
The heterogeneity of CAFs and immune cell populations in the tumor microenvironment of pancreatic adenocarcinoma

 ,
, Abstract
Over the past decade, researchers have identified and characterized the diverse cell populations within the tumor microenvironment of pancreatic cancer. The interplay between these cells in the TME either promotes or inhibits the malignant behavior of pancreatic cancer cells. Cancer-associated fibroblasts, previously thought to be one main subset, can now be broadly subclassified into three main types: inflammatory, myofibroblastic, and antigen-presenting, with the former and the latter two exerting pro-tumoral and anti-tumoral functions, respectively. Myeloid cells include myeloid-derived suppressor cells and tumor-associated macrophages. Myeloid-derived suppressor cells can be further divided into polymorphonuclear and monocytic and exhibit pro-tumoral activities. Tumor-associated macrophages exhibit M1 (anti-tumoral) or M2 (pro-tumoral) phenotypes, which are present in a dynamic fashion between the two phenotypes. Other constituents of the immune make-up of the tumor microenvironment include T and B cells and less described subsets which include natural killer cells, γδ T cells, and group 2 innate lymphoid cells. This review provides an overview of the studies that lead to the discovery of those cellular populations and highlights the recent efforts to utilize them as therapeutic targets in pancreatic cancer.
Keywords
INTRODUCTION
Pancreatic adenocarcinoma (PDAC) is the fourth leading cause of cancer-related deaths and is expected to be the second cause by 2030[1,2]. Multiple factors contribute to the dismal prognosis of PDAC, including resistance to chemotherapy and propensity for early metastasis[3]. Mutations in four main genes support the evolution of PDAC, which activate mutations in Kras and loss-of-function mutations in TP53, P16/CDKN2A, and SMAD4/DPC4[3]. Current standard chemotherapeutic regimens, such as FOLFIRINOX and gemcitabine/nab-paclitaxel, only modestly improve survival[4]. Additionally, immunotherapy, which has revolutionized the treatment of patients with other malignancies, has been largely ineffective in pancreatic cancer[5]. The exception is a small subset of patients with microsatellite instability-high disease, but those only account for approximately 1% of all patients[6].
One of the key features of PDAC that support its chemoresistance is its desmoplastic stroma which prevents the delivery of chemotherapeutic agents[7]. Moreover, its tumor microenvironment (TME) comprises a number of non-neoplastic cell populations, such as cancer-associated fibroblasts and suppressive immune cells, that prevent effector immune cell infiltration[3]. Novel research over the last few years, coupled with advances in technology such as single-cell sequencing, has improved our understanding of TME and the many processes that occur both intra- and intertumorally, and has provided the space to test innovative treatment combinations that provide the basis for future efforts in diseases that have traditionally been very difficult to treat[3]. This review highlights the different immune cell populations in the TME of PDAC.
CANCER-ASSOCIATED FIBROBLASTS
Cancer-associated fibroblasts (CAFs) are central to the development of the non-neoplastic components of the TME[8]. Although not immune cells, CAFs influence the interplay between cancer cells and immune cells in the TME either through secreted factors or direct cell-cell interactions[8]. Initially thought to exclusively arise from PSCs, studies now suggest that CAFs can arise from different populations, including: PSCs[9], circulating bone marrow-derived cells called fibrocytes[10], adipose-derived mesenchymal stem cells (MSCs)[11], and epithelial and endothelial cells through epithelial- and endothelial-to-mesenchymal transition[12,13]. The old concept that CAFs are solely tumor-promoting has been challenged by recent efforts to target the stromal aspect of PDAC[14]. Evidence suggests that subpopulations of CAFs restrain tumor growth rather than promote it[14], explaining why non-selective targeting of CAFs results in conflicting outcomes[14]. For example, depleting alpha-smooth muscle actin (α-SMA)-positive cells reduced desmoplasia in samples from PKT and KPC mice but resulted in highly undifferentiated tumors, further diminished intratumoral blood vessels with evident hypoxic necrosis, and shorter survival[14]. Deletion of type I collagen in αSMA-positive myofibroblasts augmented immune suppression and resulted in a more aggressive cancer phenotype[15,16]. Similar findings were observed when treated with an inhibitor of Smoothened, which is overexpressed on CAFs, or neoplastic cell deletion of Sonic Hedgehog 1, a major mediator of desmoplasia[17,18]. These findings explain the poor survival outcomes observed in a phase I/II trial of sonic hedgehog inhibitor, IPI-926, in combination with chemotherapy[19].
Single-cell RNA sequencing by Elyada, Hosein, and others shows heterogeneity in the CAF population that is present within the stroma of PDAC and confirms the presence of at least three different types of CAFs: myofibroblastic, inflammatory, and antigen-presenting [Figure 1][20-24]. These distinct CAF populations are stimulated through different pathways/cytokines, play different roles in PDAC, and reside in specific areas in the TME relative to cancer cells[20-24].
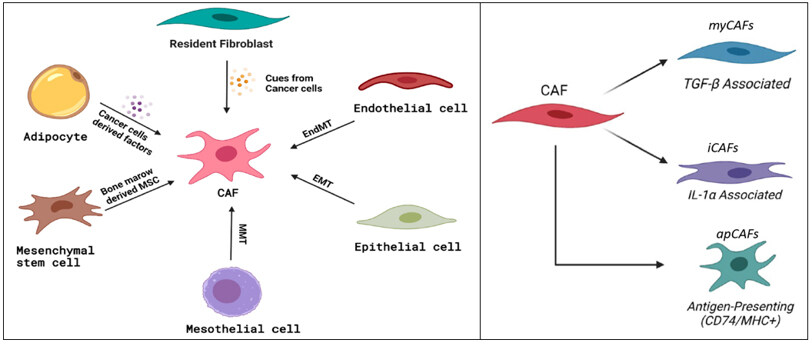
Figure 1. Origins (left) and differentiation (right) of cancer-associated fibroblasts. MSC: Mesenchymal stem cell; EndMt: endothelial-to-mesenchymal transition; EMT: epithelial-to-mesenchymal transition; MMT: mesenchymal-to-mesenchymal transition; CAF: cancer-associated fibroblast; myCAFs: myofibroblastic cancer-associated fibroblasts; iCAFs: inflammatory cancer-associated fibroblasts; apCAFs: antigen-presenting cancer-associated fibroblasts[8]. (Adopted from Herting et al., Cancer Metastasis Rev. 2021; doi:10.1007/s10555-021-09988-w).
Myofibroblastic CAFs
Myofibroblastic CAFs (myCAFs), reside in the periglandular region and have an alpha-smooth muscle actin (α-SMA)high interleukin-6 (IL-6)low phenotype[25]. They are stimulated via the TGF-β/SMAD2/3 pathway when they come in direct contact with cancer cells which explains their localization to the periglandular region[25]. myCAFs have upregulated pathways involved in smooth muscle contraction, focal adhesion, extracellular matrix (ECM) organization, and collagen formation[20]. They express upregulated connective tissue growth factor and collagen type I alpha 1 (Col1α1), which are stimulated by actin alpha 2 and transforming growth factor-β (TGF-β)[25]. Protein activity analysis showed activated proteins in myCAFs include Twist family BHLH transcription factor 1 (TWIST1), in addition to TGF-β1 and SMAD family member 2 (SMAD2), as well as the contractile proteins transgelin, the myosin light chain 9, tropomyosins 1 and 2, the ECM modulators matrix metallopeptidase 11 and periostin, and the homeobox transcription factor HOPX[20]. Lastly, Wnt signaling molecules (WNT2, WNT5A), which are implicated in myocyte differentiation and fibrosis, are overexpressed in myCAFs[20].
