cancer development, through the use of preclinical models, it has been demonstrated that different subpopulations of neutrophils modulate cancer metastasis[123]. High numbers of neutrophils, compared to lymphocytes in peripheral blood, have been associated with poor prognosis for patients with primary brain tumors or metastases to the brain[124], and in the microenvironment of a tumor they have been shown to support tumor growth, partly as a result of their polarization by IL-17+ γδ T cells[125]. It has been reported that they are attracted via G-CSF[124] or by CXCL8 secreted by microglia[73] around brain metastases and infiltrate the sites of metastasis prior to the tumor cells, to create an immunosuppressive, tumor promoting milieu through upregulation of Arg1 and other immunosuppressive factors. The neutrophil-like CD11b+Gr1+ myeloid cells promote the formation of a pre-metastatic niche within the brain through secretion of proinflammatory cytokines, such as S100A8, S100A9, and SAA3[126,127]. Activated neutrophils release S100A8 and S100A9, which form heterodimers to support this mechanism across systems[128]. Once the initial metastatic niche has been formed, additional immunosuppressive neutrophils are recruited through the function of phosphorylated EZH2[124].
Although it is not entirely understood why the inflammatory cascades are triggered in the CNS upon the circulation of cancer cells in the blood stream and their extravasation into the brain, it has been reported recently that the fact that the circulating cancer cells must first arrest in brain microvessels prior to colonizing the brain leads to local platelet activation and the formation of microthrombi, which could initiate tissue inflammatory processes[129].
Metastatic cancer stem cells and their interaction with the CNS niches: to allow for the seeding of metastatic cancer cell, a small population of cancer stem cells support the initial expansion of the cells at the secondary site. Periostin, present in the stroma of the primary breast tumor, is induced by infiltrating cancer cells in the secondary target organ to initiate colonization[130]. Part of the colonization process involves shifting of energy generation towards the infiltrating cells. For example, cancer cell-secreted circulating miR-122 suppresses CNS-resident cell glucose uptake by downregulating the glycolytic enzyme pyruvate kinase[131].
THE INFLUENCE OF EXOSOMES AND EXOSOME CARGO
Exosomes are vesicles ranging in size from 30-100 nm. Released from donor cells via exocytosis, they contain functional biomolecules such as proteins, lipids, RNA, and DNA. Through endocytosis, exosomes enter recipient cells, and the contained biomolecules are released[132-139]. Tumor derived exosomes have the ability to direct metastasis formation in specific organs based on the exosomal integrins (ITG) present. These integrins bind in a tissue-specific fashion, which initiates the formation of the pre-metastatic niche. Breast cancer derived exosomes that had preferential localization to the liver, lungs, or brain were found to express unique integrins depending on their fate. The liver-tropic exosomes expressed ITGβ5, lung-tropic exosomes expressed ITGα6, and brain-tropic exosomes expressed ITGβ3. ITGs bind a number of ECM molecules and common signaling proteins. The expressed integrins bind neighboring cells, such as Kupffer cells in the liver and epithelial cells in the lungs, at the sites of future metastasis which allows for exosome uptake. It was shown that the liver- and lung-tropic exosomes were able to increase the rate of metastases to the respective organs; however, this study was not extended to examine the brain-tropic exosomes[64]. The ability of exosomes to drive metastasis in a site-specific manner suggests that they may contain cargo that helps to promote the formation of a premetastatic niche in a target organ.
A study by Fong et al.[131] reported that breast cancer cell-secreted vesicles containing miR-122 downregulated pyruvate kinase in astrocytes and neurons, thereby decreasing their glucose uptake. The level of ATP in the miR-122 expressing cell line was not significantly changed, suggesting that an alternate metabolic pathway was used to meet energy needs. In the cells of the CNS microenvironment, the level of ATP was not measured so it remains unknown if they are affected by the decrease in glucose uptake or if they are able to overcome it by means of another metabolic pathway. It was suggested that this suppression of glucose consumption by resident cell types increased the availability of the nutrient for the cancer cells once they arrive. Interestingly, miR-122 antagonism reduced breast cancer metastasis to the brain (and the lungs) in a mouse xenograft model[131]. This study was one of the first to show modulation of metabolic environment to support cancer cell seeding and growth in the pre-metastatic niche.
