
Bioactive lipids and cancer metastasis to bone
 0
0Abstract
Bioactive lipids constitute a large family of molecules considered as inflammatory mediators. Among them, lysophosphatidic acid (LPA), sphingosine 1-phosphate (S1P), and eicosanoids (prostanoids such as PGE2 and leukotrienes such as LTB4, LTC4, and LTD4) play a central role in the pathophysiology of several inflammatory diseases. However, it has long been known that these bioactive lipids are also involved in cancer, mainly because of their ability to control the pro-inflammatory microenvironment of tumors as well as their ability to act directly on tumor cells promoting cell proliferation, migration, and survival. Recently, there has been increased interest in determining how these lipid mediators orchestrate tumor development and metastasis. Bone metastases result from a complex dialogue between tumor cells and bone cells. Recent findings demonstrate that all these bioactive lipids can profoundly affect bone metabolism by acting positively or negatively on both osteoblasts and osteoclasts. This review gives an overview of previous findings demonstrating direct involvement of LPA, S1P, and PGE2 in bone metastasis. This review also emphasizes the recent findings that characterize the activity of these bioactive lipids directly on bone cells and how these activities could be integrated into the complex molecular mechanisms leading to bone metastasis formation and progression.
Keywords
INTRODUCTION
Bone is a highly dynamically regulated tissue which undergoes continuous homeostatic and reactive remodeling by the coordinated action of bone cells, namely osteoblasts, osteoclasts, and osteocytes, and controlled by endocrine factors, immune cells, and mechanical forces. Sir Paget’s “seed and soil” theory was the first still valid concept explaining why certain types of cancers have remarkably high propensity to form bone metastases such as breast and prostate cancers[1]. During the 1990s, works released from Greg Mundy’s lab led to the development of an additional theory leading to the notion of the “vicious cycle” established at the sites of bone metastases, reflecting the reciprocal stimulation between tumor growth and bone resorption[2]. The vicious cycle theory is also still valid with even more clinical impact, as it is the current target of the best systemic therapies against bone metastases that use anti-resorptive agents (i.e., bisphosphonates and denosumab)[3]. Unfortunately, even under these best standards of care, patients with bone metastases still have limited overall survival, indicating the existence of additional molecular mechanisms. Bioactive lipids are widely present in the organism and affect almost all vital systems. In this review, we focus on two lysophospholipids, lysophosphatidic acid (LPA) and sphingosine 1-phosphate (S1P), and two classes of eicosanoids, prostanoids and leukotrienes, because all of these molecules can regulate cancer progression including bone metastasis as well as important biological processes directly related to bone such as skeletal development, mineralization, regulation of bone mass and homeostasis, osteoblast-osteoclast coupling, and bone resorption and formation.
LYSOPHOSPHATIDIC ACID
Structure, synthesis, and receptors
LPA is the simplest phospholipid which is composed of a glycerophosphate backbone linked to a single fatty acid chain. LPA can be synthesized from two pathways. In the first one, phospholipid precursors such as phosphatidylcholine (PC), phosphatidylserine (PS), or phosphatidylethanolamine (PE) can be converted to lysophosphatidylcholine (LPC), lysophosphatidylserine (LPS), and lysophosphatidylethanolamine (LPE), respectively, through the action of phosphatidylserine-specific phospholipase A1 or secretory phospholipase A2 such as in blood platelets[4,5] [Figure 1]. LPC appears to be the most abundant LPA precursor in the blood. These precursors can then be converted to LPA by a lysophospholipase D (LysoPLD). LPA can also be generated by a distinct mechanism, namely the acylation of glycerol-3-phosphate by glycerophosphate acyltransferase and the phosphorylation of monoacylglycerol by monoacylglycerol kinase[6]. LPA level is tightly tuned by degradation mechanisms mediated by different classes of enzymes such as lipid phosphate phosphatases (LPPs)[7] or LPA acyltransferase[8,9].
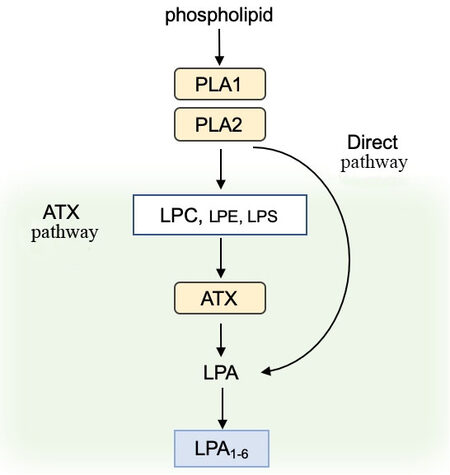
Figure 1. Overview of LPA biosynthesis and receptors. Phospholipase A1 or A2 (PLA1 and PLA2) catabolize membrane phospholipids producing LPA either directly or through the release of LPA precursors (LPC, LPE, and LPS) that are metabolized in LPA by autotaxin (ATX) due to its lysophospholipase D activity. LPA activates six specific G protein-coupled receptors (LPA1-6). LPC: Lysophosphatidylcholine; LPE: lysophosphatidylethanolamine; LPS: lysophosphatidylserine; LPA: lysophosphatidic acid.
Autotaxin (ATX or ENPP2) is responsible for LPA levels in the blood, as evidenced by LPA concentrations in serum that are half normal values in Enpp2+/- heterozygous mice[10]. As a unique member of the family of the seven ectonucleotide pyrophosphatase/phosphodiesterase (ENPP) that hydrolyze pyrophosphate and phosphodiester bonds in nucleotides and their derivatives[11], ATX possess a LysoPLD activity, allowing the hydrolysis of LPC and other lipid precursors to produce LPA. ATX is present at high concentration in the blood circulation[12]. However, the origin of ATX in blood remains to be determined. Nevertheless, adipose tissue is likely one of the main sources, as revealed in aP2-Cre/Enpp2fl/fl mice presenting 30% decrease in LPA levels in plasma[13]. ATX is also highly expressed in the brain and lymphatic high endothelial venules[14]. Although ATX is capable of hydrolyzing nucleotides in vitro, its biological function relies on its lysoPLD activity, as the apparent affinity for LPC is 10 times higher than that of nucleotides[15] and as extracellular nucleotides are rare.
LPA activates a series of six different G-protein coupled receptors (LPA1-6). Overall, LPA receptors link to all types of heterotrimeric G proteins, Gα12/13, Gαq/11, Gαi/o, and Gαs, but, due to specific interactions, LPA receptors can have redundant, synergic, or opposite actions. LPA1 is the most ubiquitous LPA receptor in adults, with prevalence in brain, heart, testis, ovary, prostate, colon, thymus, and pancreas. Eukaryotic cells frequently express multiple LPA receptors. Mature osteoclasts express LPA1, LPA2, LPA4, LPA5, and
Role of LPA on bone cells
LPA is produced at the bone site, as we previously showed in the context of bone metastasis [Figure 2]. Through its action on both cancer cells and osteoclasts, LPA promotes the progression of osteolytic lesions[19]. LPA plays a key role during bone development. However, origin of LPA in bone is still incompletely understood, although adipocytes[13] and osteoblasts[20] are potential sources that may account for bone homeostasis and bone metastasis. Lpar1-/- mice revealed growth retardation due multiple alteration at levels of both the central nervous and the musculoskeletal systems, including inhibition of chondrocyte proliferation, defects in endochondral ossification, and a low bone mass phenotype linked to decreased osteoblast activity[21]. On the other hand, Lpar4-/- animals show increased bone formation[22], suggesting that LPA1 and LPA4 may have opposite functions in the control of bone formation during development. LPA4 activates the Gαs pathway, whereas LPA1 signals through Gαi, resulting in inverse control of adenylated cyclase activation and cAMP accumulation. LPA1 is expressed in chondrocytes and all bone cells including osteoblasts, osteoclasts, and osteocytes[21,23]. As a result, the bone phenotype of Lpar1-/- mice is likely due to a combination of numerous cellular defects that even individually may impact bone remodeling. To elucidate the physiological role of LPA1 expressed by osteoblasts, we generated conditional knockout mice for Lpar1 in osteoblastic cell lineage (Lpar1-△Ob)[17]. These mice revealed reduced bone mineralization and increase in the areas of both osteocytic lacunaes and osteocyte apoptosis. We have known since the release of Karin lab’s work that LPA can promote dendrite outgrowth in MLO-Y4 osteocyte cell line[24]. Osteocyte network was remarkedly reduced in the cortical bone of Lpar1-△Ob mice and dendrite connections were drastically reduced in Lpar1-△Ob osteocytes in vitro in response to fibroblast growth factor 2. These data reveal that, by acting on osteoblastic LPA1, LPA produced in the bone environment controls bone quality via bone mineralization and osteocyte function [Figure 2].
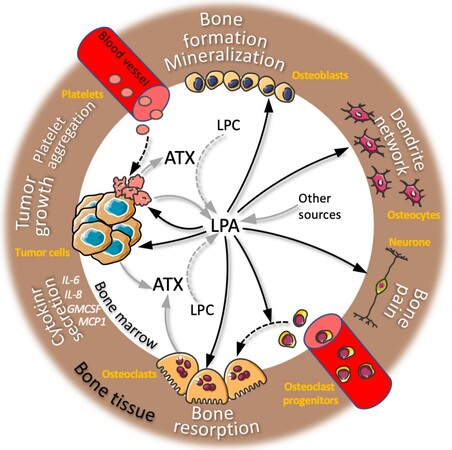
Figure 2. LPA activity in bone metastasis microenvironment: overview of actions of lysophosphatidic acid (LPA) on bone cells, cancer cells, and neurons. Black arrows indicate LPA activity on different cell types from the bone microenvironment (names in orange) resulting in multiple biological functions (text in white). Grey arrows indicate LPA or autotaxin (ATX) cell secretion. Dotted black arrows indicate cell differentiation and platelet aggregation. Dotted grey arrows indicate LPC catabolism by ATX. LPC: Lysophosphatidylcholine.
LPA also acts on osteoclasts by promoting survival and cytoskeleton rearrangement[23], which involves c-SRC signaling and phosphorylation of Thyroid Hormone Receptor Interactor 6 that drive sealing zone formation and bone resorption[25]. Furthermore, we demonstrated that LPA is mandatory at least in vitro for macrophage colony-stimulating factor/receptor activator of nuclear factor-kappa B (NF-κB) (RANK)-ligand (RANK-L)-induced osteoclastogenesis. Both genetic ablation and pharmacological inhibition of LPA1 remarkably alter mineral matrix resorption of mature osteoclasts and prevent ovariectomy-induced bone loss[16,26]. These results demonstrate that, under pathophysiological context, the production of LPA in bone might profoundly affect osteoclast function. Mice with global deletion of Lpar1 revealed remarkable resistance to bone destruction in an arthritis model induced by type II collagen injection[27], confirming the LPA-dependence of osteoclast function here under an inflammation context. This study showed decreased infiltration of macrophages and differentiation into Th17, but not Th1 or Th2, which were also suppressed under pharmacological inhibition of LPA1. Thus, in the context of bone metastasis, in addition to its action on osteoclasts by stimulating osteolysis, LPA could also promote the recruitment of immune cells that may contribute to the progression of metastases. Furthermore, we recently showed that mature osteoclasts produce functionally active ATX, thereby highlighting these cells as an additional source of LPA in bone[28]. Intriguingly, although osteoclast-derived ATX was revealed to be dispensable for bone development as well as in the classical pathological model of bone loss induced by ovariectomy, we found that autocrine activity of ATX on osteoclasts is required for systemic bone loss and bone erosion induced by inflammation, as observed in tumor necrosis factor (TNF)-transgenic(tg) mice, or after mouse treatment with LPS as well as in the arthritic mouse model using K/BxN-serum transfer[28]. In the context of bone metastasis, treatment with BMP22 of mice harboring pre-established bone metastases from the ATX-null MDA-BO2 breast cancer cells significantly reduced the progression of osteolytic lesions[29]. This clearly demonstrates that non-tumoral ATX controls osteoclastic bone resorption. It is of course possible that ATX produced by different types of cells from the bone environment could act in a paracrine manner on osteoclasts; however, as we found under inflammatory conditions, osteoclast-derived ATX might act in an autocrine manner to stimulate malignant osteolysis. However, this assertion requires further investigation.
