
Combined effects of oncolytic vaccinia virus and dendritic cells on the progression of melanoma B16-F10 in mice
 0
0Abstract
Aim: We aimed to test the hypothesis that loading of dendritic cells (DCs) with both viral and tumor-specific antigens would enhance the efficacy antitumor DC-based therapy applied simultaneously with oncolytic virus.
Methods: Vaccinia virus LIVP/GFP and melanoma B16-F10 were used in this study. DCs were pulsed with various combinations of viral and tumor-associated antigens. The maturation status of DCs was verified by expression of the markers CD80, CD86, and CCR7 and assessment of IL-6, TNF-α, and IL-12 secretion. The most efficient combination of antigens for DC loading was selected based on the analysis of the cytotoxic activity of T lymphocytes. Combination therapy using vaccinia virus LIVP/GFP and DCs pulsed with viral and tumor-specific antigens was administered to the B16-F10 melanoma/mouse C57Bl tumor model.
Results: We found that loading of DCs with viral antigens, or with a combination of viral and tumor antigens, resulted in similar levels of expression of DC maturation markers. The maximal in vitro cytotoxicity against virus-infected and non-infected B16 melanoma cells exhibited T lymphocytes activated by DCs loaded with the heat inactivated lysate of vaccinia virus LIVP/GFP infected tumor cell. The results show that the combination of vaccinia virus LIVP/GFP and DCs loaded with both tumor and viral antigens inhibit tumor growth of B16-F10 murine melanoma by more than two-fold.
Conclusions: Combination therapy with oncolytic vaccinia virus LIVP/GFP and tumor/virus antigen-loaded DCs limited the growth of established melanoma B16-F10, but no synergistic antitumor effects were observed. We propose that optimization of the therapy regimen could enhance the efficiency of combination therapy.
Keywords
INTRODUCTION
The efficacy of dendritic cell (DC)-based cancer immunotherapies has been demonstrated using tumor models and in clinical trials[1-3]. Over the past few decades, numerous experimental studies have improved our understanding of DC functioning in the context of cancer and suggested novel strategies to achieve effective and long-term anti-tumor immune responses. The results of in vivo studies demonstrate the great antitumor potential of DC-based cancer immunotherapy. Unfortunately, DC-based antitumor vaccines have failed to generate significant clinical benefits, and the limited success of DC-based therapy might be associated with different factors, including the need to optimize maturation cocktails to promote the immunostimulatory activity of DCs[4,5]. Indeed, DCs as professional antigen-presenting cells activate antitumor adaptive immune responses by efficient processing and cross-presentation of tumor antigens to CD8+ T cells followed by activation of tumor-specific cytotoxic T lymphocytes (CTLs). However, it is known that the tumor microenvironment (TME) can suppress the maturation and functions of DCs and, as a consequence, reduce the effectiveness of DC-based vaccines. Hence, it is very important to force DCs to overcome the negative effects of the TME to activate the appropriate tumor-specific cytotoxic T-lymphocyte response. Among the varied approaches to reducing the immunosuppressive effects of the TME, oncolytic virus-based therapy is one of the promising methods that has already been proven effective in anti-cancer virotherapy[6,7]. A combined immunotherapeutic approach based on DC-based cancer vaccines and oncolytic viruses (OVs) is supposed to have increased effectiveness due to the synergistic action of DCs and OVs[8]. On the one hand, immunosuppression in the TME provides effective virus replication in tumor cells, resulting in direct lysis of the cancer cells. OV-induced immunogenic death of tumor cells leads to the release of “danger signals” at the site of tumor localization and promotes more rapid maturation of DCs and priming of tumor-specific T cells, enhancing the antitumor activity of DCs in the TME[9]. On the other hand, the DCs activated against viral proteins and tumor antigens will enhance the effectiveness of virotherapy for “cleaning up” cancer non-lysed infected tumor cells. The utilization of different combinations of tumor-associated and virus antigens can potentially enhance the efficacy of DCs in the generation of an anti-tumor immune response during oncolytic virotherapy.
Previously, we showed that vaccinia virus LIVP/GFP could provide efficient tumor cell growth control in the B16-F10 melanoma model with activation of virus-specific immune responses[10]. In addition, we reported that single prophylactic vaccination with DCs loaded with different antigens was efficient at treating highly aggressive metastatic tumors, Lewis lung carcinoma, and B16-F10[11]. Taking into account our previous results, we studied the antitumor potential of oncolytic vaccinia virus and DC-based therapy as a component of combination therapy against melanoma B16-F10. Our results demonstrate that T lymphocytes primed with DCs loaded with both tumor and viral antigens exhibited significantly higher cytotoxic activity against B16-F10 cells in comparison with T lymphocytes primed with DCs loaded with either tumor or viral antigens. However, in vivo combination therapy including OV and DCs loaded with tumor/viral antigens was as effective as monotherapy with virus or DCs, and no enhancement of antitumor activity was observed.
METHODS
Cell lines and virus
Mouse melanoma B16-F10 cells were obtained from the Culture Collection of the Institute of Cytology, St. Petersburg, Russia. B16-F10 cells were cultured in IMDM medium supplemented with 10% fetal calf serum (FCS) and antibiotic-antimycotic solution (100 U/mL penicillin G, 100 units/mL streptomycin, and
Mice
Six- to eight-week-old female C57Bl/6 mice obtained from of State Research Center of Virology and Biotechnology VECTOR were kept in the vivarium conditions of the Institute of Chemical Biology and Fundamental Medicine, Siberian Branch of the Russian Academy of Sciences, with a natural light regime on a standard diet for laboratory animals [GOST (State Standard) R 50258 92] in accordance with the recommendations for the proper use and care of laboratory animals (ECC Directive 2010/63/EU), as well as the guidelines for good laboratory practice in pre-clinical studies of the Russian State Standards (R 51000.3-96 and 51000.4-96). All experiments were carried out in accordance with relevant national and international guidelines. The experimental protocols were approved by the Committee on the Ethics of Animal Experiments with the Institute of Cytology and Genetics of SB RAS (Novosibirsk, Russia) (protocol No. 52 from 23 May 2019).
