
Anti-angiogenic drugs in cancer therapeutics: a review of the latest preclinical and clinical studies of anti-angiogenic agents with anticancer potential


Abstract
Cancer is a group of diseases with significant morbidity and mortality. In cancer cells, where energy requirements are exceptionally high, angiogenesis, which is the sprouting of new blood vessels from pre-existing ones, is an important process for tumour survival and progression. Hence, extensive research in recent years focuses on the discovery of new anticancer drugs that target angiogenesis. Several methodologies have been developed preclinically, including the inhibition of pro-angiogenic factors and their receptors via micromolecular agents or monoclonal antibodies and the inhibition of other compensatory pathways beyond the traditional angiogenic ones. The purpose of the literature review is to present new anticancer drugs that target the process of angiogenesis and have been under preclinical or clinical investigation during the last five years. Many new anticancer drugs targeting angiogenesis are identified in the literature. The results of the in vitro and in vivo evaluation of these drugs show that, apart from inhibiting angiogenesis, they also affect cancer cell proliferation and tumour growth. Recent clinical studies show that these drugs increase the overall or disease-free survival of patients, even those with persistent, chemotherapy-resistant and metastatic types of cancer, although treatment-related side effects are not uncommon. Drugs that target the process of angiogenesis are likely to be the future of anticancer therapy, especially in cases where more traditional treatments do not produce the desired results and where combination regimens of anti-angiogenic agents with standard chemotherapeutics increase patient survival.
Keywords
INTRODUCTION
Cancer comprises one of the main diseases worldwide, accounting for millions of deaths. In 2018 alone, 18 million new cancer cases were recorded[1]. Due to the prevalence of this disease, the design, identification and evaluation of novel anticancer agents is currently an active field of research. Therefore, several drug targets are explored, but particular research focuses on the inhibition of angiogenesis for the treatment of different cancer subtypes.
Angiogenesis is a complex physiological process controlled by cell secreted factors, which coordinate the functions of endothelial and smooth muscle cells mainly to repair damaged blood vessels. New blood vessels are formed via two subsequent processes, vasculogenesis and angiogenesis. Angiogenesis takes place after vasculogenesis is completed in embryos, and during angiogenesis, the pre-existing blood vessels undergo disintegration or sprouting of their endothelial cells (ECs) to form the new vessels. In this way, the vascular tree can be expanded to enhance wound healing and improve organ perfusion via lateral vascular formation[2-4].
Angiogenesis also participates in pathologies and most importantly, in cancer. Tumour cells seek to attract or create blood vessels in order to sustain a continuous supply of nutrients and oxygen. Angiogenesis allows the tumour to grow beyond the size allowed by the simple diffusion of oxygen from the nearest blood vessel, which is limited to a size of 1-2 mm3 according to Folkman’s theory[5]. In addition, direct contact with the vascular network allows the tumour to metastasise and spread beyond the primary site[6]. However, cancer vessels are not similar to pre-existing ones, but have abnormal configurations of their surrounding cells, leaks and various malfunctions[7]. Therefore, these differences may serve as targets for new anticancer drugs.
STEPS INVOLVED IN TUMOUR ANGIOGENESIS
The main steps towards angiogenesis include the activation of ECs by pro-angiogenic stimuli, the production of proteases [matrix metalloproteinases (MMPs)] for the degradation of the perivascular extracellular matrix and basement membrane, the proliferation and migration of ECs towards the angiogenic stimuli, the formation of new vascular tubules, the anastomosis of the newly formed tubes, the synthesis of new basement membrane and the incorporation of smooth muscle cells and pericytes for the maturation of the vessels [Figure 1][2,8].
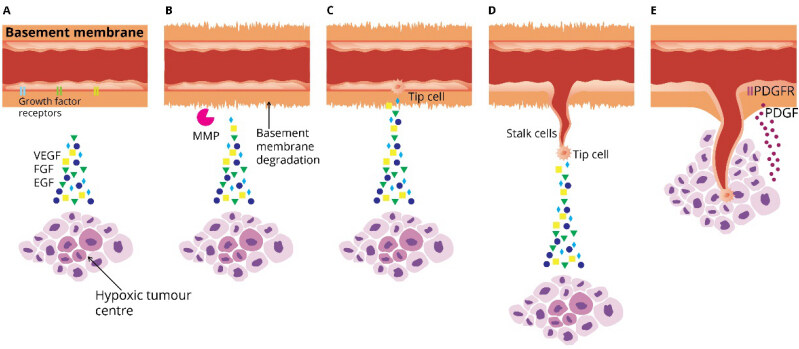
Figure 1. Steps of angiogenesis. (A) The hypoxic tumour core induces the production of hypoxia-inducible factor-1 and the consequent release of pro-angiogenic factors from tumour cells. (B) Hypoxia upregulates matrix metalloproteinase production, leading to basement membrane and perivascular extracellular matrix degradation. (C) Pro-angiogenic factors activate the endothelial cells of adjacent vessels, and a tip cell migrates along the angiogenic factor gradient. (D) This tip cell is followed by highly proliferative stalk cells, which form the new vascular tubule. (E) Platelet-derived growth factor stimulates the recruitment of pericytes and smooth muscle cells (not shown here) and the vessel matures, allowing for blood flow that stimulates further tumour growth. VEGF: Vascular endothelial growth factor; FGF: fibroblast growth factor; EGF: epidermal growth factor; MMP: matrix metalloproteinase; PDGF: platelet-derived growth factor; PDGFR: platelet-derived growth factor receptor.
The process of angiogenesis is mainly triggered by hypoxia, as well as various other stimuli such as acidic pH, hypoglycaemia, hypertension, mechanical stress, chronic inflammation and oxidative stress, which can either initiate or accelerate angiogenesis. Tissues that are sensitive to hypoxia release hypoxia-inducible factor 1 (HIF-1), which is responsible for activating the transcription of other pro-angiogenic factors. HIF-1 binds the hypoxia response element sequence within gene promoters and, thus, regulates the transcription of these factors[9].
PRO-ANGIOGENIC FACTORS
Several factors have been identified as drivers of angiogenesis, which involve pro-angiogenic and growth factors, as well as chemokines. They can be released by both tumour cells and the surrounding cells of the tumour microenvironment upon different stimuli, as extensively discussed in these excellent review articles[10-12]. These pro-angiogenic factors include vascular endothelial growth factor (VEGF)[13], basic fibroblast growth factor (bFGF)[14], angiogenin (ANG)[15], transforming growth factor (TGF)[16,17], tumour necrosis factor (TNF)[18], platelet-derived growth factor (PDGF)[19], granulocyte and granulocyte/macrophage colony-stimulating factors (G-CSF and GM-CSF)[20-22], placental growth factor (PGF)[23], interleukin-8 (IL-8)[24], hepatocyte growth factor[25] and epidermal growth factor (EGF)[26,27], among others.
The role of tumour microenvironment in neovascularisation is also currently being explored. As such, extracellular vesicles (EV) were recently identified as key mediators of neoangiogenesis via a purinergic receptor-namely P2XR4 (P2X purinoceptor 4). The proteins contained in EV from sarcoma cells were found to be able to activate pathways in human umbilical vein endothelial cells (HUVECs), where the new vessel formation was found related to processes such as rapid mitochondrial activation, elevated extracellular ATP (adenosine triphosphate) and trafficking of P2XR4 to the cell membrane. The transportation of P2XR4 to the cell membrane was particularly important for the successful formation of stable branching vascular networks, which was due to proteins and chemokines of EV, such as Del-1 (developmentally regulated endothelial locus-1). Therefore, the tumour microenvironment seems to actively promote angiogenesis, for example, through the P2XR4 of cancerous EV[28].
In growing cancers, ECs are highly active due to the release of the abovementioned factors, such as EGF, oestrogens, bFGF, IL-8, prostaglandin E1 and E2, TNF-α and VEGF, which can activate EC proliferation and migration when the production of anti-angiogenic agents is reduced[29]. Nevertheless, this redundancy of pro-angiogenic factors may explain the current non-optimal efficacy of angiogenic inhibitors[8].
Vascular endothelial growth factor
VEGF is the main pro-angiogenic factor. This family includes the proteins VEGF-A, VEGF-B, VEGF-C, VEGF-D, PGF, VEGF-E and svVEGF. With the exception of the last two members, the other five are present in mammalian genomes, including humans[30,31]. The receptors associated with VEGF [vascular endothelial growth factor receptors (VEGFRs)] are typical tyrosine kinase receptors that have an extracellular domain for ligand binding, a transmembrane domain and a cytoplasmic domain comprising the tyrosine kinase moiety. There are three receptors in this family: VEGFR1, VEGFR2 and VEGFR3[30,32]. VEGF is involved in angiogenesis by promoting blood vessel permeability and vasodilation[2]. It also induces and regulates the differentiation of endothelial progenitor cells and vascular repair, with connexin 43 (Cx43) playing an important role in this regulation. VEGF has been found to induce Cx43 protein expression and thus promote Cx43-mediated intercellular communication between neighbouring ECs[33].
INHIBITORS OF ANGIOGENESIS
Endogenous direct angiogenic inhibitors
Furthermore, there are endogenous inhibitors of angiogenesis, which interfere with the pro-angiogenic factors during the steps of angiogenesis described above Figure 1, thus dysregulating the formation of new blood vessels or destroying pre-existing vessels. These endogenous inhibitors directly target ECs in the sprouting vessels, namely their proliferation and migration, preventing them from being stimulated by pro-angiogenic factors. They are proteins or protein fragments that are produced endogenously, usually by the extracellular matrix, and limit angiogenesis. Vascular homeostasis is thus ingeniously maintained by the delicate balance between pro-angiogenic and anti-angiogenic factors[34,35].
More than 40 endogenous inhibitors have been identified over the years, including angiostatin, endostatin, β-arrestin, thrombospondin 1 and 2, endorepellin, fibulin, canstatin and tumstatin, which are released during the proteolysis of the extracellular matrix. These factors inhibit EC proliferation and migration in response to a range of pro-angiogenic factors, including VEGF, bFGF, IL-8 and PDGF. Additionally, there are endogenous inhibitors that are derived from several cells, such as IFN-α (interferon-α), various interleukins except for IL-8 (namely IL-1β, IL-4, IL-12 and IL-18) and other factors including tissue inhibitors of metalloproteinases, numerous microRNAs and 2-methoxyestradiol. The list continues to grow with new factors constantly emerging, for example, isthmin 1 and multimerin-2[36-41].
Therefore, these endogenous direct angiogenic inhibitors are crucial in maintaining the angiogenic balance and can determine the rate of new blood vessel formation in both health and disease. As such, the approach of increasing endogenous angiogenic inhibitors may be considered as a generally safe long-term anticancer therapy.
Exogenous indirect angiogenic inhibitors
Similarly, drugs that inhibit angiogenesis indirectly by targeting either cancer cells or other stromal cells associated with the tumour and the cancerous vessels, can prevent the formation of new blood vessels, so that the growth of tumours will be halted but not completely eliminated. These indirect inhibitors can downregulate the expression or action of pro-angiogenic agents such as VEGF or EGFR (epidermal growth factor receptor). However, monotherapies against angiogenesis have not yet had the expected efficacy; hence, combination therapies with conventional chemotherapeutic drugs are instead preferred[8,42-44].
For example, classic chemotherapeutic drugs such as paclitaxel and cyclophosphamide have shown good anti-angiogenic activity, usually by interfering with the cytoskeleton and migration of ECs in cancerous vessels[45-48]. Gefitinib (ZD1839, Iressa®) is a small molecular weight EGFR tyrosine kinase inhibitor (TKI) that has shown anti-angiogenic effects in colon (SW480, CaCo2), breast (ZR-75-1, MCF-7), ovary (OVCAR-3) and stomach (KATO III, N87) cancers. These cells co-express TGF-α and EGFR as pro-angiogenic agents[49,50]. Tyrosine kinase inhibitors have been a quickly growing group of anti-angiogenic drugs that target single or multiple pro-angiogenic receptors such as VEGFR, EGFR, FGFR and PDGFR[51,52]. New TKIs are constantly added to the group, for example, CM082 (VEGFR inhibitor)[53] and DW14383 (pan-FGFR inhibitor)[54].