Role of LPA in cancer and bone metastasis
Autotaxin’s name was originally attributed due to its action on melanoma cells as a new autocrine factor stimulating tumor cell invasion[30]. The ATX/LPA axis was later recognized as a major regulator of tumorigenesis through its action on cancer cell proliferation and survival, as well as through the promotion of angiogenesis and the control of the metastatic cascade by stimulating cell migration and invasion[31,32]. Increased LPA levels are found in different types of cancers[33-35]. Endogenous expression of ATX in 4T1 mouse carcinoma cells as well as high ATX expression in human MDA-BO2-ATX transfected breast cancer cells provide a higher propensity to these cells of generating bone metastases[26]. The main reason is likely to reside in the fact that these cells express high levels of LPA1 receptor, resulting in high sensitivity to LPA stimulation. By activating LPA1, LPA promotes cell motility and proliferation, but the prominent action of LPA/LPA1 signaling in the context of bone metastasis might be linked to the increase in the secretion of growth factors such as vascular endothelial growth factor (VEGF)[36,37] and cytokines [such as interleukin (IL)-6, IL-8, granulocyte-macrophage colony-stimulating factor, and monocyte chemoattractant protein 1] that are known to affect bone remodeling. Osteomimetism was shown to characterize breast cancer bone-seeking cells[38]. Gene signature of LPA signaling was not associated with osteomimetism. However, cancer cells and bone cells (osteoblasts and osteocytes) share the characteristics that, by activating LPA1 receptor, LPA also stimulates the secretion of IL-6 and IL-8 [chemokine (C-X-C motif) ligand 15 protein in mouse][39,40]. Overexpression of LPA1 in MDA-BO2/LPA1 transfected cells dramatically increases the extent of osteolytic lesion areas, whereas pharmacological inhibition of this receptor using Ki16425 and Debio 0719, two LPA1/3 antagonists, reduced cytokine production and the progression of osteolytic bone metastasis. Intriguingly, endogenous expression of ATX was revealed to be dispensable for cancer cells to metastasize to bone, as shown by the above-mentioned MDA-BO2 cells that do not express ATX but which are a sub-clone of MDA-MB-231 cells isolated in vivo for their exclusive bone tropism[41].
High ATX expression in primary tumors is frequently associated with a poor prognosis in cancer patients[42-44]. Intriguingly, the expression of ATX directly by tumor cells is often low, as found in most breast cancer cell lines. It has been well established by Brindley et al.[45] that tumor microenvironment is a major source of ATX[45]. We showed that challenging mature osteoclasts with inflammatory molecules such as TNF and LPS upregulates the secretion of ATX through a NF-κB pathway[28]. In addition, inflammatory cytokines and chemokines released by breast and thyroid cancer cells have been shown to induce the expression of ATX in tumor-associated fibroblasts and adipocytes[46]. Furthermore, during our study on the role of non-tumoral ATX in the formation of bone metastases, we found that blood platelets uptake circulating ATX that is naturally present in the bloodstream and store ATX in their granular compartments, which is eventually released under tumor cell-induced platelet aggregation[29]. Non-tumoral ATX released by platelets is functionally active as it catalyzes the production of LPA, which ultimately acts on cancer cells to promote survival, invasion, and bone metastasis[19,29]. Overall, ATX present in the tumor environment appears as a major promoter of tumor growth and metastasis. Therefore, a new paradigm is emerging at the level of bone metastasis where multiple vicious cycles establish and interconnect together. The first well-known vicious cycle was characterized in 1997 by T Guise, establishing the cross talk between tumor growth and bone resorption[47] [Figure 2]. We showed that the production and activity of LPA following tumor cell-induced platelet aggregation constitute a second vicious cycle between tumor cells and blood platelets taking place at the bone metastasis site[19]. Non-tumoral ATX released by activated platelets is likely to contribute to this bone resorption-independent vicious cycle. Furthermore, by acting on fibroblasts, adipocytes, and, potentially, osteoclasts, tumor cell-derived inflammatory cytokines might contribute to an additional vicious cycle through the secretion of ATX.
Role of LPA in bone pain
Bone pain is the most common complication of bone metastases for cancer patients[48]. LPA is well known to promote neuropathic pain[31], but pain occurring in the context of bone metastasis depends on different mechanisms; as shown primarily by Yoneda et al.[49], the release of protons from vacuolar resorption pits during osteoclastic resorption create acidic microenvironments that stimulate sensory nociceptive neurons that innervate bone. This is likely to explain why anti-resorptive agents, such as bisphosphonates and denosumab, efficiently reduce bone pain in bone metastasis patients[3]. Although LPA might indirectly promote metastasis-induced bone pain through the stimulation of osteoclast activity, it might also have a direct action. Using an osteosarcoma-induced bone pain model in rat, Zhao et al.[50] showed that, two weeks after cancer cell implantation in vivo, sural C-fibers become more sensitive to LPA stimulation, an effect that was blocked after treatment with VPC32183, an antagonist of LPA1 receptor. The analysis of dorsal root ganglion neurons showed increased LPA1receptor expression two weeks after cancer cell injection into the rat tibia[51]. In this context, bone pain was regulated by LPA through LPA1 signaling in dorsal root ganglion neurons, potentiating potential vanilloid 1 (TRPV1) receptors via protein kinase Cε pathway. In addition, in this rat model, mechanical allodynia and thermal hyperalgesia were alleviated under treatment with VPC32183[51]. Furthermore, using paw withdrawal threshold and flinching behavior assays,
SPHINGOSINE 1-PHOSPHATE
Structure, synthesis, and receptors
Sphingolipids are another class of phospholipids that are a part of cell membrane. They comprise a polar head and two non-polar tail domains. Sphingosine, a long chain amino-alcohol, is an integral component of this class of lipids[53]. Sphingolipid metabolism involves three interconnected pathways, namely the de novo synthesis pathway, salvage pathway, and sphingomyelinase (SMase) pathway, all of which generate ceramide from complex lipids that is eventually converted into sphingosine and S1P[54,55]. These pathways were initially thought to be autonomous of each other in ceramide generation, but the metabolites synthesized in these three metabolic pathways are highly reversible, non-distinguishable, and hence interdependent.
Ceramide produced by these pathways is converted into sphingosine and eventually S1P by the concatenated effect of enzymes such as ceramidase and functionally redundant sphingosine kinases 1 and 2 (SphK1 and SphK2), respectively[56] [Figure 3]. This process is also reversible and ceramide synthase and sphingosine phosphate phosphatase can revert sphingosine into ceramide and S1P to sphingosine, respectively. S1P can be irreversibly degraded by S1P lyase (SPL) present on the cytosolic side of the endoplasmic reticulum to form non-sphingolipid products: phospho-ethanolamine and hexadecenal (a fatty aldehyde). While these enzymes are intracellular, LPPs reside on the cell membrane and keep extracellular S1P in check[57,58]. Although minimal roles of intracellular S1P signaling have been reported, the major biological functions of S1P (such as embryonic and postnatal vascular development, vascular integrity and tone, hematopoiesis and trafficking of immune and stem cells, and platelet formation and activation in addition to bone homeostasis) are suggested to be receptor dependent and necessitate an efficient export of S1P into the blood and lymph where the levels of S1P are higher as compared to the tissue parenchyma[58-60]. The two bona fide transporters of S1P identified thus far are spinster homolog 2 (Spns2) that is expressed on lymphatic and blood endothelial cells and major facilitator superfamily transporter 2b that exports S1P from erythrocytes and activated platelets[61], therefore making these cells “sources of S1P”. This exported S1P needs chaperons to promote aqueous solubility of S1P, increase resistance against degradation and dephosphorylation, and accelerate release of S1P from cellular sources, and they may alter receptor selectivity and signaling bias[59,62,63]. These chaperons include high-density lipoprotein bound to apolipoprotein M, albumin, low-density lipoprotein, very low-density lipoprotein, and the recently identified chaperon apolipoprotein A4[64,65]. While blood chaperones are well described, less is known about what chaperones S1P associates within the lymph and interstitial fluids.
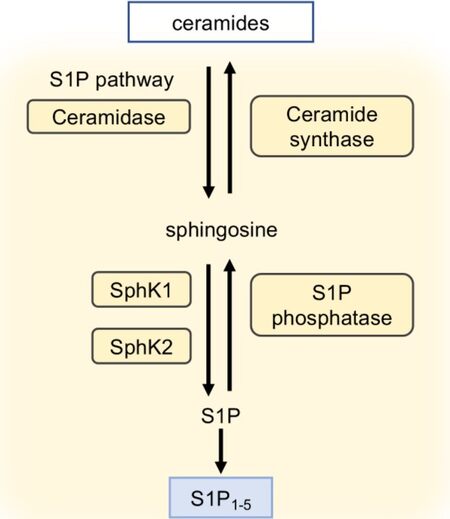
Figure 3. Overview of sphingosine 1-phosphate (S1P) biosynthesis and receptors. Ceramides are converted into sphingosine under the action of ceramidase. Sphingosine is then metabolized in S1P by the action of two sphingosine kinases (SphK1 and SphK2). All biosynthesis steps are reversible in cells. S1P activates five specific G protein-coupled receptors (S1P1-5).
Highly regulated local differences in the synthesis, export, and intracellular and extracellular degradation of S1P lead to marked differences in its abundance among blood (~1 µM), lymph (~0.1 µM), and the tissue parenchyma (< 1 nM), as well as the formation of local S1P gradients within tissues. Sensing of this S1P gradient by the receptors drives biological processes such as immune cell trafficking and vascular homeostasis[58,66,67]. Factors such as presence of de novo synthesis machinery, lack of degradation machinery, and expression of S1P transporters govern whether a cell can be deemed as a source of S1P.
S1P binds to and activates a family of cognate G-protein coupled receptors, S1P1-5. Due to overlap and divergence in Gα subunit selectivity, S1P receptors can act in synergy or in opposition to each other[68,69]. Notable examples include antagonistic activities of the exclusively Gαi-coupled S1P1 and the predominantly Gα12/13-coupled S1P2 and sometimes synergistic activities of S1P1 and S1P3, which can also couple to Gαi. S1P1 is the most widely expressed receptor, which is well studied for its role in maintaining vascular, immune, and bone homeostasis. S1PR1, S1PR2, and S1PR3 transcripts in primary osteoblasts and S1PR1 and S1PR2 in osteoclasts have been detected with negligible or null quantity of S1PR4 and S1PR5, but this could be attributed to poor sensitivity of the tools available to detect these receptors rather than the actual absence of these receptors.
Role of S1P on bone cells
Initially, the S1P contribution to bone homeostasis has been mainly assigned to bone remodeling with particular emphasis on possible roles in the circulation of osteoclast progenitors[70-72]. Accordingly, a S1P gradient between blood and bone marrow has been proposed that chemoattracts osteoclast progenitors away from the bone microenvironment (and hence fewer of them differentiating to mature osteoclasts) towards blood through S1P1. Accordingly, mice lacking S1P1 in CD11b+ cells were mildly osteopenic.
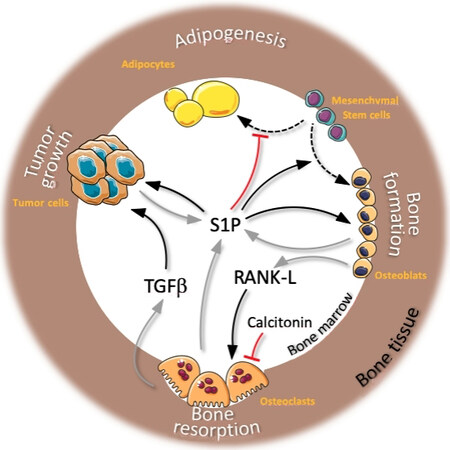
Figure 4. Sphingosine 1-phosphate (S1P) activity in bone metastasis microenvironment: overview of actions of S1P on bone cells, cancer cells, and adipocytes. Black arrows indicate activities of S1P, receptor-activated NF-κB ligand (RANK-L), and tumor growth factor β (TGFβ) on different cell types from the bone microenvironment (names in orange) resulting in multiple biological functions (text in white). Grey arrows indicate S1P and RANK-L secretions and TGFβ release from resorbed bone matrix. Dotted black arrows indicate cell differentiation (adipogenesis and osteoblastogenesis). Red lines indicate S1P and Calcitonin inhibitory activities.