Inflammatory CAFs
Inflammatory CAFs (iCAFs), reside near the tumor periphery, express an α-SMAlow IL-6high phenotype, and are stimulated by the IL-1/JAK-STAT3 pathway[20,25,26]. Interestingly, while TGFβ promotes the transformation of fibroblasts to myCAFs, it inhibits their transformation to iCAFs by downregulating interleukin-1 (IL-1) receptor expression[26], and it is hypothesized that myCAFs and iCAFs are interchangeable based on their location and their exposure to cancer cells[26]. Although iCAFs lack an activated TGF-β program, they show a differential activation of TGFβ receptors TGFBR2 and TGFBR3[20]. This expression may indicate a negative feedback loop arising from the absence of TGF-β signaling in iCAFs[20]. iCAFs have enriched expression of inflammatory pathways such as IFNγ response, TNF/NF-κB, IL2/STAT5, IL6/JAK/STAT3, nuclear factor kappa B (NF-κB) pathway, and others that are known to regulate immune response[20]. iCAFs overexpress platelet-derived growth factor receptor alpha (PDGFRA), IL-6, Interleukin 8 (IL-8) and chemokines such as CXCL1, CXCL2, CCL2, and CXCL12[20]. In addition, they exhibit strong expression of complement factor D (CFD) and matrix proteins such as lamin A/C and dermatopontin (DPT)[20]. Additionally, the hypoxia regulator HIF1α and the redox regulators Nrf2 and superoxide dismutase 2 are active in iCAFs, suggesting a potential role for iCAFs in oxidative stress relief[20]. Intriguingly, iCAFs also express the hyaluronan synthases (HAS1, HAS2), the enzymes responsible for the synthesis of hyaluronan, which is a major component of the ECM regarded as a significant barrier to the treatment of PDAC[20]. Efforts targeting hyaluronan in PDAC include the use of pegylated hyaluronidase in combination with chemotherapy[27,28]. Although early results were promising, later-phase trials showed no survival benefit[27,28].
Antigen-presenting CAFS
Like iCAFs, antigen-presenting CAFs (apCAFs), also reside in the periphery of tumor in areas of extensive desmoplasia and are likely regulated by interferon gamma (IFNγ) signaling[20]. apCAFs express genes that belong to the major histocompatibility complex (MHC) class II family, such as histocompatibility 2, class II antigen A, alpha (H2-Aa) and beta 1 (H2-Ab1), that encode the alpha and beta chains of MHC class II, and CD74 that encodes the invariant chain[20]. Additionally, they express unique markers such as Serum Amyloid A3, which is regarded as a pro-tumorigenic factor in pancreatic CAFs, and Secretory Leukocyte Peptidase Inhibitor, which was previously identified as a pro-inflammatory gene in dysplastic skin fibroblasts[20]. Pathways upregulated in apCAFs include those involved in antigen presentation and processing, fatty acid metabolism, MYC and MTORC1 signaling[20]. Proteins upregulated in apCAfs include other regulators of immune activity such as Bcam (CD239) and F11r (CD321), members of the immunoglobulin superfamily, and interferon regulatory factor 5, which is an Interferon-regulating protein[20]. apCAFs exhibit a higher activity for Stat1, which is known to mediate MHCII expression in response to IFNγ[20]. apCAFs have the capacity to present a model antigen to CD4+ T cells, but they lack the costimulatory molecules needed to induce T cell proliferation. Therefore, it is likely that the MHC class II expressed by apCAFs serves as a decoy receptor to deactivate CD4+ T cells by inducing either anergy or differentiation into regulatory T cells (Tregs), and in that, contributes to an immune suppressive TME[20].
MYELOID CELLS
One of the abundant immune cells that play a crucial role in the progression of PDAC is myeloid cells[29]. Of those, myeloid-derived suppressor cells (MDSCs) and tumor-associated macrophages (TAMs) are some of the most studied types[29]. Although with similar phenotypes and surface markers, MDSCs and TAMs are two distinct populations [Figure 2][29]. Sophisticated cross-talk between these cells and tumor cells collectively leads to resistance to chemotherapy, inhibition of effector T cell, and promotion of tumor invasiveness and metastases.
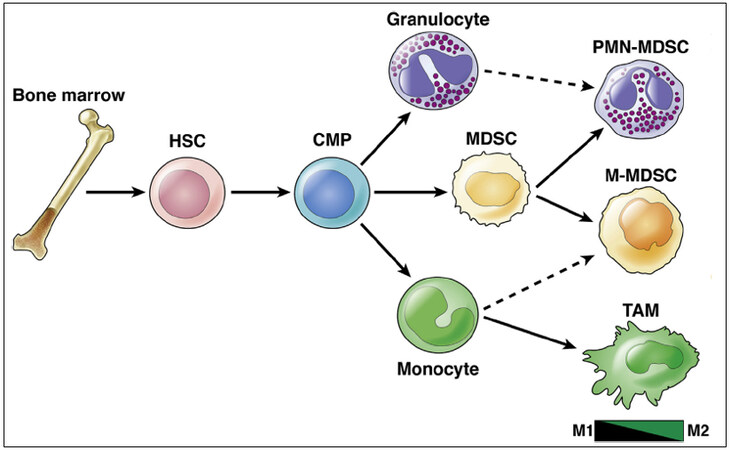
Figure 2. Myeloid cell lineage differentiation and markers. HSC: Hematopoietic stem cells; CMP: common myeloid progenitors; MDSC: myeloid-derived suppressor cell; TAM: tumor-associated macrophage; PMN-MDSC: polymorphonuclear MDSC; M-MDSC: monocytic MDSC[29].
MDSCs
MDSCs are characterized by an immature, immune suppressive phenotype[30]. MDSCs are classified into two subsets: 1- polymorphonuclear-MDSC (PMN-MDSC) that share markers expressed on neutrophils, and 2- monocytic-MDSC (M-MDSC) that share markers expressed on monocytes and macrophages[6,29]. A third subpopulation of MDSCs exists, representing a common progenitor of MDSCs, referred to as early-stage MDSC or eMDSC[29,31,32]. This subpopulation has yet to be functionally evaluated in PDAC[29]. MDSCs in PDAC are bone marrow-derived, and their formation is driven by kras, tumor-derived granulocyte-macrophage colony-stimulating factor (GM-CSF), granulocyte colony-stimulating factor (G-CSF), and macrophage colony-stimulating factor (M-CSF)[30]. Although PMN-MDSCs are more abundant than M-MDSCs, M-MDSCs are more immune suppressive[30,33]. Phenotypic differences between murine and human MDSCs exist[31,32]. For example, murine MDSCs were originally described as CD11b+ and Gr-1+[31,32]. Gr-1 is composed of a subunit from the Ly6 family and further classifies cells into M-MDSCs, which are CD11b+Ly6G-Ly6Chi, or PMN-MDSCs, which are CD11b+Ly6G+Ly6Clo[31,32]. Other markers suggested for more effective identification of murine MDSCs include CD84, CD244, fatty acid transporter protein 2 (FATP2) and CD36[31,32]. On the other hand, human MDSCs are characterized by CD11b+CD14+CD15-HLA-DRlo/- for M-MDSCs and CD11b+CD14-CD15+HLA-DR- for PMN-MDSCs[31,32]. The low expression of HLA-DR distinguishes M-MDSCs from monocytes[31,32]. CD33 can be used instead of CD11b, as it is expressed in high levels in M-MDSCs and dimly in PMN-MDSCs[31,32]. Additionally, CD15 or CD66b can be used as a marker for PMN-MDSCs[31,32].
