
Is the EGFR pathway relevant for the pathogenesis but not for treatment of acute myeloid leukemia?

Abstract
Despite intense research and the development of several new chemotherapeutics, the prognosis for specific subsets of acute myeloid leukemia (AML) has not improved significantly. Thus, the investigation of signaling pathways associated with the pathogenesis and progression of AML has become a source for the discovery of more effective treatments. The epidermal growth factor receptor (EGFR) belongs to the HER family of tyrosine kinase (TK) receptors and is involved in the progression of a variety of solid tumors. Although the expression of members of the HER family appears to be limited to epithelial tissues and derived neoplasms, there is evidence demonstrating their role in hematopoiesis and hematological neoplasms. In AML, preclinical studies and two anecdotal cases of response to EGFR TK inhibitors (TKI) supported the EGFR signaling pathway as a potential therapeutic target. Indeed, the presence of EGFR ligands in the bone marrow microenvironment has been shown to play pathological and regenerative/protective roles in AML. However, data reporting the expression of EGFR in AML remain controversial and the EGFR pathway inhibition in AML patients has demonstrated limited clinical significance. Further studies are required to determine the relevance of the EGFR pathway in AML biology and which patients may benefit from using EGFR TKI or other drugs that target TK receptors.
Keywords
INTRODUCTION
Epidermal growth factor receptor (EGFR) is a type of tyrosine kinase (TK) receptor recognized as an important player in the tumor biology of many solid neoplasms, making it a key target for therapeutic strategies, especially in non-small cell lung cancer (NSCLC). EGFR TK inhibitors (TKI) have demonstrable efficacy in the treatment of patients with EGFR-mutated NSCLC with a favorable impact on progression-free survival, but not in the subgroup of EGFR wild-type tumors[1,2]. In contrast, EGFR mutations are rarely found in AML[3,4], while their expression has been reported to be associated with poor prognostic outcomes[5]. Accordingly, in vitro, ex vivo, and in vivo results demonstrate that EGFR TKI induces terminal differentiation and cell death of AML cells[2,6-8]. Moreover, in the bone marrow microenvironment, interactions between leukemia stem/progenitor cells (LSPCs) and immune cells promoted the expansion of leukemia cell subsets, which was associated with the upregulation of interleukin (IL)-3 gene expression in LSPCs [Figure 1][9,10]. Of note, Radpour et al.[11] showed that, in favorable-risk AML, IL-3 gene expression in CD34+ LSPCs was positively correlated with EGF/EGFR gene expression in CD8+ T cells. Furthermore, this finding was functionally validated in a co-culture system with CD8+ T cells and CD34+ LSPC. Accordingly, CD8+ T cells promoted the expansion of LSPC by increasing IL-3 mRNA and protein. On the other hand, IL-3 gene expression and cell proliferation were reduced in LSPC in the presence of the neutralizing antibody to EGFR[11]. Together, these data support the idea that EGFR TKI could be a promising alternative therapy to treat AML. However, EGFR expression is low or absent in most AML samples and cell lines[2,5,12], indicating that the anti-proliferative and pro-apoptotic effects of EGFR TKI may occur by EGFR-independent mechanisms, through interaction with other TK receptors[2,6,7,13,14]. Among them, preclinical data identify SYK (Spleen tyrosine kinase)[15], Lyn[16], other TK of the Src family kinases (SFKs)[17], signal transducer and activator of transcription 5, and Janus kinase 2[2] as off-targets of the EGFR TKI erlotinib and gefitinib in AML cells. However, it needs to be pointed out that no difference in survival or objective response was observed in clinical trials of patients with AML in which EGFR TKI was administered[18-20].
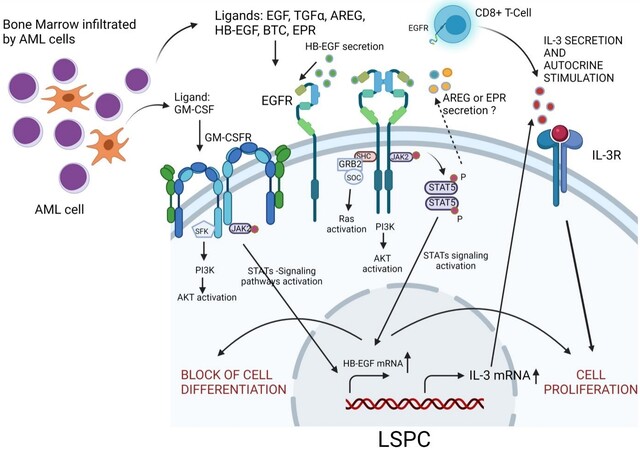
Figure 1. Schematic representation of the EGFR signaling axis and its most important downstream targets in the AML microenvironment. EGFR ligands in the bone marrow microenvironment phosphorylate EGFR receptors on the surface of LSPC or CD8+ T cells, leading to the production and release of IL-3. In turn, IL-3 induces proliferation signals in LSPCs, promoting the expansion of AML cells. Direct arrows represent direct interactions, and the dotted arrow represents indirect effects. AML: Acute myeloid leukemia; LSPCs: leukemia stem/progenitor cells; IL-3: interleukin-3; EGFR: epidermal growth factor receptor; EGF: epidermal growth factor; TGF-α: transforming growth factor-alpha; AREG: amphiregulin; HB-EGF: EGF-like heparin-binding factor; BTC: betacellulin; EPR: epiregulin; SFK: src family kinases; PI3K: phosphatidylinositol 3-kinase; AKT: protein kinase B; JAK2: Janus-activated kinase 2; JAK: Janus-activated kinase; STAT5: signal transducer and activator of transcription 5. Image created using BioRender.com.