Other reports propose additional exosomal cargo that modulates the ability of breast cancer cells to metastasize to the brain. MiR19a-enriched exosomes are secreted by astrocytes and taken up by tumor cells. The uptake results in reduced expression of PTEN. Suppression of miR19a significantly decreased breast to brain metastasis[140]. In a recent study by Arnold et al.[141] live cell imaging and microarray data revealed that Tubulin Tyrosine Ligase Like 4 (TTLL4) expression correlated with brain metastasis by altering extracellular vesicle homeostasis and allowing the breast cancer cells to adhere to endothelial cells and increasing the compromise of the BBB. In some studies, the compromise of BBB was not necessary, as tumor-derived exosomes could be transferred into the CNS parenchyma by transcytosis[142]. Beyond aligning and adhering to blood vessels, Lu et al.[143] reported that lncRNA GS1-600G8.5 was responsible for compromising the BBB by decreasing the expressing of tight junction proteins, including ZO-1, Claudin-5, and N-cadherin. Exosomes containing cell migration-inducing and hyaluronan-binding protein (CEMIP) facilitated metastasis specifically to the brain, as opposed to other organs. Uptake of CEMIP by parenchymal microglia and by endothelial cells induced perivascular inflammation and local vascular remodeling[144].
CONCLUSION
Conditioning of the premetastatic niche is vital for metastatic colony formation. Exosomes originating from the primary tumor have organ specific characteristics and can dictate the site of metastasis. Exosomes and the cargo they carry may cause the initial priming of the premetastatic niche. Further crosstalk between exosomes or circulating tumor cells with the resident cells of the target tissue around the premetastatic niche allows for initial changes to be made to the microenvironment. M2-like activated microglia not only release pro-tumoral cytokines, but they also decrease their innate immune function which allows circulating tumor cells to enter the CNS. The secretion of heparanase from activated astrocytes degrades the endothelial basement membranes which further aids metastatic cell invasion. The extracellular matrix of the premetastatic niche is affected by pericytes which secrete factors that have chemoattractant, adhesion-, and proliferation-enhancing effects on circulating tumor cells. Endothelial cells aid in the process of extravasation through the upregulation of adhesion molecules on the cell surface. Exosomes carrying miR-122 are able to interact with astrocytes and neurons of the premetastatic niche and may change local cellular metabolism[145], resulting in a decrease in glucose uptake from the environment. This excess glucose can then be utilized by invading metastatic tumor cells which require a larger amount of glucose to meet the metabolic demands.
Metastasis is the leading cause of breast cancer related death with metastasis to the brain having the worst prognosis. While molecular subtype and histological grade can allow for predictions on likelihood of metastasis as well as metastatic sites to be made, secondary metastatic colonies would not be able to form without initial priming of the premetastatic niche. Further understanding of the components of the premetastatic niche and how they work in concert is vital for more accurate predictions of metastatic location and formation and may provide therapeutic targets.
DECLARATIONS
Acknowledgements
We would like to thank members of the Tsirka lab and especially Dr. Kaitlyn Thompson for advice and suggestions for this review.
Author’s contributions
Co-wrote the review: Malone K, Tsirka SE
Availability of data and materials
Not applicable.
Financial support and sponsorship
Support was provided by T32GM0075186 (MKM) and TRO-Walk-for-Beauty Foundation Research Award (SET).
Conflict of interest
Both authors declared that there are no conflicts of interest.
Ethical approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Copyright
©The Author(s) 2021.
REFERENCES
1. Cancer Facts & Figures 2020. Atlanta, GA: American Cancer Society; 2020.
2. National Cancer Institute. Cancer Stat Facts: Female Breast Cancer. Available from: https://seer.cancer.gov/statfacts/html/breast.html. [Last accessed on 27 May 2021].
3. Weigelt B, Peterse JL, van 't Veer LJ. Breast cancer metastasis: markers and models. Nat Rev Cancer 2005;5:591-602.
5. Chen W, Hoffmann AD, Liu H, Liu X. Organotropism: new insights into molecular mechanisms of breast cancer metastasis. NPJ Precis Oncol 2018;2:4.
6. Engel J, Eckel R, Aydemir U, et al. Determinants and prognoses of locoregional and distant progression in breast cancer. Int J Radiat Oncol Biol Phys 2003;55:1186-95.
7. Niwińska A, Murawska M, Pogoda K. Breast cancer brain metastases: differences in survival depending on biological subtype, RPA RTOG prognostic class and systemic treatment after whole-brain radiotherapy (WBRT). Ann Oncol 2010;21:942-8.
8. Chen MT, Sun HF, Zhao Y, et al. Comparison of patterns and prognosis among distant metastatic breast cancer patients by age groups: a SEER population-based analysis. Sci Rep 2017;7:9254.
