
Clinical implications of the molecular characterization of intraductal papillary mucinous neoplasms of the pancreas

Abstract
Intraductal papillary mucinous neoplasm (IPMN) is a pre-malignant, mucin-producing epithelial lesion arising from pancreatic ducts. Observational reports define IPMN behavior as ranging from indolent, asymptomatic lesions to dysplasia that sometimes degenerate into pancreatic adenocarcinoma. The goal of IPMN management is risk-reducing surgery for high-risk cysts and observation of the remainder. Discriminating high- from low-risk IPMN disease still relies on imaging and clinical cyst characteristics. Here, we review the accepted classification of IPMN including the most common histological subtypes, their clinical features, and currently-accepted high-risk phenotypes. We then deeply examine the known molecular landscape of IPMN, which has largely been derived from post-resection analysis. This includes those gene variants unique to IPMN, chiefly GNAS and RNF43, but also examines the overlap between IPMN and conventional pancreatic adenocarcinoma. Utilizing molecular markers in the clinical setting relies on endoscopically-obtained cyst fluid and presumes that it accurately represents the molecular characteristics of the cystic epithelium. We synthesize existing data on mutational analysis from IPMN cyst fluid and consider the benefits and proper role of current commercially-available cyst fluid molecular analysis kits. We conclude that carefully interpreted molecular analysis of resected IPMN tissue reveals insights into its biology and natural history while cyst fluid analysis offers prognostication and data to guide treatment decisions. However, knowledge gaps remain, especially in characterizing IPMN molecular heterogeneity, time to progression, and correlating cyst fluid genotype data with surveillance strategies. As such, substantial additional research is required before the promise of true molecular guidance of IPMN management can be realized.
Keywords
INTRODUCTION
Intraductal papillary mucinous neoplasm (IPMN) is defined as a grossly visible, mucin-producing epithelial neoplasm arising from pancreatic ducts[1,2]. Although long recognized as a pathologic entity under various terms such as mucinous duct ectasia, mucin hypersecreting tumor, or intraductal papillary mucinous tumors, IPMN was first codified as a unique entity by the World Health Organization in 1996[3]. Following adoption of this standard definition, knowledge of IPMN including its characteristics and natural history has greatly expanded. Initial efforts focused on describing the histopathologic features of IPMN and the prevalence of associated invasive pancreatic adenocarcinoma. Determining the proper timing for surveillance of IPMNs and surgical intervention remain ongoing challenges. Recently, improving tools for genomic analysis has allowed a much deeper understanding of the underlying biology of these pre-malignant lesions. However, the molecular origin of IPMN and its implication for IPMN behavior remain poorly understood.
Postmortem analysis demonstrates that cystic masses in the pancreas are exceedingly common and strongly correlate with age[4]. Due to the increased use, availability, and quality of cross-sectional diagnostic imaging, more pancreatic cystic lesions are being diagnosed in patients with unclear clinical relevance[5-7]. In patients without a history of pancreatitis, IPMN is by far the most common incidentally-diagnosed cystic lesion of the pancreas. IPMN is, by definition, a premalignant lesion, in which pancreatic adenocarcinoma can arise within these cysts. However, the majority of IPMNs do not progress to invasive disease or even high-grade dysplasia; thus, there are no screening recommendations for detection of IPMN in standard risk individuals[8,9]. As most IPMNs are incidentally discovered, the vast majority are asymptomatic at the time of diagnosis. In such patients, recommendations are individualized and can involve several modalities, including imaging, endoscopy, and rarely, immediate surgery. Molecular analysis of IPMN can occur following surgical resection or via endoscopic sampling. Surgical sampling is advantageous as cyst tissue is readily available and can be directly examined; however, this requires resection. In the non-operative setting, endoscopic ultrasound (EUS) can be used to locate the pancreatic lesion and fine needle aspiration (FNA) can obtain cyst endothelial tissue in select cases. However, EUS-guided aspiration is most commonly used to obtain cyst fluid. Biochemical analysis of aspirated pancreatic cyst fluid has long been utilized to discriminate mucinous vs. serous lesions[10] but modern tools have allowed molecular assays as well. While cyst fluid of many IPMNs is aspirated, as discussed below, the resulting personalized molecular data infrequently alter treatment plans in the current era. This is in part because of an incomplete understanding of IPMN biology and malignant potential[5,11,12], as well as limited molecular analysis tools and their associated costs. The role of molecular analysis in IPMN management is an area of ongoing investigation and continues to rapidly evolve. This manuscript will review the progress in defining IPMN as a clinical entity, the current molecular understanding of the disease, and available means to assess these findings and their clinical utility in patients with IPMN.
OVERVIEW OF IPMN MANAGEMENT
Current guidelines for management are based on three decades of accrued data on the correlation of clinical and radiographic features with IPMN natural history. The goal of IPMN management is to prevent progression to overt pancreatic adenocarcinoma while avoiding unnecessary surgery or overly burdensome surveillance. Recent technological advances allowing rapid genetic characterization of cyst fluid have been utilized for research purposes and are beginning to be used in the clinical setting as well. Commercial assays for cyst fluid genetic analysis are now in widespread use[13-17] although evidence supporting routine employment is lacking. Despite these advances, management principles remain focused on cross-sectional imaging and endoscopic cyst characteristics. While many groups have offered guidelines, the most widely accepted international consensus recommendations can be summarized [Figure 1][18-20]. In addition, areas where input from molecular analysis may be particularly useful have been indicated.
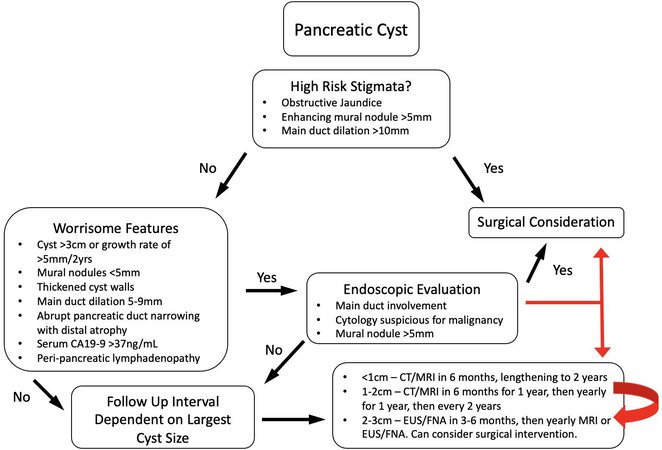
Figure 1. Summary of clinical management of pancreatic cysts. Adapted from Tanaka et al.[20] Pancreatology 2017. Current clinical management guidelines for pancreatic cystic lesions summarized in a flow chart. Red arrows denote specific areas in the flow chart where novel molecular data may be able to assist in clinical decision making.
These strategies have greatly clarified and improved the management of IPMN, but some patients still develop late-stage pancreatic adenocarcinoma despite careful surveillance. Moreover, 60%-80% of patients submitted for resection lack high-grade dysplasia or early invasive disease[21], implying surgery may not have been necessary at that time. Finally, most IPMNs seem to convey a field effect on the pancreas with multifocal disease and recurrence being the norm. Thus, even patients undergoing successful surgery still require ongoing observation. These features greatly complicate IPMN treatment and surveillance. It is hoped that better molecular understanding of IPMN will clarify these questions and guide improved management strategies in the future. This work reviews the molecular characterization of IPMN, mutational information from cyst fluid and resection specimen analysis, and the clinical implications of IPMN in the context of histologic subtype. Furthermore, it discusses commercially available genetic analysis kits and their utility and niche in clinical decision-making.
CONVENTIONAL CLASSIFICATION OF IPMN
Several classification schemes have been used to describe IPMN: anatomic examination, histology, spectrum of dysplasia and others. Together these classification systems are used in clinical guidelines and are commonly reported in the IPMN literature but can be incongruent. As a result, correlating these classifications with natural history, prognosis, and malignant potential of IPMN is challenging and inconsistent[15,22-24]. Molecular designations are increasingly being recognized but are yet to be comprehensively integrated into classifying IPMN. Importantly, recent studies have consistently found IPMN to be a heterogeneous lesion, with multiple geographically and genetically distinct regions residing within a single cyst or group of cysts. It seems likely that this contributes to the inconsistencies earlier studies observed between IPMN classification and behavior[25,26].
Anatomic classification - main duct, branch duct, or mixed
Macroscopic examination is the basis for classifying IPMN as a main duct (MD), branch duct (BD), or mixed lesion[5]. This has been consistently recognized as an important clinical factor from the earliest guidelines[27] to the present day[19,20]. In resected IPMN specimens, early studies reported a higher risk of malignancy among main duct lesions (31%-70%) as opposed to branch-duct lesions (3%-25%)[5,11,12,28]. Risks in mixed IPMN are generally considered analogous to main duct IPMN[29,30]. Sub-group analysis of BD-IPMN has greatly informed current management strategies[31-33] with significantly increased rates of invasive adenocarcinoma found in BD-IPMNs with (1) cyst size greater than 3 cm; (2) presence of a mural nodule; and (3) associated dilation of the main pancreatic duct (i.e., mixed IPMN)[31].