Tumor cell lysate preparation for DC loading
B16-F10 cells were seeded at a density of 4 × 106 cells/mL in IMDM10 and cultured at 37 °C in 5% CO2 for 16-24 h. Then, cells were harvested, washed with phosphate-buffered saline (PBS), and frozen at -80 °C followed by thawing at 37 °C. The freeze-thaw cycles were repeated three times. The lysate was treated with ultrasound and passed through a 0.22 μm syringe filter (TPP, Switzerland). Protein concentration was analyzed with the Qubit Protein Assay Kit (Invitrogen, USA).
Preparation of the lysate of infected tumor cells for DC loading
B16-F10 tumor cells were cultured as described previously. Then, cells were washed with PBS and incubated with VV [at a multiplicity of infection (MOI) of 0.5 PFU/cell] for 1 h. The medium containing the virus was removed, and the cells were cultured in 10 mL IMDM supplemented with 2% FCS and 1% antibiotic-antimycotic solution at 37 °C in 5% CO2 for 16-24 h. Further steps of lysate preparation were described above. After that, the lysate of infected tumor cells was inactivated at 56 °C for 2 h[13].
Generation of bone-marrow-derived DCs
Bone marrow-derived DCs were generated from bone marrow cells isolated from the femur and tibia of female 7-8-week-old wild-type C57Bl/6 mice by exposing the bone marrow and flushing out the cells with PBS using a sterile syringe. The harvested cells were cultured in R10 medium (RPMI medium, 10% FCS, 1% antibiotic-antimycotic solution, 1% HEPES, 50 mM beta-mercaptoethanol, 2 mM Glutamine, and 1 mM sodium pyruvate). The culture medium was supplemented with 50 ng/mL recombinant murine IL-4 and
Flow cytometry analysis of DC maturation
For the analysis of the status of DC maturation, the DCs were stained with fluorophore-conjugated monoclonal antibodies recognizing CCR7, CD80, or CD86 (Life Technologies, USA) at 4 °C for 1 h. After washing twice in PBS, the cells were fixed in 4% formaldehyde in PBS. Flow cytometry was performed using a NovoCyte flow cytometer (ACEA Biosciences, USA), and the data were analyzed with NovoExpress software (ACEA Biosciences, USA). The experiments were performed in duplicate.
Measurement of cytokine levels
The levels of TNF-α, IFN-γ, IL-6, and IL-12 in the medium of cultured DCs or the serum of experimental mice were measured using mouse IL-6 and mouse TNF-α colorimetric ELISA kits (Biolegend, USA), as well as mouse IL-12 and mouse IFN-γ colorimetric ELISA kits (Thermo Scientific, USA), according to the manufacturer’s protocols. Absorbance was measured at 450 and 570 nm using a plate-reading spectrophotometer Multiskan RC (Thermo Lab Systems, USA). The experiments were performed in triplicate.
Preparation of murine splenocytes and analysis of cytotoxicity of T lymphocytes ex vivo
The cytotoxic effect of T lymphocytes on uninfected B16-F10 cells or cells infected with the VV was measured using microelectronic biosensor technology on the xCELLigence real-time device (ACEA Biosciences Inc., USA). The xCELLigence system is a technological approach that performs real-time cell analysis (RTCA) of cell culture. The cellular index (CI) is an indicator of the electrical potential that reflects the status of cells (viability, degree of adhesion, and growth dynamics). CI was calculated automatically based on the changes in resistance parameters during the interaction of cells with electrodes on microelectronic plates (E-plates).
Mice were implanted with dendritic cells activated by various combinations of the antigens (L, iL, LPS, or hiVV). After seven days, mice were euthanized; spleens were removed and placed in 3 mL R10. Excised spleens were ground through a 40 μm cell strainer using the plunger; cells were washed with PBS and centrifuged at 200 g for 5 min. Then, splenocytes were resuspended in 200 μL of PBS and 800 μL of ACK Lysis Buffer (Thermo Fisher Scientific, USA) and incubated at 20 °C for 5 min. After that, cells were washed with PBS and resuspended in 1 mL R10 for cell counting. Splenocytes were cultured at a concentration of
In vivo antitumor effects of combination oncolytic virus/DC immunotherapy
B16-F10 tumor cells (1.8 × 105/100 μL in IMDM) were subcutaneously (s/c) injected into the abdominal right flank of 6-8-week-old female C57Bl/6 mice. When the tumor volumes reached 20-25 mm3 (Day 7 or 8 after cell injection), mice were randomized and assigned to one of the experimental groups. Tumor-bearing mice were s/c injected with DCs (1 × 105/100 μL IMDM/mice) loaded with different antigens followed by peritumoral injection of VV (5 × 107 PFU/100 μL IMDM) (combination therapy) or IMDM only (DC-based monotherapy) two days after DC administration. In parallel, one group of mice received treatment with VV only (viral monotherapy). Control animals were injected with 100 μL IMDM only. Tumor growth was monitored every other day using a digital caliper, and tumor volume was calculated with the following formula: volume = 0.5 × L × W2, where L is length and W is width.