9. Wu Q, Li J, Zhu S, et al. Breast cancer subtypes predict the preferential site of distant metastases: a SEER based study. Oncotarget 2017;8:27990-6.
10. Gong Y, Liu YR, Ji P, Hu X, Shao ZM. Impact of molecular subtypes on metastatic breast cancer patients: a SEER population-based study. Sci Rep 2017;7:45411.
11. Gu Y, Wu G, Zou X, Huang P, Yi L. Prognostic value of site-specific metastases and surgery in de novo stage IV triple-negative breast cancer: a population-based analysis. Med Sci Monit 2020;26:e920432.
12. Zeeshan R, Mutahir Z. Cancer metastasis - tricks of the trade. Bosn J Basic Med Sci 2017;17:172-82.
13. Obenauf AC, Massagué J. Surviving at a distance: organ-specific metastasis. Trends Cancer 2015;1:76-91.
14. Steeg PS, Camphausen KA, Smith QR. Brain metastases as preventive and therapeutic targets. Nat Rev Cancer 2011;11:352-63.
15. Massagué J, Ganesh K. Metastasis-initiating cells and ecosystems. Cancer Discov 2021;11:971-994.
16. Pereira ER, Kedrin D, Seano G, et al. Lymph node metastases can invade local blood vessels, exit the node, and colonize distant organs in mice. Science 2018;359:1403-7.
17. Heitz F, Harter P, Lueck HJ, et al. Triple-negative and HER2-overexpressing breast cancers exhibit an elevated risk and an earlier occurrence of cerebral metastases. Eur J Cancer 2009;45:2792-8.
18. Heitz F, Rochon J, Harter P, et al. Cerebral metastases in metastatic breast cancer: disease-specific risk factors and survival. Ann Oncol 2011;22:1571-81.
19. Miller KD, Weathers T, Haney LG, et al. Occult central nervous system involvement in patients with metastatic breast cancer: prevalence, predictive factors and impact on overall survival. Ann Oncol 2003;14:1072-7.
20. Niikura N, Hayashi N, Masuda N, et al. Treatment outcomes and prognostic factors for patients with brain metastases from breast cancer of each subtype: a multicenter retrospective analysis. Breast Cancer Res Treat 2014;147:103-12.
21. Pestalozzi BC, Zahrieh D, Price KN, et al. International Breast Cancer Study Group (IBCSG). Identifying breast cancer patients at risk for Central Nervous System (CNS) metastases in trials of the International Breast Cancer Study Group (IBCSG). Ann Oncol 2006;17:935-44.
22. Weil RJ, Palmieri DC, Bronder JL, Stark AM, Steeg PS. Breast cancer metastasis to the central nervous system. Am J Pathol 2005;167:913-20.
23. Kennecke H, Yerushalmi R, Woods R, et al. Metastatic behavior of breast cancer subtypes. J Clin Oncol 2010;28:3271-7.
24. Smid M, Wang Y, Zhang Y, et al. Subtypes of breast cancer show preferential site of relapse. Cancer Res 2008;68:3108-14.
26. Arciero CA, Guo Y, Jiang R, et al. ER(+)/HER2(+) breast cancer has different metastatic patterns and better survival than ER(-)/HER2(+) breast cancer. Clin Breast Cancer 2019;19:236-45.
27. Carter CL, Allen C, Henson DE. Relation of tumor size, lymph node status, and survival in 24,740 breast cancer cases. Cancer 1989;63:181-7.
28. Koscielny S, Tubiana M, Lê MG, et al. Breast cancer: relationship between the size of the primary tumour and the probability of metastatic dissemination. Br J Cancer 1984;49:709-15.
29. Ross JS, Fletcher JA, Linette GP, et al. The Her-2/neu gene and protein in breast cancer 2003: biomarker and target of therapy. Oncologist 2003;8:307-25.
30. Hellman S. Karnofsky Memorial Lecture. Natural history of small breast cancers. J Clin Oncol 1994;12:2229-34.
31. Rosen PP, Groshen S, Saigo PE, Kinne DW, Hellman S. Pathological prognostic factors in stage I (T1N0M0) and stage II (T1N1M0) breast carcinoma: a study of 644 patients with median follow-up of 18 years. J Clin Oncol 1989;7:1239-51.
32. Page DL. Prognosis and breast cancer. Recognition of lethal and favorable prognostic types. Am J Surg Pathol 1991;15:334-49.