Another group of anti-angiogenic drugs includes monoclonal antibodies against pro-angiogenic factors. Bevacizumab (Avastin®), for example, is a recombinant humanised monoclonal antibody against VEGF-A, which leads to starvation and consequent growth inhibition of cancer cells[55,56]. This drug is often used in combination with other chemotherapeutics to increase efficacy, such as irinotecan[57], leucovorin and fluorouracil[58,59], carboplatin and paclitaxel[60,61] or platinum-based chemotherapy[62,63]. Other similar examples include ramucirumab (Cyramza®)[64], panitumumab (Vectibix®)[65] and cetuximab (Erbitux®)[66]. Finally, there are therapies that target other angiogenesis-related factors, such as MMPs and Hsp90 (heat shock protein 90), and tumour-associated cells such as stromal cells and bone marrow-derived myeloid cells[35].
Resistance mechanisms to anti-angiogenic therapies
Although it has been believed for many years that the spread of cancer cells and the growth of localised tumours beyond a certain size requires local angiogenesis, recent studies have reported that tumours in the brain[67,68], lung[69] and liver[70,71] can co-opt and grow along pre-existing vessels without causing new vessel sprouting (growth via co-option of pre-existing host vessels). Normal cells near the tumour may also support a pro-angiogenic environment[72]. This process of vessel co-option can also sustain the growth of distant metastases emerging from cancers such as melanomas and liver cancers[73,74]. This alternative to angiogenesis co-optive growth pattern can be a mechanism of primary and adaptive resistance against anti-angiogenic therapy[75,76].
There are also other mechanisms involved in anti-angiogenic therapy resistance, which occur at several time points following the treatment. For example, immediate events happening within minutes to a few hours after the treatment involve angiogenic redundancy, glycosylation of key angiogenic receptors, metabolic adaptation and incomplete autophagy after lysosomal sequestration of the anti-angiogenic drug. Angiogenic redundancy mainly relates to VEGF-A/VEGFR2 signalling pathway, where the other pro-angiogenic factors mentioned above such as FGF and PDGF and their receptors/signalling pathways compensate for the inhibition of VEGF-A/VEGFR2, thus leading to alternative ways of EC activation[77,78]. Glycosylation events at key angiogenic receptors such as VEGFR2 enhance the ligand-independent activation of the receptor, thus prolonging the localisation of the receptor at the cell surface and its ligand-independent activation[79]. Metabolic adaptation is driven by tumour hypoxia, where cancer cells switch their energy production from classic oxidative phosphorylation to glycolysis, thereby sustaining tumour growth even under hypoxic conditions[80]. Moreover, autophagy with lysosomal sequestration of the drug enables cancer cells to overcome the treatment-related adaptation of the microenvironment, enhancing their survival, especially under hypoxic conditions[81,82]. For example, this was the case for sunitinib, which had limited therapeutic effects due to lysosomal sequestration, thereby removing the drug from its cytoplasmic targets[83,84]. In addition, the cleavage and phagocytosis of VEGFR2 in ECs drive anti-angiogenic treatment resistance[85].
A few days after the anti-angiogenic treatment, the recruitment of several cells into the tumour also plays a significant role in the development of resistance. As such, cancer-associated fibroblasts and pericytes drive the recruitment of progenitor ECs and the production of MMPs, conferring resistance[86-88]. Additionally, the heterogeneity of tumour ECs can contribute to the observed resistance through the overexpression of pro-angiogenic factors and genes[89-92].
Finally, within months following anti-angiogenic therapy, mechanisms involved in angiogenic dormancy[93], induction of cancer stem cells[94,95], induction of lymphangiogenesis[96] and vessel co-option, as mentioned above, help tumours adopt neovascularisation processes and initiate metastasis with adverse outcomes for patients [Figure 2].
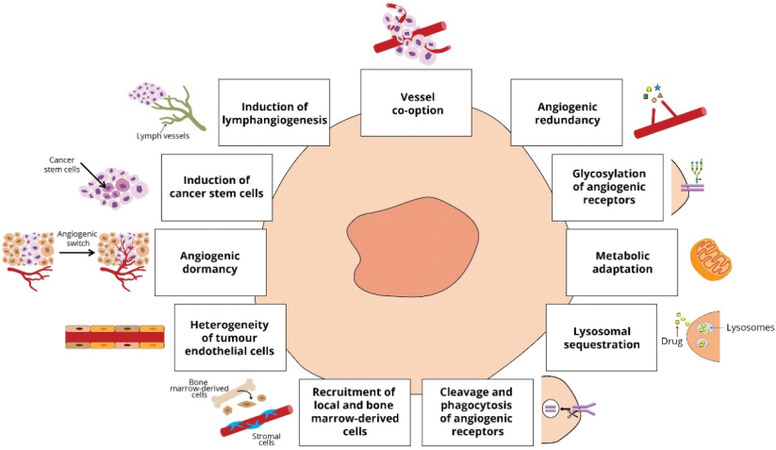
Figure 2. Resistance mechanisms to anti-angiogenic therapies.
These resistance mechanisms have been extensively reviewed[97-99] and have been used for the design of novel anticancer strategies that can overcome this resistance. These strategies include the combined inhibition of pro-angiogenic factors such as VEGF-A and angiopoietin-2[100,101], the simultaneous inhibition of FGF2 and VEGF-A in preclinical models of head and neck squamous cell carcinoma[102] or of VEGFRs and FGFRs in endometrial cancer[103], the combinatorial treatment of sorafenib and autophagy inhibitors[104], the dual inhibition of angiogenesis and metabolic adaptation[105,106] and the inhibition of PDGFR by imatinib and sunitinib in combination with anti-VEGFR[107], among others, which have shown promising anticancer efficacy on experimental tumours[108,109].
As can be understood from the literature, angiogenesis is a necessary process for the growth and survival of tumours. For this reason, ongoing anticancer research focuses on finding agents that inhibit cancerous angiogenesis. In recent years, in fact, several new drugs have been developed which are being tested for their efficacy and safety in ongoing preclinical and clinical trials. Therefore, it is important to summarise the results of these recent studies and to compare the drugs to each other. This knowledge will help anticancer research move forward, as emerging therapies and promising clinical agents are highlighted in this review to point out future directions of this field.
UPDATES ON ANTI-ANGIOGENIC THERAPIES-PRECLINICAL STUDIES
Multiple receptor tyrosine kinase inhibitors
Ogasawara et al.[110] studied the effect of lenvatinib, a multiple receptor TKI of vascular endothelial growth factor receptors (VEGFR1-3), fibroblast growth factor receptors (FGFR1-4), KIT (tyrosine-protein kinase KIT or mast/stem cell growth factor receptor), platelet-derived growth factor receptor α (PDGFRα) and RET (rearranged during transfection)[111], on hepatocellular carcinoma (HCC). The in vitro experiments of this study focused on the treatment of eleven HCC cell lines and two combined hepatocellular/cholangiocarcinoma cell lines with 0-30 μM lenvatinib. The results show that lenvatinib induced dose- and time-dependent growth suppression of HCC cell lines, with no signs of apoptosis. Cell lines expressing FGFR1, -2, -3 and -4, FGF19, FRS2α (fibroblast growth factor receptor substrate 2 alpha) and RET showed the greatest response to treatment. In addition, two HCC cell lines were implanted subcutaneously in mice, which were then treated with 3, 10 or 30 mg/kg/day lenvatinib for 14 days. In the in vivo study, lenvatinib-treated mice showed dose-dependent inhibition of tumour growth. In addition, a decrease in blood vessel density and an increase in necrosis were observed in mice, but again no signs of apoptosis were observed. Therefore, this study demonstrates the antiproliferative and anti-angiogenic effects of lenvatinib on liver cancer cells both in vitro and in vivo, indicating that it could be a promising treatment for patients with HCC. Indeed, lenvatinib is currently used in patients with advanced HCC, and, although the clinical benefits remain limited[112], lenvatinib also seems to delay functional deterioration in these patients, helping with the preservation of health-related quality of life during treatment[113].
In another more recent study on lenvatinib, Jin et al.[114] potentiated the antiproliferative effects of lenvatinib by combining it with gefitinib-an inhibitor of EGFR[115]. This drug combination resulted in synthetic lethality against HCC both in vitro and in vivo in patient-derived HCC tumours in mice. The mechanistic studies showed that gefitinib was able to abrogate the feedback activation of the EGFR-PAK2 [P21 (RAC1) activated kinase 2]-ERK5 (extracellular signal-regulated kinase 5) signalling pathway that was induced by the inhibition of FGFR by lenvatinib. The combination treatment was subsequently administered to 12 patients unresponsive to lenvatinib with advanced HCC, where a better clinical response was achieved, proposing this regimen as a novel strategy for advanced HCC patients with overexpression of EGFR.
Anlotinib is a multiple kinase inhibitor that has shown efficacy against various types of cancer[116,117]. To date, no effective treatment has been found for patients with poorly differentiated papillary thyroid cancer (PTC) or anaplastic thyroid cancer (ATC)[118], a recent study aimed to investigate the effect of anlotinib against thyroid cancer in vitro and in vivo[119]. The results show growth inhibition of ATC and PTC cells in vitro with IC50 values of 3.02-5.42 µM, an action attributed to abnormal spindle assembly, cell cycle arrest at the G2/M phase and the activation of TP53 (tumour protein p53). In addition, anlotinib inhibited cell migration, as well as the in vivo growth of thyroid tumours transplanted to mice. Thus, this study shows that anlotinib has anticancer activity and could be considered as an effective therapeutic approach for patients with advanced thyroid carcinoma.
For more effective delivery of anlotinib, Gao et al.[120] created an anlotinib-containing hydrogel and studied its anticancer effects and safety compared to anlotinib both in vitro and against Lewis lung cancer (LLC) transplanted to a mouse model. The hydrogel was prepared by encapsulating anlotinib with hyaluronic acid-tyramine (HA-Tyr) conjugates (AL-HA-Tyr). The in vitro investigation showed that AL-HA-Tyr successfully suppressed the angiogenic capacity and proliferation of HUVECs and LLC cells, respectively. In addition, inhibition of invasion and migration of HUVECs and LLC cells was observed. In the in vivo studies, LLC mouse models were treated with oral saline solution, oral anlotinib or intratumoral injection of HA-Tyr or AL-HA-Tyr. AL-HA-Tyr reduced visceral toxicity and downregulated Ki67 and VEGF-A in cancer cells, increasing mouse survival. Thus, this study proposes a more effective anlotinib delivery strategy in order to reduce the systemic toxicity of anlotinib and potentiate its efficacy.
Goff et al.[121] hypothesised that dual inhibition of VEGFR and the Axl receptor tyrosine kinase would be even more effective against carcinogenesis, as many human tumours overexpress Axl[122]. At the same time, this molecule is involved in pathways that contribute to tumour growth, angiogenesis and metastasis[123,124]. One of the double inhibitors developed, R916562, showed comparable activity to sunitinib against breast cancer and renal cell carcinoma xenograft models. For breast cancer, treatment with R916562 resulted in a 79% decrease in tumour growth in vitro, while oral administration of 85 and 125 mg/kg for 21 days resulted in 69% and 83% inhibition of tumour growth, respectively, in a mouse tumour xenograft model, compared to 84% inhibition observed with sunitinib at 80 mg/kg. In the renal carcinoma model, a dose of 85 mg/kg R916562 resulted in 80% inhibition of angiogenesis, compared to 85% with sunitinib at 80 mg/kg. Thus, it appears that dual inhibition of Axl receptor and VEGFR may be an effective anticancer treatment comparable to the known anti-angiogenic agent sunitinib.
FGFR inhibitors
Previously reported data show that FGF promotes angiogenic signalling in HCC through the activation of the VEGF pathway[125-127]. Moreover, FGF seems to contribute to the resistance of cancer cells to VEGF/VEGFR targeting agents. Therefore, infigratinib-a pan-FGFR kinase inhibitor-was explored for its potential to overcome HCC resistance to VEGF-targeting drugs, such as bevacizumab[128]. This study evaluated the anticancer activity of these two factors in HCC both as monotherapies and in combination, at doses of 20 mg/kg infigratinib and 5 mg/kg bevacizumab. The combination regimen inhibited tumour growth, as well as invasion and metastasis to the lungs in vivo. Mice bearing high-FGFR HCC tumours showed an additional prolongation in overall survival (OS). More specifically, HCC13-0109 mice (high FGFR2/3 expression) treated with infigratinib or bevacizumab died on Days 56 and 66, respectively, while those receiving the combination of the two drugs survived up to Day 120. Furthermore, HCC06-0606 mice (high FGFR2/3/4 expression but undetectable FGF19) treated with infigratinib or bevacizumab died on Days 60 and 90, respectively, while those receiving the combination lived up to Day 130. Other results reveal downregulation of FGFR2-4, p-FRS-2, p-ERK1/2 and related factors, along with the upregulation of p27. Hence, this study shows that the combination of infigratinib and bevacizumab can be an effective treatment for HCC patients with high FGFR expression, while FGFR2/3 expression could emerge as a biomarker for patient response to such therapy.