Indeed, we and others demonstrated numerous effects of S1P on osteoblast behavior instead: (1) osteoclast-secreted S1P stimulated osteoblast production of RANK-L and bone morphogenic protein 6 and promoted osteoblast differentiation from mesenchymal stem cells[72,76]; (2) deletion of the bone matrix-degrading enzyme cathepsin K in osteoclast increased osteoblastic bone formation by inducing Sphk1 activity and S1P production[77]; and (3) the hormone calcitonin induced bone formation by osteoblast through S1P3 and did so by controlling osteoclast S1P secretion though regulation of the S1P transporter Spns2[71]. These studies have established S1P as a bona fide trophic factor for osteoblasts as well as a novel factor in the functional coupling of osteoclasts and osteoblasts [Figure 4].
Nevertheless, despite all the molecular insights, the most relevant question of all remains: What exactly are the consequences of pharmacological or genetic alteration of endogenous S1P for bone health and diseases in vivo, and which S1P receptors are responsible? This is particularly relevant if S1P-based drugs were to be considered for the therapy of osteoporosis or any other bone-related diseases. Studies in preclinical models of human diseases have shown that interventions that elevate whole body S1P concentration successfully corrected age-related osteoporosis, osteoporosis caused by estrogen deficiency, and the severe genetic osteoporosis caused by osteoprotegerin deficiency (a genetic model of human juvenile Paget disease)[75]. At least two major mechanisms have been identified behind the potent osteoanabolic actions of S1P. The first involves an osteoclast suppressive action of osteoblasts, where S1P/S1P2 signaling stimulated osteoblasts to produce Wnt oncogene analog 5 (Wnt5a) and osteoprotegerin through a p38-dependent glycogen synthase kinase-3 (GSK3)/β-catenin pathway, thus inhibiting both osteoclastogenesis and osteoclast activity. The second mechanism was even more fundamental: S1P had profound effects on the lineage commitment of the common osteoblast/adipocyte stem cell precursor by tilting the differentiation equilibrium in favor of osteoblastogenesis at the expense of adipogenesis [Figure 4]. This occurred by S1P activating the ostoblastogenic transcription factors osterix and Wnt5a and suppressing major adipogenic ones such as peroxisome proliferator-activated receptor gamma and CCAAT/enhancer-binding protein alpha. Accordingly, mice with high S1P had not only a higher bone mass but also less adipose tissue[75].
In humans, data on bone health or disease in relation to S1P are scarce. Several small observational studies have negatively associated blood S1P and certain parameters of osteoporosis and have even linked S1P to increased risk of bone fracture[78]. In over 4000 participants of the SHIP-Trend population study, we also found an inverse relation of plasma S1P to QUS-based bone stiffness as surrogate for classical bone mineral density but uncovered a positive association of plasma S1P with clinical bone formation markers[75]. This indicated that high S1P is not necessarily causally detrimental for bone mass but, instead, may serve as a counter-regulatory measure to boost decreasing bone quality.
As altering S1P metabolism by SPL inhibition has undesired side effects, S1P2 agonists have already been successfully used instead to correct osteoporosis caused by estrogen deficiency in mice[79], thereby opening new avenues for osteoporosis treatment in men. Considering the lack of drugs aimed at stimulating new bone formation rather that inhibiting its degradation (virtually all osteoporosis drugs are anti-resorptive in nature), S1P-targeting drugs may become part of the therapeutic quest for the osteoanabolic “Holy Grail” in the treatment of osteoporosis. On a broader perspective, any therapy successful at increasing new bone growth will certainly find applications not only in the treatment of primary and secondary osteoporosis but also in the therapy of rare genetic skeletal diseases, bone trauma and bone regeneration medicine, osteo-prosthetics, and bone-related tumor and metastasis medicine.
Rolls of S1P in cancer and bone metastasis
SphK/S1P metabolic pathway has been thoroughly investigated because of its implication in all stages of tumorigenesis, in cancer cell dissemination, and in the onset and development of metastasis[56,80]. Interestingly, the exact role of SphK/S1P/S1P receptor signaling in cancer-derived bone metastasis remains somehow unexplored; only a few studies propose the imbalance of S1P metabolism as a central driver of bone metastasis.
Recently, the analysis of tumor transcriptome of 3999 breast cancer patients showed that an increase in SPHK1, a suppression of SPHK2, and an increase in SPNS2 and S1PR1 were associated with high score tumors. All of these protein partners are implicated in the production and secretion of S1P and the promotion of angiogenesis[81]. S1PR1 expression was indeed elevated in different models of human breast cancer with bone metastatic potential such as luminal and basal/triple-negative subtypes of breast cancer cell lines and several breast tumors. Furthermore, breast cancer tumors with a correlation between IL-22R1 and S1PR1 expression were more prone to cause bone metastases[82]. S1PR3 expression and S1P secretion were significantly increased in MDA-MB-231 subline 1833, derived from a breast cancer-derived bone metastasis[83]. SphK1 expression was increased by TGF-β and associated to the metastatic potential of MDA-MB-231 breast cancer cell model[84] [Figure 4].
Up to a third of advanced renal cell carcinoma patients will also develop osteolytic bone metastases[85]. SphK/S1P signaling has been implicated in several aspects of the pathophysiology of renal cell carcinoma, notably in the regulation of tumor hypoxia and angiogenesis[86,87]. Moreover, upregulation of SphK1 expression was correlated to sunitinib resistance and poor prognosis of a large cohort of renal cell carcinoma patients[88]. An antibody directed against S1P (sphingomab/sonepcizumab) was proposed as a new therapy for resistant renal cell carcinoma after encouraging results in mouse models[89]. Finally, a phase 2 study of the effect of sonepcizumab in patients with advanced renal cell carcinoma who have previously failed up to three therapies, including VEGF and/or mammalian target of rapamycin inhibitors, was conducted. This study did not accomplish its primary endpoint (two-month progression-free survival of the disease), but a median overall survival of 21.7 months was monitored. Moreover, treatment with sonepcizumab was considered as safe[90].
Osteoblastic bone metastases are currently found in lung cancer patients and prostate cancer patients with advanced disease. Osteoblasts actively synthetize bone in localized zones such as ribs, clavicles, or spinal vertebrae. Newly synthetized bone is extremely fragile, generally provoking fractures and therefore intensive pain. In the case of lung cancer, the five S1P receptors were expressed in several non-small and small lung cancer-derived cell lines. Exogenous S1P had a pro-metastatic effect in these lung cancer cell models, increasing cell migration and their chemotactic responsiveness to different growth factors, especially HGF[91]. SphK/S1P metabolic pathway has been extensively studied in the case of prostate cancer[92,93]. Importantly, SphK1 activity and expression were increased in human prostate cancer resection specimens and correlated with higher prostate specific antigen levels, higher tumor volumes, and disease relapse[94]. There is only one study reporting the implication of S1P metabolic pathway in prostate cancer-derived bone metastasis. SphK1 activity was significantly increased in murine and human osteoblastic cell models. Osteoblasts triggered proliferation of different prostate cancer cell lines through extracellular secretion of S1P. In addition, osteoblast-derived S1P was able to induce chemo - or radioresistance of prostate cancer cell models. Furthermore, blockade of SphK1 inhibited the proliferative effects of osteoblasts on prostate cancer cells[95].
Finally, mixed bone metastases have zones of both active bone resorption and active bone formation. As mentioned above, a small population of breast cancer patients can suffer from mixed bone metastasis, and they can be found in, for example, colorectal cancer, which is the most common cause of death in these patients. SphK/S1P signaling has been extensively described to have oncogenic roles in colorectal cancer or colitis-associated colorectal cancer[96,97]. SphK1 activity was important for the promotion of metastasis in colorectal cancer[98]. SphK1 expression was indeed significantly increased in patients, and an elevated SphK1 was an independent predictor of distal metastasis[99]. Other components of S1P signaling such as irreversibly S1P-degrading enzyme, SPL[100] and S1P transporter, and Spns2[101] or S1P receptors such as S1P2[102] or
PROSTAGLANDINS/LEUKOTRIENES
Prostaglandins and leukotrienes belong to the super-family of eicosanoids. This family is composed of bioactive lipids that all derive from arachidonic acid (AA). AA is released from cellular membranes predominantly by cytosolic phospholipase A2α activity. Then, AA is processed by cyclooxygenases (COXs) to form prostanoids, including prostaglandins (PGs), prostacyclin (PGI2), and thromboxane A2 (TXA2), and by 5-lipoxygenase (5-LOX) to form leukotriene B4 (LTB4) and the cysteinyl leukotrienes (CysLTs):
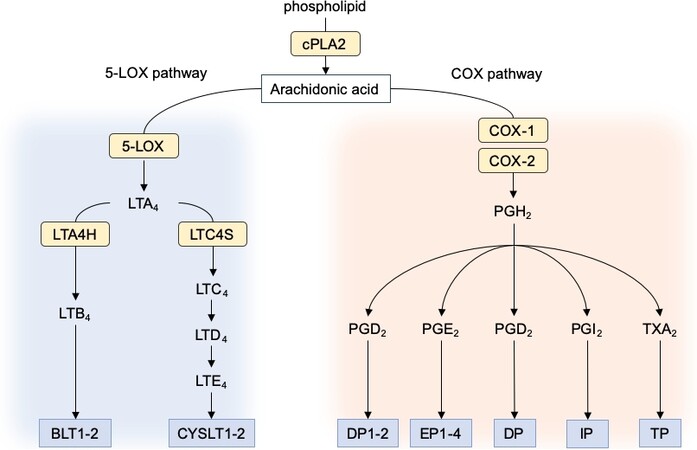
Figure 5. Overview of eicosanoid biosynthesis and receptor signaling. Arachidonic acid (AA) is liberated from the cellular membranes by cytoplasmic phospholipase A2 (cPLA2). Free AA can be metabolized by two main enzymes: cyclooxygenases (COXs) to form various prostanoids and 5-lipoxygenase (5-LOX) to form leukotrienes. In the COX pathway, the intermediate PGH2 is sequentially metabolized into prostaglandins and thromboxanes by specific prostaglandins and thromboxane synthases. In the 5-LOX pathway, AA is converted into the unstable leukotriene A4 (LTA4), which is subsequently converted to leukotriene B4 by LTA4 hydrolase (LTA4H) or to cysteinyl leukotriene LTC4 by LTC4 synthase (LTC4S). Each of the prostaglandins and leukotrienes exerts its biological effects through its cognate G protein-coupled receptor.
Structure, synthesis, and receptors
Prostaglandins are generated by the action of prostaglandin G/H synthases, colloquially known as COXs, bifunctional enzymes that contain both cyclooxygenase and peroxidase activity and exist as two distinct isoforms referred to as COX-1 and COX-2. COX isoenzymes catalyze the conversion of arachidonic acid to prostanoids, which include TXA2 and four different PGs: PGD2, PGE2, PGF2α, and PGI2. COX-1 is expressed constitutively in most cells. It is the major source of prostanoids with housekeeping functions, whereas COX-2, which is induced by inflammatory stimuli, hormones, and growth factors, is the most important source of prostanoids in inflammation and cancer[104]. Prostaglandins signal in an autocrine or paracrine manner through G-protein-coupled receptors (GPCRs) designated as DP1/2 for PGD2, EP1/2/3/4 for PGE2, FP for PGF2α, IP for PGI2, and TP for TXA2[105] [Figure 5].
Leukotrienes (LT) are generated by the 5-LOX pathway in certain types of leukocytes, such as granulocytes (neutrophils, eosinophils, and basophils) and monocytes/macrophages. 5-LOX, in conjunction with 5-LOX-activating protein (FLAP), generates the unstable intermediate leukotriene A4 (LTA4). Depending on the cellular enzymes present, LTA4 can be either converted to dihydroxy-LT LTB4 by LTA4 hydrolase or conjugated with glutathione by cysteinyl leukotriene C4 synthase (LTC4S) to generate LTC4. Subsequently, LTC4 is exported out of the cell via multidrug resistance-associated proteins 1 and 4 and further processed into cysteinyl leukotriene D4 (LTD4) and cysteinyl leukotriene E4 (LTE4). LTB4 and LTD4 are the most potent leukotrienes[106]. LTs exert their biological effects by binding to two sets of GPCRs present at the cell surface: BLT1/2 for LTB4 and CYSLT1/2 for the CysLTs. Chemotaxis, one of the principal effects of LTB4, occurs via activation of the BLT1 receptor subtype, which is the high-affinity LTB4 receptor[107]. BLT2 exhibits low affinity to LTB4 and responds in addition to various eicosanoids[108]. CYSLT1 has a high affinity for CysLTs that is higher for LTD4 than LTC4, whereas CYSLT2 has a lower overall affinity that is equal for LTC4 and LTD4[109,110]. BLT1 and CysLT1 expression is restricted to myeloid cells, whereas BLT2 and CysLT2 are expressed in a wide variety of cells [Figure 5].