As noted above, resection is advocated for MD-IPMN in acceptable surgical candidates[18,19,34], which sometimes entails total pancreatectomy. Management of BD-IPMN remains more nuanced. The previously described “high risk stigmata” and “worrisome features” based on imaging and endoscopic findings drive decision-making in these patients[20]. The Pancreatic Surgery Consortium clarified the relative risks of these possible IPMN features in 2018, with the presence of jaundice most predictive of high-risk (i.e., high-grade dysplasia or invasive) lesions (57/58), while cyst size > 3.0 cm, mural nodule, pain symptoms, and weight loss were also associated with high-risk lesions to a lesser degree[35]. Another study reported the presence of a radiographic mural nodule was the most predictive feature of invasive disease[36].
Histology and dysplasia distinction
Histological analysis of resected IPMN has defined four primary distinct subtypes: gastric, intestinal, pancreatobiliary, and oncocytic[37-42]. The appearance of the epithelial cyst lining on microscopy and differential mucin protein expression profiles form the basis of histologic subtype differentiation[22,37,43] [Table 1]. Although conventional pathology reporting often conveys a ubiquitous histologic subtype to a particular IPMN, multiregional examination has revealed that multiple histologic subtypes can exist within a single lesion[25,38]. As described below, this can have molecular implications as well. Additionally, some have suggested the oncocytic subtype is a distinct entity from conventional IPMN[44,45].
Immunohistochemical staining profiles of IPMN subtypes
A substantial number of studies have attempted to risk-stratify IPMN lesions by their histological subtype[46-51]. A systematic review by Koh et al.[52] reviewed 14 studies with 1617 unique patients with IPMN that consisted of 900 IPMNs with noninvasive features and 717 with invasive disease. They found that pancreatobiliary subtype IPMN has the highest likelihood of (1) an invasive component (67.9%); (2) presence of a mural nodule (56.6%); and (3) tumor recurrence (46.3%). Conversely, gastric-subtype IPMN had the lowest likelihood of an invasive component (10.2%) or tumor recurrence (9.4%).
The most important driver of patient survivorship with regards to IPMN is the degree of dysplasia[53]. Conventionally, IPMN is understood as a premalignant lesion that progresses from low-grade to high-grade dysplasia to invasive pancreatic carcinoma[25,54]. Historically, dysplasia was classified as low-, moderate-, or high-grade. However, international consensus has now simplified this to low- or high-grade dysplasia only[55,56], with most lesions previously characterized as moderate relegated to the low-grade dysplasia group. Those patients with invasive IPMN experience a pronounced decrement in overall survival[53]. While the overall 5-year survival of resected IPMN-associated adenocarcinoma (42%) is superior to that of pancreatic adenocarcinoma not associated with IPMN (19%)[57], this is misleading as survival is closely correlated with the invasive histologic subtype seen arising from the parent IPMN. The vast majority are of two subtypes: tubular or colloid carcinoma. A third variant, anaplastic carcinoma is rarely observed. The 5-year survival of IPMN-derived colloid carcinoma is markedly improved compared to tubular carcinoma, 57%-83% vs. 24%-37%, respectively[5,57]. The survival for patients with IPMN-derived tubular adenocarcinoma parallels those with conventional pancreatic adenocarcinoma not associated with IPMN[58]. Tubular carcinoma is associated with the pancreatobiliary and gastric IPMN subtypes, whereas colloid carcinoma is associated with the intestinal subtype[50,59]. The pancreatobiliary subtype has the highest incidence of harboring an invasive component and is strongly associated with the more aggressive tubular carcinoma. To investigate these differences in biological behavior several studies have investigated the mutational landscape of IPMN[24,60-63].
MOLECULAR CLASSIFICATION OF INTRADUCTAL PAPILLARY MUCINOUS NEOPLASMS
Molecular analysis is a broad term for evaluating human tissues via analysis of DNA or RNA transcripts, or indirect analysis of their downstream products including proteins and other effector molecules. With regards to IPMN, there is general agreement that molecular analysis potentially holds key insights into the natural course of IPMN including defining disparate subtype behavior and crucial factors leading to malignant degeneration. Conventional pathologic analysis as described above has been critically important in defining IPMN and generating the current clinical guidelines for management and there is substantial enthusiasm that molecular techniques will serve to further these efforts.
There are two major categories of genetic material for molecular analysis: pancreatic cyst fluid and direct tissue analysis. Cyst fluid, which can be obtained via endoscopic modalities, is unique as it can risk-stratify lesions thereby informing surveillance strategies and aiding selection of patients for surgical resection. Direct tissue analysis is more robust, but is often limited to those patients who have undergone surgery. This can inform future risk and clinical decision-making; however, the greatest value of this analysis is better elucidation of the underlying biology of IPMN. Much of our current understanding of IPMN molecular biology comes from direct analysis of resected IPMN.
The landscape of somatic mutations in pancreatic ductal adenocarcinoma (PDAC) has been carefully defined by numerous investigators over the past three decades. It is therefore unsurprising that the typical variants present in conventional PDAC are regularly observed at varying frequencies in IPMN, given its status as a precursor to PDAC [Table 2]. The hallmark genes most commonly mutated in PDAC, Kirsten rat sarcoma virus (KRAS), tumor protein 53 (TP53), cyclin-dependent kinase inhibitor 2A (CDKN2A), and deleted in pancreatic cancer-4 (DPC4, more commonly SMAD-4), can be all observed in IPMN as well. Initial investigations into the mutational landscape of IPMN sought to characterize these mutations and assess for the presence of recurrent variants private to IPMN to define the substantial differences in behavior between IPMN, IPMN-associated PDAC, and conventional PDAC (not associated with IPMN).
Comparison of common somatic mutations in IPMN and conventional PDAC
| Gene | IPMN, no/low-grade dysplasia | IPMN, high-grade dysplasia | IPMN-associated PDAC | Conventional PDAC |
| KRAS | 43%-89%[13,66] | 32%-71%[13,66] | 61%[13,66,76] | 92%-100%[67,145,146] |
| GNAS | 40%-90%[13,61,65,66] | 42%-72%[13,61,65,66] | 33%-61%[13,61,65,66,76] | < 5%[67] |
| RNF43 | 10%-11%[66] | 25%-75%[66] | 18%[66,76] | < 5%[67] |
| CDKN2A/p16 | < 5%[13,66] | 0%-16%[13,66] | 5%[13,66,76] | 82%-98%[147,148] |
| TP53 | < 5%[13,66] | 18%-21%[13,66] | 21%[13,66,76] | 50%-75%[149] |
| SMAD-4/DPC-4 | < 5%[13,66] | < 5%[13,66] | 15%[13,66,76] | 90%[150,151] |
Somatic variants unique to intraductal papillary mucinous neoplasms
The current molecular understanding of IPMN as a unique genetic entity emerged in 2011 with two seminal manuscripts that performed whole exome analysis of multiple cystic lesions of the pancreas, including IPMN[64] and targeted analysis of IPMN cyst fluid[61]. These complementary studies identified the PDAC-related mutations noted above, but also defined variants unique to IPMN. Specifically, frequent recurrent mutations were found in GNAS complex locus (GNAS) and ring finger protein 43 (RNF43) in IPMN with other unique IPMN-specific variants occurring with much less consistency. Subsequent studies have confirmed that mutations in GNAS are the second-most common variant in IPMN after KRAS, being observed in 41%-75% of all IPMN[61,65,66]. This is histology-specific, with > 90%-100% of intestinal-subtype IPMN but only 40%-70% of other histologic subtypes demonstrating mutations in GNAS[61,65]. GNAS mutations are uncommonly reported in conventional PDAC[67] but are present in 33% of resected PDAC cases associated with IPMN[68]. GNAS encodes a subunit of guanine nucleotide-binding protein that, when activated, leads to cell growth and proliferation[69-71]. The dominant recurring mutation found in IPMN is at R201 with R201C/H accounting for most reported variants. Additional R201 or Q227 variants have been reported, but common to all these variants is constitutive activation of GNAS by reducing the rate of GTP hydrolysis[24,67]. While these mutations have been observed in various neoplastic lesions ranging from osseous fibrous dysplasia to growth hormone-secreting adenomas of the pituitary, within the digestive tract they cluster in mucinous and pre-malignant neoplasms, such as pyloric gland adenoma, appendiceal mucinous neoplasms, and IPMN of the pancreas and biliary tree[72]. As such, it appears that GNAS is an early mutation in IPMN tumorigenesis and may synergize with other molecular changes to promote transformation but is unlikely to lead to invasive PDAC in isolation[72]. No GNAS-directed therapies are currently clinically relevant with regards to IPMN or PDAC.