Additionally, to reduce any potential resistance of FGFR1-3-dependant HCC to the long-term treatment with infigratinib, another combination of infigratinib with the CDK4/6 (cyclin-dependent kinase 4/6) inhibitor ribociclib was tested. Indeed, this combination was found effective in providing long-lasting tumour growth inhibition, reduced cell differentiation and reduced drug resistance[129].
VEGFR2-targeted inhibitors and antibodies
In a recent study, Lu et al.[130] developed a fully human anti-VEGFR-2-AF antibody (Ab) and tested its activity in in vitro and in vivo models of prostate cancer and leukaemia. Indeed, the results show that anti-VEGFR-2-AF binds effectively to VEGFR2 Ig (immunoglobulin)-like domain 3, thus inhibiting its interaction with VEGF-A. This led to in vitro inhibition of angiogenesis and Ab-dependent cell cytotoxicity and complement activation. By testing the Ab activity in a mouse prostate cancer model, the treated mice showed reduced tumour growth and angiogenesis comparable with the FDA (Food and Drug Administration)-approved ramucirumab. In addition, the combination therapy of anti-VEGFR-2-AF with docetaxel was even more effective in vivo against prostate cancer. Anti-VEGFR-2-AF treatment was also applied to mice with HL-60 leukaemia. These mice showed increased survival and inhibition of leukemic cell metastasis to the ovaries and lymph nodes compared to ramucirumab. Therefore, this study shows that the novel Ab anti-VEGFR-2-AF is a potentially effective treatment for prostate cancer, leukaemia and possibly other types of cancer that overexpress VEGFR2.
A novel VEGFR2 inhibitor, YLL545, was recently developed against triple-negative breast cancer (TNBC)[131]. This inhibitor was proved particularly effective in reducing angiogenesis, inducing a 67% reduction in capillary growth in vitro and 95% in vivo in embryonic angiogenesis assays in zebrafish and Matrigel plug assays in mice. Moreover, 70% in vitro and 50% in vivo tumour growth reduction was observed in MDA-MB-231 xenograft models. Other in vitro results report inhibition of cancer cell proliferation, migration and invasion. The effects of YLL545 were higher than or equal to those of sorafenib, a known inhibitor of angiogenic factors such as VEGFR1, VEGFR2, VEGFR3, PDGFR-β, KIT and RET[132]. Furthermore, YLL545 inhibited the phosphorylation and activation of several VEGFR2-related downstream signalling factors, such as STAT3 (signal transducer and activator of transcription 3) and ERK1/2. The results of this study suggest that the VEGFR2 inhibitor YLL545 is a potentially useful anticancer drug.
VEGFR2-independent inhibitors and antibodies
The observed resistance against clinically used anti-VEGF therapies limits the efficacy of such therapies. This resistance could be attributed to an independent pathway of angiogenesis that can compensate for the inhibition of the VEGF-related cascade, which is mediated by 2-(ω-carboxyethyl)pyrrole (CEP) in a TLR2 (Toll-like receptor 2)-dependent manner[133]. An antibody against CEP was, thus, constructed and tested for its anti-angiogenic properties both as a monotherapy and in combination with bevacizumab[134]. Antibody treatment caused an 18% reduction in tumour growth in a colon cancer model and a 33.2% reduction in a glioblastoma model, also inducing hypoxia in these tumours. Comparing the efficacy of the combination therapy with bevacizumab monotherapy at 2.5 mg/kg, the combined treatment showed a 28.9% higher growth inhibition in the glioblastoma model, suggesting a synergy between anti-CEP and anti-VEGF therapies. Thus, multiple targeting of angiogenic pathways may increase the efficacy of anticancer therapies.
Another recent study[135] tested the anticancer efficacy of a combination of four active ingredients [astragaloside IV, α-solanine, neferine and 2,3,5,6-tetramethylpyrazine (SANT)] isolated from traditional Chinese plants against TNBC. Overexpression of heparanase (HPSE) is often found in breast cancer tissues with the potential to enhance carcinogenesis and affect the process of autophagy in cancer cells[136-138]. HPSE upregulation was associated with poor outcomes in these patients. Administration of the SANT combination resulted in in vitro inhibition of cancer cell proliferation and migration and increased the rate of autophagy in TNBC cells. Moreover, the in vivo study demonstrated the anti-angiogenic activity of SANT, as the treated mice showed a 57% reduction in vascular density in the tumour. Several genes were affected, and the proteomics studies found that SANT downregulated HB-EGF (heparin-binding EGF-like growth factor), thrombospondin-2, amphiregulin, leptin, IGFBP-9 (insulin-like growth factor-binding protein 9), EGF, coagulation factor III and MMP-9 (matrix metalloproteinase 9), while increasing the levels of serpin E1 and platelet factor 4, proteins that play an important role in angiogenesis.
Finally, a novel Hsp90 inhibitor, AT-533, was recently developed against breast cancer[139]. Hsp90 has been shown to play an important role in oncogenesis, as it regulates the stabilisation and activation of many proteins that are essential for cell survival and tumour growth[140,141]. The results of the study show that AT-533 suppressed capillary formation, as well as the migration and invasion of HUVECs more efficiently than another Hsp90 inhibitor, 17-AAG. Further studying the mechanism of action of AT-533, this inhibitor appeared to suppress the activation of VEGFR2 and downstream proteins in HUVEC, including Akt (protein kinase B)/mTOR (mammalian target of rapamycin)/p70S6K (ribosomal protein S6 kinase beta-1), ERK1/2 and FAK (focal adhesion kinase); inhibited HIF-1α/VEGF signalling under hypoxia; and was cytotoxic to breast cancer cells. In an in vivo breast cancer model, AT-533 inhibited tumour growth and angiogenesis through the HIF-1α/VEGF/VEGFR2 signalling pathway. Thus, these results suggest that AT-533 may be a new drug for the treatment of breast cancer.
UPDATES ON ANTI-ANGIOGENIC THERAPIES-CLINICAL TRIALS
Multiple receptor tyrosine kinase inhibitors
In 2018, a phase II clinical trial[117] involving 166 patients with refractory metastatic soft tissue sarcoma (after receiving anthracycline-based chemotherapy, naïve from angiogenesis inhibitors and with at least one measurable lesion according to RECIST 1.1) was conducted to investigate the efficacy of anlotinib (12 mg/day for two weeks, abstinence for one week). After 12 weeks of treatment, 68% of patients had no further tumour progression. More specifically, the median progression-free survival (PFS) was 5.6 months, while the median OS was 12 months. The most common grade 3 or higher side effects associated with anlotinib treatment were hypertension, increased blood triglyceride levels and pneumothorax. No deaths were reported due to anlotinib treatment. Thus, anlotinib proved effective in increasing patient survival, while no significant adverse effects were reported.
In a different setting, a multicentre, double-blind, randomised phase III clinical trial that evaluated the efficacy and safety of anlotinib (12 mg/day) in 439 patients with advanced non-small-cell lung cancer (NSCLC) was also recently conducted[116], based on previous encouraging results of a phase II trial[142]. Patients progressing after second-line or further treatment and receiving anlotinib had significantly longer OS than the placebo group, 9.6 and 6.3 months, respectively. In addition, a significant increase was observed in the PFS (5.4 and 1.4 months, respectively), the objective response rate and the disease control rate for anlotinib-treated patients. Common grade 3 or higher side effects included hypertension and hyponatremia. Therefore, the results of this study show that anlotinib is a well-tolerated further treatment, which results in a significant improvement in patients with advanced NSCLC.
Surufatinib is a small molecular inhibitor of VEGFR1, VEGFR2, VEGFR3 and FGFR1[143]. A randomised, double-blind, placebo-controlled, phase III study was conducted to investigate the efficacy and safety of this drug in patients with extrapancreatic neuroendocrine tumours[144]. The study included 198 adult patients [after receiving up to two kinds of previous systemic regimens and with unresectable or metastatic, well-differentiated, extrapancreatic neuroendocrine tumours of Eastern Cooperative Oncology Group (ECOG) performance status of 0 or 1] who were randomised to the surufatinib group (300 mg) or the placebo group. The median PFS in the surufatinib group was 9.2 months, and 3.8 months in the placebo group. The most common grade 3 or higher side effects associated with surufatinib treatment were hypertension and proteinuria. Overall, 25% of patients receiving surufatinib experienced serious treatment-related adverse effects and three deaths were reported compared to one death in the placebo group. Hence, surufatinib significantly increases the PFS of patients with progressive, advanced, well-differentiated extrapancreatic neuroendocrine tumours, while having an acceptable toxicity profile.
In a similar multicentre, randomised, double-blind, placebo-controlled, phase III trial, surufatinib was evaluated for its efficacy and safety in patients with advanced pancreatic neuroendocrine tumours[145]. The study included 172 adult patients (after receiving up to two kinds of previous systemic regimens for advanced disease and with progressive, advanced, well-differentiated pancreatic neuroendocrine tumours of ECOG performance status of 0 or 1) who were randomised to the surufatinib (300 mg) or placebo group, both administered orally once daily in successive four-week treatment cycles. The median PFS in the surufatinib group was 10.9 months, and 3.7 months in the placebo group. The most common grade 3 or higher side effects associated with surufatinib treatment were hypertension, proteinuria and hypertriglyceridemia. Overall, 22% of patients receiving surufatinib experienced serious treatment-related adverse effects and three deaths were reported, two due to adverse effects and one due to disease progression. Therefore, surufatinib significantly increases the PFS of patients with pancreatic neuroendocrine tumours, as similarly reported for extrapancreatic neuroendocrine tumours. The drug has significant toxicity, which, however, is acceptable when compared to its overall benefit.
Based on the above trials, surufatinib received its first approval in China for the treatment of late-stage, well-differentiated, extrapancreatic neuroendocrine tumours in December 2020[146]. Surufatinib is currently in preregistration in China for pancreatic neuroendocrine tumours and in the USA for pancreatic and extrapancreatic neuroendocrine tumours, while other ongoing clinical trials are investigating its benefit for other solid tumours, including thyroid cancer, biliary tract carcinoma and soft tissue sarcoma.
Another recent phase Ib/II clinical trial was conducted[147], where the maximum tolerated dose of the lenvatinib co-administered with pembrolizumab (200 mg intravenous administration every three weeks) was determined in 137 patients, and the initial results of drug efficacy were evaluated in 124 patients with advanced solid tumours such as metastatic renal cell carcinoma (RCC), endometrial cancer, squamous cell carcinoma of the head and neck (SCCHN), melanoma, NSCLC and urothelial cancer. The highest dose of lenvatinib (24 mg daily) plus pembrolizumab was associated with two grade 3 toxicities (arthralgia and fatigue), so the recommended phase II dose of lenvatinib was established at 20 mg daily plus pembrolizumab. The objective response rate at Week 24 for each type of cancer was 63% in RCC, 52% in endometrial cancer, 36% in SCCHN, 48% in melanoma, 33% in NSCLC and 25% in urothelial cancer. The most common side effects reported were fatigue, diarrhoea, hypertension and hypothyroidism. Thus, the combination of lenvatinib with pembrolizumab in patients with advanced solid tumours shows good efficacy, while the side effects observed are manageable.
Previous studies have shown that the combination of PD-1 (programmed cell death protein 1) checkpoint inhibitors with VEGF-pathway TKIs leads to increased cytotoxicity and decreased cancer cell growth, but it is also related to severe side effects[148]. Based on this, Atkins et al.[149] conducted a phase Ib clinical trial investigating the safety and preliminary efficacy of the combination of axitinib (5 mg orally, twice daily), which is more selective than others, with pembrolizumab (2 mg/kg intravenously, every three weeks) against previously untreated, advanced renal cell carcinoma. Overall, 73% of the 52 patients in the study (predominantly clear cell subtype, primary tumour resected, at least one measurable lesion, ECOG performance status 0-1) showed an objective response to treatment with axitinib and pembrolizumab. However, more than half of the patients (65%) had grade 3 or higher treatment-related adverse effects, including hypertension, diarrhoea, fatigue and elevated alanine aminotransferase levels. Thus, the combination therapy of axitinib and pembrolizumab appears to be a relatively well-tolerated treatment, with significant efficacy against treatment-naïve advanced renal cell carcinoma.