Statistical analysis
The data are reported as mean ± SEM. Statistical analysis was performed with GraphPad Prism software version 9.01 (La Jolla, CA, USA) and OriginPro software version 9.8 (OriginLab Corporation, USA). Statistical analyses were performed using one-way ANOVA. Values with P < 0.05 were considered to be statistically significant.
RESULTS
Treatment of immature BMDCs with different combinations of tumor-associated and viral antigens did not induce upregulation of costimulatory molecules and cytokine production
In the first stage, the efficiency of the expression of DC maturation markers was evaluated upon infection of cells with the VV. According to the literature data, DC infection with VV increases the expression of CD86 but downregulates that of CD80[15]. In our experiments, the level of CD80 expression depended on the viral dose used for infection: it was 1.8-fold higher when DCs were infected with a dose of MOI 1 PFU in comparison with a dose of MOI 10 PFU [Figure 1A]. The expression level of CD86 did not change upon DC infection with different doses of the virus. For loading/activation of DCs, not only a live virus but also a hiVV was used. DCs incubated with hiVV are characterized by the highest expression levels of CD80 and CD86 in comparison with infection with live VV; therefore, hiVV was used for DC loading/maturation in experiments [Figure 1A].
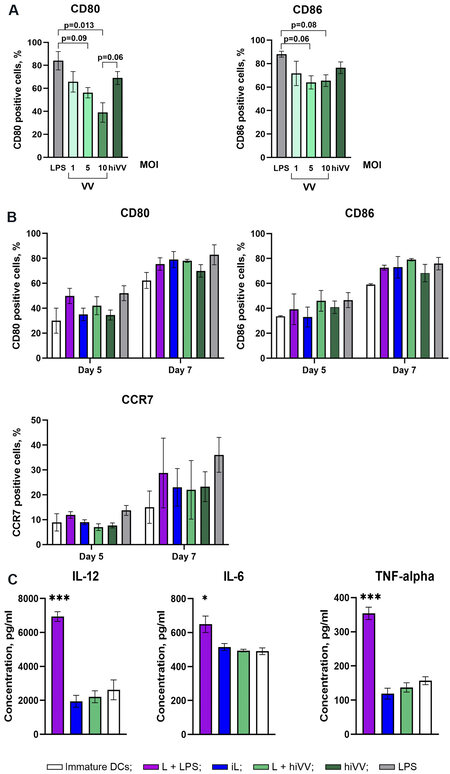
Figure 1. Influence of different sources of tumor and/or viral antigens on the expression of costimulatory/CCR7 molecules of DCs and their cytokine profile. (A) The number of CD80- and CD86-positive DCs in the cell population after treatment with LPS, live VV (VV), or heat-inactivated VV (hiVV) for 24 h. (B) The number of CD80-, CD86-, and CCR7-positive DCs generated five or seven days after treatment with tumor and/or viral antigens: L + LPS in violet; iL in blue; L + hiVV in green; hiVV in dark green; LPS in grey; negative control (immature DCs) in white. (C) The level of IL-12, IL-6, and TNF-α in the conditioned media of DCs loaded with tumor/viral antigens after 24 h of incubation [experimental group color set is the same as in (B)]. Data are presented as mean ± S.E.M.*P < 0.05, ***P < 0.001. DCs: Dendritic cells; VV: vaccinia virus; LPS: lipopolysaccharide.
The following antigens were used for DC loading/activation: the tumor cell lysate (L) in combination with LPS (L + LPS) or in combination with hiVV and infected cell lysate (iL). It is known that functionally active DCs should receive signals stimulating the maturation of DCs in addition to loading with tumor antigens[16]. Otherwise, immature DCs actively exert tolerogenic functions, induce regulatory T cells, and suppress the antitumor T cellular immune response[17]. Among different maturation stimulators of DCs, TLR4 agonist LPS is the most available and widely used agent[18-20]. We also used LPS to stimulate maturation of unloaded DCs as a positive control [Figure 1A] or tumor lysate-loaded DCs in experiments ex vivo [Figure 2] and in vivo [Figure 3]. Viral antigens in the form of hiVV or infected cell lysate (iL) contained pathogen-associated molecular patterns (PAMPs) and should activate maturation of DCs without any additional agents.
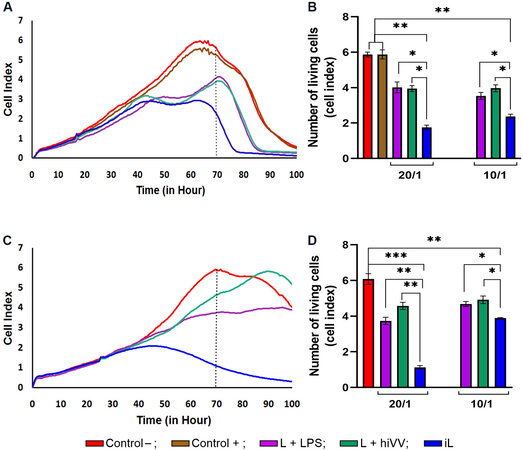
Figure 2. The cytotoxicity of mouse splenocytes primed in vivo with tumor/viral antigen-loaded DCs with respect to B16 melanoma cells. The viability of B16 melanoma cells was monitored in real time with an xCELLigence system. (A, C) The viability of infected (A) or uninfected (C) B16 cells incubated alone (control) or with primed splenocytes at an effector-to-target cell ratio of 20/1 is shown as an RTCA graph. (B, D) Bar diagrams represent the cytotoxicity of primed splenocytes against infected (B) or uninfected (D) B16 melanoma cells at an effector-to-target cell ratio of 20/1 or 10/1, corresponding to the 70 h time point in (A, C). Control- (intact B16 cells) in red; control+ (live VV infected B16 cells) in brown; DCs loaded with L + LPS in violet; with iL in blue; and with L + hiVV in green. Data are presented as mean ± S.E.M. *P < 0.05, **P < 0.01, ***P < 0.001. DCs: Dendritic cells; VV: vaccinia virus; RTCA: real-time cell analysis; hiVV: heat-inactivated vaccinia virus.