The second-most commonly mutated gene specific to non-invasive IPMN is RNF43, with studies of resected IPMN reporting 14%-75% carrying an RNF43 variant[24,26,61,64,66,73]. RNF43 is a ubiquitin E3 ligase that targets cytosolic frizzled receptors (FZD) for ubiquitination and degradation. FZD is the upstream regulator of both canonical and non-canonical Wnt pathways[74]. Similar to colon cancer, RNF43 inactivating mutations and subsequent loss of heterozygosity reduces FZD ubiquitination and upregulates Wnt signaling thereby promoting tumorigenesis[74]. Unlike GNAS, hotspot mutations are not observed in RNF43 and inactivation can occur via a combination of frameshift indels, missense, and nonsense mutations[66,73]. Moreover, intronic, epigenetic, and post-translational changes also play a role in reducing the RNF43 regulation of the Wnt pathway. This finding was highlighted in one study of 57 resected IPMN lesions where a somatic RNF43 mutation was found in only 14% of cases but decreased RNF43 protein expression was observed in 29.5%[73]. Antibodies for FZD receptors are currently under investigational development and could be potential therapeutic targets in cancers with RNF43 mutations[75].
Numerous additional somatic mutations at a lower frequency have been reported in sequencing studies of resected IPMN tissue. While their prevalence is too low to be regarded as critical for generic IPMN development, these infrequent mutations may hold insight into sub-populations of IPMN that could become clinically relevant and thus warrant reporting and further investigation. Mutations in ATM and SF3B1 have been reported in 5%-17% in resected IPMN[66,76]. ATM is a cell cycle regulator through modulation of DNA damage repair pathways, while SF3B1 is involved in RNA splicing. RNA splicing has recently been implicated in a crucial mechanism of progression certain cases of PDAC[77] and presents an interesting target for investigation in IPMN. Additional rare mutations occurring in < 4% of IPMN include variants in known oncogenic genes such as CTNNB1, STK11, and CDH1. Interestingly, CTNNB1 mutation is also highly prevalent in another cystic pancreatic lesion, solid-pseudopapillary neoplasm[64], but this entity is distinct from IPMN. As the unifying molecular mechanisms of IPMN progression are elucidated, further investigation into these rarer aberrations will be critical in incrementally improving clinical management by detecting new therapeutic targets.
Somatic variants common to intraductal papillary mucinous neoplasms and invasive pancreatic cancer
Most commonly observed mutations in IPMN mirror those seen in conventional PDAC. The frequency of these variants in IPMN appears to correlate with their prevalence in PDAC and the degree of dysplasia within an IPMN lesion [Table 2][24]. As such, KRAS variants are the most common somatic mutations found in IPMN, occurring in 50%-80% of lesions[61-63,78]. This correlates to the near-ubiquitous presence of mutated KRAS in invasive PDAC[67,79,80]. Overall, landscape studies of all PDAC cases demonstrate that GNAS is mutated or overexpressed in 6%-11%; presumably, this represents a substantial portion of those cases of PDAC derived from IPMN[67,81,82].
RAS is a monomeric, G-family proto-oncogene involved in regulation of cell proliferation, differentiation, and survival. Of its three isoforms, KRAS is the most frequently mutated gene in human cancer[83,84]. In IPMN, KRAS is also commonly mutated with reports varying from 40%-89%[13,66,76]. Early literature suggested a positive correlation between the frequency of KRAS mutation and grade of dysplasia[63,80]. However, larger and more recent sequencing studies examining IPMN heterogeneity demonstrate the rate of KRAS mutation may not correlate closely with grade of dysplasia. Intracystic heterogeneity has been a long-recognized but understudied phenomenon in IPMN in that a particular cyst may have multiple regions with varying degrees of dysplasia or histology. This heterogeneity extends to the genetic level and the prevalence of KRAS mutations in IPMN makes it the ideal marker to study this phenomenon. Recently, in situ hybridization has been utilized to demonstrate disparate KRAS variants from spatially distinct areas within a single IPMN lesion[25,60,85]. The most common loci for KRAS mutations in IPMN are in exon 1 (G12x, G13x, Q61x)[86] and are identical to those reported in conventional PDAC and many other cancers. All of these are activating mutations with Q61x mutations conveying a favorable prognosis[67]. Circulating tumor DNA with KRASG12D mutation is associated with early distant metastasis and poor outcomes in resected PDAC[87]. Despite considerable research, no approved KRAS-targeting therapies exist[88,89] although novel approaches have been recently reported. This includes exosomes impregnated with KRAS targeting siRNA[90], small molecules specifically targeting KRASG12C mutations[91], and KRASG12D knockouts using the CRISPR/Cas-9 system, but none of these have progressed beyond the investigational stage[92].
The tumor suppressor TP53 has also be implicated as a driver mutation in PDAC[67,76,93,94]. TP53 is an essential regulator involved in cell growth, protection against mutation accumulation, and suppression of oncogene activation[95]. Sequencing of resected IPMN reveals that TP53 mutations are more common in high grade dysplasia and invasive IPMN (15%-20%) as compared with low grade IPMN (0%-5%)[13,66]. An immunohistochemistry (IHC) study of 206 resected pancreas lesions including precursor lesions (IPMN and PanIN), corroborates this finding by reporting abnormal TP53 staining in 0% low grade dysplastic lesions, 42% high grade dysplastic lesions, and 68% invasive ductal carcinomas[37]. It has recently been shown that TP53 function can be moderated via alternate mechanisms such aberrations in transcript splicing[96]. Whether this occurs in IPMN and IPMN-associated PDAC has not yet been investigated. Like KRAS, no TP53-directed therapies are currently in clinical use.
CDKN2A (also known as p16) is a tumor suppressor gene that is recurrently mutated in PDAC[67,94,97] and has also been reported in IPMN[13,66]. CDKN2A is a cell cycle regulator involved in the transition from G1 to S phase[98]. Sequencing data from resected IPMN specimens reveal CDKN2A mutation rate of 0%-18%, with an increasing frequency in lesions with high grade dysplasia, mirroring TP53 and KRAS variants[13,66]. Interestingly, IHC studies have reported a much more frequent loss of CDKN2A expression: 50%-100% of high-grade dysplasia and invasive IPMN, and 10%-51% in low-grade dysplasia IPMN[24,99-101]. The mechanism of this expression loss is by epigenetic silencing through hypermethylation of CDKN2A promoters in 80% of IPMN lesions, again with an increasing prevalence with higher grades of dysplasia[102,103]. Multiple pharmaceutical agents targeting methylation are in clinical use, but thus far have been largely limited to hematologic malignancies. Some clinical trials with epigenetic drugs as potentiators of cytotoxic chemotherapy have been performed in PDAC, but the results have been modest.
SMAD-4 is a highly conserved signal transduction protein in the transforming growth factor Beta (TGF-β) pathway where it functions as a tumor suppressor by inhibiting epithelial cell growth[104]. Homozygous deletions have been reported in 30% of PDAC[105], while only 3% of IPMNs were reported to have mutations in SMAD-4[13]. Notably, SMAD-4 loss in PDAC is associated with a poor prognosis[106]. IHC of resected IPMN tissue revealed conserved expression in non-neoplastic and low-grade dysplastic tissue with loss of expression in high-grade dysplasia and IPMN-associated invasive PDAC[37,100]. Although one study reported SMAD-4 loss of heterozygosity of 80%-90% of PDAC and 22% in IPMN, IHC may again be more informative as only ~50% of PDAC and very few IPMNs actually demonstrate loss of SMAD-4 expression[107]. In total, it appears that SMAD-4 inactivation is rare in IPMN but more common in invasive disease and usually occurs via deletion, rather than a silencing mutation or other mechanisms.