Role of prostaglandins/leukotrienes in bone cells
Prostaglandins, particularly PGE2, are major regulators of bone metabolism. However, their mechanisms of action are complex as they can stimulate both bone resorption and formation and auto-amplify their effects by inducing COX-2 expression [Figure 6]. In bone, PGE2 is primarily produced by osteoblasts and contributes indirectly to osteoclastic bone resorption through the upregulation of RANK-L in osteoblasts, leading to stimulated osteoclastogenesis and bone resorption[111]. Many cytokines and growth factors known to potentiate bone resorption, such as IL-1 and IL-6, enhance PGE2 production by osteoblasts. In contrast, IL-4 and IL-13 inhibit bone resorption by suppressing PGE2 production[112]. Analysis of the four EP receptor-deficient mice revealed that PGE2 stimulates bone resorption in the cultured calvariae through the EP4-cAMP signaling pathway[113]. EP4 expression on mouse osteoblasts is required for osteoclast formation[114]. This suggests that PGE2-induced RANK-L expression is mediated through EP4 [Figure 6]. In addition, PGE2, synergistically with RANK-L and M-CSF, directly stimulates the differentiation of mouse osteoclast precursors into osteoclasts through EP2 and EP4, which are downregulated in mature osteoclasts[115,116]. This direct effect of PGE2 on human osteoclasts is however controversial[112]. Paradoxically, prostaglandins also have inhibitory effects on bone formation. PGE2 is known to inhibit bone resorption in vitro[111] and increase overall bone mass in vivo[117]. The anabolic effect of PGE2 is notably mediated by the stimulation of osteoblastic differentiation via EP4 and possibly EP2[118]. PGE2 has also been implicated in several bone-resorptive inflammatory disorders such as osteoarthritis, osteoporosis, and periodontitis[112]. PGE2 and COX-2 promote bone formation in response to mechanical loading and endogenous PGE2 participates in the recovery from osteoporosis and bone fracture[119].
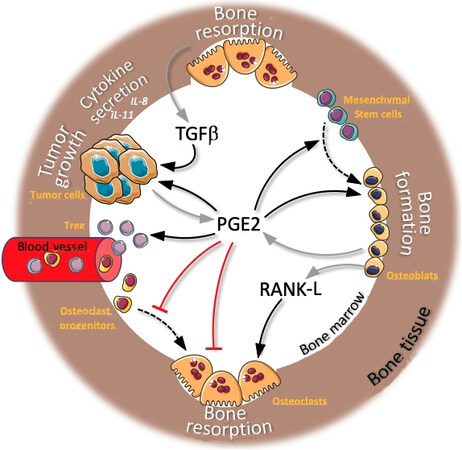
Figure 6. Prostaglandin E2 (PGE2) activity in bone metastasis microenvironment: overview of actions of PGE2 on bone cells, cancer cells, and blood cells. Black arrows indicate activities of PGE2, receptor-activated NF-κB ligand (RANK-L), and tumor growth factor β (TGFβ) on different cell types from the bone microenvironment (names in orange) resulting in multiple biological functions (text in white). Grey arrows indicate PGE2 and RANK-L secretions and TGFβ release from resorbed bone matrix. Dotted black arrows indicate cell differentiation (adipogenesis and osteoclastogenesis). Red lines indicate PGE2 inhibitory activity on osteoclast differentiation and activity.
Global deletion of 5-LOX in mice increases bone mass and protects against bone loss in the ovariectomy mouse model[120], although high-fat diet-induced bone loss was increased in 5-LOX-/- mice[121], suggesting an interaction between estrogen and leukotriene pathways. Both LTB4 and LTD4, in association with RANK-L, promote osteoclast differentiation and bone resorption[122]. Thus, 5-LOX metabolites may also act as regulators of bone metabolism. LTB4, but not CysLTs, is responsible for Aggregatibacter actinomycetemcomitans-induced bone loss in mice[123]. By promoting osteoclastic bone resorption, LTB4 may thereby inhibit bone formation. Osteoclasts express BLT1, but not BLT2, and produce LTB4, suggesting that LTB4/BLT1 signaling increases osteoclastic activity through autocrine and paracrine pathways[112]. LTB4 is involved in the induction of pain and bone damage in rheumatoid arthritis[124]. CysLTs also regulate osteoclast differentiation via CYSLT1 receptor, supporting the role of CysLT in bone loss[125,126]. Importantly, LTD4-CYSLT1 signaling activates the mitogen-activated protein kinases (MAPK)/extracellular signal-regulated kinases (Erk)/c-Jun N-terminal kinase/p38 pathway, which is a key pathway for regulating osteoclast differentiation and bone resorption. This may explain why the CYSLT1 antagonist montelukast inhibits M-CSF- and RANK-L-induced osteoclast differentiation[127]. In addition, montelukast prevents the association of RANK and TNF receptor associated factor 6 and suppresses reactive oxygen species generation, which are known to contribute to the pathophysiological development of osteoclastogenesis[128]. LTD4/CYSLT1 signaling also plays a role in inducing cellular senescence signaling in osteoblasts, a mechanism that has been described to control bone metastasis[129]. Montelukast also inhibits MMP13 expression in osteoblast, a key factor of proteolytic degradation of membrane-linked RANK-L and extracellular matrix components[130]. LTD4 induces chemotaxis and migration of CD34+ bone marrow progenitors and induces Erk/MAPK signaling in chronic lymphocytic leukemia (CLL) cells, suggesting that CysLTs may contribute to bone marrow accumulation and homing of CLL cells[131,132].
Role of prostaglandins/leukotrienes in cancer and bone metastasis
Prostaglandins
Although negligibly expressed in normal cells, COX-2 is overexpressed in many types of cancers such as lung, colorectal, breast, prostate, skin, and hepatic cancers[133]. COX-2 overexpression increases the rate of cancer recurrence[134] and reduces survival in patients[135,136]. The use of non-steroidal anti-inflammatory drugs, which inhibit PGE2 production, may reduce cancer risk of metastasis[137]. Expression of COX-2 in tumors probably occurs as an early event[138] and is related to cancer cell resistance to chemo- and radiotherapy[139]. COX-2 exerts most of its functions on tumor cells through PGE2, and elevated urinary PGE2 metabolites serve as a biomarker to predict pancreatic cancer risk[140]. PGE2 promotes tumor growth through autocrine and paracrine mechanisms by activating EP receptors present in both cancer and stromal cells. The variability of EP receptors expressed by cancer cells may influence cell response to PGE2. Typically, EP1 and EP4 are associated with tumor cell migration, invasion, and metastasis, whereas EP2 is associated with angiogenesis and immunosuppression. PGE2 supports survival and proliferation of cancer cells by upregulating Bcl-2 and epidermal growth factor receptor (EGFR). Moreover, PGE2 is an important player in tumor microenvironment, where it suppresses antitumor immunity[141]. Many in vivo studies have revealed the role of PGE2 in colon cancer carcinogenesis and progression[142-144] and showed that microsomal prostaglandin E synthase-1 (mPGES-1) deletion suppresses intestinal and breast cancer progression[145,146]. Although clinical and in vitro studies have shown a potential for the use of COX-2 inhibitors to prevent and treat malignant diseases, toxicities due to global prostanoid suppression have limited their use[147]. For this reason, EP receptors antagonists are now considered more relevant with some under clinical investigation[133] [Figure 6].
Blockade of prostaglandin synthesis with aspirin, in combination with APT102, an ADPase, decreases breast cancer and melanoma bone metastasis in mice[55,148]. B16 melanoma bone metastasis is also suppressed in mPGES-1-deficient mice, suggesting that prostaglandins may drive tumor invasion and metastasis[149]. Bone seeking cells express COX-2 and produce PGE2. TGF-β released from resorbed bone matrix enhances COX-2 expression in cancer cells, leading to a vicious cycle between bone resorption and tumor growth[150]. In addition, PGE2 also indirectly contributes to bone resorption by stimulating the secretion of pro-osteoclastic cytokines (IL-8 and IL-11) by cancer cells[151,152]. PGE2/EP4 signaling upregulates matrix metallopeptidase (MMP)-2, MMP-9, RANK-L, and Runt-related transcription factor 2 (RUNX2) and contributes to prostate cancer cell proliferation and invasion via the cAMP-protein kinase A/phosphoinositide 3-kinase (PI3K)-Akt signaling pathway[153]. Overexpression of COX-2 in TM40D mouse breast cancer cells increased bone metastasis by recruiting Treg cells through their EP2 and/or EP4 receptors[154], thus providing a favorable metastatic environment against immune system surveillance. Nevertheless, in an immunocompromised mouse model, the COX-2 inhibitor MF-tricyclic inhibited bone metastasis in mice inoculated with human breast cancer cells, suggesting a direct antitumor action of COX-2 inhibitor and/or indirect action on bone cells[155]. Blockade of PGE2-EP4 signaling in mice was shown to decrease bone metastasis, in part via the abrogation of PGE2-induced RANK-L expression in osteoblasts[156].
Leukotrienes
5-LOX is overexpressed in tissue samples of patients with bladder, breast, esophageal, kidney, oral, pancreatic, and prostate cancer, as well as in established cancer cell lines[157-161]. 5-LOX expression is present during the early neoplastic changes in cancers such as in pancreatic cancer, well before progression to invasive disease[158], supporting the role of 5-LOX in early stages of carcinogenesis. Furthermore, 5-LOX correlates with tumor stage and lymph node metastasis in colorectal adenocarcinomas[158]. Leukotrienes may modulate the initiation, progression, and metastasis of tumors through regulating the proliferation, apoptosis, migration, and invasion of cancer cells. Addition of 5-LOX expression products to tumor cells led to increased cell proliferation and activation of anti-apoptotic pathways[162]. CYSLT1 receptor expression negatively correlates with survival of patients with prostate, breast, and colon cancers and metastatic uveal melanoma[105]. Conversely, low expression of CYSLT2 is associated with poor prognosis in colon cancer. In this context, CYSLT2 signaling leads to terminal differentiation of colon carcinoma cells and growth inhibition[163]. LTC4S overexpression is found in patients with chronic myeloid leukemia, and high LTC4S levels are correlated with tumor aggressiveness in prostate cancer[164,165]. Activation of LTD4/CYSLT1 signaling has been shown to promote cell proliferation and survival through multiple parallel pathways, including GSK3β/β-catenin, PKC/Raf/ERK1, and ERK2 signaling[166,167]. LTD4 increases cancer cell survival by upregulating COX-2 expression and PGE2 production[168], indicating a crosstalk between CysLTs and the prostaglandins pathways. LTD4/CYSLT1 promotes migration and invasion in colon cancer cells by regulating MMP-9 expression[169], whereas CysLT2 signaling reduces cancer cell migration[163], suggesting opposing roles of these receptors in cancer cell motility. LTB4 levels are increased in human colon and prostate cancers[163]. Inhibition of LTB4 synthesis reduced colon cancer cell growth in patient-derived xenograft mouse model and inhibited the burden of esophageal adenocarcinoma in a rat model[170,171]. Although in vivo LTB4 may have both direct and indirect pro-tumor action, in vitro LTB4/BLT1 signaling directly stimulates colon cancer cell growth and survival through MAPK/ERK- and PI3K-Akt-dependent pathways[172,173]. LTB4, by acting on BLT2, increases mesenchymal markers and promotes epithelial-to-mesenchymal transition in several human cancer cell lines[174,175]. BLT1 appears to also affect cancer progression through immune modulation. Notably, PD-1 blockade fails to reduce melanoma growth in BLT1-/-mice due to deficiency in T cell infiltrations[176]. This might contribute to the escape of cancer patients to checkpoint inhibitor therapies. Although pharmacological leukotriene inhibitors have demonstrated promising cytotoxic and anti-proliferative effects on cancer cell lines and in animal models[162], few clinical trials have been conducted. LY293111, a well-tolerated inhibitor of BLT1, showed no significant difference in short-term survival in patients with pancreatic cancer and non-small cell lung cancer[177,178]. Conversely, cysteinyl leukotriene receptor antagonists (LTRAs), such as montelukast and zafirlukast, which are widely prescribed anti-asthmatic drugs, show more promising chemopreventive effects. A large epidemiological study on asthmatic patients showed that LTRA treatments decreased the risk of cancer development, especially lung, breast, colorectal, and liver cancers[179]. CYSLT1 antagonists also confer promising chemopreventive effects in preclinical models[180-184].