33. Laura S, Coombs NJ, Ung O, Boyages J. Tumour size as a predictor of axillary node metastases in patients with breast cancer. ANZ J Surg 2006;76:1002-6.
34. Sivaramakrishna R, Gordon R. Detection of breast cancer at a smaller size can reduce the likelihood of metastatic spread: a quantitative analysis. Acad Radiol 1997;4:8-12.
35. Caswell DR, Swanton C. The role of tumour heterogeneity and clonal cooperativity in metastasis, immune evasion and clinical outcome. BMC Med 2017;15:133.
37. Fidler IJ. The pathogenesis of cancer metastasis: the 'seed and soil' hypothesis revisited. Nat Rev Cancer 2003;3:453-8.
39. Sopik V, Narod SA. The relationship between tumour size, nodal status and distant metastases: on the origins of breast cancer. Breast Cancer Res Treat 2018;170:647-56.
40. Narod SA, Sopik V. Is invasion a necessary step for metastases in breast cancer? Breast Cancer Res 2018;169:9-23.
41. Schlicker A, Ellappalayam A, Beumer IJ, et al. Investigating the concordance in molecular subtypes of primary colorectal tumors and their matched synchronous liver metastasis. Int J Cancer 2020;147:2303-15.
42. Priedigkeit N, Hartmaier RJ, Chen Y, et al. Intrinsic subtype switching and acquired ERBB2/HER2 amplifications and mutations in breast cancer brain metastases. JAMA Oncol 2017;3:666-71.
43. Brastianos PK, Carter SL, Santagata S, et al. Genomic characterization of brain metastases reveals branched evolution and potential therapeutic targets. Cancer Discov 2015;5:1164-77.
44. Wei S, Siegal GP. Surviving at a distant site: The organotropism of metastatic breast cancer. Semin Diagn Pathol 2018;35:108-11.
45. Oehrlich NE, Spineli LM, Papendorf F, Park-Simon TW. Clinical outcome of brain metastases differs significantly among breast cancer subtypes. Oncol Lett 2017;14:194-200.
46. Kim YJ, Kim JS, Kim IA. Molecular subtype predicts incidence and prognosis of brain metastasis from breast cancer in SEER database. J Cancer Res Clin Oncol 2018;144:1803-16.
47. Barnholtz-Sloan JS, Sloan AE, Davis FG, Vigneau FD, Lai P, Sawaya RE. Incidence proportions of brain metastases in patients diagnosed (1973 to 2001) in the Metropolitan Detroit Cancer Surveillance System. J Clin Oncol 2004;22:2865-72.
48. Sakhuja S, Deveaux A, Wilson LE, et al. Patterns of de-novo metastasis and breast cancer-specific mortality by race and molecular subtype in the SEER population-based dataset. Breast Cancer Res Treat 2021;186:509-18.
49. Malladi S, Macalinao DG, Jin X, et al. Metastatic latency and immune evasion through autocrine inhibition of WNT. Cell 2016;165:45-60.
50. Risson E, Nobre AR, Maguer-Satta V, Aguirre-Ghiso JA. The current paradigm and challenges ahead for the dormancy of disseminated tumor cells. Nat Cancer 2020;1:672-80.
51. Baram T, Rubinstein-Achiasaf L, Ben-Yaakov H, Ben-Baruch A. Inflammation-driven breast tumor cell plasticity: stemness/EMT, therapy resistance and dormancy. Front Oncol 2020;10:614468.
52. Bos PD, Zhang XH, Nadal C, et al. Genes that mediate breast cancer metastasis to the brain. Nature 2009;459:1005-9.
53. Maiti A, Hait NC. Autophagy-mediated tumor cell survival and progression of breast cancer metastasis to the brain. J Cancer 2021;12:954-64.
54. Hebert JD, Myers SA, Naba A, et al. Proteomic profiling of the ECM of xenograft breast cancer metastases in different organs reveals distinct metastatic niches. Cancer Res 2020;80:1475-85.
55. Semenza GL. Hypoxia-inducible factors: coupling glucose metabolism and redox regulation with induction of the breast cancer stem cell phenotype. EMBO J 2017;36:252-9.
56. Schild T, Low V, Blenis J, Gomes AP. Unique metabolic adaptations dictate distal organ-specific metastatic colonization. Cancer Cell 2018;33:347-54.
