
Combining oncolytic virus and radiation therapy for cancer management

 ,
, Abstract
Cancer has caused a tremendous burden in developing countries. Oncolytic virus (OV) therapy is an emerging modality with the potential to be a single or combination agent with radiation therapy (RT). Following entry of OV to the cell, OV will replicate and assemble before exiting from tumor cells. Construction of OV can be done by modifying the capsid, genome, and chemical material of viruses. Irradiation will induce double-strand breaks, and further integration of OV with DNA damage response pathway will interact with the MRE11-Rad50-Nbs1 complex to regulate the mobilization of E4 open reading frame 6, protein phosphatase 2A, poly(ADP-ribose) polymerase, apoptosis-inducing factor, and topoisomerase-IIβ-binding protein 1. Degradation of DNA-dependent protein kinase catalytic subunits via human simplex virus-1-infected cell polypeptide 0 will inhibit DNA repair. OV and RT have a synergistic interaction to cause viral oncolysis and upregulation of immune response. In the clinical setting, most studies have demonstrated that OV is a safe treatment with less toxicity. Moreover, OV + RT resulted in longer median survival (62.4 vs. 37.7 weeks) in malignant glioma.
Keywords
INTRODUCTION
Having been increasing for the past decades, the incidence and mortality of cancer have caused a tremendous burden in our world. Based on the International Agency for Research on Cancer (IARC) GLOBOCAN database, in 2018, more than 18 million new cases and 9.5 million deaths were attributable to cancer in the world. This number increased from the previous 2012 GLOBOCAN database when 14.1 million cases and 8.2 million deaths were detected in the world. As an integral part of Asia, where the highest incidence and mortality reside, developing countries such as India, Indonesia, and Thailand were also facing a high disease burden, with 1.324 million, 396.9 thousand, and 190.6 thousand cases in 2020. Furthermore, the numbers of deaths in India, Indonesia, and Thailand were 851.6 thousand, 396.9 thousand, and 124.8 thousand, respectively. Based on the estimation from IARC, the worldwide incidence and deaths due to cancer will increase by 63.4% and 71.5% to 29.5 and 16.38 million cases by 2040 (compared to 2018)[1,2] As the trend of cancer burden is increasing, further measures should be taken to fight the disease.
Surgery, chemotherapy, and radiation therapy are the three main treatments for cancer, especially for solid tumors. However, those treatments may only be partially effective with short-term and long-term toxicities[3]. Radiation therapy (RT) is a modality for cancer management by using high-energy radiation sources such as photons, charged particles, and protons to alter cancer cells’ metabolism, disrupt the tumor microenvironment (TME), and reduce cancer cell survival. RT can be tailored and delivered internally or externally to the location of interest. Usually, the RT fraction is given as 1.5 Gray (Gy)-2 Gray (Gy), 5 days/week for 5 weeks-7 weeks. Recent improvements in technologies have enhanced RT delivery from 2D techniques to more effective techniques such as 3D-conformal RT (3D-CRT), intensity-modulated radiotherapy, and stereotactic radiotherapy (SRT). Based on the RT utilization rate, it is predicted that 52% of cancer patients have to be treated with RT[3,4].
Recently, due to the increasing demand for tumor-selective therapies which can treat tumors without having significant toxicities, many novel approaches are emerging.[5] One of the rapidly growing treatment strategies is immunotherapy. The human immune system is well known for its ability to recognize and kill cancer cells; thus, modulating the immune system within a patient’s body via immunotherapy gives another possible therapeutic strategy to fight cancer[4,6]. Oncolytic virus (OV), as part of immunotherapy, is an emerging modality with the potential to be a single or combination agent for cancer treatment[7]. OV can be engineered for selective replication in tumor tissues but not normal non-neoplastic host cells. After further replication, the antitumoral effect can be elicited through direct infection, tumor cell lysis, or via the immune system. This method of immune therapy enables specific localization of tumors with reduced side effects[8]. When combined with the currently developed cancer treatment modalities such as radiation therapy, OV has a synergistic interaction which may result in a better antitumoral response[7]. This review discusses the role of combining oncolytic virus and radiation therapy in cancer management.
HISTORY AND TYPE OF ONCOLYTIC VIRUSES
The history of OV therapy started from multiple anecdotal reports dated 3000 years ago in ancient Egypt, where concomitant infection with high fever resulted in tumor spontaneous disappearance[9]. In the late 1700s, vaccination against viruses was utilized in daily life. Further application of viruses for cancer therapy was explored in the early 1900s, where concurrent natural viral infection resulted in cancer remission. There are reports that a 42-year-old woman and a 4-year-old boy with leukemia demonstrated disease remission subsequent to infection by presumed influenza and chickenpox, respectively. However, leukemia relapsed and progressed rapidly after one month of remission, with death as its endpoint. Following that, several observations suggested that viruses may have the ability to kill cancer cells selectively and effectively, but alterations were required. In the 1950s, when viral cell culture advanced significantly for in vitro and animal model research, a study showed that the administration of the encephalitis virus could shrink sarcoma, but the mice models died from fatal encephalitis. As chemotherapy and radiotherapy advanced rapidly in that era, the interest in OV went into hiatus until the 1990s, when genetic engineering made viral genomic alteration possible[10,11].
OV are one of the methods for combating cancer by using attenuated viruses to infect and generate/boost immune responses towards tumor cells. In most OV therapy, genetic modification is performed on viruses to increase virulence for tumor cells but not non-neoplastic normal cells[12]. Some of the potential viruses for OV therapy with the related type of cancers trial availability are summarized in Table 1.
| Virus | Herpes simplex virus | Vaccinia virus | Newcastle disease virus | Adenovirus | Reovirus |
| Family | Herpesviridae | Poxviridae | Paramyxoviridae | Adenoviridae | Reoviridae |
| Genome | dsDNA | dsDNA | ssRNA | dsDNA | dsRNA |
| Site of replication | Nucleus | Cytoplasm | Cytoplasm | Nucleus | Cytoplasm |
| Envelope status | Enveloped | Enveloped | Enveloped | Non-enveloped | Non-enveloped |
| Name of viruses in clinical development | T-VEC | Peca-Vec | MEDI5395 | ONCOS-102, TILT-123, DNX-2440, LOAd703 | Reolysin |
| Transgene | GM-CSF | GM-CSF | GM-CSF | GM-CSF, IL-2, TNF-α OX40L trimerized CD40L, 4-1BBL | None |
| Cancer selectivity | Viral gamma 34.5 gene, thymidine kinase deletion, tumor-selective promoters, targeting microRNA | Deletion of thymidine kinase, ribonucleotide reductase, growth factor of virus, and viral B18R gene | Naturally IFN sensitive | Partial deletion of viral control for gene expression (viral gene E1A) through targeting microRNA or tumor-selective promoters | Naturally IFN sensitive |
| Type of cancer available | Melanoma, head and neck, pancreatic, GBM, breast, hepatocellular carcinoma | Head and neck, melanoma, lung, breast, hepatocellular carcinoma, colorectal cancer | GBM | Head and neck, GBM, breast, prostate, ovarian, colorectal, bladder cancer | Head and neck, pancreatic, melanoma, ovarian, NSCLC, glioma, sarcoma, colorectal cancer |
CONSTRUCTION OF ONCOLYTIC VIRUSES
Natural viral tropism towards cancer but not normal cells has been discovered in some types of viruses where the sustainability of cancer growth is maintained for viral infection and replication purposes. These viruses can selectively bind and infect overexpressed surface receptors on cancer cells. Some examples are found in squamous cell carcinoma, where elevated HSV-1 infection and cytotoxicity will lead to the increasing level of nectin-1 (cell surface adhesion molecule); several human cancers such as hepatocellular carcinoma, colorectal cancer, ovarian cancer, and breast cancer also express CD46 surface receptor, which may be utilized by measles virus for cellular entry. Deficiency of anti-viral defense in cancer cells, such as abnormality of IFN pathways and protein kinase R activity, which play a role in protein synthesis and prevention of viral replication, also lead to increasing susceptibility towards viral infection[10].