Although the role of 5-LOX metabolites in carcinogenesis is well established both in vitro and in murine models, few studies have focused on tumor progression and metastasis. MK591, a 5-LOX inhibitor, inhibits in vitro invasion of C4-2B human prostate cancer cells[99]. Increased resorption in bone explants induced by the breast-cancer cell lines MDA-MB-231 and MCF-7 was inhibited by blocking prostaglandin and leukotriene synthesis[185]. These findings suggest that prostaglandins, as well as leukotrienes, may drive osteolytic bone lesions. Unexpectedly, 5-LOX deletion in an immunocompetent orthotopic model of lung cancer resulted in increased primary tumor growth and metastasis, suggesting an antitumorigenic role for some 5-LOX products[186]. Given the crosstalk between the different prostanoid pathways and their possible opposing effects, caution should be taken in targeting these pathways in cancer.
CONCLUSIONS
Inflammation plays a prominent role in carcinogenesis and metastasis[187]. Multiples reports have demonstrated during the past decade that inflammation can promote tumor initiation, cancer cell growth, and metastasis. Remarkably, chemotherapy, radiation therapy, and surgery frequently result in the production of pro-inflammatory and pro-angiogenic factors that significantly reduce or even counteract the efficacy of those anti-cancer therapies[188-195]. Increased knowledge of the tumor microenvironment composition has brought the concept of tumors as inflammatory sites that do not heal. Beyond their direct action on both cancer cells and bone cells, LPA, S1P, and eicosanoids also promote inflammation at the bone metastatic site by inducing the production of pro-inflammatory cytokines and the recruitment of immune cells. Therefore, therapeutic strategies combining anti-inflammatory drugs and blockers of bioactive lipids deserve future investigations. However, the recent success of immunotherapies against various types of cancers has shifted the paradigm that mobilizing the immune system is beneficial against cancer. Meanwhile, loss of resolution of inflammation has recently emerged as a new mechanism of cancer pathogenesis[196-202]. This suggests that resolution of inflammation rather than blocking it might be a better therapeutic approach[203,204]. Resolution of inflammation relates to the clearance of cellular debris by macrophages resulting in reduced localized pro-inflammatory cytokines[205]. Many specialized pro-resolving mediators (SPMs) have been characterized based on their endogenous inhibitory action of inflammation such as resolvins, lipoxins, protectins, and maresins[205]. Failure of resolution vs. inflammation in carcinogenesis has been reviewed in detail[206]. Interestingly, lipoxin A4 (LXA4), which is also a bioactive lipid derived from AA following combined activities of 5-LOX and 12-LOX[207], inhibited osteoclast differentiation and resorption activity[208]. As expected, in vivo, LXA4 treatment prevented ovariectomy-induced bone loss in mice. Interestingly, serum levels of pro-osteoclastic cytokines (TNF-α, IL-1β, IL-6, and RANKL) were significantly reduced in ovariectomized animals treated with LXA4. These results support the hypothesis for direct inhibition of osteoclasts in vivo by LXA4. They also reveal an additional indirect osteoclastic action of LXA4 via reducing the production of pro-osteoclastic mediators. Therefore, LXA4 appears as a potential new tool for the treatment of osteoporosis as well as bone metastasis. Although the number of studies directly linking bioactive lipid/receptor axes to bone metastasis remains limited, the constant presence of these lipids in the tumor microenvironment as well as their potent direct activity on bone cells suggest a still underestimated action of these molecules among the complex integrated molecular mechanisms responsible for bone metastases progression. Even under the best standard of care, current targeted bone metastasis therapies are still provided in a palliative purpose with unfortunately no long-term curative benefit, highlighting the dramatic need for additional strategies. In this context, targeting bioactive lipids in combination with SPMs may potentially lead to the development of novel therapeutic approaches that undoubtedly deserve future investigations.
DECLARATIONS
Author’s contributionsMade substantial contributions to writing and correcting the manuscript: Saier L, Niazi H, Brizuela L, Levkau B, Peyruchaud O
Availability of data and materialsNot applicable.
Financial support and sponsorshipThis review was supported by grants from the INSERM (Peyruchaud O), the Université Claude Bernard Lyon 1 (Peyruchaud O), and the ANR grant BoneTAX (Grant No. ANR-20-CE14-0036-01) (Peyruchaud O). Saier L was the recipient of a fellowship from the French Ministère de l’Enseignement Supérieur et de la Recherche. Niazi H was supported by Higher Education Commission of Pakistan. This work was funded in part by the Else Kröner-Fresenius-Stiftung (Levkau B).
Conflicts of interestAll authors declared that there are no conflicts of interest.
Ethical approval and consent to participateNot applicable.
Consent for publicationNot applicable.
Copyright© The Author(s) 2021.
REFERENCES
2. Mundy GR. Metastasis to bone: causes, consequences and therapeutic opportunities. Nat Rev Cancer 2002;2:584-93.
4. Aoki J, Inoue A, Okudaira S. Two pathways for lysophosphatidic acid production. Biochim Biophys Acta 2008;1781:513-8.
5. Yung YC, Stoddard NC, Chun J. LPA receptor signaling: pharmacology, physiology, and pathophysiology. J Lipid Res 2014;55:1192-214.
6. Bektas M, Payne SG, Liu H, Goparaju S, Milstien S, Spiegel S. A novel acylglycerol kinase that produces lysophosphatidic acid modulates cross talk with EGFR in prostate cancer cells. J Cell Biol 2005;169:801-11.
7. Tomsig JL, Snyder AH, Berdyshev EV, et al. Lipid phosphate phosphohydrolase type 1 (LPP1) degrades extracellular lysophosphatidic acid in vivo. Biochem J 2009;419:611-8.
8. Saba JD. Lysophospholipids in development: Miles apart and edging in. J Cell Biochem 2004;92:967-92.
9. Pagès C, Simon M, Valet P, Saulnier-blache JS. Lysophosphatidic acid synthesis and release. Prostaglandins Other Lipid Mediat 2001;64:1-10.
10. van Meeteren LA, Ruurs P, Stortelers C, et al. Autotaxin, a secreted lysophospholipase D, is essential for blood vessel formation during development. Mol Cell Biol 2006;26:5015-22.
11. Stefan C, Jansen S, Bollen M. NPP-type ectophosphodiesterases: unity in diversity. Trends Biochem Sci 2005;30:542-50.
12. Nikitopoulou I, Oikonomou N, Karouzakis E, et al. Autotaxin expression from synovial fibroblasts is essential for the pathogenesis of modeled arthritis. J Exp Med 2012;209:925-33.
13. Dusaulcy R, Rancoule C, Grès S, et al. Adipose-specific disruption of autotaxin enhances nutritional fattening and reduces plasma lysophosphatidic acid. J Lipid Res 2011;52:1247-55.
14. Kanda H, Newton R, Klein R, Morita Y, Gunn MD, Rosen SD. Autotaxin, an ectoenzyme that produces lysophosphatidic acid, promotes the entry of lymphocytes into secondary lymphoid organs. Nat Immunol 2008;9:415-23.
15. van Meeteren LA, Ruurs P, Christodoulou E, et al. Inhibition of autotaxin by lysophosphatidic acid and sphingosine 1-phosphate. J Biol Chem 2005;280:21155-61.
16. David M, Machuca-Gayet I, Kikuta J, et al. Lysophosphatidic acid receptor type 1 (LPA1) plays a functional role in osteoclast differentiation and bone resorption activity. J Biol Chem 2014;289:6551-64.
17. Alioli CA, Demesmay L, Laurencin-Dalacieux S, et al. Expression of the type 1 lysophosphatidic acid receptor in osteoblastic cell lineage controls both bone mineralization and osteocyte specification. Biochim Biophys Acta Mol Cell Biol Lipids 2020;1865:158715.
18. Pasternack SM, von Kügelgen I, Al Aboud K, et al. G protein-coupled receptor P2Y5 and its ligand LPA are involved in maintenance of human hair growth. Nat Genet 2008;40:329-34.
19. Boucharaba A, Serre CM, Grès S, et al. Platelet-derived lysophosphatidic acid supports the progression of osteolytic bone metastases in breast cancer. J Clin Invest 2004;114:1714-25.
20. Blackburn J, Mansell JP. The emerging role of lysophosphatidic acid (LPA) in skeletal biology. Bone 2012;50:756-62.
21. Gennero I, Laurencin-Dalicieux S, Conte-Auriol F, et al. Absence of the lysophosphatidic acid receptor LPA1 results in abnormal bone development and decreased bone mass. Bone 2011;49:395-403.
22. Liu YB, Kharode Y, Bodine PV, Yaworsky PJ, Robinson JA, Billiard J. LPA induces osteoblast differentiation through interplay of two receptors: LPA1 and LPA4. J Cell Biochem 2010;109:794-800.
23. Lapierre DM, Tanabe N, Pereverzev A, et al. Lysophosphatidic acid signals through multiple receptors in osteoclasts to elevate cytosolic calcium concentration, evoke retraction, and promote cell survival. J Biol Chem 2010;285:25792-801.
24. Karagiosis SA, Karin NJ. Lysophosphatidic acid induces osteocyte dendrite outgrowth. Biochem Biophys Res Commun 2007;357:194-9.
25. McMichael BK, Meyer SM, Lee BS. c-Src-mediated phosphorylation of thyroid hormone receptor-interacting protein 6 (TRIP6) promotes osteoclast sealing zone formation. J Biol Chem 2010;285:26641-51.
26. David M, Wannecq E, Descotes F, et al. Cancer cell expression of autotaxin controls bone metastasis formation in mouse through lysophosphatidic acid-dependent activation of osteoclasts. PLoS One 2010;5:e9741.
27. Miyabe Y, Miyabe C, Iwai Y, et al. Necessity of lysophosphatidic acid receptor 1 for development of arthritis. Arthritis Rheum 2013;65:2037-47.
28. Flammier S, Peyruchaud O, Bourguillault F, et al. Osteoclast-derived autotaxin, a distinguishing factor for inflammatory bone loss. Arthritis Rheumatol 2019;71:1801-11.
29. Leblanc R, Lee SC, David M, et al. Interaction of platelet-derived autotaxin with tumor integrin αVβ3 controls metastasis of breast cancer cells to bone. Blood 2014;124:3141-50.
30. Stracke ML, Krutzsch HC, Unsworth EJ, Arestad A, Cioce V, Schiffmann E, et al. Identification, purification, and partial sequence analysis of autotaxin, a novel motility-stimulating protein. J Biol Chem 1992;267:2524-9.
31. Lin ME, Herr DR, Chun J. Lysophosphatidic acid (LPA) receptors: signaling properties and disease relevance. Prostaglandins Other Lipid Mediat 2010;91:130-8.
32. Choi JW, Herr DR, Noguchi K, et al. LPA receptors: subtypes and biological actions. Annu Rev Pharmacol Toxicol 2010;50:157-86.
33. Sedláková I, Vávrová J, Tošner J, Hanousek L. Lysophosphatidic acid (LPA)-a perspective marker in ovarian cancer. Tumour Biol 2011;32:311-6.
34. Ikeda H, Enooku K, Ohkawa R, Koike K, Yatomi Y. Plasma lysophosphatidic acid levels and hepatocellular carcinoma. Hepatology 2013;57:417-8.
35. Xu Y, Fang XJ, Casey G, Mills GB. Lysophospholipids activate ovarian and breast cancer cells. Biochem J 1995;309:933-40.
36. Lin CI, Chen CN, Huang MT, et al. Lysophosphatidic acid upregulates vascular endothelial growth factor-C and tube formation in human endothelial cells through LPA(1/3), COX-2, and NF-kappaB activation- and EGFR transactivation-dependent mechanisms. Cell Signal 2008;20:1804-14.
37. Sitohy B, Nagy JA, Dvorak HF. Anti-VEGF/VEGFR therapy for cancer: reassessing the target. Cancer Res 2012;72:1909-14.
38. Bellahcène A, Bachelier R, Detry C, Lidereau R, Clézardin P, Castronovo V. Transcriptome analysis reveals an osteoblast-like phenotype for human osteotropic breast cancer cells. Breast Cancer Res Treat 2007;101:135-48.