57. Lah TT, Novak M, Breznik B. Brain malignancies: Glioblastoma and brain metastases. Semin Cancer Biol 2020;60:262-73.
58. Elia I, Haigis MC. Metabolites and the tumour microenvironment: from cellular mechanisms to systemic metabolism. Nat Metab 2021;3:21-32.
59. Sharma MK, Seidlitz EP, Singh G. Cancer cells release glutamate via the cystine/glutamate antiporter. Biochem Biophys Res Commun 2010;391:91-5.
60. Blazquez R, Rietkötter E, Wenske B, et al. LEF1 supports metastatic brain colonization by regulating glutathione metabolism and increasing ROS resistance in breast cancer. Int J Cancer 2020;146:3170-83.
61. Gao Y, Bado I, Wang H, Zhang W, Rosen JM, Zhang XH. Metastasis organotropism: redefining the congenial soil. Dev Cell 2019;49:375-91.
62. Masui K, Cavenee WK, Mischel PS. mTORC2 dictates Warburg effect and drug resistance. Cell Cycle 2014;13:1053-4.
63. Pedrosa RMSM, Mustafa DA, Soffietti R, Kros JM. Breast cancer brain metastasis: molecular mechanisms and directions for treatment. Neuro Oncol 2018;20:1439-49.
64. Hoshino A, Costa-Silva B, Shen TL, et al. Tumour exosome integrins determine organotropic metastasis. Nature 2015;527:329-35.
65. Kaplan RN, Rafii S, Lyden D. Preparing the "soil": the premetastatic niche. Cancer Res 2006;66:11089-93.
66. Kaplan RN, Riba RD, Zacharoulis S, et al. VEGFR1-positive haematopoietic bone marrow progenitors initiate the pre-metastatic niche. Nature 2005;438:820-7.
67. Aguado BA, Bushnell GG, Rao SS, Jeruss JS, Shea LD. Engineering the pre-metastatic niche. Nat Biomed Eng 2017;1:10077.
68. Guo Y, Ji X, Liu J, et al. Effects of exosomes on pre-metastatic niche formation in tumors. Mol Cancer 2019;18:39.
69. Liu Y, Cao X. Characteristics and significance of the pre-metastatic niche. Cancer Cell 2016;30:668-81.
70. Peinado H, Zhang H, Matei IR, et al. Pre-metastatic niches: organ-specific homes for metastases. Nat Rev Cancer 2017;17:302-17.
71. Brandi J, Manfredi M, Speziali G, Gosetti F, Marengo E, Cecconi D. Proteomic approaches to decipher cancer cell secretome. Semin Cell Dev Biol 2018;78:93-101.
72. Witzel I, Oliveira-Ferrer L, Pantel K, Müller V, Wikman H. Breast cancer brain metastases: biology and new clinical perspectives. Breast Cancer Res 2016;18:8.
73. Klemm F, Maas RR, Bowman RL, et al. Interrogation of the microenvironmental landscape in brain tumors reveals disease-specific alterations of immune cells. Cell 2020;181:1643-60.e17.
74. Carvalho R, Paredes J, Ribeiro AS. Impact of breast cancer cells secretome on the brain metastatic niche remodeling. Semin Cancer Biol 2020;60:294-301.
75. Lull ME, Block ML. Microglial activation and chronic neurodegeneration. Neurotherapeutics 2010;7:354-65.
76. Boche D, Perry VH, Nicoll JA. Review: activation patterns of microglia and their identification in the human brain. Neuropathol Appl Neurobiol 2013;39:3-18.
77. Sidani M, Wyckoff J, Xue C, Segall JE, Condeelis J. Probing the microenvironment of mammary tumors using multiphoton microscopy. J Mammary Gland Biol Neoplasia 2006;11:151-63.
78. Le W, Rowe D, Xie W, Ortiz I, He Y, Appel SH. Microglial activation and dopaminergic cell injury: an in vitro model relevant to Parkinson's disease. J Neurosci 2001;21:8447-55.
79. Li R, Huang YG, Fang D, Le WD. (-)-Epigallocatechin gallate inhibits lipopolysaccharide-induced microglial activation and protects against inflammation-mediated dopaminergic neuronal injury. J Neurosci Res 2004;78:723-31.
80. Block ML, Zecca L, Hong JS. Microglia-mediated neurotoxicity: uncovering the molecular mechanisms. Nat Rev Neurosci 2007;8:57-69.
81. Butovsky O, Talpalar AE, Ben-Yaakov K, Schwartz M. Activation of microglia by aggregated beta-amyloid or lipopolysaccharide impairs MHC-II expression and renders them cytotoxic whereas IFN-gamma and IL-4 render them protective. Mol Cell Neurosci 2005;29:381-93.