The natural tropism of viruses is also affected by the type of viruses. RNA viruses have a faster tumor cell killing effect due to their replication in the cytoplasm of cancer cells. Conversely, nuclei-replicated DNA viruses have a better tumor-selective property. Comparing the presence of envelope and size of viruses, naked viruses and smaller viruses have a better tumor tropism due to their immune evasion and tumor infiltration/diffusion abilities, respectively. Nevertheless, larger viruses have better efficiency in gene insertion[8].
Besides natural tropism, recent advances have shifted towards genetically modified OV for cancer therapy. The selection of OV for certain cancers depends on the selectivity of each virus; for example, coxsackie virus has the ability to bind ICAM-1 and DAF surface molecules which are prominent in melanoma cells but not surrounding normal tissues[13]. To increase tropism and reduce adverse effects of OV, several strategies are developed, including three main modifications of capsid, genomic engineering, and chemical. There are also other interventions to increase OV effectiveness, such as interference in OV delivery efficiency; microenvironment alteration; use of cytokines/chemokines; integrating tumor-associated antigens (TAA), immune-activating ligands, or bispecific T cell engager (BiTE) molecules; and strategies to track OV[8,14].
Capsid modification is one of the methods to enhance virus-tumor binding through the insertion of peptide/protein domain into the viral capsid. Following insertion, transduction efficiencies and facilitation of OV attachment to tumor cell membranes will increase, which results in viral internalization. To enhance the tropism, several modifications have been constructed, including the construction of a random-peptide-displaying library for developing tumor target-specific vector or insertion of single-chain Fv fragments (scFv) gene. The latter showed an enhanced infection selectivity on the receptor of tumor cells, such as in the human epidermal growth factor receptor 2+ (HER2+) lung cancer animal model, where HSV-OV armed with IL-12 and HER2 inhibited the cancer growth[8].
Other types of intervention are genomic engineering and chemical modification. There are two types of genomic engineering to construct OV: knocking out the essential genes for normal cells replication and insertion of tumor-specific transcription promoters. Deletion of apoptotic cell death inhibiting genes, such as E1B55kd viral protein, which plays a role in degrading p53, resulted in increasing ONYX-015 adenovirus tumor-selective replication. Furthermore, adding tumor-specific transcription promoters such as human telomerase reverse transcriptase resulted in selective viral transcription on cancer cells. Chemical modification is an approach to integrate chemical substances such as a pH-sensitive polymer complex so that OV can specifically infect cancer cells at lower than normal cell pH conditions[8].
Oncolytic viruses can be delivered systemically or locally. Ideally, systemic delivery should be preferred to broadly distribute viruses to primary and metastatic tumors. However, the host immune system may recognize the viral particles and degrade them rapidly. To overcome this, several measures have been taken to increase tumor tropism without hampering tumor regression, including the usage of nanoparticles, liposomes, or loading viruses onto myeloid-derived suppressor cells. Intratumoral is also a feasible method for OV delivery system; however, inaccessible locations such as brain/pancreatic cancer limit the administration of OV. Image-guided OV delivery may be an alternative for intratumoral OV administration[8].
Other types of OV modifications are integration with TAA, immune-activating ligands, and BiTE molecules and a strategy to track OV. TAA provides a potent and persistent systemic antitumoral response, but, due to its variability, especially in solid tumors, and expression in healthy tissues, an ideal TAA is difficult to find. BiTE is a novel immunotherapeutic agent with the ability to activate T-cell response via CD3epsilon receptor without MHC expression. When vaccinia virus armed with BiTE molecule targets antigen EphA2 (EphA2-TEA-VV), it will interact with epithelial cell adhesion molecule (EpCAM) on lung cancer cells (EnAd-SA-EpCAM) and activate T cells, resulting in the killing of tumor cells. To track OV, incorporation of certain types of genes which may encode luciferase, green fluorescent protein (GFP), red fluorescent protein, or human sodium iodide symporter is performed on the viral genome or capsids. Once integrated, in addition to using invasive methods such as biopsy, investigators can quantify the viral replication non-invasively in vivo[8].
MECHANISM OF ACTION OF ONCOLYTIC VIRUSES
Upon injection, OV will interact with tumor cells in three phases: OV entry, replication and assembly, and exit of the cell. After binding on the host cell receptor, OV can enter the cell via endocytosis (i.e., adenovirus), genetic material direct injection into the cytoplasm (i.e., coxsackievirus), or active fusion of OV to the cell membrane (i.e., measles, mumps, and Newcastle disease virus). Another non-enveloped virus (i.e., reovirus) can bind and enter host cells, with the additional function to enhance their spread by cellular membrane fusion with adjacent cells[13].
Following their entry, OVs will replicate in the respective cells, either in the cytoplasm (most RNA viruses) or the host’s nucleus (i.e., adenovirus). OV will modify the host machinery and use it for the synthesis of viral proteins and nucleic acids. After viral assembly, OV particles will exit the cell by budding through the cell membrane or passive release after lysis of the host cell. Both methods of viral exit will kill the host cell[13].
Antitumor effects can be induced by OV through several mechanisms, including direct tumor oncolysis and activation of innate or adaptive immune responses[15]. The combination of those mechanisms will lead to OV-induced tumor cell death. Viral oncolysis may be induced by the infected cells through antigen presentation or proinflammatory signaling peptides. OV can cause lysis of cells throughout their life cycle for various reasons. Infected cells will transfer antigens (namely pathogen-associated molecular patterns) such as viral capsid, nucleic acids, or proteins to the cell surface. Moreover, the presence of the virus, cell lysis, tumor antigen, and danger-associated molecular patterns may promote antitumor immunity. Following that, an immune response will be elicited to activate JAK/STAT pathway through IFN, TNF, Toll-like receptor (TLR), and retinoic acid-inducible gene 1 cascade. Upon stimulation, positive feedback from JAK/STAT pathway to IFN will activate protein kinase R, which senses the intracellular material of the virus, stop transcription, and promote apoptosis and viral clearance[12,16].
Following infection, host pattern recognition receptors (PRR) will recognize viral PAMP through its surface or intracellular domain. Recognition by PRR is dependent on the type of viruses: single-strand RNA viruses will be recognized by retinoid acid-inducible gene-1, melanoma differentiation-associated protein 5, and TLR 7; double-strand RNA will be detected by TLR3; and DNA viruses will be sensed by cytosolic double-stranded DNA sensors. Then, chemokines and cytokines (i.e., IFN I) will activate innate immune responses [neutrophils, granulocytes, antigen-presenting cells (APC), and natural killer cells] and immune response inhibition molecules. Activation of innate immune responses will destroy tumors with a viral infection, inhibit tumor growth by releasing cytokines, and activate adaptive immunity by a viral-tumor-antigen presentation from dying tumor cells. Nevertheless, cytokines/chemokines release and innate immune responses are able to compromise the activity of OV. Recruitment of MDSC and Treg into TME will hamper tumor immune response. Therefore, it is encouraged that clinicians use immunosuppressive drugs such as IFN type 1 inhibitor or cyclophosphamide before OV administration. The administration of immunosuppressive drugs will lead to viral replication, killing of tumors, and priming of anti-TAA responses by T cells. Combining GM-CSF and histone deacetylase inhibitor herpesvirus OV showed augmentation of viral replication, oncolysis, antitumor immunity, macrophages, and CD8 cell infiltration and inhibition of viral clearance[15].
Oncolytic viruses can also induce tumor immunogenic cell death (ICD), which is subsequently associated with a potent immune response. Dying tumor cells will release damage-associated molecular patterns (DAMPs) comprised of high-mobility group box 1 (HMGB1), annexin 1, calreticulin, adenosine triphosphate (ATP), IFN type 1, and nucleic acids from cancer cells. Having sensed DAMPs, innate response and APC will be activated; tumor antigen will effectively cross-present to antitumoral human T cells. Integrating ICD-related DAMP genome will increase TAA visibility and immunogenicity upon infection. Production of X-C motif chemokine ligand 1 by OV will also enhance the recruitment of conventional type 1 dendritic cells as an APC for cancer immunity. Recent development has shown that bispecific engager molecules are able to activate cytotoxic T cells without the need for APC for MHC-1 expression[15].