MDSCs exert their immune suppressive functions through multiple mechanisms[33]. MDSCs release both reactive oxygen species (ROS) and reactive nitrogen species (RNS), which inhibit T cells fitness, proliferation and migration within the TME[33]. MDSCs produce high levels of Arginase 1 and indoleamine 2,3-dioxygenase 1, which metabolize L-arginine and L-tryptophan, respectively, and lead to effector T cell suppression[33]. Additionally, MDSCs induce T cell tolerance through the expression of inhibitory receptors such as the PD-L1 and CTLA-4 receptors, as well as sustain the development of Tregs through the CD40 engagement in the presence of interleukin-10 (IL-10) and TGF-β[34,35]. MDSCs produce HB-EGF, an EGFR ligand, which activates EGFR/MAPK signaling in tumor cells and leads to increased PD-L1 expression[36]. Activated NF-κB promotes the functions of MDSCs through the production of colony-stimulating factors such as GM-CSF, as well as the secretion of chemokines, such as CXCL1, 2, and 5 that recruit CXCR2+ MDSCs to the TME[37,38]. The frequency of tumor-infiltrating T cells correlates inversely with the presence of MDSCs[33]. MDSCs also promote tumor progression through non-immune processes, including tumor angiogenesis through the secretion of vascular endothelial growth factor (VEGF)[34].
Due to their tumor-promoting nature, restraining the expansion or functions of MDSCs is an interesting therapeutic target. Decreased tumor growth and increased effector T cell infiltration were observed in GM-CSF knock-down mouse grafts[39]. Similar results were observed through the depletion of PMN-MDSCs using a Ly6G-targeting antibody in mouse models[40]. In another mouse model, inhibition of CXCR2, which is upregulated in MDSCs and regulates their recruitment to the TME, resulted in reduced metastatic potential, synergy with immunotherapy, and improved survival[41].
TAMs
Macrophages arise from tissue-resident as well as bone marrow-derived macrophages[42]. They are linked to poor survival[43]. Tissue-resident macrophages originate from embryonic precursors or adult hematopoietic stem cell progenitors[43]. Marrow-derived macrophages are recruited into tissues by chemotactic signals[43]. The origin of TAMs has an implication on their function, as monocyte-derived TAMs have increased antigen presentation abilities, while embryonically derived TAMs have profibrotic features[44]. Macrophages are present in early pancreatic intraepithelial neoplasia (PanIN) lesions and are regarded as one of the earliest immune responses[45]. Mutation in kras ubiquitously present in these early lesions drives an epithelial-macrophage interplay that promotes a pro-tumorigenic phenotype in the TAMs[46]. Macrophages in these early PanIN lesions secrete inflammatory factors that facilitate epithelial cell growth[47]. In addition, macrophages can serve as sources of IL-6 and IL-10 that activate STAT3 and fuel tumorigenesis through direct impacts on tumor cells or infiltrating cytotoxic immune cells[47].
TAMs have different polarization states within the TME, and their function can be pro-inflammatory/anti-tumoral (M1 phenotype) or anti-inflammatory/pro-tumoral (M2 phenotype)[29]. M1 TAMs are induced via IFNγ and tumor necrosis factor α (TNFα), and they exhibit a high expression of interleukin 12 (IL-12), TNF, and inducible nitric oxide synthase[29,48]. IFNγ and TNFα are produced by antigen-presenting cells upon recognition of pathogenic signals, but they are also secreted by T-cells and natural killer (NK) cells[29,48]. Upon activation, M1 TAMs secrete pro-inflammatory cytokines (e.g., TNFα, IL1β, IL12), effector molecules (e.g., reactive nitrogen intermediates), and chemokines (e.g., CXCL9, CXCL10) to amplify and sustain their tumoricidal activity[29,48]. M2 TAMs are stimulated by interleukin 4 (IL-4) and interleukin 13 (IL-13)[42]. M2 macrophages lose their antigen presentation abilities and function instead to suppress the immune response through a variety of mechanisms. M1 TAMs reside in the periglandular region close to cancer cells, while M2 TAMs usually reside more peripherally[6]. Interestingly, when M2 TAMs are observed close to cancer cells, they are associated with worse survival[6]. While the M1/M2 classification is a useful and simplified approach to understanding the TAM population, it should be noted that TAMs exist in a fluid and dynamic state, where their polarization is part of the continuous spectrum between M1 and M2 and can change depending on cytokines and interactions with other cells in the TME[48].
Initiatives tailored to TAMs as therapeutic targets have focused on two main concepts, either reprogramming TAMs to be anti-tumor or retrieving their antigen-presenting abilities. The current review summarizes some of these initiatives. The colony-stimulating factor 1 (CSF1) polarizes TAMs toward their immunosuppressive phenotype via colony-stimulating factor 1 receptor[49]. Research on human and mouse samples shows that the TME is infiltrated with CSF1R+ macrophages[49]. Inhibiting CSF1R in KPC mouse models resulted in an increase in effector T cell infiltrations and decreased tumor burden[49]. Additionally, in syngeneic orthotopic mouse models using KC and KI cells, CSF1R blockade sensitizes tumors to immune checkpoint blockade with PD-1 or CTLA-4 antagonists[50]. Cabiralizumab, a CSF1R blocking antibody, is the subject of an ongoing phase 2 clinical trial evaluating its use in combination with nivolumab and chemotherapy in patients with advanced PDAC[51].
The CCL2/CCR2 axis is another pathway that is integral to the genesis of TAMs[43]. Human PDAC cells secrete CCL2 that recruits CCR2+ monocytes from the bone marrow to the circulation, which then differentiate into TAMs after entering the tumor tissue[43]. In orthotopic mouse models of PDAC (C57BL/6 and CCR2-/- mice), blockade of CCR2 results in retention of monocytes in the bone marrow and impaired tumor growth[43]. Synergy was observed with the addition of gemcitabine[43]. In an early phase clinical trial, the combination of chemotherapy with a CCR2 inhibitor (PF-04136309) was associated with a reduction in circulating CCR2+ monocytes and TAMs in the tumor and an increase in CD8+ T cells in patients with PDAC[52]. However, there was a lack of clinical benefit in further trials[53].
CD40, a member of the TNF receptor superfamily, is expressed broadly on immune cells, including monocytes and macrophages[54]. In KPC mouse models, treatment with the CD40 agonist FGK45 resulted in upregulation of MHC II in macrophages from the tumor and spleen, suggesting that CD40 activation reprograms TAMs to their anti-tumor phenotype[54]. Combining CD40 agonism with chemotherapy resulted in increased T cell infiltration, decreased tumor burden, and increased susceptibility to immune checkpoint therapy[54-56]. In a phase 1 trial with treatment-naïve metastatic PDAC patients, the combination of the CD40 agonist APX005M (sotigalimab) with chemotherapy was associated with a promising response rate of 58%[57].