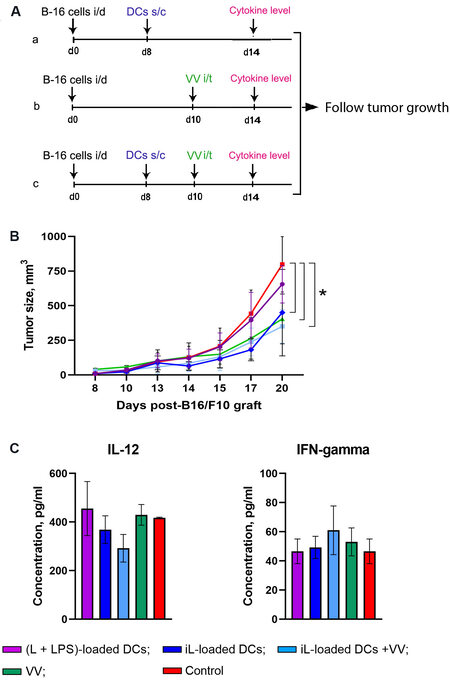
Figure 3. Antitumor effects of therapy based on DCs and oncolytic VV in a murine melanoma B16 model. (A) Experimental setup. B16-bearing C57BL/6 mice (n = 10) on Day 8 after tumor transplantation were subcutaneously (s/c) injected with antigen-loaded DCs (a); on Day 10 with VV intratumorally (i/t) administered (b); or on Day 8 DCs (s/c) followed by intratumoral injection with VV on Day 10 (c). (B) Progress curves of melanoma B16 growth. (C) The levels of cytokines in the blood serum of B16-bearing C57BL/6 mice receiving either of the monotherapies with DCs or oncolytic VV or combination therapy. (L + LPS)-loaded DCs in violet; iL-loaded DCs in dark blue; iL-loaded DCs + VV in blue; VV in green; and control in red. Data from two independent experiments are presented as mean ± S.E.M. *P < 0.05. DCs: Dendritic cells; VV: vaccinia virus; LPS: lipopolysaccharide.
Currently, to obtain mature DCs, different activation schemes are used, and the process of generation of immature DCs varies from 48 h to 7 days[21,22]. In this work, we obtained immature DCs using either fast or standard protocols, which require four or six days, respectively. The level of DC activation was assessed 24 h after the addition of appropriate antigens to DCs (on Day 5 or 7 of the DC generation process, respectively) [Figure 1B]. The maturation status of DCs was assessed by the expression of activation markers CD80 and CD86. The expression level of chemokine receptor CCR7, which is involved in the migration of DCs to the lymph nodes[23], was also evaluated.
The maximal level of CD80 expression [Figure 1B] was observed for DCs loaded with L + LPS (49.87%) generated by the fast protocol, whereas loading of DCs with other antigens caused less efficient CD80 expression [Figure 1B]. However, DCs generated by the standard six-day protocol were characterized by almost equal levels of CD80 expression regardless of the type of loaded antigen [Figure 1B].
The expression level of CD86 was the highest (46.6%) in DCs stimulated with L + hiVV (fast protocol), while, under all other experimental conditions, similar to CD80, the number of CD86+ cells did not exceed 40% [Figure 1B]. The expression level of CD86 in DCs generated by standard protocols increased by ~30% in comparison with DCs generated by the fast protocol and reached 80% for DCs stimulated with L + hiVV, while, for DCs stimulated with other antigens, CD86 expression was also high with statistically insignificant differences between samples. It should also be noted that the expression levels of activation markers generated by standard protocol were the highest but statistically insignificant in comparison with a fast protocol.
The expression of the CCR7 chemokine receptor in DCs stimulated by various antigens is shown in Figure 1B. The fast protocol of DC generation mediated low expression of the CCR7 receptor in all DC samples, and the level of CCR7 expression did not exceed 15%. DCs generated by the standard protocol were characterized by a much higher level of CCR7 expression: the number of CCR7+ cells in the DC population varied from 20% to 30%, and the maximum number of CCR7+ DCs was 28.7% in DCs activated by L + LPS. However, the statistical analysis of the obtained data did not reveal significant differences between the level of expression of the marker and the method of activation for any of the studied markers.
The analysis of the influence of different sources of tumor and/or viral antigens on the efficiency of IL-12, IL-6, and TNF-α secretion showed that, as expected, L + LPS induced the maximum level of expression of all studied cytokines [Figure 1C], while there was no increase in cytokine production in DCs activated by other antigens, in comparison with the control (immature DCs).
DCs loaded with lysate of LIVP-infected B16 melanoma cells efficiently prime antitumor T lymphocytes in vivo
In the next step, we estimated the antitumor activity of effector T lymphocyte priming by DCs loaded with tumor or viral antigens or their combination. DCs loaded with the tumor cell lysate (L) in combination with LPS (L + LPS) or hiVV (L + hiVV) and infected cell lysate (iL) were administered s/c in mice (n = 2 per group). Both infected and uninfected B16 melanoma cells (depicted as Control+ and Control-, respectively) were selected as target cells for cytotoxic T lymphocytes. We hypothesized that DC activation by an iL containing a large amount of PAMP, danger-associated molecular patterns, and tumor antigens[24] would promote the development of an effective cytotoxic immune response against not only infected tumor cells but also uninfected cells. Splenocytes (effector cells) from treated mice were added to B16-F10 melanoma cells (target cells) in effector-to-target cell ratios of 20:1 and 10:1 and incubated for 100 h. The viability of tumor cells was monitored in real-time mode by using an xCELLigence real-time device. The cytotoxicity of T lymphocytes was assessed by comparing the cell index (CI) of melanoma B16 cells in the experimental groups (DCs loaded with L + LPS, iL, or L + hiVV) with the CI for the control groups (uninfected and infected melanoma cells) [Figure 2].