82. Zhou X, Spittau B, Krieglstein K. TGFbeta signalling plays an important role in IL4-induced alternative activation of microglia. J Neuroinflammation 2012;9:210.
83. Benbenishty A, Gadrich M, Cottarelli A, et al. Prophylactic TLR9 stimulation reduces brain metastasis through microglia activation. PLoS Biol 2019;17:e2006859.
84. Pukrop T, Dehghani F, Chuang HN, et al. Microglia promote colonization of brain tissue by breast cancer cells in a Wnt-dependent way. Glia 2010;58:1477-89.
85. Xing F, Liu Y, Wu SY, et al. Loss of XIST in breast cancer activates MSN-c-Met and reprograms microglia via exosomal miRNA to promote brain metastasis. Cancer Res 2018;78:4316-30.
87. Friebel E, Kapolou K, Unger S, et al. Single-cell mapping of human brain cancer reveals tumor-specific instruction of tissue-invading leukocytes. Cell 2020;181:1626-42.e20.
88. Kloosterman DJ, Akkari L. Mapping the uncharted territories of human brain malignancies. Cell 2020;181:1454-7.
89. Guldner IH, Wang Q, Yang L, et al. CNS-native myeloid cells drive immune suppression in the brain metastatic niche through Cxcl10. Cell 2020;183:1234-48.e25.
90. Schulz M, Michels B, Niesel K, et al. Cellular and molecular changes of brain metastases-associated myeloid cells during disease progression and therapeutic response. iScience 2020;23:101178.
92. Da Silva L, Simpson PT, Smart CE, et al. HER3 and downstream pathways are involved in colonization of brain metastases from breast cancer. Breast Cancer Res 2010;12:R46.
93. Lorger M, Felding-Habermann B. Capturing changes in the brain microenvironment during initial steps of breast cancer brain metastasis. Am J Pathol 2010;176:2958-71.
94. Seike T, Fujita K, Yamakawa Y, et al. Interaction between lung cancer cells and astrocytes via specific inflammatory cytokines in the microenvironment of brain metastasis. Clin Exp Metastasis 2011;28:13-25.
95. Lin Q, Balasubramanian K, Fan D, et al. Reactive astrocytes protect melanoma cells from chemotherapy by sequestering intracellular calcium through gap junction communication channels. Neoplasia 2010;12:748-54.
96. Valiente M, Obenauf AC, Jin X, et al. Serpins promote cancer cell survival and vascular co-option in brain metastasis. Cell 2014;156:1002-16.
97. Voura EB, Ramjeesingh RA, Montgomery AM, Siu CH. Involvement of integrin alpha(v)beta(3) and cell adhesion molecule L1 in transendothelial migration of melanoma cells. Mol Biol Cell 2001;12:2699-710.
98. Silletti S, Mei F, Sheppard D, Montgomery AM. et al. Plasmin-sensitive dibasic sequences in the third fibronectin-like domain of L1-cell adhesion molecule (CAM) facilitate homomultimerization and concomitant integrin recruitment. J Cell Biol 2000;149:1485-502.
99. Xing F, Kobayashi A, Okuda H, et al. Reactive astrocytes promote the metastatic growth of breast cancer stem-like cells by activating Notch signalling in brain. EMBO Mol Med 2013;5:384-96.
100. Nam DH, Jeon HM, Kim S, et al. Activation of notch signaling in a xenograft model of brain metastasis. Clin Cancer Res 2008:14;4059-66.
101. Shen Q, Reedijk M. Notch signaling and the breast cancer microenvironment. In: Reichrath J, Reichrath S, editors. Notch signaling in embryology and cancer. Cham: Springer; 2021. p. 183-200.
102. Meurette O. Shaping of the tumor microenvironment by notch signaling. In: Birbrair A, editor. Tumor Microenvironment. Cham: Springer; 2020. p. 1-16.
103. Marchetti D, Aucoin R, Blust J, Murry B, Greiter-Wilke A. p75 neurotrophin receptor functions as a survival receptor in brain-metastatic melanoma cells. J Cell Biochem 2004;91:206-15.
104. Marchetti D, Li J, Shen R. Astrocytes contribute to the brain-metastatic specificity of melanoma cells by producing heparanase. Cancer Res 2000;60:4767-70.