A subsequent phase III clinical trial trying to assess the long-term efficacy and safety of the combination of pembrolizumab (200 mg intravenously, every three weeks for up to 35 cycles) with axitinib (5 mg orally, twice daily), showed increased efficacy of the combination treatment over sunitinib monotherapy (50 mg orally, once daily for four weeks per six-week cycle) in treatment-naive, advanced renal cell carcinoma[150]. Of the 861 registered patients with a median follow-up of 30.6 months, the pembrolizumab plus axitinib group showed higher OS and PFS than sunitinib alone, while the treatment-related side effects were the same as those previously reported in[149], and no new treatment-related deaths occurred compared to the interim analysis. Therefore, the combination treatment has a superior clinical benefit over sunitinib monotherapy and can be considered as the first-line treatment for advanced renal cell carcinoma patients.
The safety and efficacy of regorafenib (160 mg/day for three weeks, one-week abstinence) in anti-angiogenic therapy-naïve and chemotherapy-refractory advanced colorectal cancer was tested in a monocentric phase IIb clinical study with 59 registered patients[151]. Regorafenib is an oral diphenylurea multikinase inhibitor against angiogenic (VEGFR1, VEGFR2, VEGFR3 and TIE2), stromal (PDGFR-β and FGFR) and oncogenic receptor tyrosine kinases (KIT, RET and RAF), which has been established for metastatic colorectal cancer[152,153]. The median PFS achieved was 3.5 months and OS 7.4 months. The most common grade 3 or higher side effects were hypertension, hand-foot skin reaction and hypophosphataemia, with no treatment-related deaths. Hence, this study shows that regorafenib is an effective treatment for chemotherapy-refractory metastatic colorectal cancer with an acceptable safety profile.
VEGFR2 inhibitors
The overexpression of α-fetoprotein in patients with advanced HCC has been correlated with poor prognosis[154-156]. Therefore, a multicentre, randomised, double-blind, placebo-controlled, phase III trial was performed to determine the efficacy of ramucirumab (8 mg/kg intravenously every two weeks), a human IgG1 monoclonal antibody that inhibits VEGFR2, in patients with advanced HCC and α-fetoprotein concentrations of 400 ng/mL or higher (ECOG performance status of 0 or 1, previously treated with first-line sorafenib)[157]. The median follow-up was 7.6 months, during which a significant improvement in median OS (8.5 months and 7.3 months, respectively) and PFS (2.8 months and 1.6 months, respectively) was observed for the ramucirumab group (197 patients) compared to the placebo group (95 patients). However, the analysis of the results did not show a significant difference in the objective response and the median deterioration time between the two groups. Common grade 3 or higher treatment-related adverse effects were hypertension, hyponatraemia and elevated aspartate aminotransferase. Ramucirumab-related side effects were also responsible for the death of three patients. Hence, this study shows that ramucirumab is a potentially useful treatment for patients with advanced HCC and α-fetoprotein overexpression previously on sorafenib, although the treatment causes significant side effects, and further studies are needed to gain statistically significant results.
Combination of VEGF/VEGFR2 pathway inhibitors with chemotherapeutics
TAS-102, an oral nucleoside anticancer agent that combines trifluridine and tipiracil, has shown a significant OS prolongation in patients with refractory metastatic colorectal cancer[158]. The co-treatment of TAS-102 with bevacizumab has also shown promising results in colorectal cancer xenograft models compared to the corresponding monotherapies[159]. Thus, Kuboki et al.[160] conducted an open-label, single-arm, multicentre, phase I/II study to investigate the efficacy and safety of this drug combination in 25 patients with unresectable, metastatic colorectal adenocarcinoma (refractory or intolerant to standard chemotherapies, ECOG performance status of 0 or 1). The phase I results of the study determined the recommended dose for TAS-102 at 35 mg/m2 orally twice daily on Days 1-5 and 8-12 over a 28-day cycle, co-administered with intravenous infusion of bevacizumab 5 mg/kg for 30 min every two weeks. After 16 weeks of treatment, PFS was achieved in 42.9% of patients. The most common grade 3 or higher side effects observed in patients were neutropenia, leukopenia, anaemia, febrile neutropenia and thrombocytopenia. Serious adverse reactions were reported in only three patients, but no deaths were reported due to the regimen. This study shows that the combination of TAS-102 with bevacizumab has significant efficacy in patients with refractory metastatic colorectal cancer, while it is a treatment that does not present significant overall toxicity.
The aim of the phase II study by Palazzo et al.[161] was to investigate the efficacy of a neoadjuvant regimen comprising chemotherapy and anti-angiogenic treatment for inflammatory breast cancer, a rare and highly aggressive form of cancer. Patients received weekly carboplatin and paclitaxel (70 mg/m2 intravenously on Days 1, 8 and 15 every 28 days for six cycles), in combination with bevacizumab (15 mg/kg intravenously on Day 1 every three weeks for eight cycles) and metronomic cyclophosphamide (50 mg/day orally for six months). An objective response was observed in 30 patients and a pathologic complete remission (pCR) in 10 of the 34 patients. A significantly higher pCR was observed in patients with HER2 (human epidermal growth factor receptor 2)-positive tumours (57%) compared to patients with other types (20% in triple-negative type and 0% in luminal B-like/HER2-negative tumours). After five years of follow-up, disease-free survival was observed in 58% of participants, while OS reached 72%. This study showed that this neoadjuvant therapy, including chemotherapy and anti-angiogenic treatment, significantly increases disease-free survival and OS for patients with inflammatory breast cancer.
Another recent phase II study evaluated the efficacy and safety of a treatment that included apatinib (500 mg orally once daily) and etoposide (50 mg orally once daily on Days 1-14 of a 21-day cycle for up to six cycles) in patients with platinum-resistant or platinum-refractory ovarian cancer[162]. Of the 35 patients in the study, 19 showed an objective response to treatment, while the most common grade 3 or higher side effects observed were neutropenia, fatigue, anaemia and mucositis, and two patients developed severe side effects. However, no treatment-related deaths were reported. Therefore, the combination of apatinib with etoposide seemed to be significantly effective in more than half of the platinum-resistant ovarian cancer patients registered in the present study, with manageable adverse reactions.
DISCUSSION
The aim of this review article is to present the most up-to-date data on novel anti-angiogenic factors that are under development or clinical evaluation. In total, 15 different anti-angiogenic agents are included based on recent publications of the last five years, which confirms that the discovery of anti-angiogenic factors has been an active and ongoing field of research.
The obvious disadvantage of preclinical studies is that the pharmacokinetics and possible side effects of the proposed drugs are usually not studied. Indeed, a regimen may be effective in vitro and in vivo but toxic for humans and, therefore, not pursued further. In addition, we must always consider the differences between the in vivo models and humans, which may be responsible for the clinical failure of preclinically identified promising agents.
In this scope, both preclinical and clinical studies of lenvatinib and anlotinib are included to follow up on the overall efficacy of these two agents. Lenvatinib showed promising in vitro and in vivo antiproliferative and anti-angiogenic activity against liver cancer cells[110]. As proof of concept, lenvatinib showed a significant response rate in patients with metastatic renal cell carcinoma, endometrial cancer, squamous cell carcinoma of the head and neck, melanoma, non-small cell lung cancer and urothelial carcinoma, while it was not correlated with serious side effects[147].
Anlotinib was found cytotoxic against thyroid cancer cells, suppressed in vitro migration and decreased tumour growth in vivo[119]. In clinical trials, anlotinib was effective against refractory soft tissue sarcoma[117] and non-small cell lung cancer[142]. Although no treatment-related deaths were reported, several adverse reactions were observed in patients in both clinical trials. Of particular interest are the results of a recent study[120] where, to avoid anlotinib-related systemic side effects, a hydrogel containing anlotinib was manufactured, followed by an intratumoral injection to a mouse model of Lewis lung cancer. The results are particularly encouraging, giving new hope for providing targeted therapies with increased efficacy and reduced side effects via novel delivery systems.
Several other anti-angiogenic agents under preclinical development are also discussed, including YLL545 for breast cancer[131], R916562 for breast and renal cancer[121] and AT-533 for breast cancer[139]. Recently completed clinical trials with positive results are also presented, namely for monotherapies such as oral surufatinib against pancreatic and extrapancreatic neuroendocrine cancers, even though severe side effects were reported[144,145], and regorafenib against advanced colorectal cancer[151]. In addition, ramucirumab, a mAb under development, was clinically tested against advanced hepatocellular carcinoma in patients with increased α-fetoprotein levels, where it showed high efficacy[157].
Finally, the combination of chemotherapeutics with different mechanisms of action is a common practice in cancer patient care; therefore, some studies with combination regimens are also discussed here. Indicatively, bevacizumab was combined with carboplatin, paclitaxel and metronomic cyclophosphamide, where it was found useful against inflammatory breast cancer[161], and in another clinical trial, it was combined with TAS-102 against metastatic colorectal cancer, where again positive results were obtained[160]. Moreover, apatinib combined with etoposide was effective against ovarian cancer unresponsive to platinum[162], whereas an anti-PD-1 mAb combined with axitinib also showed an increased patient response, although several side effects were induced[149,150].
Summaries of the presented anti-angiogenic agents categorised into preclinical and clinical studies, along with the main findings of each study, are shown in Tables 1 and 2.