Therapeutic strategies that target the EGFR pathway have received considerable attention in other oncologic settings, but it is still unclear which patients with AML may benefit from EGFR TKI therapy. In this review, we summarize the knowledge regarding the role of the EGFR signaling pathway in AML and provide an overview of experimental and clinical studies using EGFR TKI to suppress leukemia progression.
EGFR
EGFR (also described as HER1 and ERBB1) is a transmembrane glycoprotein (170 kDa) that belongs to the HER family of TK receptors, which comprises four members (HER1 or EGFR, HER2, HER3, and HER4) and plays an important role in regulating cell proliferation, survival, differentiation, angiogenesis, and metastatic spread[21-23]. The extracellular domain of this receptor can bind to seven official different types of ligands: EGF, transforming growth factor-alpha, amphiregulin (AREG), EGF-like heparin-binding factor (HB-EGF), betacellulin, epiregulin (EPR), and epigen[24,25]. Moreover, the connective tissue growth factor (CTGF/CCN2) has been identified as a new EGFR ligand[26]. Interestingly, the affinity of EGFR can be varied, depending on the specific ligand, tissue, and physiological and pathological conditions, which cause different cellular responses[27-29]. The interactions of EGFR with its ligands induce the homodimerization or heterodimerization of the receptor with another member of the HER family. Dimerization leads to EGFR autophosphorylation at specific tyrosine residues in the intracellular domain, triggering the activation of downstream signaling pathways, such as mitogen-activated protein kinases (MAPK), phosphatidylinositol 3-kinase/protein kinase B (PI3K/AKT), and Janus-activated kinase/signal transducer and activator of transcription (JAK-STAT)[24,25].
The aberrant expression and/or mutation of EGFR, as well as the continuous stimulation of this receptor due to the greater presence of its ligands in the tumor microenvironment, have a direct implication in the pathogenesis and progression of cancer since they are associated with increased tumor growth, invasion, and metastasis[30]. For this reason, EGFR has been described as an important therapeutic target in several malignant neoplasms, such as pancreas, colorectal, lung, breast, and head and neck carcinomas[31]. However, in hematological malignancies, EGFR mutations are very rare[3,4,32], and its expression and clinical response to EGFR TKI in AML is still controversial.
EGFR EXPRESSION IN AML
Aberrant expression of EGFR has been associated with aggressive behavior in a wide range of solid tumors[31], but the role and pattern of EGFR expression in hematological malignancies are still not well understood. Specifically, in AML, EGFR mRNA was detectable in approximately 35% of patients and correlated with decreased overall (OS) or event-free survival[5,33]. Notably, although acute promyelocytic leukemia (APL) is the most curable form of AML in adults, the EGFR gene expression in 5/29 (17.5%) of the patients was also associated with an adverse clinical outcome[34]. At the protein level, some studies pointed out the absence of EGFR expression in some AML cell lineages and primary cells[2,6,8,35]. However,
The reason EGFR expression affects the prognosis of patients with AML has not been addressed so far. Interestingly, increased HSC mobilization induced by granulocyte colony-stimulating factor (G-CSF) was achieved in healthy donors after the pharmacological inhibition of EGFR activity through activation of GTPase Cdc42 (cell division control protein-42)[36]. In addition, in a chemotherapy-induced myelosuppression model, the combination of G-CSF and EGF was synergistic for regeneration of the bone marrow compared to either G-CSF or EGF alone, and EGF increased G-CSF receptor expression following exposure to 5-fluorouracil. Conversely, G-CSF treatment increased both EGFR and phosphorylation of EGFR in hematopoietic stem/progenitor cells. Considering that in AML sensitization of leukemic cells with hematopoietic growth factors may enhance the cytotoxicity of chemotherapy, the association of G-CSF and EGFR TKI may be beneficial[37]. Accordingly, a Japanese nationwide retrospective analysis of the outcome of cord blood transplantation for AML showed that the addition of G-CSF-combined cytarabine to a total body irradiation plus cyclophosphamide conditioning regimen resulted in a significantly better disease-free survival and OS and a reduced relapse rate[38]. It is conceivable that the association of EGFR TKI to G-CSF could increase the sensitization of leukemic cells to cytotoxic therapy and further improve the outcome of HCT in AML.