Adaptive immunity, including T cells, will be activated and navigate towards a successful antitumor response. There are four steps to how OV helps T cells to attack tumor cells: efficient priming of TAA, trafficking to and infiltration of T cells to the tumor, avoiding immunosuppression in TME, and recognition, engagement, and lysing of tumor cells. PAMP and DAMP released from OV infection will be loaded and lead the maturation process of dendritic cells (APC). APC will circulate to the nearest draining lymph nodes for priming and induction of tumor-specific T cell immunity. Then, primed T cells must travel to tumor sites through the trafficking and infiltration process. This process is mediated by type 1 IFN, which stimulates the production of chemokines such as CXC-chemokine ligands 9-11. OV can be engineered to produce T cell chemo-attractants, which can increase the production of chemokines by host cells. Induction of TNF and IL-1β by OV also leads to the expression of adhesion molecules on endothelial and blood cells (selectin and integrin). Adhesion molecules are important for T cell extravasation from the vasculature. After arriving at the site of tumor cells, T cells have to overcome the immunosuppressive environment secreted by tumor-associated macrophages (TAM) and MDSC. Genetic modification of OV may deplete MDSC by expressing certain enzymes (e.g., prostaglandin-inactivating enzyme 15-hydroxyprostaglandin dehydrogenase). After conquering the immunosuppressive environment, OV will help the T cells in tumor engagement by inducing the production of type I interferon and subsequent expression of MHC class I and II on tumor cells/APC. Those responses will help T cells recognize, engage with, and lyse the tumor cells. Recent studies have shown that genetic modification of OV [namely, bispecific T cell engagers (BiTEs)] can mediate direct tumor-T cell engagement. Binding of CD3 on T cell surface and target antigen on cancer cells through BiTEs will allow direct tumor-T cell-mediated immune response[17].
COMBINATION OF ONCOLYTIC VIRUS AND RADIATION THERAPY IN CANCER
Irradiation will induce chemical bond breakage and macromolecular structure disruption. The random deposit of energy will lead to damage to all molecules in a cell. Some molecules with rapid turnover (such as water, mRNA, and protein) will experience the least damage, but other molecules with limited turnover (such as DNA) will undergo heavy damage, including permanent and lethal DNA damage. Some types of DNA damage are base damage, single-strand break, double-strand break (DSB), and cross-link break. Among those, DSB is the most lethal lesion[18].
Following DSB, cells will respond with two main mechanisms to prevent chromosome or chromatid damage: homologous recombination (HR) and non-homologous end joining (NHEJ). Sister chromatid is used by HR as a template for DSB repair, restoring the original DNA sequence. In NHEJ, two DNA DSBs are joined without using the homologous template, resulting in a higher risk of error, which may cause permanent DNA changes[18]. HR usually occurs in the G2/S phase, where template chromatid is visible. On the contrary, NHEJ occurs in the phase where no sister chromatid is available as a template (G1 phase). Throughout the process of HR and NHEJ, the MRE11-Rad50-Nbs1 (MRN) complex plays a pivotal role as a sensor for DSB[19,20].
During HR, DSB will be sensed by the MRN complex and double-stranded DNA will be processed into single strands. Following that, MRN will recruit and activate ataxia-telangiectasia mutated (ATM), which recruits downstream repair proteins [i.e., BRCA1 and 2, Rad family of proteins, and replication protein A (RPA)]. Rad51 then mediates homologous strand invasion to sister chromatid for DNA replication. After that, DNA synthesis and gap-filling at the break site occur and Rad54 comes to facilitate DNA end release and annealing[21,22].
In NHEJ, ATM and Rad3-related (ATR), as part of the phosphatidylinositol-3-kinase-related kinase, are initially recruited to the damage site. Following that, Ku70/80 facilitates the recognition of damaged strands and recruited DNA-dependent protein kinase catalytic subunit (DNA-PKC). Autophosphorylation and release of DNA-PKC from DNA will subsequently attract DNA ligase IV and X-ray repair cross-complementing group to come for final repair and processing[19].
Interaction between radiation and viruses is shown by multiple virus types. Human T-cell leukemia virus type 1 Tax protein has the ability to decrease DNA damage-induced apoptosis via Chk2, inhibit Chk1 kinase and G2 arrest, and activate the DNA-PK pathway until saturated and damage response impaired[23,24]. HIV-1 may express Tat protein which plays a role as a radiosensitizer, downregulate DNA-PKcs, and inhibit DNA repair[25]. Radiosensitivity of human epithelial cells to radiation is also promoted when Epstein-Barr virus-related latent membrane protein 1 inhibits p53 and DNA repair[26,27].
The capability of interaction in the DNA damage response (DDR) pathway has been shown by several OV, such as reovirus, adenovirus, and HSV-1. Reovirus has the ability to downregulate DNA repair genes [i.e., poly(ADP-ribosyl)-transferase, xeroderma pigmentosum complementing, and DNA polymerase alpha][28]. Due to its double-stranded genomes, DDR can recognize adenovirus as DSB and activate the NHEJ pathway to further integrate viral to the host genome, forming long concatemeric adenoviral molecules. The MRN complex may come to the sites of viral replication, but through proteasome-dependent degradation, Mre11 and Rad50 will be downregulated and the MRN complex will be relocated away from the viral replication center via E4 open reading frame 6 (E4orf3). Activation of E4orf6 (found in human colorectal carcinoma and glioblastoma cell lines) will degrade MRN complex and inhibit protein phosphatase 2A, resulting in no γH2AX dephosphorylation. Prolonged phosphorylation of H2AX following radiation will induce activation of poly(ADP-ribose) polymerase and translocate apoptosis-inducing factor (AIF) to the nucleus. Translocation of AIF to the nucleus is important for radiosensitization by E4orf6[29-32]. Another radiosensitivity mechanism of adeno OV is related to the stabilization of replication fork and DNA repair. Adenoviral protein will interact with ATR kinase and topoisomerase-IIβ-binding protein 1, which results in inhibition of cellular changes and phosphorylation of cell division cycle 25 phosphatases[19,33,34].
Regarding HSV-1, irradiation of glioblastoma cells will degrade DNA-PKcs via the expression of ICP0 (immediate-early protein). Degraded DNA-PKcs can inhibit DNA repair and increase HSV replication. ICP0 protein expressed by HSV-1 may also radiosensitize human glioblastoma cells. ICP0 may redistribute ATR interacting protein into virus-induced, chaperone-enriched domain, resulting in sequestration of RPA away from cellular DNA DSB[35-37].
Radiotherapy and OV have a synergistic interaction in stimulating immune responses. Radiotherapy is known to stimulate an immunosuppressive effect through regulatory T cells, tumor-associated macrophages, MDSC, and downregulation of cytokine released from dendritic cells[38-40]. Nevertheless, augmentation of immune response may also be elicited by radiotherapy via killing of suppressor T cells, upregulation of surface markers for cytotoxic T cells interaction, infiltration of immune cells to tumors, activation of dendritic cells, antigen presentation within the tumor and draining lymph nodes, production of proinflammatory cytokines and chemokines, and induction of immunogenic cell death. Even though it is known that radiotherapy may induce an immune response, the exact relation between the dose-fractionation scheme and the specific immune-related response is yet to be discovered[41-51].
As mentioned in the previous section, OV may stimulate danger response, associated with inflammasome complexes and type I interferon. Moreover, immunogenic cell death may also be elicited after infection of tumor cells by EnAd, a chimeric oncolytic adenovirus[52]. Irradiation upregulates adenovirus early gene expression and impairs DDR, releasing EnAd particle, which may spread the virus cytotoxicity behavior and cause cell death[53]. Another study on prostate cancer xenograft model treated with Ad5/3-D24-hTNF alpha and radiotherapy showed a significant reduction in tumor growth. This result may be due to a significant increase in ATP, calreticulin, and HMGB1 release, which play a role in immunogenic cell death[19,54]. Additionally, γ34.5-deleted HSV and radiotherapy also showed a synergistic effect via GADD34 expression[55].