In addition to the many functions in PDAC, myeloid cells play a role in establishing a pre-metastatic niche and promoting metastatic disease[58]. This is carried out either by 1- the physical translocation of myeloid cells to organs outside of the pancreas and direct cross-talks with host organs or by 2- tumor-derived exosomes which can carry out these changes more distally[29]. A body of evidence now shows that MDSCs colonize distal organs prior to tumor cells[58]. Granulin secreted from macrophages recruited to the liver activates myCAFs and creates a permissible environment for tumor growth and survival[59]. Lastly, upregulated STAT3 in hepatocytes recruits myeloid cells to the liver by secreted serum amyloid A1 and A2, leading to liver fibrosis[58]. This results in increased susceptibility of the liver to tumor seeding and growth[58]. Ablation of the IL6/STAT3/serum amyloid A pathway resulted in fewer recruited MDSCs which translated to the prevention of metastatic dissemination[58].
LYMPHOCYTES
T cells
The T cell population in PDAC TME encompasses different subpopulations with distinguished spatial distributions and roles that can be immune stimulating or suppressive, the former of which is largely restrained by tumor cells and the other constituents of the TME [Figure 3]. Cytotoxic or effector CD8+ T cells represent a small subset and are confined to the tumor periphery[60]. A high cytotoxic T cell infiltration of tumors is associated with improved survival[60]. A growing body of literature shows that cytotoxic T cells strongly express immune checkpoint markers, such as PD-1, VISTA, and TIGIT, and are exposed to their ligands expressed on immune suppressive stromal cells, which make them susceptible to inhibition by different mechanisms[61-63]. Additionally, cytotoxic T cells are the subject of immunosuppressive cytokines produced by tumor and stromal cells, such as IL-6, TGFβ1, CXCL12, and IL-10[61-63]. Cytotoxic T cell function is further inhibited by the relative scarcity of antigen-presenting cells within the TME and insufficient tumor antigenicity[60,64]. Lastly, the dense stroma surrounding tumors acts as a physical barrier for the recruitment of cytotoxic T cells[16,65]. An abundant component of the extracellular matrix is fibrillar collagens, such as type I and III collagen[16,65]. The major producer of fibrillar collagens are CAFs, and tumor cells produce a small amount of CAFs[66].
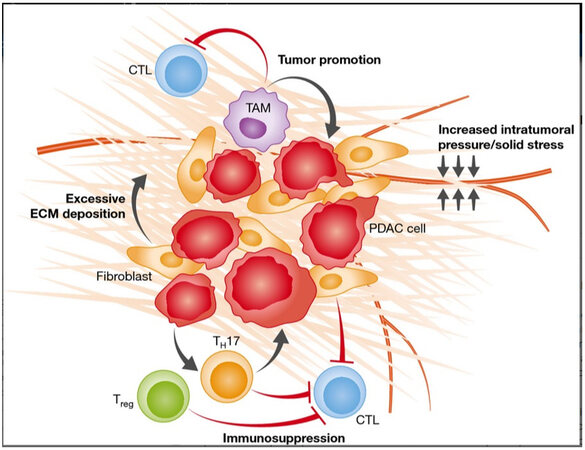
Figure 3. T lymphocytes within the TME of PDAC. Regulatory T cell (Tregs) and CD4+ helper 17 (Th17) cells. Along with tumor-associated macrophages, they inhibit cytotoxic T lymphocytes. Additionally, the dense stroma confers elevated intratumoral pressures resulting in vascular compression, which reduces the recruitment of cytotoxic T lymphocytes to the TME. CTL: cytotoxic T lymphocyte; TAM: tumor-associate macrophage; ECM: extracellular matrix; PDAC: pancreatic adenocarcinoma; Treg: regulatory T lymphocyte. TH17, CD4+ helper 17 lymphocyte. (Adopted from Carr et al., EMBO Mol Med. 2016;8:80-2. doi:10.15252/emmm.201505948).
Initiatives to enhance the activity of cytotoxic T cells are ongoing[67]. This includes a phase I/II trial that is being conducted by our group (NCT04191421)[67]. In this trial, patients with metastatic, treatment-refractory disease receive spartalizumab, a checkpoint inhibitor of PD-1, and siltuximab, which inhibits IL-6, in an attempt to sensitize these tumors to PD-1 blockade[67].
Studies on human as well as mouse models (KPC and others) showed that blockade of the CXCR4/CXCL12 pathway resulted in increased T cell infiltration and synergized with immune checkpoint inhibitors[68-70]. Combining the CXCR4 blocker BL-8040 (motixafortide) with chemo- and immunotherapy showed a promising signal in patients with metastatic PDAC in the phase II trial COMBAT[68].
CD4+ T cells include Tregs and CD4+ helper 17 (Th17), both of which exert tumor-promoting functions[71]. Tregs express a CD3+CD4+CD25+FoxP3+ phenotype and are associated with poor prognosis[72,73]. Tregs communicate intimately with MDSCs primarily through cell-cell interactions to promote their survival and promote an immunosuppressive TME[74]. Interestingly, depletion of Tregs in KC;Foxp3DTR and KC;CD4-/- mice resulted in an increase of MDSCs within tumors and pronounced tumor growth[74]. These results highlight the effects of one cell population on the rest of the whole population.
Th17 cells produce IL-17, a crucial cytokine in the development and progression of PDAC[75,76]. Oncogenic Kras drives the expression of IL-17 receptor on PanIN lesions, and IL-17 from lymphocytes promotes tumor progression and induces cancer stemness through pathways such as IL-6/STAT3 and NF-κB[75,76]. Recent efforts showed that CD11b+CD103- dendritic cells produce IL-23 and TGF-β and induce CD4+FOXP3-IL10+IL-17+IFNγ+ T cells[77]. Identifying additional interactions these cells have with other constituents of the TME can be exploited as therapeutic targets.
B cells
Contrary to other immune cells in the TME of PDAC, B lymphocytes are some of the least studied cell populations and their role in the development of PDAC remains controversial[78,79]. Research shows the presence of B cells in the early stages of PanIN lesions[78]. Recent work shows that B cells can be found in two main locations in PDAC[80]: 1- scattered infiltrating the tumor-stromal surface and 2- organized in ectopic lymph-node like structures called tertiary lymphoid tissue (TLT)[80]. After affinity maturation of CD20+ naïve B cells in the germinal centers of TLT, circulating plasmablasts are produced[81]. Those plasmablasts have been shown to be the most potent subset of B cells to stimulate collagen production by CAFs, and they show high expression of genes involved in fibroblast activation and proliferation, including the collagen genes COL1A1 and COL1A2, insulin-like growth factor-1 (IGF-1), and lysyl oxidase homolog 2 (LOXL2)[81]. A high density of B cells in TLT is associated with improved survival[80]. Interestingly, in a mouse model devoid of TLT, depletion of B cells using an anti-CD20 antibody restored anti-tumor immunity[80]. Therefore, the prognostic relevance and anti-tumor potential of B cells rely on their spatial organization within PDAC[80]. The spatial localization of T and B lymphocytes within TLT is crucial for the recruitment of tumor-infiltrating T cells[80]. B cells appear to be regulated by follicular helper T cells[80]. Interestingly, depletion of B cells may prevent the formation of TLT, which might be detrimental in PDAC as the tumors are deprived of effector cell localization and anti-tumor immune response[78].