EGFR LIGANDS EXPRESSION IN AML
The mRNA expression of AREG[39], HB-EGF[40], and EREG[41] genes, whose translational products are EGFR ligands, were previously detected in AML samples, but their prognostic relevance has not been defined. The AREG expression pattern has been recognized to be useful to distinguish AML from B-cell lymphoblastic leukemia[39]. Similarly, HB-EGF is expressed in human myeloid and T, but not B, lymphoid cell lines[40]. Moreover, the main differentially expressed gene between arginase-resistant and -sensitive AML is EREG, which encodes the protein EPR[41]. Indeed, EREG overexpression in solid tumors was associated with cancer cell proliferation via EGFR/MAPK/PI3K/AKT signaling[42]. However, more investigation is necessary to understand the biological implications of EREG signaling in AML.
Wu et al.[43] evaluated the urine of 18 patients with APL and found that they had significantly elevated levels of EGF when compared to healthy patients. Besides, lower and higher levels of EGF excretion were associated with complete remission and clinical recurrence, respectively, in patients with APL. Thus, these authors suggested that quantifying EGF levels may serve as a means of monitoring this disease. However, the significant correlation between EGF and creatinine detected in the urine did not have a direct relationship with EGF levels in blood serum or plasma[44]. It suggests that EGF is mainly derived from kidney biosynthesis secretion and that the levels of EGF in the urine are probably a reflection of treatment with ATRA rather than that released by the APL cells themselves. In the bone marrow microenvironment, CD8+ T cells promoted LSPCs expansion via upregulation of IL-3 through EGF/EGFR signaling in favorable-risk, but not intermediate- or adverse-risk, AML[11]. In vitro, the pro-proliferative effect of CD8+ T cells on LSPCs was abrogated in the presence of anti-EGFR antibodies[11]. The differential response of LSPC cells to the CD8+ T stimuli suggests that LSPCs in favorable-risk AML cases display a higher dependence on the microenvironment to expand [Figure 1]. We found that EGF bone marrow plasma levels were lower in APL patients when compared to healthy control subjects. In addition, AREG was detected in 5/17 APL bone marrow plasma samples and absent in 20 control samples. However, the significance of these ligands in APL development remains to be determined once our in vivo data show that gefitinib treatment did not provide additional survival benefit in a transgenic mouse model of APL. (de Almeida LY and Rego EM, own unpublished observation, 15 June 2021).
EGFR ligands exert distinct functions depending on the biological context and on what type of cell they originate. Ramadan et al.[45] experimentally demonstrated that a specific T cell subtype [T9IL-33 (interleukin-9-producing T cells activated via the ST2-IL-33 pathway)], responsible for graft-vs.-leukemia reactivity, express high levels of AREG, and the AREG blockade on T cells increased graft-vs.-host disease severity in vivo, but it did not affect their ability to kill myeloid leukemia cells in vitro. In chronic myeloid leukemia (CML), CML cell-derived exosomes carrying AREG activated the EGFR signaling in stromal cells, which in turn secreted IL-8, stimulating the proliferation of leukemic cells[46]. Furthermore, in the context of myelosuppression, EGF promoted survival and regeneration of hematopoietic stem cells following irradiation[47], thereby providing a beneficial improvement for myeloid reconstitution. Together, these findings suggest that EGFR ligands have important pathological and protective functions in myeloid leukemogenesis.