It was demonstrated that CTL activated with DC-loaded iL (CTLs-DC/iL) exhibited the maximum cytotoxic activity (71% of infected B16 cells were lysed) compared to CTLs primed against only tumor antigens (CTLs-DC/L + LPS) and tumor/viral antigens (CTLs-DC/ L + hiVV) at an effector-to-target cell ratio of 20:1 with respect to infected B16 cells [Figure 2A]. Overall, 61% of the population of infected B16 cells were destroyed by CTLs-DC/iL at an effector-to-target cell ratio of 10:1 in comparison with 40% and 33% for the CTLs-DC/L + LPS and CTLs-DC/ L + hiVV groups, accordingly. The observed effects were strengthened when non-infected melanoma B16 cells were used as target cells. Surprisingly, the lysate of infected B16 cells was the most efficient source of antigens and mediated activation of CTLs that killed up to 82% of non-infected tumor cells (iL; Figure 2B) at the effector-to-target cell ratios of 20:1, while cell lysis ranged from 31.5% to 17.4% for the CTLs-DC/L + LPS and CTLs-DC/ L + hiVV groups, accordingly [Figure 2D]. The obtained data suggest that CTLs-DC/iL should efficiently inhibit melanoma B16-F10 in vivo during DC-based monotherapy and immunotherapy combined with oncolytic viruses.
Therapeutic efficacy of combination therapy with DCs loaded with tumor/viral antigens and oncolytic vaccinia virus in an established melanoma B16-F10 tumor model in vivo
First, to compare the therapeutic efficacy of DC monotherapy in mice with established B16-F10 melanoma tumors, DCs loaded with L + LPS or iL were subcutaneously injected on Day 8 after tumor transplantation
To evaluate in vivo the therapeutic efficacy of VV in combination with DCs, melanoma bearing C57BL/6 mice were s/c injected with iL-loaded DCs on Day 8 after tumor transplantation followed by intratumoral injection of VV on Day 10 [Figure 3A and B]. The experimental treatments included: (1) i/t PBS only (the control); (2) i/t VV only (5 × 107 viral particle/injection); and (3) a combination of s/c iL-loaded DCs
DISCUSSION
Melanoma is one of the most aggressive forms of skin cancer, metastasizing to various vital organs and responding to both chemotherapeutic and radiological treatment. Currently, DC-based vaccines are frequently used for melanoma treatment. Since their first application, many clinical trials with DC-based therapy have been conducted in melanoma patients, resulting in an 8.5% objective response rate[25]. Being professional antigen presentation cells, DCs present tumor antigens to naïve T lymphocytes and activate the adaptive antitumor immune response. Moreover, it was demonstrated that activation of antitumor CTL is a key mechanism of elimination of tumor cells upon DC-based immunotherapy[26-28]. Although the second generation of anti-melanoma DC-based vaccines has already been developed, immunotherapy with DCs does not lead to the complete elimination of tumors[29].
The aim of our study was to select a combination of tumor/viral antigens for DC loading that should efficiently mediate DC maturation and induction of CTLs against tumor melanoma cells. We also planned to use these DCs as part of the combination therapy of melanoma with vaccinia virus. Previously, we showed that total tumor RNA used as a source of tumor antigens for DC loading stimulated significantly higher antitumor effect in a metastatic melanoma model in comparison with tumor cell lysate[11,30-32]. However, in this work, protein antigens, namely lysate of tumor cells and/or viral particles, were selected as a source of antigens for DC loading, since it was reasonable to evaluate the immunogenic properties of DCs loaded with a “natural” source of tumor/viral antigens presented in tumor node after virotherapy and use a mixture of tumor and viral antigens in the same form. Moreover, in numerous works, DCs loaded with tumor cell lysates, proteins, or apoptotic bodies have been shown to activate antitumor cytotoxic T lymphocytes, effectively reduce tumor size, and increase the survival of tumor-bearing animals in various mouse tumor models[33-36].
Firstly, we evaluated the effects of various stimuli, including LPS and VV, on DC maturation. It is known that vaccinia virus encodes for a number of immune evasion proteins[37,38] and proteins inhibiting the expression of cytokines[39] that finally suppress the immune response. On the other hand, proteins of vaccinia virus[40] and the Western Reserve strain of vaccinia virus[15] were demonstrated to activate DCs. We showed that infection of DCs with VV led to upregulation of the expression of CD80 at a low MOI, wherein the expression of this marker decreases with increasing viral dosage. At the same time, the expression of CD86 did not respond to the infection and remained on the same level regardless of the MOI used. Previously, it was shown that treatment of DCs with heat-inactivated or UV-inactivated vaccinia virus Ankara resulted in induction of IFN production via the cGAS-STING cytosolic DNA-sensing pathway[41]. Indeed, in our case, heat-inactivated VV more efficiently activated the expression of maturation markers in comparison with the live virus [Figure 1A], but the differences between the samples for CD80 (VV at MOI = 1 and hiVV) and CD86 were not statistically significant. It is interesting that LPS used as a positive control effectively stimulated CD80 and CD86 expression, amounting to almost 80% and 90% positive cells in the DC population, respectively.