105. Nakajima M, Irimura T, Nicolson GL. Heparanases and tumor metastasis. J Cell Biochem 1988;36:157-67.
106. Vlodavsky I, Elkin M, Ilan N. Impact of heparanase and the tumor microenvironment on cancer metastasis and angiogenesis: basic aspects and clinical applications. Rambam Maimonides Med J 2011;2:e0019.
107. Alhusban L, Ayoub NM, Alhusban A. ProBDNF is a novel mediator of the interaction between MDA-MB-231 breast cancer cells and brain microvascular endothelial cells. Curr Mol Med 2020; doi: 10.2174/1566524020666201120142717.
108. Denkins Y, Reiland J, Roy M, et al. Brain metastases in melanoma: roles of neurotrophins. Neuro Oncol 2004;6:154-65.
109. Louie E, Chen XF, Coomes A, Ji K, Tsirka S, Chen EI. Neurotrophin-3 modulates breast cancer cells and the microenvironment to promote the growth of breast cancer brain metastasis. Oncogene 2013;32:4064-77.
110. Priego N, Zhu L, Monteiro C, et al. STAT3 labels a subpopulation of reactive astrocytes required for brain metastasis. Nat Med 2018;24:1024-35.
111. Hosonaga M, Saya H, Arima Y. Molecular and cellular mechanisms underlying brain metastasis of breast cancer. Cancer Metastasis Rev 2020;39:711-20.
112. Bergers G, Song S. The role of pericytes in blood-vessel formation and maintenance. Neuro Oncol 2005;7:452-64.
113. Cheng L, Huang Z, Zhou W, et al. Glioblastoma stem cells generate vascular pericytes to support vessel function and tumor growth. Cell 2013;153:139-52.
114. Cooke VG, LeBleu VS, Keskin D, et al. Pericyte depletion results in hypoxia-associated epithelial-to-mesenchymal transition and metastasis mediated by met signaling pathway. Cancer Cell 2012;21:66-81.
115. Keskin D, Kim J, Cooke VG, et al. Targeting vascular pericytes in hypoxic tumors increases lung metastasis via angiopoietin-2. Cell Rep 2015;10:1066-81.
116. Molnár K, Mészáros Á, Fazakas C, et al. Pericyte-secreted IGF2 promotes breast cancer brain metastasis formation. Mol Oncol 2020;14:2040-57.
117. Lyle LT, Lockman PR, Adkins CE, et al. Alterations in pericyte subpopulations are associated with elevated blood-tumor barrier permeability in experimental brain metastasis of breast cancer. Clin Cancer Res 2016;22:5287-99.
118. Zeng Q, Michael IP, Zhang P, et al. Synaptic proximity enables NMDAR signalling to promote brain metastasis. Nature 2019;573:526-31.
119. Schulz M, Salamero-Boix A, Niesel K, Alekseeva T, Sevenich L. Microenvironmental regulation of tumor progression and therapeutic response in brain metastasis. Front Immunol 2019;10:1713.
120. Soto MS, Serres S, Anthony DC, Sibson NR. Functional role of endothelial adhesion molecules in the early stages of brain metastasis. Neuro Oncol 2014;16:540-51.
122. Mantovani A, Cassatella MA, Costantini C, Jaillon S. Neutrophils in the activation and regulation of innate and adaptive immunity. Nat Rev Immunol 2011;11:519-31.
123. Wu M, Ma M, Tan Z, Zheng H, Liu X. Neutrophil: a new player in metastatic cancers. Front Immunol 2020;11:565165.
124. Zhang L, Yao J, Wei Y, et al. Blocking immunosuppressive neutrophils deters pY696-EZH2-driven brain metastases. Sci Transl Med 2020;12:eaaz5387.
125. Hyun YM, Seo SU, Choi WS, et al. Endogenous DEL-1 restrains melanoma lung metastasis by limiting myeloid cell-associated lung inflammation. Sci Adv 2020;6:eabc4882.
126. Liu Y, Kosaka A, Ikeura M, et al. Premetastatic soil and prevention of breast cancer brain metastasis. Neuro Oncol 2013;15:891-903.
127. Tomonobu N, Kinoshita R, Sakaguchi M. et al. S100 soil sensor receptors and molecular targeting therapy against them in cancer metastasis. Transl Oncol 2020;13:100753.
128. Sinha P, Okoro C, Foell D, Freeze HH, Ostrand-Rosenberg S, Srikrishna G. Proinflammatory S100 proteins regulate the accumulation of myeloid-derived suppressor cells. J Immunol 2008;181:4666-75.