THE ROLE OF EGFR TKI IN THE TREATMENT OF AML
Since EGFR activation is involved in cancer progression, a variety of drugs targeting this signaling pathway have been developed and proven to be effective in the treatment of solid tumors and, particularly, in NSCLC[48]. The main classes of FDA-approved drugs that act on the EGFR pathway are monoclonal antibodies (e.g., cetuximab and panitumumab), which bind to the EGFR extracellular domain competing with endogenous ligands, and the TKI (e.g., gefitinib, erlotinib, and afatinib), which block the intracellular domain of this receptor, impairing the activation of cell signaling cascades that promote the proliferation of malignant cells[49-51]. EGFR-based therapies were also tested in leukemic settings, demonstrating to induce apoptosis and differentiation of AML cell lines and primary blasts at low micromolar concentrations that are achievable in clinical practice, as summarized in Table 1. Noteworthy, unlike healthy CD34+ cells, erlotinib particularly induced AML-derived CD34+ cells apoptosis in a time- and dose-dependent manner[2]. In addition, a complete remission of AML in two adult patients with concurrent NSCLC was obtained after treatment with erlotinib[35,52]. Although EGFR gene expression may be clinically useful for predicting AML outcomes[5,33,34], the receptor was not detected in the blasts of these patients, suggesting that erlotinib probably acted via EGFR-independent mechanisms. Thus, based on the controversial data regarding EGFR expression in AML samples, further studies analyzed the cross-pharmacological interactions of EGFR TKI to predict potential off-targets. In this regard, preclinical data demonstrate that erlotinib induces apoptosis in AML cells by inhibiting JAK-2 and STAT-5[2] and reduced the activation of SFK and mTOR signaling pathways[17], which are constitutively activated in AML blasts. In addition, proteomic and genetic approaches identified SYK as a gefitinib target for AML differentiation in vitro[15]. It is particularly noteworthy that increased expression of phosphorylated SYK in AML samples, especially in those harboring internal tandem duplication mutations in the FLT3 gene (FLT3-ITD), was related to unfavorable clinical outcomes[53,54]. Recently, Cao et al.[16] demonstrated that erlotinib plays a dual role targeting FLT3 in FLT3-ITD mutant AML cells and SYK and Lyn, another TK associated with leukemia proliferation, in FLT3-ITD negative AML cells, thereby showing a potential advantage of erlotinib use to overcome the cellular heterogeneity that exists in AML.
Effects of EGFR inhibitors in AML
| Study | Sample | Study design | Type of EGFR inhibitor | Result |
| Boehrer et al.[2], 2008 | P39, KG-1, HL-60 EGFR negative AML cell lines CD34+ EGFR negative AML primary blasts SCID mice inoculated with KG-1 cells | In vitro and in vivo treatments | Erlotinib | Off-target effects: differentiation, cell cycle arrest, and apoptosis Apoptosis Reduced tumor growth |
| Sun et al.[5], 2012 | HL-60, HEL, Molt-4, and Hut78 EGFR negative AML cell lines K562 and CEM EGFR positive AML cell lines EGFR negative and positive AML primary cells | In vitro treatment | Cetuximab (monoclonal antibody anti-EGFR) | EGFR positive AML cells are responsive to the cytotoxicity of cetuximab |
| Stegmaier et al.[6], 2005 | HL-60, Kasumi-1, U937 EGFR negative AML cell lines AML primary blasts | In vitro treatment | Gefitinib | Off-target effects: cell differentiation Cell viability inhibition and differentiation |
| Boehrer et al.[7], 2008 | P39, MOLM-13, MV4-11, U937, HL-60, KG-1 MDS (myelodysplastic syndrome)/AML cell lines CD34+ EGFR negative MDS/AML primary blasts | In vitro treatment | Erlotinib and gefitinib | Off-target effects: cell viability inhibition, differentiation, and apoptosis |
| Lindhagen et al.[8], 2008 | AML primary blasts and MV4-11 EGFR negative AML cell lines | In vitro treatment with gefitinib alone or combined to standard antileukemic drugs | Gefitinib alone or combined to standard antileukemic drugs | Off-target effects: apoptosis via caspase-3 pathway Synergistic interaction with etoposide Additive interactions with doxorubicin, cytarabine, and cisplatin |
| Miranda et al.[13], 2008 | HL-60, NB4, U937 AML cell lines | In vitro treatment | Gefitinib alone or combined to all-trans retinoic acid (ATRA) | Gefitinib enhanced ATRA-induced cell differentiation MEK/ERK pathway is potentially involved in the process of AML differentiation induced by ATRA/gefitinib |
| Noh et al.[14], 2010 | NB4 AML cell line | In vitro treatment | Gefitinib and arsenic trioxide (ATO) | Gefitinib enhanced ATO-induced cell differentiation and reactive oxygen species (ROS) generation ERK pathway is required for gefitinib enhancement of ATO-induced cell differentiation P38 MAPK pathway is potentially involved in the process of AML differentiation induced by ATO/gefitinib |
| Hahn et al.[15], 2009 | HL-60 AML cell line | Mass spectrometry and RNAi screening | Gefitinib | Syk was identified as a target for gefitinib-induced cell differentiation Gefitinib inhibits Syk phosphorylation |
| Cao et al.[16], 2020 | MV4-11 and KG-1 AML cell lines | In vitro and in vivo treatments | Erlotinib | Erlotinib inhibits the in vitro growth of MV4-11 and KG-1 cells via targeting FLT3 and Lyn, respectively Erlotinib inhibits the in vivo growth of MV4-11 cells |