IMPLICATIONS OF THE SOMATIC MUTATIONAL LANDSCAPE OF IPMN
As in other cancers, the accumulation of driver mutations is implicated in the development of IPMN-associated invasive PDAC. However, the reality is much more complex than the archetypal progression from normal epithelium to adenoma and carcinoma as classically described in colorectal cancers[108,109]. Sequencing analysis of spatially distinct regions within resected IPMN specimens has identified considerable heterogeneity at the molecular level. Tan et al.[66] demonstrated that KRAS-wild type high-grade dysplasia can exist in an IPMN with KRAS-mutant low-grade dysplasia. Despite this, KRAS mutations often appear to be one of the earliest IPMN driver mutations during progression to invasive PDAC, as phylogenetic and whole-exome sequencing data do not reveal any shared mutations in surrounding tissue that precedes KRAS[25,76]. In addition, using an in-situ hybridization approach, heterogeneity in individual KRAS clones can be demonstrated in a single patient[25,76]. In metastatic untreated PDAC, the metastatic lesions contained identical driver mutations as compared with the primary lesion with heterogeneity in subsequent passenger variants[110]. Synthesizing these data is challenging but some conclusions can be derived. The diverse mutational landscape present in non-dysplastic or low-grade IPMN with increasing uniformity in advanced PDAC suggests clonal selection is a hallmark of IPMN malignant transformation [Figure 2]. The recognition of TP53 and SMAD-4 as late-occurring mutations often associated with high-grade or invasive disease, has broadened our understanding of worrisome features. Using resected tissue specimens to characterize molecular differences of low-grade vs. high-grade and invasive IPMN informs the interpretation of cyst fluid analysis and cyst wall biopsy. Appreciation of IPMN as a heterogeneous lesion through multi-focal analysis offers context in interpreting earlier single biopsy studies and has important implications for the natural history of low-grade IPMN. It also implies that variants of KRAS or GNAS alone are likely not sufficient to drive the development of invasive disease in IPMN. Interestingly, polyclonality observed in precancerous lesions of other organ systems has been attributed to environmental carcinogen exposure[111-113] resulting in a field defect to the affected tissue. This has not been demonstrated experimentally in the pancreas but may be an area for future inquiry. Lastly, the oncocytic histology subtype has recently been scrutinized based on recent molecular data. Sequencing studies of oncocytic IPMN reveal that KRAS and GNAS mutations are not present and RNF43 mutations are rare, suggesting that the mutational landscape and biological behavior of these lesions is dissimilar to other IPMN sub-types[44,45]. It is possible that oncocytic-subtype IPMN may be reclassified in the future as a distinct pathologic entity separate from IPMN.
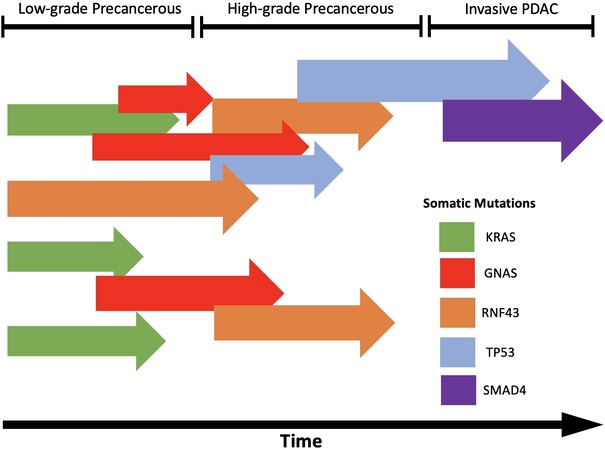
Figure 2. Schematic of clonal expansion of IPMN to invasive PDAC. Adapted from Fischer et al.[25] Intraductal Papillary Mucinous Neoplasms Arise from Multiple Independent Clones, Each with Distinct Mutations, Gastroenterology 2019. This schematic illustrates multiple precancerous cells undergoing clonal expansion with in an IPMN. Clones with distinct mutations in known driver genes (KRAS & GNAS), may acquire further mutations and vie for dominance while displaying increasing levels of dysplasia. Eventually enough key mutations accumulate, and a single clonal becomes dominate and displays invasive behavior.
Heritable causes of pancreatic cancer & IPMN
While accounting for 2%-10% of pancreatic cancer[114-117], three heritable pathways for developing pancreatic cancer have been discussed: (1) Hereditary tumor predisposition syndromes; (2) tumor syndromes stemming from chronic inflammation; and (3) familial pancreatic cancer (FPC). FPC is defined as at least two first-degree relatives with PDAC, which are not at increased risk from other syndromes[117]. There is a body of literature establishing a link between numerous specific germline mutations and pancreatic cancer: ATM, BRCA1/2, CDKN2A, MLH1, MSH2, PALB2, PMS2, PRSS1, and STK11[118-125].
The literature base regarding heritable IPMN is more limited; however, in a 2019 study of resected IPMN tissue Skaro et al.[125] found that 7.3% of patients carried germline mutations in one of the 94 genes captured in the TruSight Cancer probe, and 2.9% of patients carried a germline mutation specifically associated with pancreatic cancer. They also found that patients with IPMN and a concurrent invasive cancer were more likely to have pancreatic-specific germline mutations, than patients with IPMN alone[125]. This study suggests that there may be a link between FPC and IPMN development; however, currently there is a paucity of data on the topic.
CLINICAL IMPLICATIONS OF IPMN MOLECULAR ANALYSIS
Despite years of study, the management of IPMN remains a major challenge for clinicians. As the vast majority IPMNs fall into a low or intermediate risk category, determining optimal need and timing for surgical intervention is difficult. Once reaching intermediate risk, usually due to questionable worrisome features or increased size on imaging, the next evaluation typically includes endoscopy with ultrasound and fine needle biopsy. Thoughtful interpretation of pancreatic cyst fluid analysis, in the context of the genetic and pathologic studies discussed above, offers a relevant diagnostic tool with the potential for personalized risk-stratification and informing clinical decisions.
Cyst fluid molecular analysis
Clinical cytology studies from EUS-FNA samples of IPMN lesions have historically been heavily factored into clinical decision-making when available. However, low cellularity, low interobserver reliability, and indeterminate results often obscure the clinical picture[126,127]. For instance, when cytology reveals high-grade dysplasia, it is strongly supportive of proceeding with resection. However, such clarity is rare. The more typical result is a paucicellular specimen or visualization of “atypical” cells, which should not be interpreted as dysplastic. Aspiration via fiberoptic endoscopic instruments invariably results in atypical cellular morphology and these findings more often complicate rather than simplify management. Instead, studies have looked to other markers to assist in risk-stratifying pancreatic cystic lesions. A number of studies have proven that molecular analysis of IPMN cyst fluid is technically feasible, allowing for genetic and epigenetic analysis[128-133]. These have investigated cytopathology, proteomics with particular focus on mucin expression[134], biochemical analysis including levels of CEA[10,135] and Das-1[136], telomere status[137,138], miRNA[130,132], and mutations in GNAS, KRAS, RNF43, SMAD4, and TP53[139,140].
In a study investigating EUS fine needle aspiration of pancreatic cyst fluid, KRAS/GNAS mutations were detected via genetic analysis of cell-free DNA (cfDNA) in 56/56 IPMN. Consistent with sequencing data discussed above, this study found that the combination of KRAS/GNAS mutation and an alteration in TP53/PIK3CA/PTEN had an 89% sensitivity and 100% specificity for high risk IPMN[140]. A major criticism regarding genetic analysis of cyst fluid aspiration involves the volume of cyst fluid required to obtain enough DNA for sequencing, sometimes 1 mL or more[126,128]. A novel “through the needle biopsy” technique was developed to capture cyst wall and allow for a richer histological evaluation and provide more material for genetic analysis[141].
To date miR-216, Das-1, and combination panel of tumor suppressor and proto-oncogenes mutations have shown promise in differentiating benign tumors from IPMN with high-grade dysplasia or invasive components[130,136,138,140]. However, the majority of these studies are retrospective, limiting their applicability in the prospective setting. At present, no clinical trials have been opened utilizing molecular markers in comparing cyst management strategies [a current ongoing multi-institutional trial does compare high- vs. low-intensity surveillance regimens, but this utilizes clinical features only (ECOG-ACRIN EA2185)]. Also, and perhaps more importantly, none of these studies have final tissue pathology from surgical resection to correlate with the cyst fluid marker. Thus, it is impossible to truly ascribe a high- or low-risk designation to a particular marker. The first publication from the prospective ZYSTEUS trial[85] sought to overcome that concern and reported that the 12 patients who underwent fine needle aspiration of their IPMN all yielded either KRAS or GNAS mutations. The aspirate was fractionated into a cellular component and a liquid component, but the liquid component did not yield cfDNA of sufficient quality for analysis in 25% of patients, often due to viscosity. As this prospective trial continues to mature and accrue more patients it may offer important insights into the utility and practicality of using genetic analysis of cyst fluid for risk stratification and clinical decision making in the management of patients with IPMN.
To unify existing molecular knowledge and assist practicing clinicians in risk management of cystic pancreatic lesions, commercial molecular diagnostic kits have recently become widely available. These kits generally require aspirated cyst fluid (~600 µL) which, in combination with clinical factors, is subjected to a targeted molecular analysis. A panel of oncogenes and tumor suppressor gene variants associated with high-risk lesions is assessed as well as DNA quantity/quality. These kits signify the first attempt at personalized management of cystic lesions of the pancreas. This approach has revolutionized other tumor types, with OncotypeDx in breast cancer staging and treatment being the archetypal example[142,143]. Clearly, much work remains, but several clinical scenarios regarding IPMN management could benefit from molecular analysis. For example, asymptomatic patients with low-risk imaging but a concerning mutation profile may benefit from more aggressive intervention. However, a frail patient with a clinically-concerning IPMN but found to have a reassuring molecular profile may be best served by close surveillance.