The combination of OV and external beam radiation therapy may induce viral oncolysis. There are four mechanisms involved in this response: viral receptor upregulation and increased viral uptake, viral gene expression, upregulation of specific transgene under the control of the radio-inducible promoter and increased viral replication. Radiation can exert cellular expression of Dynamin 2, which leads to internalization of adenovirus when bound to coxsackie-adenoviral receptor/integrin as an adenoviral cellular receptor[56,57]. An increase in GFP-positive cells when irradiation is given before viral infection has also been shown[58].
Radiation-induced viral gene expression is observed during in vitro studies on the measles virus, which expressed human carcinoembryonic antigen as the reporter gene[59]. This gene expression may be useful for further augmentation of viral vector therapeutic efficacy, as shown in radiation-induced NIS gene expression that enhanced antitumor activity[60-62].
Radio-inducible promoter has the ability to control the expression of a specific transgene, which affects therapeutic efficacy. Adenovirus encoding TNF-α and early growth response (Egr)-1 radio-inducible promoter, when injected intratumoral to irradiated cells, resulted in significantly higher TNF-α concentration after 7 days-21 days[63]. It has been shown that a 2 Gy radiation dose is effective for the activation of transgene expression[64]. The studies of transgene upregulation using radio-inducible promoters also showed a positive result with an excellent safety profile in various tumors, including glioma and soft tissue sarcoma[65,66].
Radiation-induced viral replication is dependent on the cell line. Viral replication is enhanced in some OV, such as adenoviruses in prostate, lung, and glioblastoma models[67-71]. Other reports failed to show increased viral replication (i.e., reovirus in melanoma and colorectal cancer)[72]. Nevertheless, a study using HSV and radiation showed that, even though viral replication did not occur, a synergistic effect between OV and radiation was observed[73]. Therefore, viral replication is not compulsory for the antitumor effect[72,73]. Figure 1 illustrates the combined mechanism of oncolytic virus and radiation therapy.
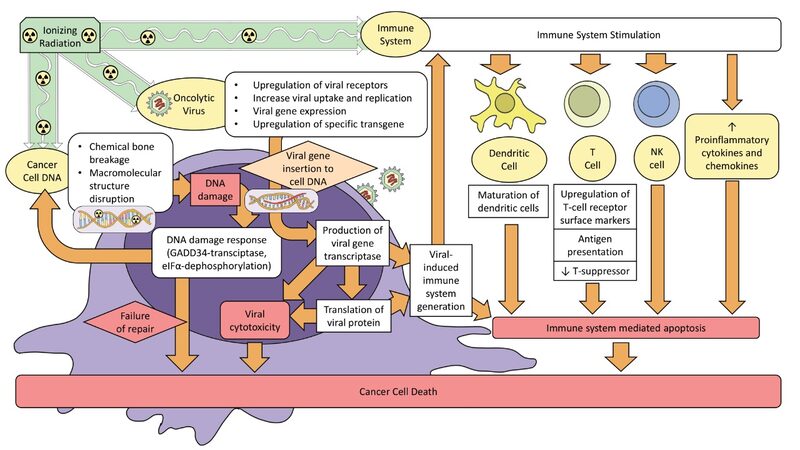
Figure 1. Combined mechanism of oncolytic virus and radiation therapy.
CLINICAL DATA ON COMBINATIONS OF ONCOLYTIC VIRUS AND RADIATION THERAPY
Since the era of genetic engineering in the 1990s, multiple studies on OV have been established[10]. Two types of OV are discussed in the European Society of Medical Oncology Handbook of Immuno-Oncology: talimogene laherparepvec (T-VEC) and coxsackievirus A21 (CVA21). T-VEC is the first U.S. Food and Drug Administration (FDA) approved OV, modified from HSV with additional expression of GM-CSF. T-VEC was shown to be beneficial for the treatment of unresectable malignant melanoma. In addition, CVA21, although yet to be approved by FDA, has exhibited promising clinical responses in different types of solid cancers[74].
In the field of radiation oncology, limited trials were found regarding the combination of OV and radiation therapy. Several studies are summarized in Table 2. All studies in the table are phase 1-3, but only two are phase 3 trials. Most trials used adenovirus, followed by two trials using HSV and one trial using reovirus. Regarding the tumor types, OV was used in trials for various cancers, including prostate, non-small cell lung cancer (NSCLC), sarcoma, breast cancer, colorectal cancer, head and neck cancer, and glioblastoma. All trials showed positive results with tolerable OV-related adverse events. Moreover, intervention with OV led to prolonged survival and better disease control. Immonen et al.[75] showed that a combination of AdvHSV-tk and standard treatment for malignant glioma resulted in longer median survival than the control treatment (62.4 vs. 37.7 weeks, P = 0.0095). Another study by Freytag et al.[77,78,79] also resulted in a 42% reduction of biopsy positivity of prostate cancer in the OV and radiation therapy group two years after intervention[65,66,75-76,80-83].
Clinical studies on oncolytic viruses and radiation therapy
| No. | Author (year) | Study design | Endpoint(s) | Patients/subjects | Intervention | Outcome |
| 1 | Swisher et al. | Phase 2 trial | To assess the possibility and apoptosis induction mechanisms after gene transfer of adenoviral/Ad-p53 (INGN 201) and RT. | 19 patients with nonmetastatic NSCLC are not eligible for chemoradiation/surgery. | Ad-p53 (INGN 201) was injected intratumor on three different days (Days 1, 18, and 32) in an outpatient setting; RT (30 2 Gy) was delivered concomitantly over 6 weeks to the primary tumor and mediastinal lymph nodes. | Common adverse events were grade 1/2 fevers and chills (79 and 53%). 3-month follow-up biopsies: no viable tumor (63% patients), viable tumor (16% patients), and the rest not assessed. |
| 2 | Freytag et al. | Phase 1 trial | To establish the DLT after the administration of OV concurrent with conventional-dose 3D-CRT and increasing duration of 5-FC + vGCV prodrug therapy. Moreover, transgene expression and assessment of therapeutic response are demonstrated by prostate biopsy and serum PSA level. | 15 patients with prostate cancer with adenocarcinoma subtype (stage T1c to T4). | A single intraprostatic injection of replication-capable Ad5-CD/TKrep adenovirus particles, followed by valganciclovir prodrug therapy and 5-fluorocytosine for 1 (Cohorts 1-3), 2 (Cohort 4), or 3 (Cohort 5) weeks along with 70-74 Gy 3D-CRT, 2 Gy per fraction. | There were no DLT and intervention-related adverse reactions. Transgene expression persisted in the prostate for up to 3 weeks after the OV injection. The average PSA half-life in subjects who received the prodrug therapy for more than 1 week was significantly shorter than 1 week (0.6 vs. 2.0 months; P < 0.02), faster than the previous report for subjects who were given 3D-CRT conventional-dose only (2.4 months). In the intermediate-risk group, 0/12 patients were positive at 1-year followed up prostate biopsy. |
| 3 | Senzer et al. | Phase 1 trial | Evaluation of safety, tolerability, feasibility, and antitumor activity of intratumoral injection of TNFerade (adenovector) in parallel with RT. | 36 cancer patients (pancreatic, NSCLC, breast, colorectal, head and neck, esophagus, bladder, sarcoma biliary, primary liver, melanoma, and adrenal). | TNFerade was given via intratumoral injection twice weekly (Weeks 1-2), then once weekly (Weeks 3-6). 1.8-2.0 Gy/day of radiation was given until a total dose of 20-66.6 Gy. | The most common toxicities included fever, injection site pain, and chills (22%, 19%, and 19%, respectively). No grade 3-4 toxicities were observed. 70% of 30 patients demonstrated CR, PR, or MR at 4 weeks post-treatment. |
| 4 | Mundt et al. | Phase 1 trial | To evaluate the safety of concomitant TNFerade and radiotherapy. | 14 patients with soft tissue sarcoma of the limb (liposarcoma, leiomyosarcoma, chondrosarcoma, and spindle cell sarcoma malignant fibrous histiocytoma). | 3 escalating dose levels of TNFerade were given via intratumoral injections, twice a week (Week 1) then once a week (Week 2-5) throughout single daily fraction of radiation therapy All patients received daily concomitant RT in 1.8-2.0 Gy per fraction for a total dose of 36-50.4 Gy. | No DLT was observed. The most common adverse events were grade 1-2 chills, fever, fatigue, and flu-like symptoms (50%, 43%, 36%, and 21%, respectively); 85% of subjects showed an objective tumor response with 2 CR, 9 PR, and 1 SD. |