39. Wu X, Ma Y, Chen H, et al. Lysophosphatidic acid induces interleukin-6 and CXCL15 secretion from MLO-Y4 cells through activation of the LPA1 receptor and PKCθ signaling pathway. Int Immunopharmacol 2019;74:105664.
40. Aki Y, Kondo A, Nakamura H, Togari A. Lysophosphatidic acid-stimulated interleukin-6 and -8 synthesis through LPA1 receptors on human osteoblasts. Arch Oral Biol 2008;53:207-13.
41. Peyruchaud O, Winding B, Pécheur I, Serre CM, Delmas P, Clézardin P. Early detection of bone metastases in a murine model using fluorescent human breast cancer cells: application to the use of the bisphosphonate zoledronic acid in the treatment of osteolytic lesions. J Bone Miner Res 2001;16:2027-34.
42. Masuda A, Nakamura K, Izutsu K, et al. Serum autotaxin measurement in haematological malignancies: a promising marker for follicular lymphoma. Br J Haematol 2008;143:60-70.
43. Nakai Y, Ikeda H, Nakamura K, et al. Specific increase in serum autotaxin activity in patients with pancreatic cancer. Clin Biochem 2011;44:576-81.
44. Zhang G, Zhao Z, Xu S, Ni L, Wang X. Expression of autotaxin mRNA in human hepatocellular carcinoma. Chin Med J (Engl) 1999;112:330-2.
45. Benesch MG, Tang X, Dewald J, et al. Tumor-induced inflammation in mammary adipose tissue stimulates a vicious cycle of autotaxin expression and breast cancer progression. FASEB J 2015;29:3990-4000.
46. Benesch MG, Ko YM, Tang X, et al. Autotaxin is an inflammatory mediator and therapeutic target in thyroid cancer. Endocr Relat Cancer 2015;22:593-607.
47. Guise TA, Yin JJ, Taylor SD, et al. Evidence for a causal role of parathyroid hormone-related protein in the pathogenesis of human breast cancer-mediated osteolysis. J Clin Invest 1996;98:1544-9.
48. Aielli F, Ponzetti M, Rucci N. Bone metastasis pain, from the bench to the bedside. Int J Mol Sci 2019;20:280.
49. Yoneda T, Hata K, Nakanishi M, et al. Involvement of acidic microenvironment in the pathophysiology of cancer-associated bone pain. Bone 2011;48:100-5.
50. Zhao J, Pan HL, Li TT, Zhang YQ, Wei JY, Zhao ZQ. The sensitization of peripheral C-fibers to lysophosphatidic acid in bone cancer pain. Life Sci 2010;87:120-5.
51. Pan HL, Zhang YQ, Zhao ZQ. Involvement of lysophosphatidic acid in bone cancer pain by potentiation of TRPV1 via PKCε pathway in dorsal root ganglion neurons. Mol Pain 2010;6:85.
52. Wu JX, Yuan XM, Wang Q, Wei W, Xu MY. Rho/ROCK acts downstream of lysophosphatidic acid receptor 1 in modulating P2X3 receptor-mediated bone cancer pain in rats. Mol Pain 2016;12:174480691664492.
54. Hannun YA, Obeid LM. Principles of bioactive lipid signalling: lessons from sphingolipids. Nat Rev Mol Cell Biol 2008;9:139-50.
55. Kitatani K, Idkowiak-Baldys J, Hannun YA. The sphingolipid salvage pathway in ceramide metabolism and signaling. Cell Signal 2008;20:1010-8.
56. Hannun YA, Obeid LM. Sphingolipids and their metabolism in physiology and disease. Nat Rev Mol Cell Biol 2018;19:175-91.
57. Ramos-Perez WD, Fang V, Escalante-Alcalde D, Cammer M, Schwab SR. A map of the distribution of sphingosine 1-phosphate in the spleen. Nat Immunol 2015;16:1245-52.
58. Schwab SR, Pereira JP, Matloubian M, Xu Y, Huang Y, Cyster JG. Lymphocyte sequestration through S1P lyase inhibition and disruption of S1P gradients. Science 2005;309:1735-9.
59. Yanagida K, Hla T. Vascular and immunobiology of the circulatory sphingosine 1-phosphate gradient. Annu Rev Physiol 2017;79:67-91.
60. Sartawi Z, Schipani E, Ryan KB, Waeber C. Sphingosine 1-phosphate (S1P) signalling: Role in bone biology and potential therapeutic target for bone repair. Pharmacol Res 2017;125:232-45.
61. Vu TM, Ishizu AN, Foo JC, et al. Mfsd2b is essential for the sphingosine-1-phosphate export in erythrocytes and platelets. Nature 2017;550:524-8.
62. Proia RL, Hla T. Emerging biology of sphingosine-1-phosphate: its role in pathogenesis and therapy. J Clin Invest 2015;125:1379-87.
63. Thuy AV, Reimann CM, Hemdan NY, Gräler MH. Sphingosine 1-phosphate in blood: function, metabolism, and fate. Cell Physiol Biochem 2014;34:158-71.
64. Christoffersen C, Obinata H, Kumaraswamy SB, et al. Endothelium-protective sphingosine-1-phosphate provided by HDL-associated apolipoprotein M. Proc Natl Acad Sci U S A 2011;108:9613-8.
65. Obinata H, Kuo A, Wada Y, et al. Identification of ApoA4 as a sphingosine 1-phosphate chaperone in ApoM- and albumin-deficient mice. J Lipid Res 2019;60:1912-21.
66. Cyster JG, Schwab SR. Sphingosine-1-phosphate and lymphocyte egress from lymphoid organs. Annu Rev Immunol 2012;30:69-94.
67. Hla T, Venkataraman K, Michaud J. The vascular S1P gradient-cellular sources and biological significance. Biochim Biophys Acta 2008;1781:477-82.
68. Chun J, Hla T, Lynch KR, Spiegel S, Moolenaar WH. International Union of Basic and Clinical Pharmacology. LXXVIII. Lysophospholipid receptor nomenclature. Pharmacol Rev 2010;62:579-87.
69. Rosen H, Stevens RC, Hanson M, Roberts E, Oldstone MB. Sphingosine-1-phosphate and its receptors: structure, signaling, and influence. Annu Rev Biochem 2013;82:637-62.
70. Ishii M, Kikuta J. Sphingosine-1-phosphate signaling controlling osteoclasts and bone homeostasis. Biochim Biophys Acta 2013;1831:223-7.
71. Keller J, Catala-Lehnen P, Huebner AK, et al. Calcitonin controls bone formation by inhibiting the release of sphingosine 1-phosphate from osteoclasts. Nat Commun 2014;5:5215.
72. Ryu J, Kim HJ, Chang EJ, Huang H, Banno Y, Kim HH. Sphingosine 1-phosphate as a regulator of osteoclast differentiation and osteoclast-osteoblast coupling. EMBO J 2006;25:5840-51.
73. Ishii M, Kikuta J, Shimazu Y, Meier-Schellersheim M, Germain RN. Chemorepulsion by blood S1P regulates osteoclast precursor mobilization and bone remodeling in vivo. J Exp Med 2010;207:2793-8.
74. Ishii T, Shimazu Y, Nishiyama I, Kikuta J, Ishii M. The role of sphingosine 1-phosphate in migration of osteoclast precursors; an application of intravital two-photon microscopy. Mol Cells 2011;31:399-403.
75. Weske S, Vaidya M, Reese A, et al. Targeting sphingosine-1-phosphate lyase as an anabolic therapy for bone loss. Nat Med 2018;24:667-78.
76. Pederson L, Ruan M, Westendorf JJ, Khosla S, Oursler MJ. Regulation of bone formation by osteoclasts involves Wnt/BMP signaling and the chemokine sphingosine-1-phosphate. Proc Natl Acad Sci U S A 2008;105:20764-9.
77. Lotinun S, Kiviranta R, Matsubara T, et al. Osteoclast-specific cathepsin K deletion stimulates S1P-dependent bone formation. J Clin Invest 2013;123:666-81.
78. Bae SJ, Lee SH, Ahn SH, Kim HM, Kim BJ, Koh JM. The circulating sphingosine-1-phosphate level predicts incident fracture in postmenopausal women: a 3.5-year follow-up observation study. Osteoporos Int 2016;27:2533-41.
79. Weske S, Vaidya M, von Wnuck Lipinski K, et al. Agonist-induced activation of the S1P receptor 2 constitutes a novel osteoanabolic therapy for the treatment of osteoporosis in mice. Bone 2019;125:1-7.
80. Ogretmen B. Sphingolipid metabolism in cancer signalling and therapy. Nat Rev Cancer 2018;18:33-50.
81. Oshi M, Newman S, Tokumaru Y, et al. Intra-tumoral angiogenesis is associated with inflammation, immune reaction and metastatic recurrence in breast cancer. Int J Mol Sci 2020;21:6708.
82. Kim EY, Choi B, Kim JE, Park SO, Kim SM, Chang EJ. Interleukin-22 mediates the chemotactic migration of breast cancer cells and macrophage infiltration of the bone microenvironment by potentiating S1P/SIPR signaling. Cells 2020;9:131.
83. Filipenko I, Schwalm S, Reali L, et al. Upregulation of the S1P(3) receptor in metastatic breast cancer cells increases migration and invasion by induction of PGE(2) and EP(2)/EP(4) activation. Biochimica et biophysica acta 2016;1861:1840-51.
84. Stayrook KR, Mack JK, Cerabona D, et al. TGFβ-mediated induction of SphK1 as a potential determinant in human MDA-MB-231 breast cancer cell bone metastasis. Bonekey Rep 2015;4:719.
86. Bouquerel P, Gstalder C, Müller D, et al. Essential role for SphK1/S1P signaling to regulate hypoxia-inducible factor 2α expression and activity in cancer. Oncogenesis 2016;5:e209.
87. Gstalder C, Ader I, Cuvillier O. FTY720 (Fingolimod) Inhibits HIF1 and HIF2 Signaling, Promotes Vascular Remodeling, and Chemosensitizes in Renal Cell Carcinoma Animal Model. Mol Cancer Ther 2016;15:2465-74.
88. Xu Y, Dong B, Wang J, Zhang J, Xue W, Huang Y. Sphingosine kinase 1 overexpression contributes to sunitinib resistance in clear cell renal cell carcinoma. Oncoimmunology 2018;7:e1502130.
89. Zhang L, Wang X, Bullock AJ, et al. Anti-S1P Antibody as a Novel Therapeutic Strategy for VEGFR TKI-Resistant Renal Cancer. Clin Cancer Res 2015;21:1925-34.
90. Pal SK, Drabkin HA, Reeves JA, et al. A phase 2 study of the sphingosine-1-phosphate antibody sonepcizumab in patients with metastatic renal cell carcinoma. Cancer 2017;123:576-82.
91. Schneider G, Sellers ZP, Bujko K, Kakar SS, Kucia M, Ratajczak MZ. Novel pleiotropic effects of bioactive phospholipids in human lung cancer metastasis. Oncotarget 2017;8:58247-63.
92. Ader I, Gstalder C, Bouquerel P, et al. Neutralizing S1P inhibits intratumoral hypoxia, induces vascular remodelling and sensitizes to chemotherapy in prostate cancer. Oncotarget 2015;6:13803-21.
93. Brizuela L, Ader I, Mazerolles C, Bocquet M, Malavaud B, Cuvillier O. First evidence of sphingosine 1-phosphate lyase protein expression and activity downregulation in human neoplasm: implication for resistance to therapeutics in prostate cancer. Mol Cancer Ther 2012;11:1841-51.
94. Malavaud B, Pchejetski D, Mazerolles C, et al. Sphingosine kinase-1 activity and expression in human prostate cancer resection specimens. Eur J Cancer 2010;46:3417-24.
95. Brizuela L, Martin C, Jeannot P, et al. Osteoblast-derived sphingosine 1-phosphate to induce proliferation and confer resistance to therapeutics to bone metastasis-derived prostate cancer cells. Mol Oncol 2014;8:1181-95.
96. Bao Y, Guo Y, Zhang C, Fan F, Yang W. Sphingosine Kinase 1 and Sphingosine-1-Phosphate Signaling in Colorectal Cancer. Int J Mol Sci 2017;18:2109.
97. Grbčić P, Sedić M. Sphingosine 1-Phosphate Signaling and Metabolism in Chemoprevention and Chemoresistance in Colon Cancer. Molecules 2020;25:2436.