| Boehrer et al.[17], 2011 | KG-1, KG-1a, MOLM-13, and HL-60 AML cell lines | In vitro treatment | Erlotinib alone or combined to rapamycin | Synergistic interaction in reducing the proliferation of AML cells by decreasing the constitutive activation of SRC family kinases (SFK) |
| Deangelo et al.[18], 2014 | 18 (11 relapsed) AML patients negative for FLT3-ITD mutation with a median age of 72 (range 57-84 years) | Phase II prospective non-randomized clinical trial | Gefitinib | No patients had objective responses 1 patient had a prolonged stable disease (16 months) |
| Abou Dalle et al.[19], 2018 | 29 relapsed/refractory AML patients with a median age of 67 (range 20-83 years) | Pilot phase II prospective non-randomized clinical trial | Erlotinib | 26 patients (90%) discontinued therapy due to disease progression 2 patients discontinued therapy due to adverse events 2 patients had > 50% reduction in bone marrow blasts 1 patient achieved complete remission |
| Sayar et al.[20], 2015 | 11 de novo AML patients (2 with relapsed/refractory disease and 9 with previously untreated AML) with a median age of 76 (range 60-85 years) | Pilot prospective non-randomized clinical trial | Erlotinib | 2 patients had > 50% reduction in circulating blasts followed by disease progression 9 patients had disease progression without any response No immunophenotypic evidence of cell differentiation |
| Chan and Pilichowska[35], 2007 | 68-year-old male AML patient with concurrent non-small cell lung cancer | Case report | Erlotinib | Complete remission of AML Blasts negative for EGFR |
| Pitini et al.[52], 2008 | 64-year-old male AML patient with concurrent non-small cell lung cancer | Case report | Erlotinib | Normal blood count and absence of circulating blasts after 3 months of erlotinib therapy Less than 2% blasts in bone marrow after 7 months of erlotinib therapy |
| Lainey et al.[55], 2013 | SKM1, MOLM-13, KG-1, Kasumi-1, HL-60, and MV4-11 AML cell lines Primary MDS and AML cells | In vitro treatment | Erlotinib alone or combined to azacytidine | Synergistic cytotoxic and antiproliferative effects |
| Lainey et al.[56], 2013 | HL-60 and MOLM-13 AML cell lines CD34+ AML primary blasts | In vitro treatment | Erlotinib/Gefitinib alone or combined to ATRA or vitamin D (VD) | Synergistic pro-differentiation, cytotoxic and antiproliferative effects P38 MAPK and SFK pathways are potentially involved in the process of AML differentiation induced by erlotinib |
| Lainey et al.[57], 2012 | KG-1 AML cells CD34+ AML primary blasts | In vitro treatment | Erlotinib/Gefitinib alone or combined to standard antileukemic drugs | Increased chemosensitization of AML cells to standard antileukemic agents by limiting drug export via ATP binding cassette (ABC) transporters |
| Thepot et al.[58], 2014 | 30 MDS/AML patients with a median age of 77.5 (range 53-86 years) | Phase I/II prospective non-randomized clinical trial | Erlotinib treatment after resistance to azacytidine | 1 patient achieved complete remission 4 patients had hematological improvement 12 patients discontinued therapy due to early death (n = 5), disease progression (n = 2), toxicity (n = 4) and consent withdrawal (n = 1) 7patients had stable disease Median overall survival of 7 months |
In addition to the use of EGFR TKI as single agents, the combination with other drugs has shown synergistic interactions in AML [Table 1]. Erlotinib plus azacytidine, an inhibitor of DNA methyltransferases, increased the cell cycle arrest and apoptosis in AML cell lines[55]. In addition, gefitinib plus ATRA or arsenic trioxide potentiated the differentiation of APL and non-APL AML cells in vitro[13,14]. Corroborating these results, Lainey et al.[56] reported that erlotinib and gefitinib acted synergistically when associated with ATRA and vitamin D, increasing the expression of differentiation markers CD11b and CD14 in AML cells. In this context, the inhibition of drug efflux via ABC transporters by erlotinib and gefitinib is among the molecular mechanisms underlying the increase of AML chemosensitization[57].
Despite the strong experimental evidence that EGFR inhibitors have an anti-leukemic activity in AML, erlotinib and gefitinib have shown limited clinical efficacy. Erlotinib monotherapy failed to induce differentiation[20] and avoid disease progression in 26/29 (90%) patients with refractory or relapsed AML[19], while a response rate of 20% was achieved in azacytidine-resistant patients with AML/MDS (myelodysplastic syndromes)[58]. Moreover, the use of gefitinib as a single agent in a phase II trial conducted in 18 patients with advanced AML did not promote an objective response, except for one patient who had stable disease for 16 months[18]. Similarly, in vitro treatment with gefitinib showed no difference in terms of cytotoxic activity in leukemic blasts from patients with newly diagnosed or relapsed AML[8]. Nevertheless, these studies have potential limitations. Some hypotheses that could explain the failure of EGFR TKI treatment in AML are the small number of patients, the lack of criteria for selection of patients expressing EGFR or other biomarkers suitable for EGFR TKI therapy, an inadequate dose, and drug administration only as monotherapy. Overall, the administration of gefitinib and erlotinib as single agents appears to be well-tolerated, without significant organ/systemic toxicity[18-20,51,58], encouraging further exploration of EGFR TKI in combination with other drugs already used to treat AML in larger clinical trials.