Two commercially-available kits are in widespread use in the United States: PancraGEN[16] (Interpace Diagnostics) and PancreaSeq[17] (University of Pittsburgh Medical Center). Both tests screen for > 20 mutations associated with pancreatic cystic lesions. PancreaSeq utilizes next generation sequencing, while PancraGEN uses Sanger sequencing. The mutations captured by both kits also include variants common to non-mucinous lesions, such as VHL variants in serous cystadenoma. Reports provide the relevant genomic analysis and, in combination with clinical risk factors, stratify lesions as low-, moderate-, or high-risk. As always, patient factors such as candidacy for surgery or willingness to undergo surveillance remain critical components to the shared decision-making process.
In practice, data are sparse but seem to support selective employment of these assays. In general, patients with conflicting clinical, personal, or imaging risk profiles benefit the most from the currently available molecular assays. Patients with worrisome imaging features but cyst fluid analysis questioning the diagnosis of IPMN (i.e., low CEA) are ideal candidates for molecular profiling. In such patients, the presence or absence of a GNAS or KRAS variant can serve to conclusively direct management. Routine employment of molecular techniques to patients with small, reassuring IPMNs or those with clear high-risk stigmata (such as jaundice) is not supported by evidence at this time. For example, in a patient with IPMN-associated jaundice, a reassuring molecular profile would not alter recommendations for intervention. Judicious use of these tests is also indicated as they are costly and sometimes result in a substantial out-of-pocket expense to the patient in the United States. Guidelines for appropriate usage of molecular testing will be reliant on the accrual of prospective data in the future.
CONCLUSION
IPMN is the most common cystic pre-malignant pancreatic lesion. However, the natural history and molecular underpinnings of its malignant transformation have not been fully characterized and it therefore remains a challenging entity to manage. Decades of observational studies have laid the groundwork for its histopathologic classification and the current consensus towards management based on clinical and imaging risk factors. With the application of modern molecular investigative techniques to both resected surgical specimens and endoscopically-obtained cyst fluid aspirates, it is hoped that a deeper molecular understanding of IPMN can allow informed design of improved care strategies. At present, our growing knowledge of IPMN biology has begun to create new opportunities for personalized management but has also uncovered previously-unappreciated molecular complexity.
Mutations private to IPMN, defined by examination of resected IPMN tissue, hold potential in defining novel therapeutic targets to reverse, halt, or slow the process of malignant progression. These studies can also inform the post-resection risk of recurrence or synchronous pathology, and therefore help improve surveillance paradigms. In addition, cyst fluid analysis holds tremendous clinical potential to risk-stratify lesions prior to resection. Ultimately, this could assist in determining optimal treatment or surveillance regimens. First-generation commercially available genetic analysis kits are already in practice, and their optimal role in routine practice is being explored. A prospective study aimed to specifically investigate the clinical utility of molecular analysis tools in guiding clinical decision making is the next logical step. While individualized patient-specific management remains paramount, even with cyst fluid genetic analysis, the rapidly evolving field of IPMN molecular analysis promises continued future improvement and augmentation of IPMN management strategies.
DECLARATIONS
Authors’ contributionsPerformed literature review, made significant contributions to concept and design of figure/tables, and wrote the manuscript: Peters NV, Kunstman JW
Availability of data and materialsNot applicable.
Financial support and sponsorshipNone.
Conflicts of interestBoth authors declared that there are no conflicts of interest.
Ethical approval and consent to participateNot applicable.
Consent for publicationNot applicable.
Copyright© The Author(s) 2021.
REFERENCES
1. Hruban RH, Adsay NV, Albores-Saavedra J, et al. Pancreatic intraepithelial neoplasia: a new nomenclature and classification system for pancreatic duct lesions. Am J Surg Pathol 2001;25:579-86.
2. Hruban RH, Takaori K, Klimstra DS, et al. An illustrated consensus on the classification of pancreatic intraepithelial neoplasia and intraductal papillary mucinous neoplasms. Am J Surg Pathol 2004;28:977-87.
3. Kloppel G, Solcia E, Longnecker DS, Capella C, Sobin LH. . Histological typing of tumours of the exocrine pancreas. 2nd ed. Science & Business Media, Germany: Springer; 1996.
4. Kimura W, Nagai H, Kuroda A, Muto T, Esaki Y. Analysis of small cystic lesions of the pancreas. Int J Pancreatol 1995;18:197-206.
5. Sohn TA, Yeo CJ, Cameron JL, et al. Intraductal papillary mucinous neoplasms of the pancreas: an updated experience. Ann Surg 2004;239:788-97; discussion 797.
6. Klibansky DA, Reid-Lombardo KM, Gordon SR, Gardner TB. The clinical relevance of the increasing incidence of intraductal papillary mucinous neoplasm. Clin Gastroenterol Hepatol 2012;10:555-8.
7. Yamada M, Kozuka S, Yamao K, Nakazawa S, Naitoh Y, Tsukamoto Y. Mucin-producing tumor of the pancreas. Cancer 1991;68:159-68.
8. Owens DK, Davidson KW, Krist AH, et al. Screening for pancreatic cancer: US preventive services task force reaffirmation recommendation statement. Jama 2019;322:438-44.
9. Chari ST, Kelly K, Hollingsworth MA, et al. Early detection of sporadic pancreatic cancer: summative review. Pancreas 2015;44:693-712.
10. Brugge WR, Lewandrowski K, Lee-Lewandrowski E, et al. Diagnosis of pancreatic cystic neoplasms: a report of the cooperative pancreatic cyst study. Gastroenterology 2004;126:1330-6.
11. Schmidt CM, White PB, Waters JA, et al. Intraductal papillary mucinous neoplasms: predictors of malignant and invasive pathology. Ann Surg 2007;246:644-51; discussion 651.
12. Salvia R, Fernández-del Castillo C, Bassi C, et al. Main-duct intraductal papillary mucinous neoplasms of the pancreas: clinical predictors of malignancy and long-term survival following resection. Ann Surg 2004;239:678-85; discussion 685.
13. Amato E, Molin MD, Mafficini A, et al. Targeted next-generation sequencing of cancer genes dissects the molecular profiles of intraductal papillary neoplasms of the pancreas. J Pathol 2014;233:217-27.
14. Fritz S, Fernandez-del Castillo C, Mino-Kenudson M, et al. Global genomic analysis of intraductal papillary mucinous neoplasms of the pancreas reveals significant molecular differences compared to ductal adenocarcinoma. Ann Surg 2009;249:440-47.
15. Moris D, Damaskos C, Spartalis E, et al. Updates and Critical Evaluation on Novel Biomarkers for the Malignant Progression of Intraductal Papillary Mucinous Neoplasms of the Pancreas. Anticancer Res 2017;37:2185-94.
16. Biosciences I. PancraGEN. https://pancragen.com/. Published 2020. [Last accessed on 19 May 2021].
17. Lab UMaGP. PancreaSeq. https://mgp.upmc.com/Home/Test/PanSeq_details. Published 2021. [Last accessed on 19 May 2021].
18. Khalid A, Brugge W. ACG practice guidelines for the diagnosis and management of neoplastic pancreatic cysts. Am J Gastroenterol 2007;102:2339-49.
19. Study Group on Cystic Tumours of the Pancreas. European evidence-based guidelines on pancreatic cystic neoplasms. Gut 2018;67:789-804.
20. Tanaka M, Fernández-Del Castillo C, Kamisawa T, et al. Revisions of international consensus Fukuoka guidelines for the management of IPMN of the pancreas. Pancreatology 2017;17:738-53.
21. Valsangkar NP, Morales-Oyarvide V, Thayer SP, et al. 851 resected cystic tumors of the pancreas: a 33-year experience at the Massachusetts General Hospital. Surgery 2012;152:S4-12.
22. Adsay NV, Merati K, Basturk O, et al. Pathologically and biologically distinct types of epithelium in intraductal papillary mucinous neoplasms: delineation of an "intestinal" pathway of carcinogenesis in the pancreas. Am J Surg Pathol 2004;28:839-48.
23. Lüttges J, Zamboni G, Longnecker D, Klöppel G. The immunohistochemical mucin expression pattern distinguishes different types of intraductal papillary mucinous neoplasms of the pancreas and determines their relationship to mucinous noncystic carcinoma and ductal adenocarcinoma. Am J Surg Pathol 2001;25:942-8.
24. Fischer CG, Wood LD. From somatic mutation to early detection: insights from molecular characterization of pancreatic cancer precursor lesions. J Pathol 2018;246:395-404.
25. Fischer CG, Beleva Guthrie V, Braxton AM, et al. Intraductal Papillary Mucinous Neoplasms Arise From Multiple Independent Clones, Each With Distinct Mutations. Gastroenterology 2019;157:1123-37.e1122.
26. Felsenstein M, Noë M, Masica DL, et al. IPMNs with co-occurring invasive cancers: neighbours but not always relatives. Gut 2018;67:1652-62.
27. Tanaka M, Chari S, Adsay V, et al. International consensus guidelines for management of intraductal papillary mucinous neoplasms and mucinous cystic neoplasms of the pancreas. Pancreatology 2006;6:17-32.