98. Liu SQ, Xu CY, Wu WH, et al. Sphingosine kinase 1 promotes the metastasis of colorectal cancer by inducing the epithelialmesenchymal transition mediated by the FAK/AKT/MMPs axis. Int J Oncol 2019;54:41-52.
99. Bae GE, DO SI, Kim K, Park JH, Cho S, Kim HS. Increased Sphingosine Kinase 1 Expression Predicts Distant Metastasis and Poor Outcome in Patients With Colorectal Cancer. Anticancer Res 2019;39:663-70.
100. Schwiebs A, Herrero San Juan M, Schmidt KG, et al. Cancer-induced inflammation and inflammation-induced cancer in colon: a role for S1P lyase. Oncogene 2019;38:4788-803.
101. Gu X, Jiang Y, Xue W, et al. SPNS2 promotes the malignancy of colorectal cancer cells via regulating Akt and ERK pathway. Clin Exp Pharmacol Physiol 2019;46:861-71.
102. Zhang YH, Cui SX, Wan SB, Wu SH, Qu XJ. Increased S1P induces S1PR2 internalization to blunt the sensitivity of colorectal cancer to 5-fluorouracil via promoting intracellular uracil generation. Acta Pharmacol Sin 2021;42:460-9.
103. Olesch C, Sirait-Fischer E, Berkefeld M, et al. S1PR4 ablation reduces tumor growth and improves chemotherapy via CD8+ T cell expansion. J Clin Invest 2020;130:5461-76.
104. Mitchell JA, Warner TD. Cyclo-oxygenase-2: pharmacology, physiology, biochemistry and relevance to NSAID therapy. Br J Pharmacol 1999;128:1121-32.
107. Yokomizo T, Izumi T, Chang K, Takuwa Y, Shimizu T. A G-protein-coupled receptor for leukotriene B4 that mediates chemotaxis. Nature 1997;387:620-4.
108. Bäck M, Powell WS, Dahlén SE, et al. Update on leukotriene, lipoxin and oxoeicosanoid receptors: IUPHAR Review 7. Br J Pharmacol 2014;171:3551-74.
109. Lynch KR, O'Neill GP, Liu Q, et al. Characterization of the human cysteinyl leukotriene CysLT1 receptor. Nature 1999;399:789-93.
110. Heise CE, O'Dowd BF, Figueroa DJ, et al. Characterization of the human cysteinyl leukotriene 2 receptor. J Biol Chem 2000;275:30531-6.
111. Pilbeam CC, Harrison JR, Raisz LG. Chapter 54 - Prostaglandins and Bone Metabolism. In: Bilezikian JP, Raisz LG, Rodan GA, editors. Principles of Bone Biology (Second Edition). San Diego: Academic Press; 2002. p. 979-94.
112. Hikiji H, Ishii S, Yokomizo T, Takato T, Shimizu T. A distinctive role of the leukotriene B4 receptor BLT1 in osteoclastic activity during bone loss. Proc Natl Acad Sci U S A 2009;106:21294-9.
113. Miyaura C, Inada M, Suzawa T, et al. Impaired bone resorption to prostaglandin E2 in prostaglandin E receptor EP4-knockout mice. J Biol Chem 2000;275:19819-23.
114. Sakuma Y, Tanaka K, Suda M, et al. Crucial involvement of the EP4 subtype of prostaglandin E receptor in osteoclast formation by proinflammatory cytokines and lipopolysaccharide. J Bone Miner Res 2000;15:218-27.
115. Wani MR, Fuller K, Kim NS, Choi Y, Chambers T. Prostaglandin E2 cooperates with TRANCE in osteoclast induction from hemopoietic precursors: synergistic activation of differentiation, cell spreading, and fusion. Endocrinology 1999;140:1927-35.
116. Kobayashi Y, Take I, Yamashita T, et al. Prostaglandin E2 receptors EP2 and EP4 are downregulated during differentiation of mouse osteoclasts from their precursors. J Biol Chem 2005;280:24035-42.
117. Yoshida K, Oida H, Kobayashi T, et al. Stimulation of bone formation and prevention of bone loss by prostaglandin E EP4 receptor activation. Proc Natl Acad Sci U S A 2002;99:4580-5.
118. Alander CB, Raisz LG. Effects of selective prostaglandins E2 receptor agonists on cultured calvarial murine osteoblastic cells. Prostaglandins Other Lipid Mediat 2006;81:178-83.
119. Forwood MR. Inducible cyclo-oxygenase (COX-2) mediates the induction of bone formation by mechanical loading in vivo. J Bone Miner Res 1996;11:1688-93.
120. Bonewald LF, Flynn M, Qiao M, Dallas MR, Mundy GR, Boyce BF. Mice lacking 5-lipoxygenase have increased cortical bone thickness. Adv Exp Med Biol 1997;433:299-302.
121. Le P, Kawai M, Bornstein S, DeMambro VE, Horowitz MC, Rosen CJ. A high-fat diet induces bone loss in mice lacking the Alox5 gene. Endocrinology 2012;153:6-16.
122. Moura AP, Taddei SR, Queiroz-Junior CM, et al. The relevance of leukotrienes for bone resorption induced by mechanical loading. Bone 2014;69:133-8.
123. Madeira MFM, Queiroz-Junior CM, Corrêa JD, et al. The role of 5-lipoxygenase in Aggregatibacter actinomycetemcomitans-induced alveolar bone loss. J Clin Periodontol 2017;44:793-802.
124. Zheng LX, Li KX, Hong FF, Yang SL. Pain and bone damage in rheumatoid arthritis: role of leukotriene B4. Clinical and experimental rheumatology. 2019;37:872-8.
125. Lee JM, Park H, Noh AL, et al. 5-Lipoxygenase mediates RANKL-induced osteoclast formation via the cysteinyl leukotriene receptor 1. J Immunol 2012;189:5284-92.
126. Kang JH, Lim H, Lee DS, Yim M. Montelukast inhibits RANKLinduced osteoclast formation and bone loss via CysLTR1 and P2Y12. Mol Med Rep 2018;18:2387-98.
127. Zheng C, Shi X. Cysteinyl leukotriene receptor 1 (cysLT1R) regulates osteoclast differentiation and bone resorption. Artif Cells Nanomed Biotechnol 2018;46:S64-70.
128. Lee NK, Choi YG, Baik JY, et al. A crucial role for reactive oxygen species in RANKL-induced osteoclast differentiation. Blood 2005;106:852-9.
129. Luo X, Fu Y, Loza AJ, et al. Stromal-initiated changes in the bone promote metastatic niche development. Cell Rep 2016;14:82-92.
130. Wei J, Chen S, Huang C, et al. The cysteinyl leukotriene receptor 1 (CysLT1R) antagonist montelukast suppresses matrix metalloproteinase-13 expression induced by lipopolysaccharide. Int Immunopharmacol 2018;55:193-7.
131. Boehmler AM, Drost A, Jaggy L, et al. The CysLT1 ligand leukotriene D4 supports alpha4beta1- and alpha5beta1-mediated adhesion and proliferation of CD34+ hematopoietic progenitor cells. J Immunol 2009;182:6789-98.
132. Drost AC, Seitz G, Boehmler A, et al. The G protein-coupled receptor CysLT1 mediates chemokine-like effects and prolongs survival in chronic lymphocytic leukemia. Leuk Lymphoma 2012;53:665-73.
133. Finetti F, Travelli C, Ercoli J, Colombo G, Buoso E, Trabalzini L. Prostaglandin E2 and cancer: insight into tumor progression and immunity. Biology (Basel) 2020;9:434.
134. Montezuma MAP, Fonseca FP, Benites BM, et al. COX-2 as a determinant of lower disease-free survival for patients affected by ameloblastoma. Pathol Res Pract 2018;214:907-13.
135. Sun H, Zhang X, Sun D, et al. COX-2 expression in ovarian cancer: an updated meta-analysis. Oncotarget 2017;8:88152-62.
136. Esbona K, Yi Y, Saha S, et al. The Presence of Cyclooxygenase 2, Tumor-Associated Macrophages, and Collagen Alignment as Prognostic Markers for Invasive Breast Carcinoma Patients. Am J Pathol 2018;188:559-73.
137. Rothwell PM, Wilson M, Price JF, Belch JF, Meade TW, Mehta Z. Effect of daily aspirin on risk of cancer metastasis: a study of incident cancers during randomised controlled trials. Lancet 2012;379:1591-601.
138. Charalambous MP, Maihöfner C, Bhambra U, Lightfoot T, Gooderham NJ. Colorectal Cancer Study Group. Upregulation of cyclooxygenase-2 is accompanied by increased expression of nuclear factor-kappa B and I kappa B kinase-alpha in human colorectal cancer epithelial cells. Br J Cancer 2003;88:1598-604.
139. Harris RE. Cyclooxygenase-2 (cox-2) blockade in the chemoprevention of cancers of the colon, breast, prostate, and lung. Inflammopharmacology 2009;17:55-67.
140. Cui Y, Shu XO, Li HL, et al. Prospective study of urinary prostaglandin E2 metabolite and pancreatic cancer risk. Int J Cancer 2017;141:2423-9.
141. Miao J, Lu X, Hu Y, et al. Prostaglandin E(2) and PD-1 mediated inhibition of antitumor CTL responses in the human tumor microenvironment. Oncotarget 2017;8:89802-10.
142. Hansen-Petrik MB, McEntee MF, Jull B, Shi H, Zemel MB, Whelan J. Prostaglandin E(2) protects intestinal tumors from nonsteroidal anti-inflammatory drug-induced regression in Apc(Min/+) mice. Cancer Res 2002;62:403-8.
143. Myung SJ, Rerko RM, Yan M, et al. 15-Hydroxyprostaglandin dehydrogenase is an in vivo suppressor of colon tumorigenesis. Proc Natl Acad Sci U S A 2006;103:12098-102.
144. Yan M, Myung SJ, Fink SP, et al. 15-Hydroxyprostaglandin dehydrogenase inactivation as a mechanism of resistance to celecoxib chemoprevention of colon tumors. Proc Natl Acad Sci U S A 2009;106:9409-13.
145. Nakanishi M, Menoret A, Tanaka T, et al. Selective PGE(2) suppression inhibits colon carcinogenesis and modifies local mucosal immunity. Cancer Prev Res (Phila) 2011;4:1198-208.
146. Howe LR, Subbaramaiah K, Kent CV, et al. Genetic deletion of microsomal prostaglandin E synthase-1 suppresses mouse mammary tumor growth and angiogenesis. Prostaglandins Other Lipid Mediat 2013;106:99-105.
147. O'Callaghan G, Houston A. Prostaglandin E2 and the EP receptors in malignancy: possible therapeutic targets? Br J Pharmacol 2015;172:5239-50.
148. Uluçkan O, Eagleton MC, Floyd DH, et al. APT102, a novel adpase, cooperates with aspirin to disrupt bone metastasis in mice. J Cell Biochem 2008;104:1311-23.
149. Inada M, Takita M, Yokoyama S, et al. Direct Melanoma Cell Contact Induces Stromal Cell Autocrine Prostaglandin E2-EP4 Receptor Signaling That Drives Tumor Growth, Angiogenesis, and Metastasis. J Biol Chem 2015;290:29781-93.
150. Hiraga T, Myoui A, Choi ME, Yoshikawa H, Yoneda T. Stimulation of cyclooxygenase-2 expression by bone-derived transforming growth factor-beta enhances bone metastases in breast cancer. Cancer Res 2006;66:2067-73.
151. Singh B, Berry JA, Shoher A, Lucci A. COX-2 induces IL-11 production in human breast cancer cells. J Surg Res 2006;131:267-75.
152. Singh B, Berry JA, Vincent LE, Lucci A. Involvement of IL-8 in COX-2-mediated bone metastases from breast cancer. J Surg Res 2006;134:44-51.
153. Xu S, Zhou W, Ge J, Zhang Z. Prostaglandin E2 receptor EP4 is involved in the cell growth and invasion of prostate cancer via the cAMPPKA/PI3KAkt signaling pathway. Mol Med Rep 2018;17:4702-12.
154. Karavitis J, Zhang M. COX2 regulation of breast cancer bone metastasis. Oncoimmunology 2013;2:e23129.
155. Singh B, Berry JA, Shoher A, Ayers GD, Wei C, Lucci A. COX-2 involvement in breast cancer metastasis to bone. Oncogene 2007;26:3789-96.