A subset of regulatory B cells, also referred to as B1 regs, produce IL35, which promotes tumor progression and cytotoxic T cell evasion[78]. Bregs express high levels of PD-L1 which interacts with PD-1 on cytotoxic T cells, rendering them exhausted[82,83]. Through the activity of Bruton tyrosine kinase (BTK), B cells interact with TAMs and reprogram them to the pro-tumoral M2 phenotype[84]. In studies with Ink4 2.2 and p53 2.1.1 mouse cell lines and male FVB/n mice, inhibition of BTK with ibrutinib resulted in the reprogramming of TAMs to the anti-tumoral M1 phenotype, increased cytotoxic T cell infiltration, and decreased tumor growth[84]. This provided the basis of the RESOLVE trial, in which treatment-naïve metastatic PDAC patients were randomized to ibrutinib or placebo, combined with chemotherapy[85]. Unfortunately, the combination failed to show a survival benefit[85]. This underscores that there are likely more intricate cross-talks between tumor and stromal cells and that targeting one signaling pathway is not sufficient[86]. Additionally, caution must be exercised when deducing results from preclinical models, as they might not accurately mimic human cancer[86]. One example is that orthotopic models lack TLT[80].
As mentioned above, CD40, a member of the tumor necrosis factor receptor superfamily, activates antigen-presenting cells, such as dendritic cells and B cells[57,87]. In studies on Panc02 mouse pancreatic cancer cell lines and C57BL/6 (H-2b) mice, treatment with CD40 agonistic antibody resulted in T-cell-dependent tumor regression and improved survival[46]. Synergy was observed with PD-1 blockade on similar models[88]. A phase 1 trial evaluated the safety of the CD40 agonist APX005M (sotigalimab) in combination with chemotherapy, with or without nivolumab in treatment-naïve metastatic PDAC patients[57]. The combination was safe and associated with a promising efficacy signal[57]. Other CD40 agonists are currently being tested in earlier stages of PDAC[89].
Dendritic cells and other populations
Over the last decade, dendritic cells (DCs) have gained popularity as a subject of interest in the field of PDAC, especially in the development of cancer vaccines[90]. DCs represent a diverse group of antigen-presenting cells that are able to recognize tumor antigens and generate tumor-specific immunity[91]. The presence of DCs is associated with longer survival in PDAC, both in the metastatic and early stages[92,93]. They are also associated with a greater infiltration of cytotoxic T cells[94]. However, DCs are a rare population in PDAC, have an attenuated function, and are localized to the periphery of the tumor, all of which inhibit their anti-tumor effects[95]. Several trials are capitalizing on DCs as a therapeutic target, including the use of CD40 agonisms mentioned above[96-98].
Other components of innate and adaptive immunity, including natural killer cells, γδ T cells, and other innate lymphoid cells (ILCs), have recently gained attention; however, their roles continue to be incompletely understood. A key component of innate immunity, natural killer cells (NK cells) in PDAC also have an attenuated anti-tumor function, tend to reside in the periphery of the tumor, and are associated with improved survival[99,100]. γδ T cells are a more abundant type of lymphocyte with pro-tumoral activity and their presence is associated with poor prognosis[101,102]. They reside within the tumor and suppress the anti-tumoral activities of αβ T cells[101,102]. Lastly, the innate lymphoid cells (ILCs) groups 1, 2, and 3 represent a heterogeneous group of tissue-resident cells that play conflicting roles in solids tumors[103]. In PDAC, ILC2s were shown to amplify PD-1 blockade by activating tissue-specific cancer immunity through IL-33-dependent mechanisms[104]. Targeting these subsets for the treatment of PDAC is still in its infancy. Table 1 summarizes the efforts aimed at targeting the immune populations of the TME.
CONCLUSION
Previously regarded as an “immune desert”, we now know that the TME of PDAC is enriched with immune cells that play both pro- and anti-tumoral roles and interact very intimately with tumor cells as well as the other cellular populations in the TME. Many of the pro-tumoral immune subsets are present in the very early stages of cancer development and govern the progression and metastases of PDAC through mechanisms that include producing a dense stroma, creating a niche in metastatic sites, and maintaining an immune suppressive environment that inhibits the activities of anti-tumoral populations. However, recent advances in single-cell analysis highlight several anti-tumoral subsets that are not only associated with improved survival but can also be used as therapeutic targets. Ongoing trials are studying novel dendritic cell vaccines, as well as different combinations of immune modulators with immune checkpoint inhibitors in an attempt to render “cold” tumors into “hot” tumors. Additionally, more research is urgently needed to better identify the potential role of rarer subsets, such as NK cells, as therapeutic targets.
DECLARATIONS
Authors’ contributionsContributed significantly to the generation of this manuscript: Diab M, El-Rayes BF
Availability of data and materialsNot applicable.
Financial support and sponsorshipNone.
Conflicts of interestBoth authors declared that there are no conflicts of interest.
Ethical approval and consent to participateNot applicable.
Consent for publicationNot applicable.
Copyright© The Author(s) 2022.
REFERENCES
1. Rahib L, Smith BD, Aizenberg R, Rosenzweig AB, Fleshman JM, Matrisian LM. Projecting cancer incidence and deaths to 2030: the unexpected burden of thyroid, liver, and pancreas cancers in the United States. Cancer Res 2014;74:2913-21.
3. Al-Share B, Hammad N, Diab M. Pancreatic adenocarcinoma: molecular drivers and the role of targeted therapy. Cancer Metastasis Rev 2021;40:355-71.
4. Conroy T, Desseigne F, Ychou M, et al. FOLFIRINOX versus gemcitabine for metastatic pancreatic cancer. N Engl J Med 2011;364:1817-25.
5. Singh RR, O’Reilly EM. New treatment strategies for metastatic pancreatic ductal adenocarcinoma. Drugs 2020;80:647-69.
6. Väyrynen SA, Zhang J, Yuan C, et al. Composition, spatial characteristics, and prognostic significance of myeloid cell infiltration in pancreatic cancer. Clin Cancer Res 2021;27:1069-81.
8. Vaish U, Jain T, Are AC, Dudeja V. Cancer-associated fibroblasts in pancreatic ductal adenocarcinoma: an update on heterogeneity and therapeutic targeting. Int J Mol Sci 2021;22:13408.
9. Gopurappilly R, Bhat V, Bhonde R. Pancreatic tissue resident mesenchymal stromal cell (MSC)-like cells as a source of in vitro islet neogenesis. J Cell Biochem 2013;114:2240-7.
10. Quante M, Tu SP, Tomita H, et al. Bone marrow-derived myofibroblasts contribute to the mesenchymal stem cell niche and promote tumor growth. Cancer Cell 2011;19:257-72.
11. Mathew E, Brannon AL, Del Vecchio A, et al. Mesenchymal stem cells promote pancreatic tumor growth by inducing alternative polarization of macrophages. Neoplasia 2016;18:142-51.
12. Zeisberg EM, Potenta S, Xie L, Zeisberg M, Kalluri R. Discovery of endothelial to mesenchymal transition as a source for carcinoma-associated fibroblasts. Cancer Res 2007;67:10123-8.
13. Iwano M, Plieth D, Danoff TM, Xue C, Okada H, Neilson EG. Evidence that fibroblasts derive from epithelium during tissue fibrosis. J Clin Invest 2002;110:341-50.