| 5 | Immonen et al. | Phase 3 RCT | Safety and efficacy of adenovirus with the cloning of HSV-tk (AdvHSV-tk) and GCV administration via intravenous injection. | 36 subjects (operable primary or recurring malignant glioma); AdvHSV-tk gene therapy was given to 17 subjects and control treatment (surgery with/without radiotherapy) was given to 19 subjects | AdvHSV-tk was injected directly into the healthy tissue of the wound after tumor resection in the intervention group. Subsequently, GCV was given intravenously after the gene therapy. In all groups, steroids, antiepileptics, and post-operative 60 Gy RT were given to the subjects. | The intervention group had a 65% longer median survival than the control (62.4 vs. 37.7 weeks, P = 0.0095). AdvHSV-tk intervention was well-tolerated without the occurrence of important safety issues. |
| 6 | Freytag et al. | Phase 1 trial | To establish the toxicity-related intraprostatic injection of OV + vGCV prodrug therapy, 5-FC, and IMRT. Another endpoint is to determine the presence of positive prostate needle biopsies at 6, 12, and 24 months and post-treatment PSA | 9 subjects (5 intermediate-risk and 4 high-risk prostate cancer) | A single intraprostatic injection of Ad5-yCD/mutTKSR39rep-ADP, then the subjects received 2.6 weeks (13 days, only on weekdays) of 5-FC + vGCV prodrug therapy + 74 Gy IMRT. In another cohort, two intraprostatic injections of OV were administered, followed by a subsequent 2.6 weeks of 5-FC + vGCV prodrug therapy + 74 Gy IMRT. | The investigational therapy found low toxicity (grade 1-2); there were no DLT and treatment-related serious adverse events. 7 of 8 patients were negative in the last post-treatment prostate biopsies (6 or 12 months later). |
| 7 | Harrington et al. | Phase 1/II trial | Safety and recommended schedule/dose of JS1/34.5-/47-/GM-CSF (HSV encoding GM-CSF) for future study. Antitumor responses (pathologic, radiologic, recurrence rates, and survival) were also monitored | 17 head and neck cancer subjects [oropharynx (palatine tonsil and base of tongue), supraglottis, and hypopharynx (pyriform fossa)] | JS1/34.5-/47-/GM-CSF was injected into malignant lymph node(s) on the cervical region using a scheduled and escalated dosing manner (no injections were done to mucosal disease) All patients received radiotherapy (70 Gy in 35 daily fractions) and standard-dose cisplatin (100 mg/m2 body surface area) chemotherapy | All subjects had at least one OV-related adverse event, 86% grade 1-2, one grade 3-4 observed in each subject. 82.3% of 17 patients showed tumor response by Response Evaluation Criteria in Solid Tumors, and 93% of neck dissected patients showed pathologic complete remission. HSV replication was found with a titer higher than the input dose. All subjects were seropositive at the end of the intervention. No locoregional reappearance; disease-specific survival was 82.4%, median follow-up: 29 months (19-40 months) |
| 8 | Harrington et al. | Phase 1 trial | To determine safety, feasibility, antitumor activity, and viral replication of combining intratumoral administration of reovirus type 3 Dearing (RT3D) and radiation to subjects with advanced malignancy during fractionated radiotherapy. This study will give recommendations on the dose combination schedule | 25 patients with squamous cell carcinoma of the skin, melanoma, lung, head and neck cancer, pancreas, ovarian, esophagus, colorectal, and unknown primary. | At the first stage (phase 1a): local tumor radiation to a dose of 20 Gy in 5 successive daily fractions + 2 intratumoral injections of RT3D in consecutive log dose-escalating for the cohorts consisting of 3 patients. In the second stage (phase 1b), local tumor radiation to a dose of 36 Gy (12 fractions) over 16 days in parallel with 2, 4, or 6 intratumoral RT3D doses. | No DLT was observed. No viral shedding was found on RT-PCR of blood, urine, stool, and sputum. Most of the common toxicities were grade 1-2, such as pyrexia, flu-like symptoms, vomiting, asymptomatic lymphopenia, and neutropenia. In the low-dose (5 4 Gy) radiation group, out of 7 patients, 5 had SD and 2 PR. Out of 7 patients in the high-dose (12 3 Gy) radiation group, 2 had SD and 5 had PR. |
| 9 | Freytag et al. | Phase 2/3 trial. | To determine the safety and efficacy of OAMCGT + IMRT for intermediate-risk prostate cancer. Other aims were prostate biopsy positivity at 2 years, quality of life (QOL), no apparent biochemical/clinical failure, metastases, and survival. | 44 subjects who had intermediate-risk prostate cancer. (OAMCGT+IMRT: 21 patients; IMRT: 23 patients). | The intervention group received an Ad5-yCD/mutTKSR39rep-ADP adenovirus via a single intraprostatic injection. Then, subjects in the same group were given 5-FC and vGCV orally for 2 weeks (given only during weekdays). All patients received 40 × 2 Gy of IMRT. | Significant differences in gastrointestinal or genitourinary events and quality of life among the two groups were not observed. However, neutropenia, low-grade influenza-like symptoms, transaminitis, and thrombocytopenia were more prevalent in the intervention group. Subjects in the intervention group showed 42% less biopsy positivity at 2 years compared with the other group. Biopsy positive in the intervention vs. control group: 33% vs. 58%. |
| 10 | Markert JM et al. | Phase 1 trial | To know the safety of direct inoculation of HSV/G207 into recurrent malignant gliomas followed by focal radiation to the tumor. Other outcomes were efficacy, overall survival, and radiographic and performance response HSV/G207-degree of post-treatment viral shedding and specific antibody response. | 9 GBM subjects with progressive disease despite surgery (resection or stereotactic biopsy), chemotherapy, and radiation therapy. | G207 was inoculated using stereotactic technique into 5 enhanced parts of the tumor edge for 2 min to avoid any reflux. All subjects received 5 Gy radiation therapy. | No subjects experienced serious adverse effects due to the intervention and no subjects showed any evidence of HSV encephalitis. The most common adverse events were headache, seizure, hemiparesis, and fever. 6 subjects had SD or PR. Quantitative PCR of HSV in blood serum stayed negative in all subjects. The median survival was 7.5 months (time from G207 inoculation to death). |
| 11 | Mell LK et al. | Phase 1 trial | To determine the safety of GL-ONC1 when delivered intravenously with chemoradiation to patients with primary, non-metastatic head and neck cancer. | 19 patients with stage IV head and neck carcinoma. | Patients received GL-ONC1 intravenously with concurrent chemotherapy (cisplatin 100 mg/m2 on Days 1, 22, and 43) and radiation. Patients in Cohort 1 received 3 108 pfu on Day 3; Cohort 2, 1 109 pfu; Cohort 3, 3 109 pfu; Cohort 4, 3 109 pfu on Days 3 and 8; Cohort 5, 3 109 pfu on Days 3, 8, 15, and 22. | Follow-up was done at the median of 30 months, which resulted in 7 treatment failures (3 locoregional, 3 distant, and 1 both local and distant) and 7 deaths (5 due to progression of head and neck carcinoma, 1 due to a second primary cancer of gastrointestinal origin, and 1 due to non-cancer cause). Post-treatment PET/CT at 4 months in 18 patients showed negative in 11 patients, partial response in 4 patients, positive in 3 patients, and death at 3 months post-treatment in 1 patient. PFS at 1 and 2 years was 74.4% and 64.1%, respectively. |
Currently, there are seven ongoing trials on combining radiation therapy and oncolytic virus [Table 3]. Those trials are being conducted in patients with metastatic NSCLC, triple-negative breast cancer, locally advanced rectal cancer, malignant brain tumors, head and neck cancer, and locally advanced prostate cancer. The modalities of radiation therapy are varied among studies, including conventional fractionation, single hypofractionation, stereotactic body radiation therapy, and chemoradiation. The OV vectors used in these studies are adenovirus (four studies), HSV (two studies), and vaccinia virus (one study)[84].