156. Watanabe K, Tominari T, Hirata M, et al. Abrogation of prostaglandin E-EP4 signaling in osteoblasts prevents the bone destruction induced by human prostate cancer metastases. Biochem Biophys Res Commun 2016;478:154-61.
157. Gupta S, Srivastava M, Ahmad N, Sakamoto K, Bostwick DG, Mukhtar H. Lipoxygenase-5 is overexpressed in prostate adenocarcinoma. Cancer 2001;91:737-43.
158. Knab LM, Grippo PJ, Bentrem DJ. Involvement of eicosanoids in the pathogenesis of pancreatic cancer: the roles of cyclooxygenase-2 and 5-lipoxygenase. World J Gastroenterol 2014;20:10729-39.
159. Yoshimura R, Matsuyama M, Tsuchida K, Kawahito Y, Sano H, Nakatani T. Expression of lipoxygenase in human bladder carcinoma and growth inhibition by its inhibitors. J Urol 2003;170:1994-9.
160. Hoque A, Lippman SM, Wu TT, et al. Increased 5-lipoxygenase expression and induction of apoptosis by its inhibitors in esophageal cancer: a potential target for prevention. Carcinogenesis 2005;26:785-91.
161. Matsuyama M, Yoshimura R, Mitsuhashi M, et al. 5-Lipoxygenase inhibitors attenuate growth of human renal cell carcinoma and induce apoptosis through arachidonic acid pathway. Oncol Rep 2005;14:73-9.
162. Steinhilber D, Fischer AS, Metzner J, et al. 5-lipoxygenase: underappreciated role of a pro-inflammatory enzyme in tumorigenesis. Front Pharmacol 2010;1:143.
163. Magnusson C, Ehrnström R, Olsen J, Sjölander A. An increased expression of cysteinyl leukotriene 2 receptor in colorectal adenocarcinomas correlates with high differentiation. Cancer Res 2007;67:9190-8.
164. Tornhamre S, Stenke L, Granzelius A, et al. Inverse relationship between myeloid maturation and leukotriene C4 synthase expression in normal and leukemic myelopoiesis-consistent overexpression of the enzyme in myeloid cells from patients with chronic myeloid leukemia. Exp Hematol 2003;31:122-30.
165. Ali HEA, Lung PY, Sholl AB, et al. Dysregulated gene expression predicts tumor aggressiveness in African-American prostate cancer patients. Sci Rep 2018;8:16335.
166. Paruchuri S, Hallberg B, Juhas M, Larsson C, Sjölander A. Leukotriene D(4) activates MAPK through a Ras-independent but PKCepsilon-dependent pathway in intestinal epithelial cells. J Cell Sci 2002;115:1883-93.
167. Mezhybovska M, Wikström K, Ohd JF, Sjölander A. The inflammatory mediator leukotriene D4 induces beta-catenin signaling and its association with antiapoptotic Bcl-2 in intestinal epithelial cells. J Biol Chem 2006;281:6776-84.
168. Massoumi R, Nielsen CK, Azemovic D, Sjölander A. Leukotriene D4-induced adhesion of Caco-2 cells is mediated by prostaglandin E2 and upregulation of α2β1-integrin. Exp Cell Res 2003;289:342-51.
169. Salim T, Sand-Dejmek J, Sjölander A. The inflammatory mediator leukotriene D4 induces subcellular β-catenin translocation and migration of colon cancer cells. Exp Cell Res 2014;321:255-66.
170. Chen X, Li N, Wang S, et al. Leukotriene A4 hydrolase in rat and human esophageal adenocarcinomas and inhibitory effects of bestatin. J Natl Cancer Inst 2003;95:1053-61.
171. Zhao S, Yao K, Li D, et al. Inhibition of LTA4H by bestatin in human and mouse colorectal cancer. EBioMedicine 2019;44:361-74.
172. Tong WG, Ding XZ, Talamonti MS, Bell RH, Adrian TE. LTB4 stimulates growth of human pancreatic cancer cells via MAPK and PI-3 kinase pathways. Biochem Biophys Res Commun 2005;335:949-56.
173. Ihara A, Wada K, Yoneda M, Fujisawa N, Takahashi H, Nakajima A. Blockade of leukotriene B4 signaling pathway induces apoptosis and suppresses cell proliferation in colon cancer. J Pharmacol Sci 2007;103:24-32.
174. Kim H, Choi JA, Kim JH. Ras promotes transforming growth factor-β (TGF-β)-induced epithelial-mesenchymal transition via a leukotriene B4 receptor-2-linked cascade in mammary epithelial cells. J Biol Chem 2014;289:22151-60.
175. Tian W, Jiang X, Sung YK, et al. Phenotypically Silent Bone Morphogenetic Protein Receptor 2 Mutations Predispose Rats to Inflammation-Induced Pulmonary Arterial Hypertension by Enhancing the Risk for Neointimal Transformation. Circulation 2019;140:1409-25.
176. Chheda ZS, Sharma RK, Jala VR, Luster AD, Haribabu B. Chemoattractant Receptors BLT1 and CXCR3 Regulate Antitumor Immunity by Facilitating CD8+ T Cell Migration into Tumors. J Immunol 2016;197:2016-26.
177. Edelman MJ, Watson D, Wang X, et al. Eicosanoid modulation in advanced lung cancer: cyclooxygenase-2 expression is a positive predictive factor for celecoxib + chemotherapy--Cancer and Leukemia Group B Trial 30203. J Clin Oncol 2008;26:848-55.
178. Jänne PA, Paz-Ares L, Oh Y, et al. Randomized, double-blind, phase II trial comparing gemcitabine-cisplatin plus the LTB4 antagonist LY293111 versus gemcitabine-cisplatin plus placebo in first-line non-small-cell lung cancer. J Thorac Oncol 2014;9:126-31.
179. Tsai MJ, Wu PH, Sheu CC, et al. Cysteinyl leukotriene receptor antagonists decrease cancer risk in asthma patients. Sci Rep 2016;6:23979.
180. Gunning WT, Kramer PM, Steele VE, Pereira MA. Chemoprevention by lipoxygenase and leukotriene pathway inhibitors of vinyl carbamate-induced lung tumors in mice. Cancer Res 2002;62:4199-201.
181. Nozaki M, Yoshikawa M, Ishitani K, et al. Cysteinyl leukotriene receptor antagonists inhibit tumor metastasis by inhibiting capillary permeability. Keio J Med 2010;59:10-8.
182. Jose MA, Amathi R, Sathyamurthy D, Kumar BN. Chemopreventive effect of montelukast in n-nitroso n-methyl urea induced mammary carcinogenesis in female Sprague-Dawley rats. Indian J Pharmacol 2013;45:286-8.
183. Savari S, Liu M, Zhang Y, Sime W, Sjölander A. CysLT(1)R antagonists inhibit tumor growth in a xenograft model of colon cancer. PLoS One 2013;8:e73466.
184. Satapathy SR, Sjölander A. Cysteinyl leukotriene receptor 1 promotes 5-fluorouracil resistance and resistance-derived stemness in colon cancer cells. Cancer Lett 2020;488:50-62.
185. Tumber A, Morgan HM, Meikle MC, Hill PA. Human breast-cancer cells stimulate the fusion, migration and resorptive activity of osteoclasts in bone explants. Int J Cancer 2001;91:650-3.
186. Poczobutt JM, Nguyen TT, Hanson D, et al. Deletion of 5-Lipoxygenase in the Tumor Microenvironment Promotes Lung Cancer Progression and Metastasis through Regulating T Cell Recruitment. J Immunol 2016;196:891-901.
188. Fishbein A, Wang W, Yang H, et al. Resolution of eicosanoid/cytokine storm prevents carcinogen and inflammation-initiated hepatocellular cancer progression. Proc Natl Acad Sci U S A 2020;117:21576-87.
189. Camphausen K, Moses MA, Beecken WD, Khan MK, Folkman J, O’Reilly MS. Radiation therapy to a primary tumor accelerates metastatic growth in mice. Cancer Res 2001;61:2207-11.
190. Chang J, Bhasin SS, Bielenberg DR, et al. Chemotherapy-generated cell debris stimulates colon carcinoma tumor growth via osteopontin. FASEB J 2019;33:114-25.
191. Filippou PS, Karagiannis GS. Cytokine storm during chemotherapy: a new companion diagnostic emerges? Oncotarget 2020;11:213-5.
192. Gartung A, Yang J, Sukhatme VP, et al. Suppression of chemotherapy-induced cytokine/lipid mediator surge and ovarian cancer by a dual COX-2/sEH inhibitor. Proc Natl Acad Sci U S A 2019;116:1698-703.
193. Karagiannis GS, Pastoriza JM, Wang Y, et al. Neoadjuvant chemotherapy induces breast cancer metastasis through a TMEM-mediated mechanism. Sci Transl Med 2017;9:eaan0026.
194. Shaked Y. The pro-tumorigenic host response to cancer therapies. Nat Rev Cancer 2019;19:667-85.
195. Volk-Draper L, Hall K, Griggs C, et al. Paclitaxel therapy promotes breast cancer metastasis in a TLR4-dependent manner. Cancer Res 2014;74:5421-34.
196. Sulciner ML, Serhan CN, Gilligan MM, et al. Resolvins suppress tumor growth and enhance cancer therapy. J Exp Med 2018;215:115-40.
197. Gilligan MM, Gartung A, Sulciner ML, et al. Aspirin-triggered proresolving mediators stimulate resolution in cancer. Proc Natl Acad Sci U S A 2019;116:6292-7.
198. Liu H, Zeng J, Huang W, et al. Colorectal cancer is associated with a deficiency of Lipoxin A(4), an endogenous anti-inflammatory mediator. J Cancer 2019;10:4719-30.
199. Panigrahy D, Edin ML, Lee CR, et al. Epoxyeicosanoids stimulate multiorgan metastasis and tumor dormancy escape in mice. J Clin Invest 2012;122:178-91.
200. Serhan CN, Levy BD. Resolvins in inflammation: emergence of the pro-resolving superfamily of mediators. J Clin Invest 2018;128:2657-69.
201. Sulciner ML, Gartung A, Gilligan MM, Serhan CN, Panigrahy D. Targeting lipid mediators in cancer biology. Cancer Metastasis Rev 2018;37:557-72.
202. Ye Y, Scheff NN, Bernabé D, et al. Anti-cancer and analgesic effects of resolvin D2 in oral squamous cell carcinoma. Neuropharmacology 2018;139:182-93.
203. Serhan CN. The resolution of inflammation: the devil in the flask and in the details. FASEB J 2011;25:1441-8.
205. Serhan CN. Pro-resolving lipid mediators are leads for resolution physiology. Nature 2014;510:92-101.
206. Fishbein A, Hammock BD, Serhan CN, Panigrahy D. Carcinogenesis: Failure of resolution of inflammation? Pharmacol Ther 2021;218:107670.
207. Chiang N, Serhan CN, Dahlén SE, et al. The lipoxin receptor ALX: potent ligand-specific and stereoselective actions in vivo. Pharmacol Rev 2006;58:463-87.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Saier L, Niazi H, Brizuela L, Levkau B, Peyruchaud O. Bioactive lipids and cancer metastasis to bone. J Cancer Metastasis Treat 2021;7:43. http://dx.doi.org/10.20517/2394-4722.2021.87
AMA Style
Saier L, Niazi H, Brizuela L, Levkau B, Peyruchaud O. Bioactive lipids and cancer metastasis to bone. Journal of Cancer Metastasis and Treatment. 2021; 7: 43. http://dx.doi.org/10.20517/2394-4722.2021.87
Chicago/Turabian Style
Saier, Lou, Hira Niazi, Leyre Brizuela, Bodo Levkau, Olivier Peyruchaud. 2021. "Bioactive lipids and cancer metastasis to bone" Journal of Cancer Metastasis and Treatment. 7: 43. http://dx.doi.org/10.20517/2394-4722.2021.87
ACS Style
Saier, L.; Niazi H.; Brizuela L.; Levkau B.; Peyruchaud O. Bioactive lipids and cancer metastasis to bone. J. Cancer. Metastasis. Treat. 2021, 7, 43. http://dx.doi.org/10.20517/2394-4722.2021.87
About This Article
Special Issue
Copyright

Data & Comments
Data
0

 Cite This Article 8 clicks
Cite This Article 8 clicks

 Like This Article 1
likes
Like This Article 1
likes






Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.