CONCLUSION
EGFR signaling appears to support leukemogenesis or hematopoietic regeneration, depending on the cell type and biological context in which the pathway is activated, but the complete characterization regarding the expression and function of EGFR and other EGFR TKI responsive targets in the different compartments of the leukemia microenvironment remains largely unknown. Therefore, the determination of predictive biomarkers in the AML setting is necessary to guide which patients could benefit from selective targeted TKI.
DECLARATIONS
Authors’ contributionsMade substantial contributions to conception and design of the study and performed bibliography review and writing of the manuscript: de Almeida LY, Rego EM
Availability of data and materialsNot applicable.
Financial support and sponsorshipThis work was supported by FAPESP grant# 2013/08135-2 and FAPESP fellowship for de Almeida LY (grant No. 2016/02713-2).
Conflicts of interestBoth authors declared that there are no conflicts of interest.
Ethical approval and consent to participateNot applicable.
Consent for publicationNot applicable.
Copyright© The Author(s) 2021.
REFERENCES
1. Grigoriu B, Berghmans T, Meert AP. Management of EGFR mutated nonsmall cell lung carcinoma patients. Eur Respir J 2015;45:1132-41.
2. Boehrer S, Adès L, Braun T, et al. Erlotinib exhibits antineoplastic off-target effects in AML and MDS: a preclinical study. Blood 2008;111:2170-80.
3. Project GENIE Consortium. AACR project GENIE: powering precision medicine through an international consortium. Cancer Discov 2017;7:818-31.
4. Loriaux MM, Levine RL, Tyner JW, et al. High-throughput sequence analysis of the tyrosine kinome in acute myeloid leukemia. Blood 2008;111:4788-96.
5. Sun JZ, Lu Y, Xu Y, et al. Epidermal growth factor receptor expression in acute myelogenous leukaemia is associated with clinical prognosis. Hematol Oncol 2012;30:89-97.
6. Stegmaier K, Corsello SM, Ross KN, Wong JS, Deangelo DJ, Golub TR. Gefitinib induces myeloid differentiation of acute myeloid leukemia. Blood 2005;106:2841-8.
7. Boehrer S, Adès L, Galluzzi L, et al. Erlotinib and gefitinib for the treatment of myelodysplastic syndrome and acute myeloid leukemia: a preclinical comparison. Biochem Pharmacol 2008;76:1417-25.
8. Lindhagen E, Eriksson A, Wickström M, et al. Significant cytotoxic activity in vitro of the EGFR tyrosine kinase inhibitor gefitinib in acute myeloblastic leukaemia. Eur J Haematol 2008;81:344-53.
9. Onetto-Pothier N, Aumont N, Haman A et al. IL-3 inhibits the binding of GM-CSF to AML blasts, but the two cytokines act synergistically in supporting blast proliferation. Leukemia 1990;4:329-36.
10. Nievergall E, Ramshaw HS, Yong AS, et al. Monoclonal antibody targeting of IL-3 receptor α with CSL362 effectively depletes CML progenitor and stem cells. Blood 2014;123:1218-28.
11. Radpour R, Riether C, Simillion C, Höpner S, Bruggmann R, Ochsenbein AF. CD8+ T cells expand stem and progenitor cells in favorable but not adverse risk acute myeloid leukemia. Leukemia 2019;33:2379-92.
12. Mahmud H, Kornblau SM, Ter Elst A, et al. Epidermal growth factor receptor is expressed and active in a subset of acute myeloid leukemia. J Hematol Oncol 2016;9:64.
13. Miranda MB, Duan R, Thomas SM, et al. Gefitinib potentiates myeloid cell differentiation by ATRA. Leukemia 2008;22:1624-7.
14. Noh EK, Kim H, Park MJ, et al. Gefitinib enhances arsenic trioxide (AS2O3)-induced differentiation of acute promyelocytic leukemia cell line. Leuk Res 2010;34:1501-5.
15. Hahn CK, Berchuck JE, Ross KN, et al. Proteomic and genetic approaches identify Syk as an AML target. Cancer Cell 2009;16:281-94.
16. Cao ZX, Guo CJ, Song X, et al. Erlotinib is effective against FLT3-ITD mutant AML and helps to overcome intratumoral heterogeneity via targeting FLT3 and Lyn. FASEB J 2020;34:10182-90.
17. Boehrer S, Galluzzi L, Lainey E, et al. Erlotinib antagonizes constitutive activation of SRC family kinases and mTOR in acute myeloid leukemia. Cell Cycle 2011;10:3168-75.