Ongoing clinical trials[84]
| No | Title | Disease | Intervention | Study design | Clinical trial number | Country | Status |
| 1. | SBRT and oncolytic virus therapy before pembrolizumab for metastatic TNBC and NSCLC | Metastatic NSCLC or TNBC | SBRT 30 Gy (5 6 Gy) + (ADV/HSV-tk) and Valacyclovir before pembrolizumab | Phase 2 trial | NCT03004183 | United States | Recruiting |
| 2. | Chemoradiation with Enadenotucirev as a radiosensitizer in locally advanced rectal cancer | Locally advanced rectal cancer | Enadenotucirev _ chemoradiation (capecitabine + radiotherapy 25 2 Gy) | Phase 1 trial | NCT03916510 | United Kingdom | Recruiting |
| 3. | HSV G207 alone or with a single radiation dose in children with progressive or recurrent supratentorial brain tumors | Progressive/recurrent supratentorial brain tumors | HSV G207 with or without 5 Gy single-dose radiation | Phase 1 trial | NCT02457845 | United States | Recruiting |
| 4. | HSV G207 in children with recurrent or refractory cerebellar brain tumors | Recurrent or refractory cerebellar brain tumors | HSV G207 + 5 Gy radiation to tumor | Phase 1 trial | NCT03911388 | United States | Recruiting |
| 5. | Neural stem cell-based virotherapy of newly diagnosed malignant glioma | Malignant glioma | NSC-CRAd-Survivin-pk7 + chemoradiation with/without upfront surgery | Phase 1 trial | NCT03072134 | United States | Completed, no report available |
| 6. | Phase 1 trial of Interleukin-12 gene therapy for locally advanced prostate cancer | Locally advanced prostate cancer | Ad5-yCD/mutTKSR39rep-hIL12 (adenovirus) + definitive radiotherapy | Phase 1 trial | NCT02555397 | United States | Unknown * |
DEVELOPING ONCOLYTIC VIRUS THERAPY
The development of OV therapy during the earliest period was based on coincidental findings of tumor regression in cancer patients with natural infection. Further investigation showed that certain viruses grow better in cancer than normal cells. There are four premises for OV development from certain types of viruses, with the measles virus as an example: independent clinical observation of cancer regression following wild-type infection, cell entry and cell-to-cell fusion (with potential modification of receptors), particle activation and modification of virus-host interaction for cancer-specific replication, and safety of live attenuated viruses. Although derived from normally infectious diseases with the ability for human-to-human transmission, no cases of OV transmission from patients treated with OV have been reported. The effectiveness of OVs is also related to the reengineering process based on a specific cancer-related genetic background, which resulted in targeted entry to cancer cells and the ability for enveloped exchange to increase cancer cell targeting[85]. However, further measures should be taken to enhance the tropism of OV towards cancer cells, as there might be some problems encountered such as inheritable capsid modification, hampering of genomic modification due to viral size and titers, and polymer shielding which affects chemical modification[8]. Additionally, as a measure to enhance the discovery of new OV designs, scientists are also building a repository of OV, namely OvirusTdb (https://webs.iiitd.edu.in/raghava/ovirustdb/). This database consists of 5927 records with more than 20 species of virus and 50 cell lines[86].
CONCLUSION
The burden of cancer is increasingly becoming a problem in the world. Several treatment options have emerged, but efforts are needed to lever up the effectiveness. OV is an emerging treatment modality for cancer management by using an engineered virus capable of selective replication in tumor cells. Following entry into the cell, OV will replicate and assemble before exiting from tumor cells. During its lifecycle, OV will elicit an antitumoral response through direct selective oncolysis and activation of an innate or adaptive immune response. The combination of OV and radiation therapy will have a synergistic effect which leads to viral oncolysis, impairment of DDR, and stimulation of immune response. There are several completed and ongoing trials regarding the combination of OV and radiation therapy towards different types of cancer, with adenovirus as the most commonly used vector. Those trials have shown a promising efficacy with limited adverse events. Moreover, the database of oncolytic viruses is widely available to enhance further research on OV.
DECLARATIONS
Authors’ contributionsBoth authors contributed equally in drafting the manuscript.
Availability of data and materialsNot applicable.
Financial support and sponsorshipNone.
Conflicts of interestAll authors declared that there are no conflicts of interest.
Ethical approval and consent to participateNot applicable.
Consent for publicationNot applicable.
Copyright© The Author(s) 2022.
REFERENCES
1. Torre LA, Siegel RL, Ward EM, Jemal A. Global cancer incidence and mortality rates and trends-an update. Cancer Epidemiol Biomarkers Prev 2016;25:16-27.
2. Global Cancer Observatory. International agency for research on cancer-WHO. Available from: https://gco.iarc.fr/ [Last accessed on 24 Apr 2022].
3. Baskar R, Itahana K. Radiation therapy and cancer control in developing countries: Can we save more lives? Int J Med Sci 2017;14:13-7.
4. Wang JJ, Lei KF, Han F. Tumor microenvironment: recent advances in various cancer treatments. Eur Rev Med Pharmacol Sci 2018;22:3855-64.
5. Denton NL, Chen CY, Scott TR, Cripe TP. Tumor-associated macrophages in oncolytic virotherapy: friend or foe? Biomedicines 2016;4:13.
6. Reale A, Vitiello A, Conciatori V, Parolin C, Calistri A, Palù G. Perspectives on immunotherapy via oncolytic viruses. Infect Agent Cancer 2019;14:5.
7. Touchefeu Y, Vassaux G, Harrington KJ. Oncolytic viruses in radiation oncology. Radiother Oncol 2011;99:262-70.
8. Zheng M, Huang J, Tong A, Yang H. Oncolytic viruses for cancer therapy: barriers and recent advances. Mol Ther Oncolytics 2019;15:234-47.
9. Dobosz P, Dzieciątkowski T. The intriguing history of cancer immunotherapy. Front Immunol 2019;10:2965.
10. Choi AH, O’Leary MP, Fong Y, Chen NG. From benchtop to bedside: a review of oncolytic virotherapy. Biomedicines 2016;4:18.
11. Coffin RS. From virotherapy to oncolytic immunotherapy: where are we now? Curr Opin Virol 2015;13:93-100.
12. Raja J, Ludwig JM, Gettinger SN, Schalper KA, Kim HS. Oncolytic virus immunotherapy: future prospects for oncology. J Immunother Cancer 2018;6:140.
13. Brown CW, Bell JC. Oncolytic viruses: a new weapon to fight cancer. J Med Imaging Radiat Sci 2008;39:115-27.
14. Ylösmäki E, Cerullo V. Design and application of oncolytic viruses for cancer immunotherapy. Curr Opin Biotechnol 2020;65:25-36.
15. Russell L, Peng KW, Russell SJ, Diaz RM. Oncolytic viruses: priming time for cancer immunotherapy. BioDrugs 2019;33:485-501.
16. Lawler SE, Speranza MC, Cho CF, Chiocca EA. Oncolytic viruses in cancer treatment: a review. JAMA Oncol 2017;3:841-9.
17. Twumasi-Boateng K, Pettigrew JL, Kwok YYE, Bell JC, Nelson BH. Oncolytic viruses as engineering platforms for combination immunotherapy. Nat Rev Cancer 2018;18:419-32.
18. Vens C, Koritzinsky M, Wouters BG. Irradiation-induced damage and the DNA damage response. Available from: https://books.google.com.hk/books?hl=zh-CN&lr=&id=kF0PEAAAQBAJ&oi=fnd&pg=PA9&dq=Vens+C,+Koritzinsky+M,+Wouters+BG.+Irradiation-induced+damage+and+the+DNA+damage+response.+&ots=ARYOthDq28&sig=m67oM7IkReo652u9OFrDXsG4Oic&redir_esc=y#v=onepage&q&f=false [Last accessed on 24 Apr 2022].
19. O’Cathail SM, Pokrovska TD, Maughan TS, Fisher KD, Seymour LW, Hawkins MA. Combining oncolytic adenovirus with radiation-a paradigm for the future of radiosensitization. Front Oncol 2017;7:153.