14. Özdemir BC, Pentcheva-Hoang T, Carstens JL, et al. Depletion of carcinoma-associated fibroblasts and fibrosis induces immunosuppression and accelerates pancreas cancer with reduced survival. Cancer Cell 2014;25:719-34.
15. Chen Y, Kim J, Yang S, et al. Type I collagen deletion in αSMA+ myofibroblasts augments immune suppression and accelerates progression of pancreatic cancer. Cancer Cell 2021;39:548-565.e6.
16. Bhattacharjee S, Hamberger F, Ravichandra A, et al. Tumor restriction by type I collagen opposes tumor-promoting effects of cancer-associated fibroblasts. J Clin Invest 2021;131:146987.
17. Lee JJ, Perera RM, Wang H, et al. Stromal response to Hedgehog signaling restrains pancreatic cancer progression. Proc Natl Acad Sci USA 2014;111:E3091-100.
18. Walter K, Omura N, Hong SM, et al. Overexpression of smoothened activates the sonic hedgehog signaling pathway in pancreatic cancer-associated fibroblasts. Clin Cancer Res 2010;16:1781-9.
19. Catenacci DV, Junttila MR, Karrison T, et al. Randomized phase Ib/II study of gemcitabine plus placebo or vismodegib, a hedgehog pathway inhibitor, in patients with metastatic pancreatic cancer. J Clin Oncol 2015;33:4284-92.
20. Elyada E, Bolisetty M, Laise P, et al. Cross-species single-cell analysis of pancreatic ductal adenocarcinoma reveals antigen-presenting cancer-associated fibroblasts. Cancer Discov 2019;9:1102-23.
21. Baron M, Veres A, Wolock SL, et al. A single-cell transcriptomic map of the human and mouse pancreas reveals inter- and intra-cell population structure. Cell Syst 2016;3:346-360.e4.
22. Bernard V, Semaan A, Huang J, et al. Single-cell transcriptomics of pancreatic cancer precursors demonstrates epithelial and microenvironmental heterogeneity as an early event in neoplastic progression. Clin Cancer Res 2019;25:2194-205.
23. Dominguez CX, Müller S, Keerthivasan S, et al. Single-cell RNA sequencing reveals stromal evolution into LRRC15+ myofibroblasts as a determinant of patient response to cancer immunotherapy. Cancer Discov 2020;10:232-53.
24. Hosein AN, Huang H, Wang Z, et al. Cellular heterogeneity during mouse pancreatic ductal adenocarcinoma progression at single-cell resolution. JCI Insight 2019;5:129212.
25. Öhlund D, Handly-Santana A, Biffi G, et al. Distinct populations of inflammatory fibroblasts and myofibroblasts in pancreatic cancer. J Exp Med 2017;214:579-96.
26. Biffi G, Oni TE, Spielman B, et al. IL1-induced JAK/STAT signaling is antagonized by TGFβ to shape CAF heterogeneity in pancreatic ductal adenocarcinoma. Cancer Discov 2019;9:282-301.
27. Van Cutsem E, Tempero MA, Sigal D, et al. Randomized phase III trial of pegvorhyaluronidase alfa with nab-paclitaxel plus gemcitabine for patients with hyaluronan-high metastatic pancreatic adenocarcinoma. J Clin Oncol 2020;38:3185-94.
28. Ramanathan RK, McDonough SL, Philip PA, et al. Phase IB/II randomized study of FOLFIRINOX plus pegylated recombinant human hyaluronidase versus FOLFIRINOX alone in patients with metastatic pancreatic adenocarcinoma: SWOG S1313. J Clin Oncol 2019;37:1062-9.
29. Kemp SB, Pasca di Magliano M, Crawford HC. Myeloid cell mediated immune suppression in pancreatic cancer. Cell Mol Gastroenterol Hepatol 2021;12:1531-42.
30. Sharma V, Aggarwal A, Jacob J, Sahni D. Myeloid-derived suppressor cells: bridging the gap between inflammation and pancreatic adenocarcinoma. Scand J Immunol 2021;93:e13021.
31. Bronte V, Brandau S, Chen SH, et al. Recommendations for myeloid-derived suppressor cell nomenclature and characterization standards. Nat Commun 2016;7:12150.
32. Vanhaver C, van der Bruggen P, Bruger AM. MDSC in mice and men: mechanisms of immunosuppression in cancer. J Clin Med 2021;10:2872.
33. Trovato R, Fiore A, Sartori S, et al. Immunosuppression by monocytic myeloid-derived suppressor cells in patients with pancreatic ductal carcinoma is orchestrated by STAT3. J Immunother Cancer 2019;7:255.
34. Ugel S, De Sanctis F, Mandruzzato S, Bronte V. Tumor-induced myeloid deviation: when myeloid-derived suppressor cells meet tumor-associated macrophages. J Clin Invest 2015;125:3365-76.
35. Huang B, Pan PY, Li Q, et al. Gr-1+CD115+ immature myeloid suppressor cells mediate the development of tumor-induced T regulatory cells and T-cell anergy in tumor-bearing host. Cancer Res 2006;66:1123-31.
36. Zhang Y, Velez-Delgado A, Mathew E, et al. Myeloid cells are required for PD-1/PD-L1 checkpoint activation and the establishment of an immunosuppressive environment in pancreatic cancer. Gut 2017;66:124-36.
37. Chao T, Furth EE, Vonderheide RH. CXCR2-dependent accumulation of tumor-associated neutrophils regulates T-cell immunity in pancreatic ductal adenocarcinoma. Cancer Immunol Res 2016;4:968-82.
38. Bayne LJ, Beatty GL, Jhala N, et al. Tumor-derived granulocyte-macrophage colony-stimulating factor regulates myeloid inflammation and T cell immunity in pancreatic cancer. Cancer Cell 2012;21:822-35.
39. Pylayeva-Gupta Y, Lee KE, Hajdu CH, Miller G, Bar-Sagi D. Oncogenic kras-induced GM-CSF production promotes the development of pancreatic neoplasia. Cancer Cell 2012;21:836-47.
40. Stromnes IM, Brockenbrough JS, Izeradjene K, et al. Targeted depletion of an MDSC subset unmasks pancreatic ductal adenocarcinoma to adaptive immunity. Gut 2014;63:1769-81.
41. Steele CW, Karim SA, Leach JDG, et al. CXCR2 inhibition profoundly suppresses metastases and augments immunotherapy in pancreatic ductal adenocarcinoma. Cancer Cell 2016;29:832-45.
42. Yang S, Liu Q, Liao Q. Tumor-associated macrophages in pancreatic ductal adenocarcinoma: origin, polarization, function, and reprogramming. Front Cell Dev Biol 2020;8:607209.
43. Sanford DE, Belt BA, Panni RZ, et al. Inflammatory monocyte mobilization decreases patient survival in pancreatic cancer: a role for targeting the CCL2/CCR2 axis. Clin Cancer Res 2013;19:3404-15.
44. Zhu Y, Herndon JM, Sojka DK, et al. Tissue-resident macrophages in pancreatic ductal adenocarcinoma originate from embryonic hematopoiesis and promote tumor progression. Immunity 2017;47:323-338.e6.
45. Clark CE, Hingorani SR, Mick R, Combs C, Tuveson DA, Vonderheide RH. Dynamics of the immune reaction to pancreatic cancer from inception to invasion. Cancer Res 2007;67:9518-27.