It was revealed that different sources of antigens, L + hiVV, hiVV, iL, and L + LPS, used for DC loading induced similar levels of maturation markers (CD80 and CD86) and chemokine receptor CCR7 expression [Figure 1B]. The degree of DC maturity was also assessed by the production of cytokines IL-12, IL-6, and TNF-α. Statistical analysis did not show significant differences in cytokine production by any of mature DCs except the (L + LPS)-loading DCs [Figure 1C]. It seems likely that all sets of antigens used for DC loading except LPS stimulated the maturation of DCs equally.
We further assessed the degree of CTL activation in mice after administration of DCs loaded with various antigens. We demonstrated that DCs loaded with different sources of tumor/viral antigens efficiently activated antitumor CTLs in vivo (Figure 2A and C, effector-to-target ratio 20:1). According to the obtained data, CTL activated by iL-loaded DCs exhibited the maximum cytotoxic activity against both infected
To study the combined antitumor effects of DC-based vaccines and VV virotherapy in vivo, the most efficient source of tumor/viral antigens was selected to load DCs. In vivo experiments showed that treatment of melanoma-bearing mice with iL-loaded DCs (monotherapy) resulted in retardation of tumor growth, while inhibition of tumor growth was not observed in mice treated with DCs stimulated with L + LPS [Figure 3B]. Therefore, these results confirm the efficacy of iL-loaded DCs in targeting CTL against melanoma cells. In addition, it was revealed that either DC- or VV-based monotherapy or DC + VV combination therapy exhibited similar antitumor effects in vivo: tumor size was decreased 2.3-fold in comparison with a non-treated control group. It can be assumed that antitumor effects in each group were achieved through different mechanisms: (1) DC-mediated activation of antitumor CTLs; (2) direct lysis of tumor cells by oncolytic virus; and (3) their combination. It is interesting that all tested treatment protocols resulted in retardation of tumor growth with similar efficiency, and neither an additive nor a synergistic effect of combination therapy was observed. We supposed that the treatment regimen was not optimal, and antitumor/antiviral CTLs were not activated in time. Such a therapeutic schedule was chosen based on the fact that DCs migrate to the lymph nodes within 72 h after intradermal administration[42]. However, it should be noted that an effective CTL response was observed after much longer circulation (seven days) of antigen-loaded DCs in mice after subcutaneous injection. Therefore, it is reasonable to speculate that optimization of the experimental design by prolongation of the time between antigen-loaded DC administration and viral therapy could increase the effectiveness of combination therapy for less aggressive tumors, while changing the time frame of the administration of DCs and VVs is problematic for such a rapidly growing tumor as B16-F10 melanoma. Moreover, we cannot exclude the possibility that weakening of tumor immunosuppression by direct lysis of VV-infected tumor cells was not enough to induce a strong antitumor T cellular response primed by DCs. Thus, further optimization of the treatment regimen for DC/oncolytic virus combination therapy is needed. The frequencies of vaccine administration and time interval between VV and DC administration as well as the dosage of VV and DCs can be optimized. It should also be noted that DC + VV combination therapy might be used for converting so-called “cold tumors”, characterized by scare or absent T-cell infiltration in tumor nests and surrounding stroma, into immunologically “hot” tumor. A combination of iL-loaded DCs and VV might enhance the efficiency of anti-tumor immunotherapy including adoptive T-cell response and immune checkpoints.
We also estimated the level of IL-12 and IFN-γ in the serum of mice two weeks after tumor cell implantation. IL-12, a heterodimeric cytokine produced mainly by antigen-presenting cells, regulates innate responses and determines the type of adaptive immune responses. Many of the immunoregulatory effects of IL-12 have been attributed to its ability to induce IFN-γ production. IFN-γ directly acts as a cytotoxic CD8 T-cell differentiation signal, and it is essential for the induction of cytotoxic T-cell precursor proliferation. The results obtained reveal increasing levels of IFN-γ in serum of mice treated with iL-loaded DCs, but the differences were not statistically significant [Figure 3C]. The experiments showed no significant fluctuations in the IL-12 level in the serum of mice from any group except the mice treated with iL-loaded DCs, but this fluctuation was also not statistically significant.
In conclusion, we identified a set of antigens to obtain mature DCs that activate CTLs efficiently lysing B16-F10 melanoma cells. Our results demonstrate that T lymphocytes activated with iL-loaded DCs showed the maximum cytotoxic activity. We found that monotherapy with DCs loaded with a mixture of tumor/viral antigens (iL) is more efficient in the treatment of established B16-F10 melanoma tumors in comparison with DCs stimulated only by tumor antigens (L + LPS). However, the obtained results indicate that the chosen scheme of combination therapy with DCs and VV has no advantages over VV-based or iL-loaded DCs-based monotherapy in a melanoma B16-F10 tumor model in vivo. These studies should be extended to investigate the different schemes of cooperation of DC and oncolytic virus.
DECLARATIONS
Authors’ contributionsGoncharova EP, Gamburg TA, Markov OV, Zenkova MA
Designed the overall project: Goncharova EP, Zenkova MA
Performed the experiments: Goncharova EP, Gamburg TA
Analyzed and interpreted the data: Goncharova EP, Markov OV, Zenkova MA
Wrote the manuscript: Goncharova EP, Gamburg TA, Markov OV, Zenkova MA
Availability of data and materialsThe data that support the findings of this study are available from the corresponding author upon request.
Financial support and sponsorshipThis work was supported by the Russian Foundation for Basic Research project # 18-34-20109, Russian state budget of ICBFM SB RAS project # 121031300044-5, Russian Science Foundation RSF # 19-74-30011.