129. Feinauer MJ, Schneider SW, Berghoff AS, et al. Local blood coagulation drives cancer cell arrest and brain metastasis in a mouse model. Blood 2021;137:1219-32.
130. Malanchi I, Santamaria-Martínez A, Susanto E, et al. Interactions between cancer stem cells and their niche govern metastatic colonization. Nature 2011;481:85-9.
131. Fong MY, Zhou W, Liu L, et al. Breast-cancer-secreted miR-122 reprograms glucose metabolism in premetastatic niche to promote metastasis. Nat Cell Biol 2015;17:183-94.
132. Balaj L, Lessard R, Dai L, et al. Tumour microvesicles contain retrotransposon elements and amplified oncogene sequences. Nat Commun 2011;2:180.
133. Skog J, Würdinger T, van Rijn S, et al. Glioblastoma microvesicles transport RNA and proteins that promote tumour growth and provide diagnostic biomarkers. Nat Cell Biol 2008;10:1470-6.
134. Théry C, Ostrowski M, Segura E. Membrane vesicles as conveyors of immune responses. Nat Rev Immunol 2009;9:581-93.
135. Raposo G, Stoorvogel W. Extracellular vesicles: exosomes, microvesicles, and friends. J Cell Biol 2013;200:373-83.
136. Peinado H, Lavotshkin S, Lyden D. The secreted factors responsible for pre-metastatic niche formation: old sayings and new thoughts. Semin Cancer Biol 2011;21:139-46.
137. Choi DS, Kim DK, Kim YK, Gho YS. Proteomics, transcriptomics and lipidomics of exosomes and ectosomes. Proteomics 2013;13:1554-71.
138. Valadi H, Ekström K, Bossios A, Sjöstrand M, Lee JJ, Lötvall JO. Exosome-mediated transfer of mRNAs and microRNAs is a novel mechanism of genetic exchange between cells. Nat Cell Biol 2007;9:654-9.
139. Thakur BK, Zhang H, Becker A, et al. Double-stranded DNA in exosomes: a novel biomarker in cancer detection. Cell Res 2014;24:766-9.
140. Zhang L, Zhang S, Yao J, et al. Microenvironment-induced PTEN loss by exosomal microRNA primes brain metastasis outgrowth. Nature 2015;527:100-4.
141. Arnold J, Schattschneider J, Blechner C, et al. Tubulin Tyrosine Ligase Like 4 (TTLL4) overexpression in breast cancer cells is associated with brain metastasis and alters exosome biogenesis. J Exp Clin Cancer Res 2020;39:205.
142. Morad G, Carman CV, Hagedorn EJ, et al. Tumor-derived extracellular vesicles breach the intact blood-brain barrier via transcytosis. ACS Nano 2019;13:13853-65.
143. Lu Y, Chen L, Li L, Cao Y. Exosomes derived from brain metastatic breast cancer cells destroy the blood-brain barrier by carrying lncRNA GS1-600G8.5. Biomed Res Int 2020;2020:7461727.
144. Rodrigues G, Hoshino A, Kenific CM, et al. Tumour exosomal CEMIP protein promotes cancer cell colonization in brain metastasis. Nat Cell Biol 2019;21:1403-12.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Malone K, Tsirka SE. Breast-to-brain metastasis: a focus on the pre-metastatic niche. J Cancer Metastasis Treat 2021;7:40. http://dx.doi.org/10.20517/2394-4722.2021.37
AMA Style
Malone K, Tsirka SE. Breast-to-brain metastasis: a focus on the pre-metastatic niche. Journal of Cancer Metastasis and Treatment. 2021; 7: 40. http://dx.doi.org/10.20517/2394-4722.2021.37
Chicago/Turabian Style
Malone, Kathryn, Stella E. Tsirka. 2021. "Breast-to-brain metastasis: a focus on the pre-metastatic niche" Journal of Cancer Metastasis and Treatment. 7: 40. http://dx.doi.org/10.20517/2394-4722.2021.37
ACS Style
Malone, K.; Tsirka SE. Breast-to-brain metastasis: a focus on the pre-metastatic niche. J. Cancer. Metastasis. Treat. 2021, 7, 40. http://dx.doi.org/10.20517/2394-4722.2021.37
About This Article
Special Issue
Copyright

Data & Comments
Data


 Cite This Article 31 clicks
Cite This Article 31 clicks

 Like This Article 7
likes
Like This Article 7
likes






Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.