46. Ziske C, Etzrodt PE, Eliu AS, et al. Increase of in vivo antitumoral activity by CD40L (CD154) gene transfer into pancreatic tumor cell-dendritic cell hybrids. Pancreas 2009;38:758-65.
47. Liu Q, Li Y, Niu Z, et al. Atorvastatin (Lipitor) attenuates the effects of aspirin on pancreatic cancerogenesis and the chemotherapeutic efficacy of gemcitabine on pancreatic cancer by promoting M2 polarized tumor associated macrophages. J Exp Clin Cancer Res 2016;35:33.
48. Poh AR, Ernst M. Tumor-associated macrophages in pancreatic ductal adenocarcinoma: therapeutic opportunities and clinical challenges. Cancers 2021;13:2860.
49. Candido JB, Morton JP, Bailey P, et al. CSF1R+ macrophages sustain pancreatic tumor growth through T Cell suppression and maintenance of key gene programs that define the squamous subtype. Cell Rep 2018;23:1448-60.
50. Zhu Y, Knolhoff BL, Meyer MA, et al. CSF1/CSF1R blockade reprograms tumor-infiltrating macrophages and improves response to T-cell checkpoint immunotherapy in pancreatic cancer models. Cancer Res 2014;74:5057-69.
51. Wang-gillam A, O’reilly EM, Bendell JC, et al. A randomized phase II study of cabiralizumab (cabira) + nivolumab (nivo) ± chemotherapy (chemo) in advanced pancreatic ductal adenocarcinoma (PDAC). JCO 2019;37:TPS465.
52. Nywening TM, Wang-gillam A, Sanford DE, et al. Targeting tumour-associated macrophages with CCR2 inhibition in combination with FOLFIRINOX in patients with borderline resectable and locally advanced pancreatic cancer: a single-centre, open-label, dose-finding, non-randomised, phase 1b trial. Lancet Oncol 2016;17:651-62.
53. Noel M, O’Reilly EM, Wolpin BM, et al. Phase 1b study of a small molecule antagonist of human chemokine (C-C motif) receptor 2 (PF-04136309) in combination with nab-paclitaxel/gemcitabine in first-line treatment of metastatic pancreatic ductal adenocarcinoma. Invest New Drugs 2020;38:800-11.
54. Beatty GL, Chiorean EG, Fishman MP, et al. CD40 agonists alter tumor stroma and show efficacy against pancreatic carcinoma in mice and humans. Science 2011;331:1612-6.
55. Beatty GL, Winograd R, Evans RA, et al. Exclusion of T cells from pancreatic carcinomas in mice is regulated by Ly6C(low) F4/80(+) extratumoral macrophages. Gastroenterology 2015;149:201-10.
56. Winograd R, Byrne KT, Evans RA, et al. Induction of T-cell immunity overcomes complete resistance to PD-1 and CTLA-4 blockade and improves survival in pancreatic carcinoma. Cancer Immunol Res 2015;3:399-411.
57. O’hara MH, O’reilly EM, Varadhachary G, et al. CD40 agonistic monoclonal antibody APX005M (sotigalimab) and chemotherapy, with or without nivolumab, for the treatment of metastatic pancreatic adenocarcinoma: an open-label, multicentre, phase 1b study. Lancet Oncol 2021;22:118-31.
58. Lee JW, Stone ML, Porrett PM, et al. Hepatocytes direct the formation of a pro-metastatic niche in the liver. Nature 2019;567:249-52.
59. Nielsen SR, Quaranta V, Linford A, et al. Macrophage-secreted granulin supports pancreatic cancer metastasis by inducing liver fibrosis. Nat Cell Biol 2016;18:549-60.
60. Lohneis P, Sinn M, Bischoff S, et al. Cytotoxic tumour-infiltrating T lymphocytes influence outcome in resected pancreatic ductal adenocarcinoma. Eur J Cancer 2017;83:290-301.
61. Steele NG, Carpenter ES, Kemp SB, et al. Multimodal mapping of the tumor and peripheral blood immune landscape in human pancreatic cancer. Nat Cancer 2020;1:1097-112.
62. Topcu KSB, Korucu EN, Menevse E, Kocak N, Duran T. Investigation of the effects of the toll-like receptor 4 pathway on immune checkpoint vista in pancreatic cancer. Invest New Drugs 2022;40:519-28.
63. Hou Z, Pan Y, Fei Q, et al. Prognostic significance and therapeutic potential of the immune checkpoint VISTA in pancreatic cancer. J Cancer Res Clin Oncol 2021;147:517-31.
64. Frey N, Tortola L, Egli D, et al. Loss of Rnf31 and Vps4b sensitizes pancreatic cancer to T cell-mediated killing. Nat Commun 2022;13:1804.
65. Tian C, Huang Y, Clauser KR, et al. Suppression of pancreatic ductal adenocarcinoma growth and metastasis by fibrillar collagens produced selectively by tumor cells. Nat Commun 2021;12:2328.
66. Sahai E, Astsaturov I, Cukierman E, et al. A framework for advancing our understanding of cancer-associated fibroblasts. Nat Rev Cancer 2020;20:174-86.
67. Akce M, Shaib WL, Diab M, et al. Phase Ib/II trial of siltuximab and spartalizumab in patients in metastatic pancreatic cancer. JCO 2022;40:TPS626-TPS626.
68. Bockorny B, Semenisty V, Macarulla T, et al. BL-8040, a CXCR4 antagonist, in combination with pembrolizumab and chemotherapy for pancreatic cancer: the COMBAT trial. Nat Med 2020;26:878-85.
69. Seo YD, Jiang X, Sullivan KM, et al. Mobilization of CD8+ T cells via CXCR4 blockade facilitates PD-1 checkpoint therapy in human pancreatic cancer. Clin Cancer Res 2019;25:3934-45.
70. Feig C, Jones JO, Kraman M, et al. Targeting CXCL12 from FAP-expressing carcinoma-associated fibroblasts synergizes with anti-PD-L1 immunotherapy in pancreatic cancer. Proc Natl Acad Sci USA 2013;110:20212-7.
71. Goulart MR, Stasinos K, Fincham REA, Delvecchio FR, Kocher HM. T cells in pancreatic cancer stroma. World J Gastroenterol 2021;27:7956-68.
72. Diana A, Wang LM, D’Costa Z, et al. Prognostic value, localization and correlation of PD-1/PD-L1, CD8 and FOXP3 with the desmoplastic stroma in pancreatic ductal adenocarcinoma. Oncotarget 2016;7:40992-1004.
73. Tang Y, Xu X, Guo S, et al. An increased abundance of tumor-infiltrating regulatory T cells is correlated with the progression and prognosis of pancreatic ductal adenocarcinoma. PLoS One 2014;9:e91551.
74. Zhang Y, Lazarus J, Steele NG, et al. Regulatory T-cell depletion alters the tumor microenvironment and accelerates pancreatic carcinogenesis. Cancer Discov 2020;10:422-39.
75. McAllister F, Bailey JM, Alsina J, et al. Oncogenic Kras activates a hematopoietic-to-epithelial IL-17 signaling axis in preinvasive pancreatic neoplasia. Cancer Cell 2014;25:621-37.
76. Zhang Y, Zoltan M, Riquelme E, et al. Immune cell production of interleukin 17 Induces stem cell features of pancreatic intraepithelial neoplasia cells. Gastroenterology 2018;155:210-223.e3.