28. Salvia R, Crippa S, Partelli S, et al. Differences between main-duct and branch-duct intraductal papillary mucinous neoplasms of the pancreas. World J Gastrointest Surg 2010;2:342-6.
29. Tian X, Gao H, Ma Y, Zhuang Y, Yang Y. Surgical treatment and prognosis of 96 cases of intraductal papillary mucinous neoplasms of the pancreas: a retrospective cohort study. Int J Surg 2015;13:49-53.
30. Masuda A, Arisaka Y, Hara S, et al. MUC2 expression and prevalence of high-grade dysplasia and invasive carcinoma in mixed-type intraductal papillary mucinous neoplasm of the pancreas. Pancreatology 2013;13:583-88.
31. Anand N, Sampath K, Wu BU. Cyst features and risk of malignancy in intraductal papillary mucinous neoplasms of the pancreas: a meta-analysis. Clin Gastroenterol Hepatol 2013;11:913-21; quiz e59.
32. Oyama H, Tada M, Takagi K, et al. Long-term Risk of Malignancy in Branch-Duct Intraductal Papillary Mucinous Neoplasms. Gastroenterology 2020;158:226-37.e225.
33. Ohno E, Hirooka Y, Kawashima H, et al. Natural history of pancreatic cystic lesions: A multicenter prospective observational study for evaluating the risk of pancreatic cancer. J Gastroenterol Hepato 2018;33:320-8.
34. Vege SS, Ziring B, Jain R, Moayyedi P. American gastroenterological association institute guideline on the diagnosis and management of asymptomatic neoplastic pancreatic cysts. Gastroenterology 2015;148:819-22.
35. Attiyeh MA, Fernández-Del Castillo C, Al Efishat M, et al. Development and validation of a multi-institutional preoperative nomogram for predicting grade of dysplasia in Intraductal Papillary Mucinous Neoplasms (IPMNs) of the Pancreas: a report from the pancreatic surgery consortium. Ann Surg 2018;267:157-63.
36. Lee JE, Choi SY, Min JH, et al. Determining malignant potential of intraductal papillary mucinous neoplasm of the Pancreas: CT versus MRI by using revised 2017 International Consensus Guidelines. Radiology 2019;293:134-43.
37. Furukawa T, Klöppel G, Volkan Adsay N, et al. Classification of types of intraductal papillary-mucinous neoplasm of the pancreas: a consensus study. Virchows Arch 2005;447:794-9.
38. Furukawa T, Fukushima N, Itoi T, et al. A consensus study of the grading and typing of intraductal papillary mucinous neoplasms of the pancreas. Pancreas 2019;48:480-7.
39. Klöppel G, Basturk O, Schlitter AM, Konukiewitz B, Esposito I. Intraductal neoplasms of the pancreas. Semin Diagn Pathol 2014;31:452-66.
40. Adsay NV, Conlon KC, Zee SY, Brennan MF, Klimstra DS. Intraductal papillary-mucinous neoplasms of the pancreas: an analysis of in situ and invasive carcinomas in 28 patients. Cancer 2002;94:62-77.
41. Nakamura A, Horinouchi M, Goto M, et al. New classification of pancreatic intraductal papillary-mucinous tumour by mucin expression: its relationship with potential for malignancy. J Pathol 2002;197:201-10.
42. Adsay V, Mino-Kenudson M, Furukawa T, et al. Pathologic evaluation and reporting of intraductal papillary mucinous neoplasms of the pancreas and other tumoral intraepithelial neoplasms of pancreatobiliary tract: recommendations of Verona consensus meeting. Ann Surg 2016;263:162-77.
43. Castellano-Megías VM, Andrés CI, López-Alonso G, Colina-Ruizdelgado F. Pathological features and diagnosis of intraductal papillary mucinous neoplasm of the pancreas. World J Gastrointest Oncol 2014;6:311-24.
44. Basturk O, Chung SM, Hruban RH, et al. Distinct pathways of pathogenesis of intraductal oncocytic papillary neoplasms and intraductal papillary mucinous neoplasms of the pancreas. Virchows Arch 2016;469:523-32.
45. Basturk O, Tan M, Bhanot U, et al. The oncocytic subtype is genetically distinct from other pancreatic intraductal papillary mucinous neoplasm subtypes. Mod Pathol 2016;29:1058-69.
46. Distler M, Kersting S, Niedergethmann M, et al. Pathohistological subtype predicts survival in patients with intraductal papillary mucinous neoplasm (IPMN) of the pancreas. Ann Surg 2013;258:324-30.
47. Kang MJ, Lee KB, Jang JY, Han IW, Kim SW. Evaluation of clinical meaning of histological subtypes of intraductal papillary mucinous neoplasm of the pancreas. Pancreas 2013;42:959-66.
48. Kim J, Jang KT, Mo Park S, et al. Prognostic relevance of pathologic subtypes and minimal invasion in intraductal papillary mucinous neoplasms of the pancreas. Tumour Biol 2011;32:535-42.
49. Mino-Kenudson M, Fernández-del Castillo C, Baba Y, et al. Prognosis of invasive intraductal papillary mucinous neoplasm depends on histological and precursor epithelial subtypes. Gut 2011;60:1712-20.
50. Furukawa T, Hatori T, Fujita I, et al. Prognostic relevance of morphological types of intraductal papillary mucinous neoplasms of the pancreas. Gut 2011;60:509-16.
51. Hata T, Mizuma M, Motoi F, et al. An integrated analysis of host- and tumor-derived markers for predicting high-grade dysplasia and associated invasive carcinoma of intraductal papillary mucinous neoplasms of the pancreas. Surg Today 2020;50:1039-48.
52. Koh YX, Zheng HL, Chok AY, et al. Systematic review and meta-analysis of the spectrum and outcomes of different histologic subtypes of noninvasive and invasive intraductal papillary mucinous neoplasms. Surgery 2015;157:496-509.
53. Niedergethmann M, Grützmann R, Hildenbrand R, et al. Outcome of invasive and noninvasive intraductal papillary-mucinous neoplasms of the pancreas (IPMN): a 10-year experience. World J Surg 2008;32:2253-60.
54. Reid MD, Saka B, Balci S, Goldblum AS, Adsay NV. Molecular genetics of pancreatic neoplasms and their morphologic correlates: an update on recent advances and potential diagnostic applications. Am J Clin Pathol 2014;141:168-80.
55. Nagtegaal ID, Odze RD, Klimstra D, et al. The 2019 WHO classification of tumours of the digestive system. Histopathology 2020;76:182-8.
56. Basturk O, Hong SM, Wood LD, et al. A revised classification system and recommendations from the Baltimore Consensus Meeting for neoplastic precursor lesions in the Pancreas. Am J Surg Pathol 2015;39:1730-41.
57. Poultsides GA, Reddy S, Cameron JL, et al. Histopathologic basis for the favorable survival after resection of intraductal papillary mucinous neoplasm-associated invasive adenocarcinoma of the pancreas. Ann Surg 2010;251:470-6.
58. Yopp AC, Katabi N, Janakos M, et al. Invasive carcinoma arising in intraductal papillary mucinous neoplasms of the pancreas: a matched control study with conventional pancreatic ductal adenocarcinoma. Ann Surg 2011;253:968-74.
60. Kuboki Y, Fischer CG, Beleva Guthrie V, et al. Single-cell sequencing defines genetic heterogeneity in pancreatic cancer precursor lesions. J Pathol 2019;247:347-56.
61. Wu J, Matthaei H, Maitra A, et al. Recurrent GNAS mutations define an unexpected pathway for pancreatic cyst development. Sci Transl Med 2011;3:92ra66.
62. Schönleben F, Allendorf JD, Qiu W, et al. Mutational analyses of multiple oncogenic pathways in intraductal papillary mucinous neoplasms of the pancreas. Pancreas 2008;36:168-72.
63. Satoh K, Shimosegawa T, Moriizumi S, Koizumi M, Toyota T. K-ras mutation and p53 protein accumulation in intraductal mucin-hypersecreting neoplasms of the pancreas. Pancreas 1996;12:362-8.
64. Wu J, Jiao Y, Dal Molin M, et al. Whole-exome sequencing of neoplastic cysts of the pancreas reveals recurrent mutations in components of ubiquitin-dependent pathways. Proc Natl Acad Sci U S A 2011;108:21188-93.
65. Ohtsuka T, Tomosugi T, Kimura R, et al. Clinical assessment of the GNAS mutation status in patients with intraductal papillary mucinous neoplasm of the pancreas. Surg Today 2019;49:887-93.
66. Tan MC, Basturk O, Brannon AR, et al. GNAS and KRAS Mutations Define Separate Progression Pathways in Intraductal Papillary Mucinous Neoplasm-Associated Carcinoma. J Am Coll Surg 2015;220:845-54.e1.
67. Witkiewicz AK, McMillan EA, Balaji U, et al. Whole-exome sequencing of pancreatic cancer defines genetic diversity and therapeutic targets. Nat Commun 2015;6:6744.