Summary of the anti-angiogenic agents currently being tested in pre-clinical models discussed in this article
| Anti-angiogenic agent | Target | Type of cancer | Preclinical model | Dose | Route of administration | Anticancer effect | Signalling pathway involved | Reference |
| Monotherapies | ||||||||
| Lenvatinib | VEGFR1-3 FGFR1-4 KIT PDGFRα RET | Hepatocellular carcinoma Hepatocellular/Cholangiocarcinoma | In vitro: 11 HCC cell lines and 2 hepatocellular/ cholangiocarcinoma cell lines In vivo: KYN-2 cells or HAK-1B cells in 4-week-old female BALB/c nude mice | In vitro: 0-30 μM In vivo: 3, 10, 30 mg/kg/day | Oral | In vitro: dose- and time-dependent growth suppression, no signs of apoptosis In vivo: dose-dependent inhibition of tumour growth, decrease in blood vessel density, increase in necrosis, no signs of apoptosis | VEGFR- and FGFR-signalling pathways and indirect activity Direct suppression of cell proliferation in high FGFR-expressing cells | [110] |
| Anlotinib | PDGFR RET Aurora-B EGFR FGFR VEGFR c-KIT | Papillary thyroid cancer Anaplastic thyroid cancer | In vitro: 3 papillary thyroid cancer and 3 anaplastic thyroid cancer cell lines In vivo: K1 cells in 4-week-old male BALB/c athymic nude mice | In vitro: IC50 3.02-5.42 μM In vivo: 3 mg/kg | Intraperitoneal | In vitro: abnormal spindle assembly, cell cycle arrest at G2/M, inhibition of TP53 activation, inhibition of cell migration In vivo: inhibition of tumour growth | Activation of TP53 pathway | [119] |
| Anlotinib hydrogel (AL-HA-Tyr) | PDGFR RET Aurora-B EGFR FGFR VEGFR c-KIT | Lewis lung cancer | In vitro: LLC and HUVEC cell lines In vivo: LLC cells in 4-6 week-old female C57BL/6J mice | In vitro: AL: 5 μM; HA-Tyr: 1.0 wt.% In vivo: 3 mg/kg/day | Intratumoral | In vitro: suppression of angiogenesis and proliferation in HUVEC and LLC cells, inhibition of cell cycle In vivo: reduced visceral toxicity, downregulation of Ki67 and VEGF-A, increased mouse survival | Decrease of Ki-67 and VEGF-A | [120] |
| SANT | Multiple targets | Triple-negative breast cancer | In vitro: MDA-MB-231 cells In vivo: MDA-MB-231 cells in 6-8 week-old BALB/c female nude mice | In vitro: α-solanine 12 μmol/L; neferine 20 μmol/L; astragaloside IV 30 μmol/L; 2,3,5,6-tetramethyl- pyrazine 10 μmol/L In vivo: α-sola- nine 5 mg/kg; NEF 10 mg/kg; AS-IV 15 mg/kg; TMP 5 mg/kg | Intraperitoneal | In vitro: inhibition of proliferation and migration, increased autophagy rate In vivo: reduced vascular density and tumour growth, affected gene expression | Downregulation of HB-EGF, thrombospondin-2, amphiregulin, leptin, IGFBP-9, EGF, coagulation factor III, MMP-9 Increase of serpin E1, platelet factor 4 | [135] |
| AT-533 | Hsp90 | Breast cancer | In vitro: HUVEC, MDA-MB-231, MCF-7 cell lines In vivo: 5-week-old female BALB/c nude mice | In vitro: 5, 10, 50, 75 nM In vivo: 10 mg/kg | Intraperitoneal | In vitro: decreased capillary formation, migration and invasion, decreased activation of VEGFR2, Akt/mTOR/p70S6K, ERK1/2 and FAK in HUVECs, inhibited HIF-1α/VEGF signalling under hypoxia, cytotoxic to cancer cells In vivo: inhibition of tumour growth and angiogenesis | HIF-1α/VEGF/VEGFR2 signalling pathway | [139] |
| YLL545 | VEGFR2 | Triple-negative breast cancer | In vitro: HUVEC, MDA-MB-231 cell lines In vivo: female BALB/c nude mice | In vitro: 2.5, 5, 10 In vivo: 0.625 to 1.25 μM for in vivo assays, 50 mg/kg/day for mice | Oral | In vitro: reduction in capillary growth, tumour growth, inhibition of cancer cell proliferation, migration and invasion, downregulation of ITGAV, ENG, THBS1, FN1, TEK In vivo: reduction in capillary growth, tumour growth | VEGFR2 signalling pathway and independent pathways | [131] |
| R916562 | VEGFR2 Axl | Breast cancer Renal cell carcinoma | In vivo: MDA-MB-231 human breast cancer xenograft model Caki-1 human renal carcinoma xenograft model | In vivo: breast cancer: 85 mg/kg and 125 mg/kg Renal cancer: 85 mg/kg | Oral | In vitro: decrease in tumour growth, reduction in FGF-induced neovascularization In vivo: decrease in tumour growth | VEGFR2 and Axl signalling pathways | [121] |
| Combination therapies | ||||||||
| Infigratinib + bevacizumab | Infig: pan-FGFR Bevac: VEGF-A | Hepatocellular carcinoma | HCC PDX models in 9-10 week-old male C.B-17 SCID mice, HCC13-0109 and HCC06-0606 xenograft models | Infig: 20 mg/kg Bevac: 5 mg/kg | Infig: oral Bevac: intraperitoneal | Inhibition of tumour growth, cell cycle, invasion and metastasis to the lungs, prolongation of survival | Downregulation of FGFR2-4, p-FRS-2, p-ERK1/2 and related factors, upregulation of p27 MEK/ERK and p70S6K pathways | [128] |
| Anti-VEGFR-2-AF + docetaxel | Antibody: VEGFR2 Docet: microtubules | Prostate cancer Leukemia | In vitro: HUVEC, PC-3 cell lines In vivo: prostate cancer: PC-3 cells in 6-week-old male mice Leukemia: HL-60 cells in 6-week-old NSG female mice | In vivo: anti-VEGFR-2-AF: 20 mg/kg Docet: 5 mg/kg | Intravenous | In vitro: inhibition of angiogenesis and Ab-dependent cell cytotoxicity, complement activation In vivo: reduced tumour growth and angiogenesis, a combination more effective, increased survival, inhibition of metastasis to ovaries and lymph nodes | VEGF-A/VEGFR2 signalling pathway | [130] |
| Anti-CEP antibody + bevacizumab | Antibody: CEP Bevac: VEGF-A | Colon cancer Glioblastoma | Colon cancer: CRC 174 cells in 6-8 week-old female athymic nude mice Glioblastoma: U87-MG cells in 6-8 week-old female athymic nude mice | Anti-CEP IgG: | Intraperitoneal | Reduction in tumour growth and new blood vessel formation | TLR2 signalling pathway (anti-CEP-antibody) VEGFR signalling pathway (bevacizumab) | [134] |
| Lenvatinib + gefitinib | Lenvat: VEGFR1-3 FGFR1-4 KIT PDGFRα RET Gefit: EGFR | Hepatocellular carcinoma | In vitro: 12 human and 2 mouse liver cancer cell lines In vivo: SNU449 or Huh6 cells in 6-week-old BALB/c nude mice Human HCC tumour cells in 6-8-week-old female BALB/c nude mice | In vivo: lenvat: 4 mg/kg Gefit: 80 mg/kg In clinical trial: Lenvat: 8-12 mg/day Gefit: 250 mg/day | Lenvat: oral Gefit: oral | Synthetic lethality | EGFR-PAK2-ERK5 pathway | [114] |
Summary of the anti-angiogenic agents currently in clinical trials discussed in this article
| Anti-angiogenic agent | Target | Type of cancer | Phase of clinical trial | Dose | Route of administration | Main findings | Observed side effects | Reference |
| Monotherapies | ||||||||
| Anlotinib | PDGFR RET Aurora-B EGFR FGFR VEGFR c-KIT | Refractory metastatic soft tissue sarcoma | Phase II | 12 mg/day | Oral | PFR12 weeks 68%, ORR 13%, PFS 5.6 months, OS 12 months | Hypertension, increased blood triglyceride levels, pneumothorax, no deaths | [117] |
| Anlotinib | PDGFR RET Aurora-B EGFR FGFR VEGFR c-KIT | Advanced non-small cell lung cancer | Phase III | 12 mg/day | Oral | OS 9.6 months, PFS 5.4 months, improvement in ORR and disease control rate | Hypertension, hyponatremia, no deaths | [116] |
| Surufatinib | VEGFR1-3 FGFR1 | Extrapancreatic neuroendocrine cancer | Phase III | 300 mg/day | Oral | PFS 9.2 months | Hypertension, proteinuria, serious adverse effects in 25% of patients, 3 deaths | [144] |
| Surufatinib | VEGFR1-3 FGFR1 | Advanced pancreatic neuroendocrine cancer | Phase III | 300 mg/day | Oral | PFS 10.9 months | Hypertension, proteinuria, hypertriglyceridemia, serious adverse effects in 22% of patients, 3 deaths | [145] |
| Regorafenib | VEGFR1-3 TIE2 PDGFR-β FGFR KIT RET RAF | Advanced colorectal cancer | Phase IIb | 160 mg/day | Oral | PFS 3.5 months, OS 7.4 months, week 8 PFS rate 53%, metabolic response rate 41% | Hypertension, hand-foot skin reaction, hypophosphataemia, no deaths | [151] |
| Ramucirumab | VEGFR2 | Advanced hepatocellular carcinoma | Phase III | 8 mg/kg | Intravenous | OS 8.5 months, PFS 2.8 months, no difference in objective response and deterioration time | Hypertension, hyponatraemia, elevated aspartate aminotransferase, 3 deaths | [157] |
| Combination therapies | ||||||||
| Lenvatinib + pembrolizumab | Lenvat: VEGFR1-3 FGFR1-4 KIT PDGFRα RET Pembrol: PD-1R | Metastatic renal cell carcinoma Endometrial cancer Squamous cell carcinoma of the head and neck Melanoma Non-small-cell lung cancer Urothelial cancer | Phase Ib/II | Lenvat: 20 mg/day Pembrol: 200 mg | Lenvat: oral Pembrol: intravenous | ORR24 weeks 25%-63%, PFS 4.7-19.8 months | Fatigue, diarrhea, hypertension, hypothyroidism, 2 deaths | [147] |
| Axitinib + pembrolizumab | Axit: VEGFR1-3 PDGFR c-KIT Pembrol: PD-1R | Advanced renal cell carcinoma | Phase Ib | Axit: 5 + 5 mg/day Pembrol: 2 mg/kg | Axit: oral Pembrol: intravenous | Objective response 73%, PFS 20.9 months | Hypertension, diarrhea, fatigue, elevated alanine aminotransferase levels, no deaths | [149] |
| Axitinib + pembrolizumab | Axit: VEGFR1-3 PDGFR c-KIT Pembrol: PD-1R | Advanced renal cell carcinoma | Phase III | Axit: 5 + 5 mg/day Pembrol: 200 mg | Axit: oral Pembrol: intravenous | PFS 15.4 months | Hypertension, alanine aminotransferase increase, diarrhoea, no deaths | [150] |
| TAS-102 + bevacizumab | TAS-102: thymidine phosphorylase Bevac: VEGF-A | Metastatic colorectal adenocarcinoma | Phase I/II | TAS-102: 35 mg/m2 twice daily Bevac: 5 mg/kg | TAS-102: oral Bevac: intravenous | PFS16 weeks in 42.9% of patients | Neutropenia, leukopenia, anemia, febrile neutropenia, thrombocytopenia, no deaths | [160] |
| Carboplatin + paclitaxel + bevacizumab + metronomic cyclophosphamide | Carbop: DNA Paclit: microtubules Bevac: VEGF-A Cycloph: DNA | Inflammatory breast cancer | Phase II | Carbop: AUC 2 Paclit: 70 mg/m2 Bevac: 15 mg/kg Cycloph: 50 mg/day | Carbop: intravenous Paclit: intravenous Bevac: intravenous Cycloph: oral | Objective response 88%, pCR 29%, 5-year DFS 58%, 5-year OS 72% | Neutropenia, leucopenia, infections, hypertension, paresthesias, uncomplicated febrile neutropenia, anemia, no deaths | [161] |
| Apatinib + etoposide | Apat: VEGFR2 Etop: DNA | Ovarian cancer | Phase II | Apat: 500 mg/day Etop: 50 mg/day | Apat: oral Etop: oral | Objective response 54% | Neutropenia, fatigue, anaemia, mucositis, no deaths | [162] |
It should be noted that most clinical studies found treatment-related side effects, which were mostly mild, while few deaths were reported. However, since most of the studies include patients with highly resistant or metastatic cancers that do not respond to other treatments, the overall efficacy of these therapies in prolonging patient survival outweighs the frequent side effects.
CONCLUSION AND FUTURE DIRECTIONS
Cancer is a global epidemic and the second most common cause of death. Despite the active research, it remains a condition that, in most cases, is not cured and leads to death. Observing the distinctive physiology of cancer cells, the angiogenesis pathway emerged as a promising anticancer target. Thus, in recent years, more and more researchers are aiming to develop various anti-angiogenic agents and regulate their efficacy and safety. Regarding this, the present review summarises the results of very recent preclinical and clinical studies involving monotherapies or combination therapies of anti-angiogenic agents, in order to highlight the updates in the field and encourage more related research.
Apart from these therapies, however, several other anti-angiogenic approaches are currently being explored, such as the use of natural phytochemicals as anti-angiogenic agents in cancer[163]. This strategy has recently emerged as an attractive approach, since dietary natural compounds are generally considered non-toxic, and their production/administration is easily accessible and low-cost. However, the clinical translation of dietary phytochemicals still faces challenges, as the results thus far remain limited, contradictory, inconclusive or even inexistent for some of these compounds. In this context, a significant effort is needed to improve clinical trial design, taking serious consideration of the target population, as well as the pharmacokinetic and pharmacodynamic properties of these compounds.
Other novel anti-angiogenic strategies include the exploitation of miRNAs-elucidating their role in angiogenesis and evaluating novel miRNA-mediated immunotherapies[164]. Advances in miRNA targeting and delivery strategies, such as the use of nanoparticles or cell-derived membrane vesicles for therapeutic miRNA delivery, may provide miRNA-based therapeutics as anti-angiogenic treatments.
Additionally, the use of anti-angiogenic peptides has emerged as a promising anticancer strategy, as they present several advantages, with rapid production through automated chemical synthesis and the ability to undergo structural modifications. Therefore, several anticancer peptides that have been discovered derive from nanobodies and mimotopes resources, as well as natural resources such as venoms, bacteria, fungi, sponges, animals and plants[165]. The new methods available in phage display and peptidomimetics have contributed to the development of new anti-angiogenic peptides, while the novel technologies such as pegylation, polymer-supported formulation and macromolecule conjugation can now provide these peptides with better pharmacokinetic properties and contribute to more anti-angiogenic peptide-based therapeutics.
Although all of the above treatment strategies appear to be particularly promising for the future, the issue of developing angiogenic resistance should first be addressed. The combinatorial treatments mentioned have shown excellent results and will be a key concept of future therapeutic regimens. Apart from that, further studies are needed to assess the abovementioned treatment strategies’ safety in humans. Many drugs show significant efficacy in a large proportion of patients, but they also involve side effects that, even if not serious, can affect the patient’s quality of life and adherence to the regimen. Emerging ideas about novel delivery systems of such therapies might be the way forward. In addition, the simplification of regimens should be explored, especially when combination therapies are indicated, such as oral administration or the preparation of a combination formulation to increase patient adherence.