77. Barilla RM, Diskin B, Caso RC, et al. Specialized dendritic cells induce tumor-promoting IL-10+IL-17+ FoxP3neg regulatory CD4+ T cells in pancreatic carcinoma. Nat Commun 2019;10:1424.
78. Pylayeva-Gupta Y, Das S, Handler JS, et al. IL35-producing B cells promote the development of pancreatic neoplasia. Cancer Discov 2016;6:247-55.
79. Roghanian A, Fraser C, Kleyman M, Chen J. B Cells promote pancreatic tumorigenesis. Cancer Discov 2016;6:230-2.
80. Castino GF, Cortese N, Capretti G, et al. Spatial distribution of B cells predicts prognosis in human pancreatic adenocarcinoma. Oncoimmunology 2016;5:e1085147.
81. Minici C, Rigamonti E, Lanzillotta M, et al. B lymphocytes contribute to stromal reaction in pancreatic ductal adenocarcinoma. Oncoimmunology 2020;9:1794359.
82. Tong DN, Guan J, Sun JH, et al. Characterization of B cell-mediated PD-1/PD-L1 interaction in pancreatic cancer patients. Clin Exp Pharmacol Physiol 2020;47:1342-9.
83. Khan AR, Hams E, Floudas A, Sparwasser T, Weaver CT, Fallon PG. PD-L1hi B cells are critical regulators of humoral immunity. Nat Commun 2015;6:5997.
84. Gunderson AJ, Kaneda MM, Tsujikawa T, et al. Bruton tyrosine kinase-dependent immune cell cross-talk drives pancreas cancer. Cancer Discov 2016;6:270-85.
85. Tempero M, Oh DY, Tabernero J, et al. Ibrutinib in combination with nab-paclitaxel and gemcitabine for first-line treatment of patients with metastatic pancreatic adenocarcinoma: phase III RESOLVE study. Ann Oncol 2021;32:600-8.
86. Philip PA. Targeting B cells in pancreatic adenocarcinoma: does RESOLVE resolve the question? Ann Oncol 2021;32:582-3.
87. Byrne KT, Vonderheide RH. CD40 stimulation obviates innate sensors and drives T cell immunity in cancer. Cell Rep 2016;15:2719-32.
88. Ma HS, Poudel B, Torres ER, et al. A CD40 agonist and PD-1 antagonist antibody reprogram the microenvironment of nonimmunogenic tumors to allow T-cell-mediated anticancer activity. Cancer Immunol Res 2019;7:428-42.
89. Byrne KT, Betts CB, Mick R, et al. Neoadjuvant selicrelumab, an agonist CD40 antibody, induces changes in the tumor microenvironment in patients with resectable pancreatic cancer. Clin Cancer Res 2021;27:4574-86.
90. Yang J. Dendritic cells in pancreatic cancer immunotherapy: vaccines and combination immunotherapies. Pathol Res Pract 2019;215:152691.
91. Yang J, Shangguan J, Eresen A, Li Y, Wang J, Zhang Z. Dendritic cells in pancreatic cancer immunotherapy: vaccines and combination immunotherapies. Pathol Res Pract 2019;215:152691.
92. Hirooka S. The role of circulating dendritic cells in patients with unresectable pancreatic cancer. Anticancer Res 2011;31:3827-34.
93. Yamamoto T, Yanagimoto H, Satoi S, et al. Circulating myeloid dendritic cells as prognostic factors in patients with pancreatic cancer who have undergone surgical resection. J Surg Res 2012;173:299-308.
94. Fukunaga A, Miyamoto M, Cho Y, et al. CD8+ tumor-infiltrating lymphocytes together with CD4+ tumor-infiltrating lymphocytes and dendritic cells improve the prognosis of patients with pancreatic adenocarcinoma. Pancreas 2004;28:e26-31.
95. Dallal RM, Christakos P, Lee K, Egawa S, Son YI, Lotze MT. Paucity of dendritic cells in pancreatic cancer. Surgery 2002;131:135-8.
96. Baylor college of medicine, N. Th-1 Dendritic cell immunotherapy plus standard chemotherapy for pancreatic adenocarcinoma (DECIST). Available from: https://clinicaltrials.gov/ct2/show/NCT04157127 [Last accessed on 27 September 2022].
97. Katsuda M, Miyazawa M, Ojima T, et al. A double-blind randomized comparative clinical trial to evaluate the safety and efficacy of dendritic cell vaccine loaded with WT1 peptides (TLP0-001) in combination with S-1 in patients with advanced pancreatic cancer refractory to standard chemotherapy. Trials 2019;20:242.
98. Nagai K, Adachi T, Harada H, Eguchi S, Sugiyama H, Miyazaki Y. Dendritic cell-based immunotherapy pulsed with wilms tumor 1 peptide and mucin 1 as an adjuvant therapy for pancreatic ductal adenocarcinoma after curative resection: a phase I/IIa clinical trial. Anticancer Res 2020;40:5765-76.
99. Hoshikawa M, Aoki T, Matsushita H, et al. NK cell and IFN signatures are positive prognostic biomarkers for resectable pancreatic cancer. Biochem Biophys Res Commun 2018;495:2058-65.
100. Lim SA, Kim J, Jeon S, et al. Defective localization with impaired tumor cytotoxicity contributes to the immune escape of NK cells in pancreatic cancer patients. Front Immunol 2019;10:496.
101. Dickson I. Pancreatic cancer: γδ T cells support PDAC. Nat Rev Gastroenterol Hepatol 2016;13:560.
102. Seifert AM, List J, Heiduk M, et al. Gamma-delta T cells stimulate IL-6 production by pancreatic stellate cells in pancreatic ductal adenocarcinoma. J Cancer Res Clin Oncol 2020;146:3233-40.
103. Chiossone L, Dumas PY, Vienne M, Vivier E. Natural killer cells and other innate lymphoid cells in cancer. Nat Rev Immunol 2018;18:671-88.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Diab M, El-Rayes BF. The heterogeneity of CAFs and immune cell populations in the tumor microenvironment of pancreatic adenocarcinoma. J Cancer Metastasis Treat 2022;8:42. http://dx.doi.org/10.20517/2394-4722.2022.60
AMA Style
Diab M, El-Rayes BF. The heterogeneity of CAFs and immune cell populations in the tumor microenvironment of pancreatic adenocarcinoma. Journal of Cancer Metastasis and Treatment. 2022; 8: 42. http://dx.doi.org/10.20517/2394-4722.2022.60
Chicago/Turabian Style
Diab, Maria, Bassel F. El-Rayes. 2022. "The heterogeneity of CAFs and immune cell populations in the tumor microenvironment of pancreatic adenocarcinoma" Journal of Cancer Metastasis and Treatment. 8: 42. http://dx.doi.org/10.20517/2394-4722.2022.60
ACS Style
Diab, M.; El-Rayes BF. The heterogeneity of CAFs and immune cell populations in the tumor microenvironment of pancreatic adenocarcinoma. J. Cancer. Metastasis. Treat. 2022, 8, 42. http://dx.doi.org/10.20517/2394-4722.2022.60
About This Article
Special Issue
Copyright

Data & Comments
Data


 Cite This Article 9 clicks
Cite This Article 9 clicks

 Like This Article 34
likes
Like This Article 34
likes






Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.