20. Petrini J. The cellular response to DNA double-strand breaks: defining the sensors and mediators. Trends in Cell Biology 2003;13:458-62.
21. Chaurushiya MS, Weitzman MD. Viral manipulation of DNA repair and cell cycle checkpoints. DNA Repair (Amst) 2009;8:1166-76.
22. Bugreev DV, Mazina OM, Mazin AV. Rad54 protein promotes branch migration of Holliday junctions. Nature 2006;442:590-3.
23. Park HU, Jeong JH, Chung JH, Brady JN. Human T-cell leukemia virus type 1 Tax interacts with Chk1 and attenuates DNA-damage induced G2 arrest mediated by Chk1. Oncogene 2004;23:4966-74.
24. Durkin SS, Guo X, Fryrear KA, et al. HTLV-1 Tax oncoprotein subverts the cellular DNA damage response via binding to DNA-dependent protein kinase. J Biol Chem 2008;283:36311-20.
25. Sun Y, Huang YC, Xu QZ, et al. HIV-1 Tat depresses DNA-PK(CS) expression and DNA repair, and sensitizes cells to ionizing radiation. Int J Radiat Oncol Biol Phys 2006;65:842-50.
26. Liu MT, Chen YR, Chen SC, et al. Epstein-Barr virus latent membrane protein 1 induces micronucleus formation, represses DNA repair and enhances sensitivity to DNA-damaging agents in human epithelial cells. Oncogene 2004;23:2531-9.
27. Liu MT, Chang YT, Chen SC, et al. Epstein-Barr virus latent membrane protein 1 represses p53-mediated DNA repair and transcriptional activity. Oncogene 2005;24:2635-46.
28. DeBiasi RL, Clarke P, Meintzer S, et al. Reovirus-induced alteration in expression of apoptosis and DNA repair genes with potential roles in viral pathogenesis. J Virol 2003;77:8934-47.
29. Araujo FD, Stracker TH, Carson CT, Lee DV, Weitzman MD. Adenovirus type 5 E4orf3 protein targets the Mre11 complex to cytoplasmic aggresomes. J Virol 2005;79:11382-91.
30. Stracker TH, Carson CT, Weitzman MD. Adenovirus oncoproteins inactivate the Mre11-Rad50-NBS1 DNA repair complex. Nature 2002;418:348-52.
31. Hart LS, Yannone SM, Naczki C, et al. The adenovirus E4orf6 protein inhibits DNA double strand break repair and radiosensitizes human tumor cells in an E1B-55K-independent manner. J Biol Chem 2005;280:1474-81.
32. Chowdhury D, Keogh MC, Ishii H, Peterson CL, Buratowski S, Lieberman J. gamma-H2AX dephosphorylation by protein phosphatase 2A facilitates DNA double-strand break repair. Mol Cell 2005;20:801-9.
33. Forrester NA, Sedgwick GG, Thomas A, et al. Serotype-specific inactivation of the cellular DNA damage response during adenovirus infection. J Virol 2011;85:2201-11.
34. Blackford AN, Patel RN, Forrester NA, et al. Adenovirus 12 E4orf6 inhibits ATR activation by promoting TOPBP1 degradation. Proc Natl Acad Sci U S A 2010;107:12251-6.
35. Lees-Miller SP, Long MC, Kilvert MA, Lam V, Rice SA, Spencer CA. Attenuation of DNA-dependent protein kinase activity and its catalytic subunit by the herpes simplex virus type 1 transactivator ICP0. J Virol 1996;70:7471-7.
36. Parkinson J, Lees-Miller SP, Everett RD. Herpes simplex virus type 1 immediate-early protein vmw110 induces the proteasome-dependent degradation of the catalytic subunit of DNA-dependent protein kinase. J Virol 1999;73:650-7.
37. Hadjipanayis CG, DeLuca NA. Inhibition of DNA repair by a herpes simplex virus vector enhances the radiosensitivity of human glioblastoma cells. Cancer Res 2005;65:5310-6.
38. Chiang CS, Fu SY, Wang SC, et al. Irradiation promotes an m2 macrophage phenotype in tumor hypoxia. Front Oncol 2012;2:89.
39. Vatner RE, Formenti SC. Myeloid-derived cells in tumors: effects of radiation. Semin Radiat Oncol 2015;25:18-27.
40. Gabrilovich DI, Ostrand-Rosenberg S, Bronte V. Coordinated regulation of myeloid cells by tumours. Nat Rev Immunol 2012;12:253-68.
41. Hellström KE, Hellström I, Kant JA, Tamerius JD. Regression and inhibition of sarcoma growth by interference with a radiosensitive T-cell population. J Exp Med 1978;148:799-804.
42. North RJ. Radiation-induced, immunologically mediated regression of an established tumor as an example of successful therapeutic immunomanipulation. Preferential elimination of suppressor T cells allows sustained production of effector T cells. J Exp Med 1986;164:1652-66.
43. Schaue D, Kachikwu EL, McBride WH. Cytokines in radiobiological responses: a review. Radiat Res 2012;178:505-23.
44. Lugade AA, Moran JP, Gerber SA, Rose RC, Frelinger JG, Lord EM. Local radiation therapy of B16 melanoma tumors increases the generation of tumor antigen-specific effector cells that traffic to the tumor. J Immunol 2005;174:7516-23.
45. Lee Y, Auh SL, Wang Y, et al. Therapeutic effects of ablative radiation on local tumor require CD8+ T cells: changing strategies for cancer treatment. Blood 2009;114:589-95.
46. Gupta A, Probst HC, Vuong V, et al. Radiotherapy promotes tumor-specific effector CD8+ T cells via dendritic cell activation. J Immunol 2012;189:558-66.
47. Sharabi AB, Nirschl CJ, Kochel CM, et al. Stereotactic radiation therapy augments antigen-specific PD-1-mediated antitumor immune responses via cross-presentation of tumor antigen. Cancer Immunol Res 2015;3:345-55.
48. Garnett CT, Palena C, Chakraborty M, Tsang KY, Schlom J, Hodge JW. Sublethal irradiation of human tumor cells modulates phenotype resulting in enhanced killing by cytotoxic T lymphocytes. Cancer Res 2004;64:7985-94.
49. Golden EB, Frances D, Pellicciotta I, Demaria S, Helen Barcellos-Hoff M, Formenti SC. Radiation fosters dose-dependent and chemotherapy-induced immunogenic cell death. Oncoimmunology 2014;3:e28518.
50. Apetoh L, Ghiringhelli F, Tesniere A, et al. Toll-like receptor 4-dependent contribution of the immune system to anticancer chemotherapy and radiotherapy. Nat Med 2007;13:1050-9.
51. Demaria S, Formenti SC. Radiation as an immunological adjuvant: current evidence on dose and fractionation. Front Oncol 2012;2:153.
52. Dyer A, Di Y, Calderon H, et al. Oncolytic Group B Adenovirus enadenotucirev mediates non-apoptotic cell death with membrane disruption and release of inflammatory mediators. Mol Ther Oncolytics 2017;4:18-30.
53. Pokrovska TD, Jacobus EJ, Puliyadi R, et al. External beam radiation therapy and enadenotucirev: inhibition of the DDR and mechanisms of radiation-mediated virus increase. Cancers (Basel) 2020;12:798.
54. Hirvinen M, Rajecki M, Kapanen M, et al. Immunological effects of a tumor necrosis factor alpha-armed oncolytic adenovirus. Hum Gene Ther 2015;26:134-44.
55. Ottolino-Perry K, Diallo JS, Lichty BD, Bell JC, McCart JA. Intelligent design: combination therapy with oncolytic viruses. Mol Ther 2010;18:251-63.
56. Egami T, Ohuchida K, Mizumoto K, et al. Radiation enhances adenoviral gene therapy in pancreatic cancer via activation of cytomegalovirus promoter and increased adenovirus uptake. Clin Cancer Res 2008;14:1859-67.
57. Qian J, Yang J, Dragovic AF, Abu-Isa E, Lawrence TS, Zhang M. Ionizing radiation-induced adenovirus infection is mediated by Dynamin 2. Cancer Res 2005;65:5493-7.