DECLARATIONS
Authors’ contributionsInvestigated this subject and supervised the findings of this work: Vafopoulou P
Discussed the results and contributed to the final manuscript: Vafopoulou P, Kourti M
Availability of data and materialsNot available.
Financial support and sponsorshipNone.
Conflicts of interestBoth authors declared that there are no conflicts of interest.
Ethical approval and consent to participateNot applicable.
Consent for publicationNot applicable.
Copyright© The Author(s) 2022.
REFERENCES
2. Guerra A, Belinha J, Mangir N, MacNeil S, Natal Jorge R. Simulation of the process of angiogenesis: quantification and assessment of vascular patterning in the chicken chorioallantoic membrane. Comput Biol Med 2021;136:104647.
3. Naito H, Iba T, Takakura N. Mechanisms of new blood-vessel formation and proliferative heterogeneity of endothelial cells. Int Immunol 2020;32:295-305.
5. Folkman J, Long DM Jr, Becker FF. Growth and metastasis of tumor in organ culture. Cancer 1963;16:453-67.
6. de Heer EC, Jalving M, Harris AL. HIFs, angiogenesis, and metabolism: elusive enemies in breast cancer. J Clin Invest 2020;130:5074-87.
7. Lugano R, Ramachandran M, Dimberg A. Tumor angiogenesis: causes, consequences, challenges and opportunities. Cell Mol Life Sci 2020;77:1745-70.
9. Melincovici CS, Boşca AB, Şuşman S, et al. Vascular endothelial growth factor (VEGF)-key factor in normal and pathological angiogenesis. Rom J Morphol Embryol 2018;59:455-67.
10. Bai Y, Bai L, Zhou J, Chen H, Zhang L. Sequential delivery of VEGF, FGF-2 and PDGF from the polymeric system enhance HUVECs angiogenesis in vitro and CAM angiogenesis. Cell Immunol 2018;323:19-32.
11. Armani G, Pozzi E, Pagani A, et al. The heterogeneity of cancer endothelium: the relevance of angiogenesis and endothelial progenitor cells in cancer microenvironment. Microvasc Res 2021;138:104189.
12. Huang XL, Khan MI, Wang J, et al. Role of receptor tyrosine kinases mediated signal transduction pathways in tumor growth and angiogenesis-New insight and futuristic vision. Int J Biol Macromol 2021;180:739-52.
13. Leung DW, Cachianes G, Kuang WJ, Goeddel DV, Ferrara N. Vascular endothelial growth factor is a secreted angiogenic mitogen. Science 1989;246:1306-9.
14. Montesano R, Vassalli JD, Baird A, Guillemin R, Orci L. Basic fibroblast growth factor induces angiogenesis in vitro. Proc Natl Acad Sci U S A 1986;83:7297-301.
15. Fett JW, Strydom DJ, Lobb RR, et al. Isolation and characterization of angiogenin, an angiogenic protein from human carcinoma cells. Biochemistry 1985;24:5480-6.
16. Schreiber AB, Winkler ME, Derynck R. Transforming growth factor-alpha: a more potent angiogenic mediator than epidermal growth factor. Science 1986;232:1250-3.
17. Roberts AB, Sporn MB, Assoian RK, et al. Transforming growth factor type beta: rapid induction of fibrosis and angiogenesis in vivo and stimulation of collagen formation in vitro. Proc Natl Acad Sci U S A 1986;83:4167-71.
18. Fràter-Schröder M, Risau W, Hallmann R, Gautschi P, Böhlen P. Tumor necrosis factor type alpha, a potent inhibitor of endothelial cell growth in vitro, is angiogenic in vivo. Proc Natl Acad Sci U S A 1987;84:5277-81.
19. Ishikawa F, Miyazono K, Hellman U, et al. Identification of angiogenic activity and the cloning and expression of platelet-derived endothelial cell growth factor. Nature 1989;338:557-62.
20. Bussolino F, Ziche M, Wang JM, et al. In vitro and in vivo activation of endothelial cells by colony-stimulating factors. J Clin Invest 1991;87:986-95.
21. Zhao J, Chen L, Shu B, et al. Granulocyte/macrophage colony-stimulating factor influences angiogenesis by regulating the coordinated expression of VEGF and the Ang/Tie system. PLoS One 2014;9:e92691.
22. Zheng Q, Li X, Cheng X, et al. Granulocyte-macrophage colony-stimulating factor increases tumor growth and angiogenesis directly by promoting endothelial cell function and indirectly by enhancing the mobilization and recruitment of proangiogenic granulocytes. Tumour Biol 2017;39:1010428317692232.
23. Zhou Y, Tu C, Zhao Y, Liu H, Zhang S. Placental growth factor enhances angiogenesis in human intestinal microvascular endothelial cells via PI3K/Akt pathway: potential implications of inflammation bowel disease. Biochem Biophys Res Commun 2016;470:967-74.
24. Kitadai Y, Takahashi Y, Haruma K, et al. Transfection of interleukin-8 increases angiogenesis and tumorigenesis of human gastric carcinoma cells in nude mice. Br J Cancer 1999;81:647-53.
25. Kaga T, Kawano H, Sakaguchi M, Nakazawa T, Taniyama Y, Morishita R. Hepatocyte growth factor stimulated angiogenesis without inflammation: differential actions between hepatocyte growth factor, vascular endothelial growth factor and basic fibroblast growth factor. Vascul Pharmacol 2012;57:3-9.
26. Gospodarowicz D, Bialecki H, Thakral T. The angiogenic activity of the fibroblast and epidermal growth factor. Experimental Eye Research 1979;28:501-14.
27. Yokoi A, Mccrudden KW, Huang J, et al. Human epidermal growth factor receptor signaling contributes to tumor growth via angiogenesis in her2/neu-expressing experimental Wilms’ tumor. Journal of Pediatric Surgery 2003;38:1569-73.
28. Palinski W, Monti M, Camerlingo R, et al. Lysosome purinergic receptor P2X4 regulates neoangiogenesis induced by microvesicles from sarcoma patients. Cell Death Dis 2021;12:797.
29. Pavlakovic H, Havers W, Schweigerer L. Multiple angiogenesis stimulators in a single malignancy: implications for anti-angiogenic tumour therapy. Angiogenesis 2001;4:259-62.
30. Shibuya M. Vascular endothelial growth factor (VEGF) and its receptor (VEGFR) signaling in angiogenesis: a crucial target for anti- and pro-angiogenic therapies. Genes Cancer 2011;2:1097-105.
31. Muller YA, Li B, Christinger HW, Wells JA, Cunningham BC, de Vos AM. Vascular endothelial growth factor: crystal structure and functional mapping of the kinase domain receptor binding site. Proc Natl Acad Sci U S A 1997;94:7192-7.
32. Shibuya M. Role of VEGF-flt receptor system in normal and tumor angiogenesis. Adv Cancer Res 1995;67:281-316.
33. Li L, Liu H, Xu C, et al. VEGF promotes endothelial progenitor cell differentiation and vascular repair through connexin 43. Stem Cell Res Ther 2017;8:237.
34. Zhu D, Li Y, Zhang Z, et al. Recent advances of nanotechnology-based tumor vessel-targeting strategies. J Nanobiotechnology 2021;19:435.
35. El-Kenawi AE, El-Remessy AB. Angiogenesis inhibitors in cancer therapy: mechanistic perspective on classification and treatment rationales. Br J Pharmacol 2013;170:712-29.
36. Mundel TM, Kalluri R. Type IV collagen-derived angiogenesis inhibitors. Microvasc Res 2007;74:85-9.
37. Abdollahi A, Hahnfeldt P, Maercker C, et al. Endostatin’s antiangiogenic signaling network. Molecular Cell 2004;13:649-63.
38. Ranjit PM, Anuradha C, Vishnupriya S, Girijasankar G, Girish K, Chowdary YA. Endogenous angiogenesis inhibitor endostatin: an overview. Asian Journal of Pharmaceutical and Clinical Research 2012;5:1-8. Available from: https://xueshu.baidu.com/usercenter/paper/show?paperid=cdfa6789882c0b11995e7c9aed8209c7&site=xueshu_se&hitarticle=1. [Last accessed on 26 Apr 2022].
39. Kerbel R, Folkman J. Clinical translation of angiogenesis inhibitors. Nat Rev Cancer 2002;2:727-39.
41. Rao N, Lee YF, Ge R. Novel endogenous angiogenesis inhibitors and their therapeutic potential. Acta Pharmacol Sin 2015;36:1177-90.
43. Frezzetti D, Gallo M, Maiello MR, et al. VEGF as a potential target in lung cancer. Expert Opin Ther Targets 2017;21:959-66.
44. Carmeliet P, Jain RK. Molecular mechanisms and clinical applications of angiogenesis. Nature 2011;473:298-307.
45. Xu G. Angiogenesis inhibition in the in vivo antineoplastic effect of manumycin and paclitaxel against anaplastic thyroid carcinoma. Journal of Clinical Endocrinology & Metabolism 2001;86:1769-77.
46. Xu N, Bai Y, Luo T, Duan P. Low-dose paclitaxel combined with thalidomide inhibits angiogenesis in mice bearing S180 sarcoma. Chinese Journal of Clinical Oncology 2020;47:7-11. (in Chinese) Available from: https://xueshu.baidu.com/usercenter/paper/show?paperid=1j620ed0bf510m10dv0a08y0km339097&site=xueshu_se&hitarticle=1. [Last accessed on 26 Apr 2022].
47. Zhang QY, Sun L, Wang ZH. Effect of ginsenoside Rg3 combined with cyclophosphamide on angiogenesis in rats with EMT-6 breast cancer.Chinese Journal of Clinical Rehabilitation 2004. Available from: https://xueshu.baidu.com/usercenter/paper/show?paperid=ac39f44b384ddaf8af60bee674f81bbe&site=xueshu_se&hitarticle=1. [Last accessed on 26 Apr 2022].
48. Ezoe K, Murata N, Yabuuchi A, et al. Long-term adverse effects of cyclophosphamide on follicular growth and angiogenesis in mouse ovaries. Reprod Biol 2014;14:238-42.
49. Ciardiello F, Caputo R, Bianco R, et al. Inhibition of growth factor production and angiogenesis in human cancer cells by ZD1839 (Iressa), a selective epidermal growth factor receptor tyrosine kinase inhibitor. Clin Cancer Res 2001;7:1459-65.
50. Hirata A, Ogawa S, Kometani T, et al. ZD1839 (Iressa) induces antiangiogenic effects through inhibition of epidermal growth factor receptor tyrosine kinase. Cancer Res 2002;62:2554-60.
51. Fallahi P, Ferrari SM, Galdiero MR, et al. Molecular targets of tyrosine kinase inhibitors in thyroid cancer. Semin Cancer Biol 2022;79:180-96.
52. Papadimitriou M, Papadimitriou CA. Antiangiogenic tyrosine kinase inhibitors in metastatic colorectal cancer: focusing on regorafenib. Anticancer Res 2021;41:567-82.
53. Dan H, Lei X, Huang X, Ma N, Xing Y, Shen Y. CM082, a novel VEGF receptor tyrosine kinase inhibitor, can inhibit angiogenesis in vitro and in vivo. Microvasc Res 2021;136:104146.
54. Dai MD, Wang YL, Fan J, et al. DW14383 is an irreversible pan-FGFR inhibitor that suppresses FGFR-dependent tumor growth in vitro and in vivo. Acta Pharmacol Sin 2021;42:1498-506.
55. Yang JC, Haworth L, Sherry RM, et al. A randomized trial of bevacizumab, an anti-vascular endothelial growth factor antibody, for metastatic renal cancer. N Engl J Med 2003;349:427-34.
56. Willett CG, Boucher Y, di Tomaso E, et al. Direct evidence that the VEGF-specific antibody bevacizumab has antivascular effects in human rectal cancer. Nat Med 2004;10:145-7.
57. Heinemann V, Hoff PM. Bevacizumab plus irinotecan-based regimens in the treatment of metastatic colorectal cancer. Oncology 2010;79:118-28.
58. Cui F, Chen JZ, Wan C, Chen B, Luo RC, Zheng H. Clinical research of bevacizumab in combination with irinotecan, fluorouracil and leucovorin for advanced metastatic colorectal cancer. Zhonghua Wei Chang Wai Ke Za Zhi 2009;12:374-7.