18. Deangelo DJ, Neuberg D, Amrein PC, et al. A phase II study of the EGFR inhibitor gefitinib in patients with acute myeloid leukemia. Leuk Res 2014;38:430-4.
19. Abou Dalle I, Cortes JE, Pinnamaneni P, et al. A pilot phase II study of erlotinib for the treatment of patients with relapsed/refractory acute myeloid leukemia. Acta Haematol 2018;140:30-9.
20. Sayar H, Czader M, Amin C, Cangany M, Konig H, Cripe LD. Pilot study of erlotinib in patients with acute myeloid leukemia. Leuk Res 2015;39:170-2.
21. Herbst RS. Review of epidermal growth factor receptor biology. Int J Radiat Oncol Biol Phys 2004;59:21-6.
22. Cruijsen H, Giaccone G, Hoekman K. Epidermal growth factor receptor and angiogenesis: opportunities for combined anticancer strategies. Int J Cancer 2005;117:883-8.
23. Russo A, Franchina T, Ricciardi GR, et al. Influence of EGFR mutational status on metastatic behavior in non squamous non small cell lung cancer. Oncotarget 2017;8:8717-25.
24. Jorissen R. Epidermal growth factor receptor: mechanisms of activation and signalling. Exp Cell Res 2003;284:31-53.
25. Normanno N, De Luca A, Bianco C, et al. Epidermal growth factor receptor (EGFR) signaling in cancer. Gene 2006;366:2-16.
26. Rayego-Mateos S, Rodrigues-Díez R, Morgado-Pascual JL, et al. Connective tissue growth factor is a new ligand of epidermal growth factor receptor. J Mol Cell Biol 2013;5:323-35.
27. Sanders JM, Wampole ME, Thakur ML, Wickstrom E. Molecular determinants of epidermal growth factor binding: a molecular dynamics study. PLoS One 2013;8:e54136.
28. Jones JT, Akita RW, Sliwkowski MX. Binding specificities and affinities of egf domains for ErbB receptors. FEBS Letters 1999;447:227-31.
29. Chen J, Zeng F, Forrester SJ, Eguchi S, Zhang MZ, Harris RC. Expression and function of the epidermal growth factor receptor in physiology and disease. Physiol Rev 2016;96:1025-69.
30. Sasaki T, Hiroki K, Yamashita Y. The role of epidermal growth factor receptor in cancer metastasis and microenvironment. Biomed Res Int 2013;2013:546318.
31. Modjtahedi H, Essapen S. Epidermal growth factor receptor inhibitors in cancer treatment: advances, challenges and opportunities. Anticancer Drugs 2009;20:851-5.
32. Bejar R, Stevenson K, Abdel-Wahab O, et al. Clinical effect of point mutations in myelodysplastic syndromes. N Engl J Med 2011;364:2496-506.
33. Nath S, Bhattacharyya J, Sarma PP, et al. The prognostic impact of epidermal growth factor receptor (EGFR) in patients with acute myeloid leukaemia. Indian J Hematol Blood Transfus 2020;36:749-53.
34. Nath S, Bhattacharyya J, Chandra P, Saxena R, Sazawal S, Saikia KK. Clinicopathological significance of common genetic alterations in patients with acute promyelocytic leukemia. Hematol Oncol Stem Cell Ther 2020;S1658-3876(20)30121.
35. Chan G, Pilichowska M. Complete remission in a patient with acute myelogenous leukemia treated with erlotinib for non small-cell lung cancer. Blood 2007;110:1079-80.
36. Ryan MA, Nattamai KJ, Xing E, et al. Pharmacological inhibition of EGFR signaling enhances G-CSF-induced hematopoietic stem cell mobilization. Nat Med 2010;16:1141-6.
37. Löwenberg B, van Putten W, Theobald M, et al. Dutch-Belgian Hemato-Oncology Cooperative Group; Swiss Group for Clinical Cancer Research. Effect of priming with granulocyte colony-stimulating factor on the outcome of chemotherapy for acute myeloid leukemia. N Engl J Med 2003;349:743-52.
38. Konuma T, Ooi J, Uchida N, et al. Granulocyte colony-stimulating factor combined regimen in cord blood transplantation for acute myeloid leukemia: a nationwide retrospective analysis in Japan. Haematologica 2014;99:e264-8.
39. Haouas H, Haouas S, Uzan G, Hafsia A. Identification of new markers discriminating between myeloid and lymphoid acute leukemia. Hematology 2010;15:193-203.
40. Vinante F, Rigo A, Papini E, Cassatella MA, Pizzolo G. Heparin-binding epidermal growth factor-like growth factor/diphtheria toxin receptor expression by acute myeloid leukemia cells. Blood 1999;93:1715-23.