Conflicts of interestAll authors declared that there are no conflicts of interest.
Ethical approval and consent to participate6- to 8-week-old female C57Bl/6 obtained from of State Research Center of Virology and Biotechnology VECTOR were kept in the vivarium conditions of Institute of Chemical Biology and Fundamental Medicine, Siberian Branch of the Russian Academy of Sciences, with natural light regime on a standard diet for laboratory animals [GOST (State Standard) R 50258 92] in accordance with the recommendations for the proper use and care of laboratory animals (ECC Directive 2010/63/EU), as well as the guidelines of good laboratory practice in pre-clinical studies of the Russian State Standards (R 51000.3-96 and 51000.4-96) and all experiments were carried out in accordance with relevant national and international guidelines. The experimental protocols were approved by the Committee on the Ethics of Animal Experiments with the Institute of Cytology and Genetics of SB RAS (Novosibirsk, Russia) (protocol No. 52 from 23 May 2019).
Consent for publicationNot applicable.
Copyright© The Author(s) 2022.
REFERENCES
1. Matsuo K, Yoshie O, Kitahata K, Kamei M, Hara Y, Nakayama T. Recent progress in dendritic cell-based cancer immunotherapy. Cancers (Basel) 2021;13:2495.
2. Bol KF, Schreibelt G, Rabold K, et al. The clinical application of cancer immunotherapy based on naturally circulating dendritic cells. J Immunother Cancer 2019;7:109.
3. Salah A, Wang H, Li Y, et al. Insights into dendritic cells in cancer immunotherapy: from bench to clinical applications. Front Cell Dev Biol 2021;9:686544.
4. Macri C, Dumont C, Johnston AP, Mintern JD. Targeting dendritic cells: a promising strategy to improve vaccine effectiveness. Clin Transl Immunology 2016;5:e66.
5. Lluesma S, Wolfer A, Harari A, Kandalaft LE. Cancer vaccines in ovarian cancer: how can we improve? Biomedicines 2016;4:10.
6. Lawler SE, Chiocca EA. Oncolytic virus-mediated immunotherapy: a combinatorial approach for cancer treatment. J Clin Oncol 2015;33:2812-4.
7. Pol J, Kroemer G, Galluzzi L. First oncolytic virus approved for melanoma immunotherapy. Oncoimmunology 2016;5:e1115641.
8. Pipperger L, Riepler L, Kimpel J, et al. Differential infection of murine and human dendritic cell subsets by oncolytic vesicular stomatitis virus variants. Oncoimmunology 2021;10:1959140.
9. Koske I, Rössler A, Pipperger L, et al. Oncolytic virotherapy enhances the efficacy of a cancer vaccine by modulating the tumor microenvironment. Int J Cancer 2019;145:1958-69.
10. Goncharova EP, Ruzhenkova JS, Petrov IS, Shchelkunov SN, Zenkova MA. Oncolytic virus efficiency inhibited growth of tumour cells with multiple drug resistant phenotype in vivo and in vitro. J Transl Med 2016;14:241.
11. Markov OV, Mironova NL, Sennikov SV, Vlassov VV, Zenkova MA. Prophylactic dendritic cell-based vaccines efficiently inhibit metastases in murine metastatic melanoma. PLoS One 2015;10:e0136911.
12. Petrov IS, Goncharova EP, Kolosova IV, et al. Antitumor effect of the LIVP-GFP recombinant vaccinia virus. Dokl Biol Sci 2013;451:248-52.
13. Galasso GJ, Sharp DG. Effects of heat on the infecting, antibody-absorbing, and interfering powers of vaccinia virus. J Bacteriol 1965;89:611-6.
14. Yakimovich A, Mercer J. High-content analyses of vaccinia plaque formation. Methods Mol Biol 2019;2023:237-53.
15. Salek-Ardakani S, Arens R, Flynn R, Sette A, Schoenberger SP, Croft M. Preferential use of B7.2 and not B7.1 in priming of vaccinia virus-specific CD8 T cells. J Immunol 2009;182:2909-18.
16. Morse MA, Mosca PJ, Clay TM, Lyerly HK. Dendritic cell maturation in active immunotherapy strategies. Expert Opin Biol Ther 2002;2:35-43.
17. Mahnke K, Johnson TS, Ring S, Enk AH. Tolerogenic dendritic cells and regulatory T cells: a two-way relationship. J Dermatol Sci 2007;46:159-67.
18. Matheoud D, Perié L, Hoeffel G, et al. Cross-presentation by dendritic cells from live cells induces protective immune responses in vivo. Blood 2010;115:4412-20.
19. Huck SP, Tang SC, Andrew KA, Yang J, Harper JL, Ronchese F. Activation and route of administration both determine the ability of bone marrow-derived dendritic cells to accumulate in secondary lymphoid organs and prime CD8+ T cells against tumors. Cancer Immunol Immunother 2008;57:63-71.
20. Näslund TI, Gehrmann U, Qazi KR, Karlsson MC, Gabrielsson S. Dendritic cell-derived exosomes need to activate both T and B cells to induce antitumor immunity. J Immunol 2013;190:2712-9.
21. Nazarkina ZK, Zajakina A, Laktionov PP. Maturation and antigen loading protocols influence activity of anticancer dendritic cells. Mol Biol 2018;52:222-31.
22. Skalova K, Mollova K, Michalek J. Human myeloid dendritic cells for cancer therapy: does maturation matter? Vaccine 2010;28:5153-60.
23. Penna G, Vulcano M, Sozzani S, Adorini L. Differential migration behavior and chemokine production by myeloid and plasmacytoid dendritic cells. Hum Immunol 2002;63:1164-71.