59. Kabbinavar F, Hurwitz HI, Fehrenbacher L, et al. Phase II, randomized trial comparing bevacizumab plus fluorouracil (FU)/leucovorin (LV) with FU/LV alone in patients with metastatic colorectal cancer. J Clin Oncol 2003;21:60-5.
60. Dank M, Budi L, Piko B, et al. First-line bevacizumab-paclitaxel in 220 patients with metastatic breast cancer: results from the AVAREG study. Anticancer Res 2014;34:1275-80.
61. Johnson DH, Fehrenbacher L, Novotny WF, et al. Randomized phase II trial comparing bevacizumab plus carboplatin and paclitaxel with carboplatin and paclitaxel alone in previously untreated locally advanced or metastatic non-small-cell lung cancer. J Clin Oncol 2004;22:2184-91.
62. Soria JC, Mauguen A, Reck M, et al. Meta-analysis of Bevacizumab in Advanced NSCLC Collaborative Group. Systematic review and meta-analysis of randomised, phase II/III trials adding bevacizumab to platinum-based chemotherapy as first-line treatment in patients with advanced non-small-cell lung cancer. Ann Oncol 2013;24:20-30.
63. Yu SR, Shi MQ, Xia GH. Combination of bevacizumab and platinum-based chemotherapy as first-line therapy for advanced non-squamous NSCLC. Journal of Practical Oncology 2015;30:418-22.
64. Spratlin JL, Mulder KE, Mackey JR. Ramucirumab (IMC-1121B): a novel attack on angiogenesis. Future Oncol 2010;6:1085-94.
65. Mcintyre JA, Martin L. Panitumumab: oncolytic prop inn anti-EGFR human monoclonal antibody. Drugs of the Future 2004. Available from: https://xueshu.baidu.com/usercenter/paper/show?paperid=af47c87a085e47a6dd730f339a94aa3e&site=xueshu_se&hitarticle=1. [Last accessed on 26 Apr 2022].
66. Pueyo G, Mesia R, Figueras A, et al. Cetuximab may inhibit tumor growth and angiogenesis induced by ionizing radiation: a preclinical rationale for maintenance treatment after radiotherapy. Oncologist 2010;15:976-86.
67. Bugyik E, Dezso K, Reiniger L, et al. Lack of angiogenesis in experimental brain metastases. J Neuropathol Exp Neurol 2011;70:979-91.
68. Valiente M, Obenauf AC, Jin X, et al. Serpins promote cancer cell survival and vascular co-option in brain metastasis. Cell 2014;156:1002-16.
69. Bridgeman VL, Vermeulen PB, Foo S, et al. Vessel co-option is common in human lung metastases and mediates resistance to anti-angiogenic therapy in preclinical lung metastasis models. J Pathol 2017;241:362-74.
70. Stessels F, Van den Eynden G, Van der Auwera I, et al. Breast adenocarcinoma liver metastases, in contrast to colorectal cancer liver metastases, display a non-angiogenic growth pattern that preserves the stroma and lacks hypoxia. Br J Cancer 2004;90:1429-36.
71. Vermeulen PB, Colpaert C, Salgado R, et al. Liver metastases from colorectal adenocarcinomas grow in three patterns with different angiogenesis and desmoplasia. J Pathol 2001;195:336-42.
72. Winkler F. Hostile takeover: how tumours hijack pre-existing vascular environments to thrive. J Pathol 2017;242:267-72.
73. Kuczynski EA, Yin M, Bar-Zion A, et al. Co-option of liver vessels and not sprouting angiogenesis drives acquired sorafenib resistance in hepatocellular carcinoma. J Natl Cancer Inst 2016;108:djw030.
74. Leenders WP, Küsters B, Verrijp K, et al. Antiangiogenic therapy of cerebral melanoma metastases results in sustained tumor progression via vessel co-option. Clin Cancer Res 2004;10:6222-30.
75. Frentzas S, Simoneau E, Bridgeman VL, et al. Vessel co-option mediates resistance to anti-angiogenic therapy in liver metastases. Nat Med 2016;22:1294-302.
76. Kuczynski EA, Reynolds AR. Vessel co-option and resistance to anti-angiogenic therapy. Angiogenesis 2020;23:55-74.
77. Casanovas O, Hicklin DJ, Bergers G, Hanahan D. Drug resistance by evasion of antiangiogenic targeting of VEGF signaling in late-stage pancreatic islet tumors. Cancer Cell 2005;8:299-309.
78. Ebos JM, Lee CR, Christensen JG, Mutsaers AJ, Kerbel RS. Multiple circulating proangiogenic factors induced by sunitinib malate are tumor-independent and correlate with antitumor efficacy. Proc Natl Acad Sci U S A 2007;104:17069-74.
79. Croci DO, Cerliani JP, Dalotto-Moreno T, et al. Glycosylation-dependent lectin-receptor interactions preserve angiogenesis in anti-VEGF refractory tumors. Cell 2014;156:744-58.
80. Pisarsky L, Bill R, Fagiani E, et al. Targeting metabolic symbiosis to overcome resistance to anti-angiogenic therapy. Cell Rep 2016;15:1161-74.
81. Zhai B, Hu F, Jiang X, et al. Inhibition of Akt reverses the acquired resistance to sorafenib by switching protective autophagy to autophagic cell death in hepatocellular carcinoma. Mol Cancer Ther 2014;13:1589-98.
82. Chandra A, Rick J, Yagnik G, Aghi MK. Autophagy as a mechanism for anti-angiogenic therapy resistance. Semin Cancer Biol 2020;66:75-88.
83. Giuliano S, Cormerais Y, Dufies M, et al. Resistance to sunitinib in renal clear cell carcinoma results from sequestration in lysosomes and inhibition of the autophagic flux. Autophagy 2015;11:1891-904.
84. Wu S, Huang L, Shen R, et al. Drug resistance-related sunitinib sequestration in autophagolysosomes of endothelial cells. Int J Oncol 2020;56:113-22.
85. Wen Y, Chelariu-Raicu A, Umamaheswaran S, et al. Endothelial p130cas confers resistance to anti-angiogenesis therapy. Cell Rep 2022;38:110301.
86. Tang D, Gao J, Wang S, et al. Cancer-associated fibroblasts promote angiogenesis in gastric cancer through galectin-1 expression. Tumour Biol 2016;37:1889-99.
87. Hosaka K, Yang Y, Seki T, et al. Pericyte-fibroblast transition promotes tumor growth and metastasis. Proc Natl Acad Sci U S A 2016;113:E5618-27.
88. Orlidge A, D’Amore PA. Inhibition of capillary endothelial cell growth by pericytes and smooth muscle cells. J Cell Biol 1987;105:1455-62.
89. Bussolati B, Deregibus MC, Camussi G. Characterization of molecular and functional alterations of tumor endothelial cells to design anti-angiogenic strategies. Curr Vasc Pharmacol 2010;8:220-32.
90. Bussolati B, Deambrosis I, Russo S, Deregibus MC, Camussi G. Altered angiogenesis and survival in human tumor-derived endothelial cells. FASEB J 2003;17:1159-61.
91. Hida K, Hida Y, Amin DN, et al. Tumor-associated endothelial cells with cytogenetic abnormalities. Cancer Res 2004;64:8249-55.
92. Maishi N, Annan DA, Kikuchi H, Hida Y, Hida K. Tumor endothelial heterogeneity in cancer progression. Cancers (Basel) 2019;11:1511.
93. Holmgren L, O’Reilly MS, Folkman J. Dormancy of micrometastases: balanced proliferation and apoptosis in the presence of angiogenesis suppression. Nat Med 1995;1:149-53.
94. Conley SJ, Gheordunescu E, Kakarala P, et al. Antiangiogenic agents increase breast cancer stem cells via the generation of tumor hypoxia. Proc Natl Acad Sci U S A 2012;109:2784-9.
95. Cheng L, Huang Z, Zhou W, et al. Glioblastoma stem cells generate vascular pericytes to support vessel function and tumor growth. Cell 2013;153:139-52.
96. Mohammed RA, Green A, El-Shikh S, Paish EC, Ellis IO, Martin SG. Prognostic significance of vascular endothelial cell growth factors -A, -C and -D in breast cancer and their relationship with angio- and lymphangiogenesis. Br J Cancer 2007;96:1092-100.
97. Zarrin B, Zarifi F, Vaseghi G, Javanmard SH. Acquired tumor resistance to antiangiogenic therapy: mechanisms at a glance. J Res Med Sci 2017;22:117.
98. Montemagno C, Pagès G. Resistance to anti-angiogenic therapies: a mechanism depending on the time of exposure to the drugs. Front Cell Dev Biol 2020;8:584.
99. Lopes-Coelho F, Martins F, Pereira SA, Serpa J. Anti-angiogenic therapy: current challenges and future perspectives. Int J Mol Sci 2021;22:3765.
100. Kienast Y, Klein C, Scheuer W, et al. Ang-2-VEGF-A CrossMab, a novel bispecific human IgG1 antibody blocking VEGF-A and Ang-2 functions simultaneously, mediates potent antitumor, antiangiogenic, and antimetastatic efficacy. Clin Cancer Res 2013;19:6730-40.
101. Brown JL, Cao ZA, Pinzon-Ortiz M, et al. A human monoclonal anti-ANG2 antibody leads to broad antitumor activity in combination with VEGF inhibitors and chemotherapy agents in preclinical models. Mol Cancer Ther 2010;9:145-56.
102. Gyanchandani R, Ortega Alves MV, Myers JN, Kim S. A proangiogenic signature is revealed in FGF-mediated bevacizumab-resistant head and neck squamous cell carcinoma. Mol Cancer Res 2013;11:1585-96.
103. Powell MA, Sill MW, Goodfellow PJ, et al. A phase II trial of brivanib in recurrent or persistent endometrial cancer: an NRG Oncology/Gynecologic Oncology Group Study. Gynecol Oncol 2014;135:38-43.
104. Shimizu S, Takehara T, Hikita H, et al. Inhibition of autophagy potentiates the antitumor effect of the multikinase inhibitor sorafenib in hepatocellular carcinoma. Int J Cancer 2012;131:548-57.
105. Motzer RJ, Alyasova A, Ye D, et al. Phase II trial of second-line everolimus in patients with metastatic renal cell carcinoma (RECORD-4). Ann Oncol 2016;27:441-8.
106. Bamias A, Karavasilis V, Gavalas N, et al. The combination of bevacizumab/temsirolimus after first-line anti-VEGF therapy in advanced renal-cell carcinoma: a clinical and biomarker study. Int J Clin Oncol 2019;24:411-9.
107. Pietras K, Hanahan D. A multitargeted, metronomic, and maximum-tolerated dose “chemo-switch” regimen is antiangiogenic, producing objective responses and survival benefit in a mouse model of cancer. J Clin Oncol 2005;23:939-52.
108. Ramadan WS, Zaher DM, Altaie AM, Talaat IM, Elmoselhi A. Potential therapeutic strategies for lung and breast cancers through understanding the anti-angiogenesis resistance mechanisms. Int J Mol Sci 2020;21:565.
109. Song Y, Fu Y, Xie Q, Zhu B, Wang J, Zhang B. Anti-angiogenic agents in combination with immune checkpoint inhibitors: a promising strategy for cancer treatment. Front Immunol 2020;11:1956.
110. Ogasawara S, Mihara Y, Kondo R, Kusano H, Akiba J, Yano H. Antiproliferative effect of lenvatinib on human liver cancer cell lines in vitro and in vivo. Anticancer Res 2019;39:5973-82.
111. Suyama K, Iwase H. Lenvatinib: a promising molecular targeted agent for multiple cancers. Cancer Control 2018;25:1073274818789361.
112. Kudo M, Finn RS, Qin S, et al. Lenvatinib versus sorafenib in first-line treatment of patients with unresectable hepatocellular carcinoma: a randomised phase 3 non-inferiority trial. The Lancet 2018;391:1163-73.
113. Vogel A, Qin S, Kudo M, et al. Lenvatinib versus sorafenib for first-line treatment of unresectable hepatocellular carcinoma: patient-reported outcomes from a randomised, open-label, non-inferiority, phase 3 trial. The Lancet Gastroenterology & Hepatology 2021;6:649-58.
114. Jin H, Shi Y, Lv Y, et al. EGFR activation limits the response of liver cancer to lenvatinib. Nature 2021;595:730-4.