68. Takano S, Fukasawa M, Maekawa S, et al. Deep sequencing of cancer-related genes revealed GNAS mutations to be associated with intraductal papillary mucinous neoplasms and its main pancreatic duct dilation. PLoS One 2014;9:e98718.
69. Lania AG, Mantovani G, Spada A. Mechanisms of disease: Mutations of G proteins and G-protein-coupled receptors in endocrine diseases. Nat Clin Pract Endocrinol Metab 2006;2:681-93.
70. Landis CA, Masters SB, Spada A, Pace AM, Bourne HR, Vallar L. GTPase inhibiting mutations activate the alpha chain of Gs and stimulate adenylyl cyclase in human pituitary tumours. Nature 1989;340:692-6.
71. O'Hayre M, Vázquez-Prado J, Kufareva I, et al. The emerging mutational landscape of G proteins and G-protein-coupled receptors in cancer. Nat Rev Cancer 2013;13:412-24.
72. Innamorati G, Wilkie TM, Kantheti HS, et al. The curious case of Gαs gain-of-function in neoplasia. BMC Cancer 2018;18:293.
73. Sakamoto H, Kuboki Y, Hatori T, et al. Clinicopathological significance of somatic RNF43 mutation and aberrant expression of ring finger protein 43 in intraductal papillary mucinous neoplasms of the pancreas. Mod Pathol 2015;28:261-7.
74. Koo BK, Spit M, Jordens I, et al. Tumour suppressor RNF43 is a stem-cell E3 ligase that induces endocytosis of Wnt receptors. Nature 2012;488:665-9.
75. Steinhart Z, Pavlovic Z, Chandrashekhar M, et al. Genome-wide CRISPR screens reveal a Wnt-FZD5 signaling circuit as a druggable vulnerability of RNF43-mutant pancreatic tumors. Nat Med 2017;23:60-8.
76. Noë M, Niknafs N, Fischer CG, et al. Genomic characterization of malignant progression in neoplastic pancreatic cysts. Nat Commun 2020;11:4085.
77. Escobar-Hoyos LF, Penson A, Kannan R, et al. Altered RNA Splicing by Mutant p53 Activates Oncogenic RAS Signaling in Pancreatic Cancer. Cancer Cell 2020;38:198-211.e8.
78. Kuboki Y, Shimizu K, Hatori T, et al. Molecular biomarkers for progression of intraductal papillary mucinous neoplasm of the pancreas. Pancreas 2015;44:227-35.
79. Smit VT, Boot AJ, Smits AM, Fleuren GJ, Cornelisse CJ, Bos JL. KRAS codon 12 mutations occur very frequently in pancreatic adenocarcinomas. Nucleic Acids Res 1988;16:7773-82.
80. Wood LD, Hruban RH. Pathology and molecular genetics of pancreatic neoplasms. Cancer J 2012;18:492-501.
81. Gao J, Aksoy BA, Dogrusoz U, et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal 2013;6:pl1.
82. Cerami E, Gao J, Dogrusoz U, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov 2012;2:401-4.
83. Muñoz-Maldonado C, Zimmer Y, Medová M. A comparative analysis of individual RAS mutations in cancer biology. Front Oncol 2019;9:1088.
84. Li S, Balmain A, Counter CM. A model for RAS mutation patterns in cancers: finding the sweet spot. Nat Rev Cancer 2018;18:767-77.
85. Volckmar AL, Endris V, Gaida MM, et al. Next generation sequencing of the cellular and liquid fraction of pancreatic cyst fluid supports discrimination of IPMN from pseudocysts and reveals cases with multiple mutated driver clones: First findings from the prospective ZYSTEUS biomarker study. Genes Chromosomes Cancer 2019;58:3-11.
86. Schönleben F, Qiu W, Bruckman KC, et al. BRAF and KRAS gene mutations in intraductal papillary mucinous neoplasm/carcinoma (IPMN/IPMC) of the pancreas. Cancer Lett 2007;249:242-8.
87. Guo S, Shi X, Shen J, et al. Preoperative detection of KRAS G12D mutation in ctDNA is a powerful predictor for early recurrence of resectable PDAC patients. Br J Cancer 2020;122:857-67.
88. Liu P, Wang Y, Li X. Targeting the untargetable KRAS in cancer therapy. Acta Pharm Sin B 2019;9:871-9.
89. Uprety D, Adjei AA. KRAS: From undruggable to a druggable Cancer Target. Cancer Treat Rev 2020;89:102070.
90. Kamerkar S, LeBleu VS, Sugimoto H, et al. Exosomes facilitate therapeutic targeting of oncogenic KRAS in pancreatic cancer. Nature 2017;546:498-503.
91. Christensen JG, Olson P, Briere T, Wiel C, Bergo MO. Targeting Kras(g12c) -mutant cancer with a mutation-specific inhibitor. J Intern Med 2020;288:183-91.
92. Lentsch E, Li L, Pfeffer S, et al. CRISPR/Cas9-mediated knock-out of kras(G12D) mutated pancreatic cancer cell lines. Int J Mol Sci 2019;20:5706.
93. Makohon-Moore A, Iacobuzio-Donahue CA. Pancreatic cancer biology and genetics from an evolutionary perspective. Nat Rev Cancer 2016;16:553-65.
94. Biankin AV, Waddell N, Kassahn KS, et al. Pancreatic cancer genomes reveal aberrations in axon guidance pathway genes. Nature 2012;491:399-405.
96. Escobar-Hoyos L, Knorr K, Abdel-Wahab O. Aberrant RNA Splicing in Cancer. Annu Rev Cancer Biol 2019;3:167-85.
98. Sun H, Zhang B, Li H. The Roles of Frequently Mutated Genes of Pancreatic Cancer in Regulation of Tumor Microenvironment. Technol Cancer Res Treat 2020;19:1533033820920969.
99. Furukawa T, Fujisaki R, Yoshida Y, et al. Distinct progression pathways involving the dysfunction of DUSP6/MKP-3 in pancreatic intraepithelial neoplasia and intraductal papillary-mucinous neoplasms of the pancreas. Mod Pathol 2005;18:1034-42.
100. Biankin AV, Biankin SA, Kench JG, et al. Aberrant p16(INK4A) and DPC4/Smad4 expression in intraductal papillary mucinous tumours of the pancreas is associated with invasive ductal adenocarcinoma. Gut 2002;50:861-8.
101. Mohri D, Asaoka Y, Ijichi H, et al. Different subtypes of intraductal papillary mucinous neoplasm in the pancreas have distinct pathways to pancreatic cancer progression. J Gastroenterol 2012;47:203-13.
102. Sato N, Goggins M. Epigenetic alterations in intraductal papillary mucinous neoplasms of the pancreas. J Hepatobiliary Pancreat Surg 2006;13:280-5.
103. Sato N, Ueki T, Fukushima N, et al. Aberrant methylation of CpG islands in intraductal papillary mucinous neoplasms of the pancreas. Gastroenterology 2002;123:365-72.
104. Dai JL, Turnacioglu KK, Schutte M, Sugar AY, Kern SE. Dpc4 transcriptional activation and dysfunction in cancer cells. Cancer Res 1998;58:4592-7.
105. Hahn SA, Hoque AT, Moskaluk CA, et al. Homozygous deletion map at 18q21.1 in pancreatic cancer. Cancer Res 1996;56:490-4.
106. Jiang H, He C, Geng S, et al. RhoT1 and Smad4 are correlated with lymph node metastasis and overall survival in pancreatic cancer. PLoS One 2012;7:e42234.
107. Abe T, Fukushima N, Brune K, et al. Genome-wide allelotypes of familial pancreatic adenocarcinomas and familial and sporadic intraductal papillary mucinous neoplasms. Clin Cancer Res 2007;13:6019-25.
108. Vogelstein B, Fearon ER, Hamilton SR, et al. Genetic alterations during colorectal-tumor development. N Engl J Med 1988;319:525-32.
110. Makohon-Moore AP, Zhang M, Reiter JG, et al. Limited heterogeneity of known driver gene mutations among the metastases of individual patients with pancreatic cancer. Nat Genet 2017;49:358-66.
111. Martincorena I, Roshan A, Gerstung M, et al. Tumor evolution. High burden and pervasive positive selection of somatic mutations in normal human skin. Science 2015;348:880-6.
112. Stachler MD, Taylor-Weiner A, Peng S, et al. Paired exome analysis of Barrett's esophagus and adenocarcinoma. Nat Genet 2015;47:1047-55.
113. Ross-Innes CS, Becq J, Warren A, et al. Whole-genome sequencing provides new insights into the clonal architecture of Barrett's esophagus and esophageal adenocarcinoma. Nat Genet 2015;47:1038-46.
114. Del Chiaro M, Zerbi A, Falconi M, et al. Cancer risk among the relatives of patients with pancreatic ductal adenocarcinoma. Pancreatology 2007;7:459-69.