24. Kaufman HL, Kohlhapp FJ, Zloza A. Oncolytic viruses: a new class of immunotherapy drugs. Nat Rev Drug Discov 2015;14:642-62.
25. Anguille S, Smits EL, Lion E, van Tendeloo VF, Berneman ZN. Clinical use of dendritic cells for cancer therapy. Lancet Oncol 2014;15:e257-67.
26. Tay BQ, Wright Q, Ladwa R, et al. Evolution of cancer vaccines-challenges, achievements, and future directions. Vaccines (Basel) 2021;9:535.
27. Kwak M, Leick KM, Melssen MM, Slingluff CL Jr. Vaccine strategy in melanoma. Surg Oncol Clin N Am 2019;28:337-51.
28. Franklin C, Livingstone E, Roesch A, Schilling B, Schadendorf D. Immunotherapy in melanoma: recent advances and future directions. Eur J Surg Oncol 2017;43:604-11.
29. Bulgarelli J, Tazzari M, Granato AM, et al. Dendritic cell vaccination in metastatic melanoma turns “non-T cell inflamed” into “T-cell inflamed” Tumors. Front Immunol 2019;10:2353.
30. Markov OO, Mironova NL, Maslov MA, et al. Novel cationic liposomes provide highly efficient delivery of DNA and RNA into dendritic cell progenitors and their immature offsets. J Control Release 2012;160:200-10.
31. Markov OV, Mironova NL, Shmendel EV, et al. Multicomponent mannose-containing liposomes efficiently deliver RNA in murine immature dendritic cells and provide productive anti-tumour response in murine melanoma model. J Control Release 2015;213:45-56.
32. Markov OV, Mironova NL, Shmendel EV, Maslov MA, Zenkova MA. Systemic delivery of complexes of melanoma RNA with mannosylated liposomes activates highly efficient murine melanoma-specific cytotoxic T cells in vivo. Mol Biol 2017;51:102-7.
33. Oda Y, Suzuki R, Otake S, et al. Prophylactic immunization with Bubble liposomes and ultrasound-treated dendritic cells provided a four-fold decrease in the frequency of melanoma lung metastasis. J Control Release 2012;160:362-6.
34. Pandey VK, Shankar BS, Sainis KB. G1-4 A, an arabinogalactan polysaccharide from Tinospora cordifolia increases dendritic cell immunogenicity in a murine lymphoma model. Int Immunopharmacol 2012;14:641-9.
35. Matheoud D, Baey C, Vimeux L, et al. Dendritic cells crosspresent antigens from live B16 cells more efficiently than from apoptotic cells and protect from melanoma in a therapeutic model. PLoS One 2011;6:e19104.
36. Baek S, Lee SJ, Kim MJ, Lee H. Dendritic cell (DC) vaccine in mouse lung cancer minimal residual model; comparison of monocyte-derived DC vs. hematopoietic stem cell derived-DC. Immune Netw 2012;12:269-76.
37. Yu H, Bruneau RC, Brennan G, Rothenburg S. Battle royale: innate recognition of poxviruses and viral immune evasion. Biomedicines 2021;9:765.
38. Georgana I, Sumner RP, Towers GJ, Maluquer de Motes C. Virulent poxviruses inhibit DNA sensing by preventing STING activation. J Virol 2018;92:e02145-17.
39. Di Pilato M, Mejías-Pérez E, Sorzano COS, Esteban M. Pilato , , , . Distinct roles of vaccinia virus NF-κB inhibitor proteins A52, B15, and K7 in the immune response. J Virol 2017;91:e00575-17.
40. Agrawal S, Gupta S, Agrawal A. Vaccinia virus proteins activate human dendritic cells to induce T cell responses in vitro. Vaccine 2009;27:88-92.
41. Dai P, Wang W, Yang N, et al. Intratumoral delivery of inactivated modified vaccinia virus Ankara (iMVA) induces systemic antitumor immunity via STING and Batf3-dependent dendritic cells. Sci Immunol 2017;2:eaal1713.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Goncharova EP, Gamburg TA, Markov OV, Zenkova MA. Combined effects of oncolytic vaccinia virus and dendritic cells on the progression of melanoma B16-F10 in mice. J Cancer Metastasis Treat 2022;8:10. http://dx.doi.org/10.20517/2394-4722.2021.195
AMA Style
Goncharova EP, Gamburg TA, Markov OV, Zenkova MA. Combined effects of oncolytic vaccinia virus and dendritic cells on the progression of melanoma B16-F10 in mice. Journal of Cancer Metastasis and Treatment. 2022; 8: 10. http://dx.doi.org/10.20517/2394-4722.2021.195
Chicago/Turabian Style
Goncharova, Elena P., Tatiana A. Gamburg, Oleg V. Markov, Marina A. Zenkova. 2022. "Combined effects of oncolytic vaccinia virus and dendritic cells on the progression of melanoma B16-F10 in mice" Journal of Cancer Metastasis and Treatment. 8: 10. http://dx.doi.org/10.20517/2394-4722.2021.195
ACS Style
Goncharova, EP.; Gamburg TA.; Markov OV.; Zenkova MA. Combined effects of oncolytic vaccinia virus and dendritic cells on the progression of melanoma B16-F10 in mice. J. Cancer. Metastasis. Treat. 2022, 8, 10. http://dx.doi.org/10.20517/2394-4722.2021.195
About This Article
Copyright

Data & Comments
Data
0

 Cite This Article 6 clicks
Cite This Article 6 clicks

 Like This Article 1
likes
Like This Article 1
likes






Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.