115. Yano S, Yamaguchi M, Dong RP. EGFR tyrosine kinase inhibitor “gefitinib (Iressa)” for cancer therapy. Nihon Yakurigaku Zasshi 2003;122:491-7.
116. Han B, Li K, Wang Q, et al. Effect of anlotinib as a third-line or further treatment on overall survival of patients with advanced non-small cell lung cancer: the ALTER 0303 phase 3 randomized clinical trial. JAMA Oncol 2018;4:1569-75.
117. Chi Y, Fang Z, Hong X, et al. Safety and efficacy of anlotinib, a multikinase angiogenesis inhibitor, in patients with refractory metastatic soft-tissue sarcoma. Clin Cancer Res 2018;24:5233-8.
118. Sherman SI. Advances in chemotherapy of differentiated epithelial and medullary thyroid cancers. J Clin Endocrinol Metab 2009;94:1493-9.
119. Ruan X, Shi X, Dong Q, et al. Antitumor effects of anlotinib in thyroid cancer. Endocr Relat Cancer 2019;26:153-64.
120. Gao Q, Tang S, Chen H, et al. Intratumoral injection of anlotinib hydrogel enhances antitumor effects and reduces toxicity in mouse model of lung cancer. Drug Deliv 2020;27:1524-34.
121. Goff D, Zhang J, Heckrodt T, et al. Discovery of dual Axl/VEGF-R2 inhibitors as potential anti-angiogenic and anti-metastatic drugs for cancer chemotherapy. Bioorg Med Chem Lett 2017;27:3766-71.
122. Holland SJ, Pan A, Franci C, et al. R428, a selective small molecule inhibitor of Axl kinase, blocks tumor spread and prolongs survival in models of metastatic breast cancer. Cancer Res 2010;70:1544-54.
123. Holland SJ, Powell MJ, Franci C, et al. Multiple roles for the receptor tyrosine kinase axl in tumor formation. Cancer Res 2005;65:9294-303.
125. Yoshiji H, Kuriyama S, Yoshii J, et al. Synergistic effect of basic fibroblast growth factor and vascular endothelial growth factor in murine hepatocellular carcinoma. Hepatology 2002;35:834-42.
126. Harimoto N, Taguchi K, Shirabe K, et al. The significance of fibroblast growth factor receptor 2 expression in differentiation of hepatocellular carcinoma. Oncology 2010;78:361-8.
127. Kano MR, Morishita Y, Iwata C, et al. VEGF-A and FGF-2 synergistically promote neoangiogenesis through enhancement of endogenous PDGF-B-PDGFRbeta signaling. J Cell Sci 2005;118:3759-68.
128. Le TBU, Vu TC, Ho RZW, et al. Bevacizumab augments the antitumor efficacy of infigratinib in hepatocellular carcinoma. Int J Mol Sci 2020;21:9405.
129. Prawira A, Le TBU, Vu TC, Huynh H. Ribociclib enhances infigratinib-induced cancer cell differentiation and delays resistance in FGFR-driven hepatocellular carcinoma. Liver Int 2021;41:608-20.
130. Lu RM, Chiu CY, Liu IJ, Chang YL, Liu YJ, Wu HC. Novel human Ab against vascular endothelial growth factor receptor 2 shows therapeutic potential for leukemia and prostate cancer. Cancer Sci 2019;110:3773-87.
131. Zhang J, Liu C, Shi W, et al. The novel VEGF receptor 2 inhibitor YLL545 inhibits angiogenesis and growth in breast cancer. Oncotarget 2016;7:41067-80.
133. West XZ, Malinin NL, Merkulova AA, et al. Oxidative stress induces angiogenesis by activating TLR2 with novel endogenous ligands. Nature 2010;467:972-6.
134. Wu C, Wang X, Tomko N, et al. 2-(ω-carboxyethyl)pyrrole antibody as a new inhibitor of tumor angiogenesis and growth. Anticancer Agents Med Chem 2017;17:813-20.
135. Li QW, Zhang GL, Hao CX, et al. SANT, a novel Chinese herbal monomer combination, decreasing tumor growth and angiogenesis via modulating autophagy in heparanase overexpressed triple-negative breast cancer. J Ethnopharmacol 2021;266:113430.
136. Nadir Y, Vlodavsky I, Brenner B. Heparanase, tissue factor, and cancer. Semin Thromb Hemost 2008;34:187-94.
137. Chen L, Qiu XS. Relationship between expressions of heparanase and basic fibroblast growth factor and clinicopathology of breast cancer tissues. Chinese Journal of Cancer Prevention and Treatment 2010;17:743-745+748. Available from: https://xueshu.baidu.com/usercenter/paper/show?paperid=22fd6776dcd06aea5a4e3876c132fdf9&site=xueshu_se&hitarticle=1. [Last accessed on 26 Apr 2022].
138. Gawthorpe S, Brown JE, Arif M, Nightingale P, Nevill A, Carmichael AR. Heparanase and COX-2 expression as predictors of lymph node metastasis in large, high-grade breast tumors. Anticancer Res 2014;34:2797-800.
139. Zhang PC, Liu X, Li MM, et al. AT-533, a novel Hsp90 inhibitor, inhibits breast cancer growth and HIF-1α/VEGF/VEGFR-2-mediated angiogenesis in vitro and in vivo. Biochem Pharmacol 2020;172:113771.
140. Miyata Y, Nakamoto H, Neckers L. The therapeutic target Hsp90 and cancer hallmarks. Curr Pharm Des 2013;19:347-65.
141. Calderwood SK, Gong J. Heat shock proteins promote cancer: it’s a protection racket. Trends Biochem Sci 2016;41:311-23.
142. Han B, Li K, Zhao Y, et al. Anlotinib as a third-line therapy in patients with refractory advanced non-small-cell lung cancer: a multicentre, randomised phase II trial (ALTER0302). Br J Cancer 2018;118:654-61.
143. Koumarianou A, Kaltsas G. Surufatinib-a novel oral agent for neuroendocrine tumours. Nat Rev Endocrinol 2021;17:9-10.
144. Xu J, Shen L, Zhou Z, et al. Surufatinib in advanced extrapancreatic neuroendocrine tumours (SANET-ep): a randomised, double-blind, placebo-controlled, phase 3 study. Lancet Oncol 2020;21:1500-12.
145. Xu J, Shen L, Bai C, et al. Surufatinib in advanced pancreatic neuroendocrine tumours (SANET-p): a randomised, double-blind, placebo-controlled, phase 3 study. The Lancet Oncology 2020;21:1489-99.
147. Taylor MH, Lee CH, Makker V, et al. Phase IB/II trial of lenvatinib plus pembrolizumab in patients with advanced renal cell carcinoma, endometrial cancer, and other selected advanced solid tumors. J Clin Oncol 2020;38:1154-63.
148. Yasuda S, Sho M, Yamato I, et al. Simultaneous blockade of programmed death 1 and vascular endothelial growth factor receptor 2 (VEGFR2) induces synergistic anti-tumour effect in vivo. Clin Exp Immunol 2013;172:500-6.
149. Atkins MB, Plimack ER, Puzanov I, et al. Axitinib in combination with pembrolizumab in patients with advanced renal cell cancer: a non-randomised, open-label, dose-finding, and dose-expansion phase 1b trial. The Lancet Oncology 2018;19:405-15.
150. Powles T, Plimack ER, Soulières D, et al. Pembrolizumab plus axitinib versus sunitinib monotherapy as first-line treatment of advanced renal cell carcinoma (KEYNOTE-426): extended follow-up from a randomised, open-label, phase 3 trial. The Lancet Oncology 2020;21:1563-73.
151. Riechelmann RP, Leite LS, Bariani GM, et al. Regorafenib in patients with antiangiogenic-naïve and chemotherapy-refractory advanced colorectal cancer: results from a phase IIb trial. Oncologist 2019;24:1180-7.
152. Ettrich TJ, Seufferlein T. Regorafenib. In: Martens UM, editor. Small molecules in oncology. Cham: Springer International Publishing; 2018. p. 45-56.
153. la Fouchardière C. Regorafenib in the treatment of metastatic colorectal cancer. Future Oncol 2018;14:2239-46.
154. Nakazawa T, Hidaka H, Takada J, et al. Early increase in α-fetoprotein for predicting unfavorable clinical outcomes in patients with advanced hepatocellular carcinoma treated with sorafenib. Eur J Gastroenterol Hepatol 2013;25:683-9.
155. Silva JP, Gorman RA, Berger NG, et al. The prognostic utility of baseline alpha-fetoprotein for hepatocellular carcinoma patients. J Surg Oncol 2017;116:831-40.
156. Tangkijvanich P, Anukulkarnkusol N, Suwangool P, et al. Clinical characteristics and prognosis of hepatocellular carcinoma: analysis based on serum alpha-fetoprotein levels. J Clin Gastroenterol 2000;31:302-8.
157. Zhu AX, Kang Y, Yen C, et al. Ramucirumab after sorafenib in patients with advanced hepatocellular carcinoma and increased α-fetoprotein concentrations (REACH-2): a randomised, double-blind, placebo-controlled, phase 3 trial. The Lancet Oncology 2019;20:282-96.
158. Mayer RJ, Van Cutsem E, Falcone A, et al. RECOURSE Study Group. Randomized trial of TAS-102 for refractory metastatic colorectal cancer. N Engl J Med 2015;372:1909-19.
159. Tsukihara H, Nakagawa F, Sakamoto K, et al. Efficacy of combination chemotherapy using a novel oral chemotherapeutic agent, TAS-102, together with bevacizumab, cetuximab, or panitumumab on human colorectal cancer xenografts. Oncol Rep 2015;33:2135-42.
160. Kuboki Y, Nishina T, Shinozaki E, et al. TAS-102 plus bevacizumab for patients with metastatic colorectal cancer refractory to standard therapies (C-TASK FORCE): an investigator-initiated, open-label, single-arm, multicentre, phase 1/2 study. The Lancet Oncology 2017;18:1172-81.
161. Palazzo A, Dellapasqua S, Munzone E, et al. Phase II trial of bevacizumab plus weekly paclitaxel, carboplatin, and metronomic cyclophosphamide with or without trastuzumab and endocrine therapy as preoperative treatment of inflammatory breast cancer. Clin Breast Cancer 2018;18:328-35.
162. Lan C, Wang Y, Xiong Y, et al. Apatinib combined with oral etoposide in patients with platinum-resistant or platinum-refractory ovarian cancer (AEROC): a phase 2, single-arm, prospective study. The Lancet Oncology 2018;19:1239-46.
163. Marrero AD, Quesada AR, Martínez-Poveda B, Medina MÁ. Antiangiogenic phytochemicals constituent of diet as promising candidates for chemoprevention of cancer. Antioxidants (Basel) 2022;11:302.
164. Hussen BM, Salihi A, Abdullah ST, et al. Signaling pathways modulated by miRNAs in breast cancer angiogenesis and new therapeutics. Pathol Res Pract 2022;230:153764.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Vafopoulou P, Kourti M. Anti-angiogenic drugs in cancer therapeutics: a review of the latest preclinical and clinical studies of anti-angiogenic agents with anticancer potential. J Cancer Metastasis Treat 2022;8:18. http://dx.doi.org/10.20517/2394-4722.2022.08
AMA Style
Vafopoulou P, Kourti M. Anti-angiogenic drugs in cancer therapeutics: a review of the latest preclinical and clinical studies of anti-angiogenic agents with anticancer potential. Journal of Cancer Metastasis and Treatment. 2022; 8: 18. http://dx.doi.org/10.20517/2394-4722.2022.08
Chicago/Turabian Style
Vafopoulou, Polyxeni, Malamati Kourti. 2022. "Anti-angiogenic drugs in cancer therapeutics: a review of the latest preclinical and clinical studies of anti-angiogenic agents with anticancer potential" Journal of Cancer Metastasis and Treatment. 8: 18. http://dx.doi.org/10.20517/2394-4722.2022.08
ACS Style
Vafopoulou, P.; Kourti M. Anti-angiogenic drugs in cancer therapeutics: a review of the latest preclinical and clinical studies of anti-angiogenic agents with anticancer potential. J. Cancer. Metastasis. Treat. 2022, 8, 18. http://dx.doi.org/10.20517/2394-4722.2022.08
About This Article
Copyright

Data & Comments
Data


 Cite This Article 57 clicks
Cite This Article 57 clicks

 Like This Article 14
likes
Like This Article 14
likes






Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.