115. Bartsch DK, Kress R, Sina-Frey M, et al. Prevalence of familial pancreatic cancer in Germany. Int J Cancer 2004;110:902-06.
116. Hruban RH, Canto MI, Goggins M, Schulick R, Klein AP. Update on familial pancreatic cancer. Adv Surg 2010;44:293-311.
117. Fendrich V, Langer P, Bartsch DK. Familial pancreatic cancer--status quo. Int J Colorectal Dis 2014;29:139-45.
118. Hu C, Hart SN, Bamlet WR, et al. Prevalence of Pathogenic Mutations in Cancer Predisposition Genes among Pancreatic Cancer Patients. Cancer Epidemiol Biomarkers Prev 2016;25:207-11.
119. Hu C, Hart SN, Polley EC, et al. Association between inherited germline mutations in cancer predisposition genes and risk of pancreatic cancer. Jama 2018;319:2401-9.
120. Hu C, LaDuca H, Shimelis H, et al. Multigene hereditary cancer panels reveal high-risk pancreatic cancer susceptibility genes. JCO Precis Oncol 2018;2:PO.17.00291.
121. Roberts NJ, Norris AL, Petersen GM, et al. Whole Genome Sequencing Defines the Genetic Heterogeneity of Familial Pancreatic Cancer. Cancer Discov 2016;6:166-75.
122. Grant RC, Selander I, Connor AA, et al. Prevalence of germline mutations in cancer predisposition genes in patients with pancreatic cancer. Gastroenterology 2015;148:556-64.
123. Holter S, Borgida A, Dodd A, et al. Germline BRCA Mutations in a Large Clinic-Based Cohort of Patients With Pancreatic Adenocarcinoma. J Clin Oncol 2015;33:3124-9.
124. Shindo K, Yu J, Suenaga M, et al. Deleterious Germline Mutations in Patients With Apparently Sporadic Pancreatic Adenocarcinoma. J Clin Oncol 2017;35:3382-90.
125. Skaro M, Nanda N, Gauthier C, et al. Prevalence of Germline Mutations Associated With Cancer Risk in Patients With Intraductal Papillary Mucinous Neoplasms. Gastroenterology 2019;156:1905-13.
126. Hao S, Takahashi C, Snyder RA, Parikh AA. Stratifying intraductal papillary mucinous neoplasms by cyst fluid analysis: present and future. Int J Mol Sci 2020;21:1147.
127. Shen J, Brugge WR, Dimaio CJ, Pitman MB. Molecular analysis of pancreatic cyst fluid: a comparative analysis with current practice of diagnosis. Cancer 2009;117:217-27.
128. Khalid A, Zahid M, Finkelstein SD, et al. Pancreatic cyst fluid DNA analysis in evaluating pancreatic cysts: a report of the PANDA study. Gastrointest Endosc 2009;69:1095-102.
129. Farrell JJ, Al-Haddad MA, Jackson SA, Gonda TA. Incremental value of DNA analysis in pancreatic cysts stratified by clinical risk factors. Gastrointest Endosc 2019;89:832-41.e2.
130. Farrell JJ, Toste P, Wu N, et al. Endoscopically acquired pancreatic cyst fluid microRNA 21 and 221 are associated with invasive cancer. Am J Gastroenterol 2013;108:1352-9.
131. Al-Haddad M, DeWitt J, Sherman S, et al. Performance characteristics of molecular (DNA) analysis for the diagnosis of mucinous pancreatic cysts. Gastrointest Endosc 2014;79:79-87.
132. Wang J, Paris PL, Chen J, et al. Next generation sequencing of pancreatic cyst fluid microRNAs from low grade-benign and high grade-invasive lesions. Cancer Lett 2015;356:404-9.
133. Loren D, Kowalski T, Siddiqui A, et al. Influence of integrated molecular pathology test results on real-world management decisions for patients with pancreatic cysts: analysis of data from a national registry cohort. Diagn Pathol 2016;11:5.
134. Maker AV, Katabi N, Gonen M, et al. Pancreatic cyst fluid and serum mucin levels predict dysplasia in intraductal papillary mucinous neoplasms of the pancreas. Ann Surg Oncol 2011;18:199-206.
135. Brugge WR, Lauwers GY, Sahani D, Fernandez-del Castillo C, Warshaw AL. Cystic neoplasms of the pancreas. N Engl J Med 2004;351:1218-26.
136. Das KK, Geng X, Brown JW, et al. Cross Validation of the monoclonal antibody Das-1 in identification of high-risk mucinous pancreatic cystic lesions. Gastroenterology 2019;157:720-30.e2.
137. Hashimoto Y, Murakami Y, Uemura K, et al. Telomere shortening and telomerase expression during multistage carcinogenesis of intraductal papillary mucinous neoplasms of the pancreas. J Gastrointest Surg 2008;12:17-28; discussion 28.
138. Hata T, Dal Molin M, McGregor-Das A, et al. Simple detection of telomere fusions in pancreatic cancer, intraductal papillary mucinous neoplasm, and pancreatic cyst fluid. J Mol Diagn 2018;20:46-55.
139. Springer S, Wang Y, Dal Molin M, et al. A combination of molecular markers and clinical features improve the classification of pancreatic cysts. Gastroenterology 2015;149:1501-10.
140. Singhi AD, McGrath K, Brand RE, et al. Preoperative next-generation sequencing of pancreatic cyst fluid is highly accurate in cyst classification and detection of advanced neoplasia. Gut 2018;67:2131-41.
141. Larghi A, Manfrin E, Fabbri C, et al. Interobserver agreement among expert pathologists on through-the-needle microforceps biopsy samples for evaluation of pancreatic cystic lesions. Gastrointest Endosc 2019;90:784-92.e784.
142. Giuliano AE, Connolly JL, Edge SB, et al. Breast Cancer-Major changes in the American Joint Committee on Cancer eighth edition cancer staging manual. CA Cancer J Clin 2017;67:290-303.
143. McVeigh TP, Kerin MJ. Clinical use of the Oncotype DX genomic test to guide treatment decisions for patients with invasive breast cancer. Breast Cancer (Dove Med Press) 2017;9:393-400.
144. Kobayashi M, Fujinaga Y, Ota H. Reappraisal of the Immunophenotype of Pancreatic Intraductal Papillary Mucinous Neoplasms (IPMNs)-Gastric Pyloric and Small Intestinal Immunophenotype Expression in Gastric and Intestinal Type IPMNs. Acta Histochem Cytochem 2014;47:45-57.
145. Bailey P, Chang DK, Nones K, et al. Genomic analyses identify molecular subtypes of pancreatic cancer. Nature 2016;531:47-52.
146. Grant TJ, Hua K, Singh A. Molecular Pathogenesis of Pancreatic Cancer. Prog Mol Biol Transl Sci 2016;144:241-75.
147. Caldas C, Hahn SA, da Costa LT, et al. Frequent somatic mutations and homozygous deletions of the p16 (MTS1) gene in pancreatic adenocarcinoma. Nat Genet 1994;8:27-32.
148. Schutte M, Hruban RH, Geradts J, et al. Abrogation of the Rb/p16 tumor-suppressive pathway in virtually all pancreatic carcinomas. Cancer Res 1997;57:3126-30.
149. Scarpa A, Capelli P, Mukai K, et al. Pancreatic adenocarcinomas frequently show p53 gene mutations. Am J Pathol 1993;142:1534-43.
150. Hahn SA, Schutte M, Hoque AT, et al. DPC4, a candidate tumor suppressor gene at human chromosome 18q21.1. Science 1996;271:350-3.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Peters NV, Kunstman JW. Clinical implications of the molecular characterization of intraductal papillary mucinous neoplasms of the pancreas. J Cancer Metastasis Treat 2021;7:32. http://dx.doi.org/10.20517/2394-4722.2021.67
AMA Style
Peters NV, Kunstman JW. Clinical implications of the molecular characterization of intraductal papillary mucinous neoplasms of the pancreas. Journal of Cancer Metastasis and Treatment. 2021; 7: 32. http://dx.doi.org/10.20517/2394-4722.2021.67
Chicago/Turabian Style
Peters, Nicholas V., John W. Kunstman. 2021. "Clinical implications of the molecular characterization of intraductal papillary mucinous neoplasms of the pancreas" Journal of Cancer Metastasis and Treatment. 7: 32. http://dx.doi.org/10.20517/2394-4722.2021.67
ACS Style
Peters, NV.; Kunstman JW. Clinical implications of the molecular characterization of intraductal papillary mucinous neoplasms of the pancreas. J. Cancer. Metastasis. Treat. 2021, 7, 32. http://dx.doi.org/10.20517/2394-4722.2021.67
About This Article
Copyright

Data & Comments
Data


 Cite This Article 12 clicks
Cite This Article 12 clicks

 Like This Article 16
likes
Like This Article 16
likes






Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.