58. Zhang M, Li S, Li J, Ensminger WD, Lawrence TS. Ionizing radiation increases adenovirus uptake and improves transgene expression in intrahepatic colon cancer xenografts. Molecular Therapy 2003;8:21-8.
59. Liu C, Sarkaria JN, Petell CA, et al. Combination of measles virus virotherapy and radiation therapy has synergistic activity in the treatment of glioblastoma multiforme. Clin Cancer Res 2007;13:7155-65.
60. Boland A, Ricard M, Opolon P, et al. Adenovirus-mediated transfer of the thyroid sodium/iodide symporter gene into tumors for a targeted radiotherapy. Cancer Res 2000;60:3484-92.
61. Faivre J, Clerc J, Gérolami R, et al. Long-term radioiodine retention and regression of liver cancer after sodium iodide symporter gene transfer in wistar rats. Cancer Res 2004;64:8045-51.
62. Gaut AW, Niu G, Krager KJ, Graham MM, Trask DK, Domann FE. Genetically targeted radiotherapy of head and neck squamous cell carcinoma using the sodium-iodide symporter (NIS). Head Neck 2004;26:265-71.
63. Hallahan DE, Mauceri HJ, Seung LP, et al. Spatial and temporal control of gene therapy using ionizing radiation. Nat Med 1995;1:786-91.
64. Manome Y, Kunieda T, Wen PY, Koga T, Kufe DW, Ohno T. Transgene expression in malignant glioma using a replication-defective adenoviral vector containing the Egr-1 promoter: activation by ionizing radiation or uptake of radioactive iododeoxyuridine. Hum Gene Ther 1998;9:1409-17.
65. Mundt AJ, Vijayakumar S, Nemunaitis J, et al. A Phase I trial of TNFerade biologic in patients with soft tissue sarcoma in the extremities. Clin Cancer Res 2004;10:5747-53.
66. Senzer N, Mani S, Rosemurgy A, et al. TNFerade biologic, an adenovector with a radiation-inducible promoter, carrying the human tumor necrosis factor alpha gene: a phase I study in patients with solid tumors. J Clin Oncol 2004;22:592-601.
67. Adusumilli PS, Stiles BM, Chan MK, et al. Radiation therapy potentiates effective oncolytic viral therapy in the treatment of lung cancer. Ann Thorac Surg 2005;80:409-16; discussion 416.
68. Bieler A, Mantwill K, Holzmüller R, et al. Impact of radiation therapy on the oncolytic adenovirus dl520: implications on the treatment of glioblastoma. Radiother Oncol 2008;86:419-27.
69. Chen Y, DeWeese T, Dilley J, Zhang Y, Li Y, Ramesh N, et al. CV706, a prostate cancer-specific adenovirus variant, in combination with radiotherapy produces synergistic antitumor efficacy without increasing toxicity. Cancer Res 2001;61:5453-60.
70. Dilley J, Reddy S, Ko D, Nguyen N, Rojas G, Working P, et al. Oncolytic adenovirus CG7870 in combination with radiation demonstrates synergistic enhancements of antitumor efficacy without loss of specificity. Cancer Gene Ther 2005;12:715-22.
71. Dilley J, Reddy S, Ko D, et al. Oncolytic adenovirus CG7870 in combination with radiation demonstrates synergistic enhancements of antitumor efficacy without loss of specificity. Cancer Gene Ther 2005;12:715-22.
72. Twigger K, Vidal L, White CL, et al. Enhanced in vitro and in vivo cytotoxicity of combined reovirus and radiotherapy. Clin Cancer Res 2008;14:912-23.
73. Kim SH, Wong RJ, Kooby DA, et al. Combination of mutated herpes simplex virus type 1 (G207 virus) with radiation for the treatment of squamous cell carcinoma of the head and neck. Eur J Cancer 2005;41:313-22.
74. Melief CJM, Zappasodi R, Garassino MC, Di Nicola M. Vaccines (dendritic cell vaccines, peptide vaccines, DNA vaccines, RNA vaccines, oncolytic viruses). Available from: https://ressources-aura.fr/wp-content/uploads/2020/01/2018-ESMO-Handbook-of-Immuno-Oncology.pdf [Last accessed on 24 Apr 2022].
75. Immonen A, Vapalahti M, Tyynelä K, et al. AdvHSV-tk gene therapy with intravenous ganciclovir improves survival in human malignant glioma: a randomised, controlled study. Mol Ther 2004;10:967-72.
76. Swisher SG, Roth JA, Komaki R, et al. Induction of p53-regulated genes and tumor regression in lung cancer patients after intratumoral delivery of adenoviral p53 (INGN 201) and radiation therapy. Clin Cancer Res 2003;9:93-101.
77. Freytag SO, Stricker H, Pegg J, et al. Phase I study of replication-competent Adenovirus-mediated double-suicide gene therapy in combination with conventional-dose three-dimensional conformal radiation therapy for the treatment of newly diagnosed, intermediate- to high-risk prostate cancer. Cancer Res 2003;63:7497-506.
78. Freytag SO, Movsas B, Aref I, et al. Phase I trial of replication-competent adenovirus-mediated suicide gene therapy combined with IMRT for prostate cancer. Mol Ther 2007;15:1016-23.
79. Freytag SO, Stricker H, Lu M, et al. Prospective randomized phase 2 trial of intensity modulated radiation therapy with or without oncolytic adenovirus-mediated cytotoxic gene therapy in intermediate-risk prostate cancer. Int J Radiat Oncol Biol Phys 2014;89:268-76.
80. Harrington KJ, Hingorani M, Tanay MA, et al. Phase I/II study of oncolytic HSV GM-CSF in combination with radiotherapy and cisplatin in untreated stage III/IV squamous cell cancer of the head and neck. Clin Cancer Res 2010;16:4005-15.
81. Harrington KJ, Karapanagiotou EM, Roulstone V, et al. Two-stage phase I dose-escalation study of intratumoral reovirus type 3 dearing and palliative radiotherapy in patients with advanced cancers. Clin Cancer Res 2010;16:3067-77.
82. Markert JM, Razdan SN, Kuo HC, et al. A phase 1 trial of oncolytic HSV-1, G207, given in combination with radiation for recurrent GBM demonstrates safety and radiographic responses. Mol Ther 2014;22:1048-55.
83. Mell LK, Brumund KT, Daniels GA, et al. Phase I trial of intravenous oncolytic vaccinia virus (GL-ONC1) with cisplatin and radiotherapy in patients with locoregionally advanced head and neck carcinoma. Clin Cancer Res 2017;23:5696-702.
84. U.S. National Library of Medicine. Oncolytic virus and radiation - clinicaltrials.gov. Available from: https://clinicaltrials.gov/ct2/results?cond=&term=oncolytic+virus+and+radiation&cntry=&state=&city=&dist= [Last accessed on 24 Apr 2022].
85. Cattaneo R, Russell SJ. How to develop viruses into anticancer weapons. PLoS Pathog 2017;13:e1006190.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Jayalie VF, Sekarutami SM. Combining oncolytic virus and radiation therapy for cancer management . J Cancer Metastasis Treat 2022;8:17. http://dx.doi.org/10.20517/2394-4722.2021.212
AMA Style
Jayalie VF, Sekarutami SM. Combining oncolytic virus and radiation therapy for cancer management . Journal of Cancer Metastasis and Treatment. 2022; 8: 17. http://dx.doi.org/10.20517/2394-4722.2021.212
Chicago/Turabian Style
Jayalie, Vito Filbert, Sri Mutya Sekarutami. 2022. "Combining oncolytic virus and radiation therapy for cancer management " Journal of Cancer Metastasis and Treatment. 8: 17. http://dx.doi.org/10.20517/2394-4722.2021.212
ACS Style
Jayalie, VF.; Sekarutami SM. Combining oncolytic virus and radiation therapy for cancer management . J. Cancer. Metastasis. Treat. 2022, 8, 17. http://dx.doi.org/10.20517/2394-4722.2021.212
About This Article
Copyright

Data & Comments
Data


 Cite This Article 3 clicks
Cite This Article 3 clicks

 Like This Article 9
likes
Like This Article 9
likes






Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.