41. Mussai F, Egan S, Higginbotham-Jones J, et al. Arginine dependence of acute myeloid leukemia blast proliferation: a novel therapeutic target. Blood 2015;125:2386-96.
42. Riese DJ 2nd, Cullum RL. Epiregulin: roles in normal physiology and cancer. Semin Cell Dev Biol 2014;28:49-56.
43. Wu X, Qureshi IA, Liu H, Yin J, Qian X, Ruijie X. Epidermal growth factor in acute promyelocytic leukemia treated with retinoic acid. Int J Hemato ;62:83-9.
44. Callegari C, Laborde NP, Buenaflor G, Nascimento CG, Brasel JA, Fisher DA. The source of urinary epidermal growth factor in humans. Eur J Appl Physiol Occup Physiol 1988;58:26-31.
45. Ramadan A, Griesenauer B, Adom D, et al. Specifically differentiated T cell subset promotes tumor immunity over fatal immunity. J Exp Med 2017;214:3577-96.
46. Corrado C, Saieva L, Raimondo S, Santoro A, De Leo G, Alessandro R. Chronic myelogenous leukaemia exosomes modulate bone marrow microenvironment through activation of epidermal growth factor receptor. J Cell Mol Med 2016;20:1829-39.
47. Doan PL, Himburg HA, Helms K, et al. Epidermal growth factor regulates hematopoietic regeneration after radiation injury. Nat Med 2013;19:295-304.
48. Huang L, Jiang S, Shi Y. Tyrosine kinase inhibitors for solid tumors in the past 20 years (2001-2020). J Hematol Oncol 2020;13:143.
49. Yewale C, Baradia D, Vhora I, Patil S, Misra A. Epidermal growth factor receptor targeting in cancer: a review of trends and strategies. Biomaterials 2013;34:8690-707.
50. Herbst RS, Sandler AB. Overview of the current status of human epidermal growth factor receptor inhibitors in lung cancer. Clin Lung Cancer 2004;6 Suppl 1:S7-S19.
51. Huang J, Meng L, Yang B, Sun S, Luo Z, Chen H. Safety profile of epidermal growth factor receptor tyrosine kinase inhibitors: a disproportionality analysis of FDA adverse event reporting system. Sci Rep 2020;10:4803.
52. Pitini V, Arrigo C, Altavilla G. Erlotinib in a patient with acute myelogenous leukemia and concomitant non-small-cell lung cancer. J Clin Oncol 2008;26:3645-6.
53. Boros K, Puissant A, Back M, et al. Increased SYK activity is associated with unfavorable outcome among patients with acute myeloid leukemia. Oncotarget 2015;6:25575-87.
54. Puissant A, Fenouille N, Alexe G, et al. SYK is a critical regulator of FLT3 in acute myeloid leukemia. Cancer Cell 2014;25:226-42.
55. Lainey E, Wolfromm A, Marie N, et al. Azacytidine and erlotinib exert synergistic effects against acute myeloid leukemia. Oncogene 2013;32:4331-42.
56. Lainey E, Wolfromm A, Sukkurwala AQ, et al. EGFR inhibitors exacerbate differentiation and cell cycle arrest induced by retinoic acid and vitamin D3 in acute myeloid leukemia cells. Cell Cycle 2013;12:2978-91.
57. Lainey E, Sébert M, Thépot S, et al. Erlotinib antagonizes ABC transporters in acute myeloid leukemia. Cell Cycle 2012;11:4079-92.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Almeida LY, Rego EM. Is the EGFR pathway relevant for the pathogenesis but not for treatment of acute myeloid leukemia?. J Cancer Metastasis Treat 2021;7:57. http://dx.doi.org/10.20517/2394-4722.2021.108
AMA Style
Almeida LY, Rego EM. Is the EGFR pathway relevant for the pathogenesis but not for treatment of acute myeloid leukemia?. Journal of Cancer Metastasis and Treatment. 2021; 7: 57. http://dx.doi.org/10.20517/2394-4722.2021.108
Chicago/Turabian Style
Almeida, Luciana Yamamoto de, Eduardo Magalhães Rego. 2021. "Is the EGFR pathway relevant for the pathogenesis but not for treatment of acute myeloid leukemia?" Journal of Cancer Metastasis and Treatment. 7: 57. http://dx.doi.org/10.20517/2394-4722.2021.108
ACS Style
Almeida, LY.; Rego EM. Is the EGFR pathway relevant for the pathogenesis but not for treatment of acute myeloid leukemia?. J. Cancer. Metastasis. Treat. 2021, 7, 57. http://dx.doi.org/10.20517/2394-4722.2021.108
About This Article
Special Issue
Copyright

Data & Comments
Data


 Cite This Article 10 clicks
Cite This Article 10 clicks

 Like This Article 28
likes
Like This Article 28
likes






Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.